

Abstract
With few exceptions the genetic codes of all known organisms encode the same 20 amino acids, yet all that is required to add a new building block are a unique tRNA/aminoacyl-tRNA synthetase pair, a source of the amino acid, and a unique codon that specifies the amino acid. For example, the amber nonsense codon, TAG, together with orthogonal Methanococcus jannaschii or Escherichia coli tRNA/synthetase pairs have been used to genetically encode a variety of unnatural amino acids in E. coli and yeast, respectively. However, the availability of noncoding triplet codons ultimately limits the number of amino acids encoded by any organism. Here, we report the design and generation of an orthogonal synthetase/tRNA pair derived from archaeal tRNALys sequences that efficiently and selectively incorporates an unnatural amino acid into proteins in response to the quadruplet codon, AGGA. Frameshift suppression with l-homoglutamine (hGln) does not significantly affect protein yields or cell growth rates and is mutually orthogonal with amber suppression, permitting the simultaneous incorporation of two unnatural amino acids, hGln and O-methyl-l-tyrosine, at distinct positions within myoglobin. This work suggests that neither the number of available triplet codons nor the translational machinery itself represents a significant barrier to further expansion of the genetic code.
Recently, a general method was developed that makes it possible to systematically add new amino acids to the genetic codes of bacteria and yeast (1, 6). To date, >25 unnatural amino acids have been incorporated into proteins in response to the amber nonsense codon, TAG, with high translational efficiency and fidelity. These include amino acids that can be photocrosslinked, glycosylated amino acids, chemically reactive amino acids (containing keto, alkene, or alkyne groups), redox active amino acids and fluorescent amino acids (2, 3, 5, 7–9). To genetically encode 22 or more amino acids requires additional codons that uniquely specify each new amino acid. Rare codons, or even an Escherichia coli variant in which degenerate rare codons have been deleted from the genome, represent one possible source of unique codons. Alternatively, it should be possible to use quadruplet codons and cognate suppressor tRNAs with expanded anticodon loops to encode additional amino acids.
Many examples of naturally occurring +1 frameshift suppressors exist, including UAGN (N = A, G, C, or T) suppressors derived from Su7-encoding glutamine (10), sufJ-derived suppressors of ACCN codons encoding threonine (11), and CAAA suppressors derived from tRNALys and tRNAGln (12). Moreover, genetic selections have been used to identify efficient four- and five-base codon suppressor tRNAs from large libraries of mutant tRNAs, including an E. coli tRNASerUCCU suppressor (13, 14). These frameshift suppressor tRNAs can efficiently incorporate the common 20 amino acids into proteins in vivo. Chemically aminoacylated frameshift suppressors have also been used to incorporate unnatural amino acids into proteins in in vitro translation systems in response to both four- and five-base codons (15–18). In addition, two four-base codons have been used together in a single transcript to insert two different unnatural amino acids into the same protein in vitro (19, 20).
The use of four-base codons to add amino acids to the genetic codes of bacteria or eukaryotes in vivo requires the generation of a new tRNA with an expanded anticodon loop, which is not a substrate for any endogenous synthetases and which efficiently decodes a quadruplet versus triplet codon. In addition, an aminoacyl-tRNA synthetase must be evolved that is highly selective for the new tRNA and aminoacylates it with the unnatural amino acid of interest and no endogenous amino acids. A system that permits the simultaneous incorporation of two or more unnatural amino acids into a polypeptide would most likely be achieved by using two or more orthogonal pairs of distinct origin, necessitating the development of a new four-base decoding orthogonal tRNA-synthetase pair. Here, we describe the generation of an orthogonal tRNA/synthetase pair derived from the type I lysyl-tRNA synthetase of Pyrococcus horikoshii that permits the site-specific incorporation of l-homoglutamine (hGln) in response to the four-base codon, AGGA. In combination with an O-methyl-l-tyrosine (OMeTyr)-specific synthetase derived from the Methanococcus jannaschii tyrosyl-tRNA synthetase, two different unnatural amino acids have been efficiently incorporated into distinct sites of a single polypeptide.
Methods
General Methods. Phage-resistant DH10B strain GeneHogs (Invitrogen) was used for all in vivo manipulations unless otherwise stated. Cells were propagated on LB agar plates, in 2YT liquid medium, or in GMML minimal media [1× M9 (Sigma-Aldrich M-6030)/1 mM MgSO4/0.1 mM CaCl2/8.5 mM NaCl/5 μM Fe2SO4/1% glycerol/0.3 mM leucine] as indicated. Antibiotics were used at 25 μg/ml and cells were incubated at 37°C unless otherwise noted. PCR was performed with the Expand High Fidelity PCR System (Roche Diagnostics). Restriction enzymes and T4 DNA ligase (NEB, Beverly, MA) were used for plasmid construction by using standard methods as suggested by the supplier. Antibiotics and unnatural amino acids (hGln and OMeTyr) were purchased from Sigma-Aldrich. Oligonucleotides were synthesized by Sigma-Genosys (Table 1). Premixed phosphoramidites were used during oligonucleotide synthesis for library construction to avoid sequence bias.
Table 1. Primers used in this experiment.
| vis15 | GTATCAACACCATGGTTCATTGGGCCGATTATATTG |
| ca419R | GCTATGAATTCGATCCCTCAAGTCTAAGTCGTTT |
| ca468F | CGAAAGGTCTCACATGTCTGAACAAGAAACACGG |
| ca468R | CACTAGGTCTCGAATTCAATTTCTGTGGGCGCATCG |
| ca400F | CGGAATTCGGGCCCGTAGCTCAGCCTGGTAGAGCGGCGGGCTCTAAACCCGCAGGTCG |
| ca400R | AAACTGCAGTGGCGGGCCCGGCGGGATTTGAACCCGCGACCTGCGGGTTTAGAGCC |
| ca502F | CGGAATTCGGGCCCGTAGCTCAGCCTGGTAGAGCGGCGGGCTTCCTAACCCGCAGGTCG |
| ca502R | AAACTGCAGTGGCGGGCCCGGCGGGATTTGAACCCGCGACCTGCGGGTTAGGAAGCC |
| ca514F | CGGAATTCNNNNCCGTAGCTCAGCCTGGTAGAGCGGCGGGCTTCCTAACCCGCAGGTCG |
| ca514R | AAACTGCAGTGGCNNNNCCGGCGGGATTTGAACCCGCGACCTGCGGGTTAGGAAGCC |
| ca510R | CAGTGGAATTCAGTAAGTTGGCAGCATCAC |
| ca501F | GAGTAGGTCTCTTAGGNNKCTGTTTACAGCTTATATTGTGGGC |
| ca503R | CCAGTGGTCTCGCCTAAAGTTCCCAACGTGAACGTAACCACTTG |
| ca480F | GGAGACGTCTCTGAGTTTGTTGGAATTAAGGGGCAG |
| ca480R | CGTTACGTCTCGACTCMNNCATTAAAGATAACGGAGCTTC |
| ca385N | GACAGTGGCCATACCCATAATGTGCCTGTCAAATG |
| ca303C | GGTTTCCCGGGTCAGGAGAGCGTTCACCGAC |
| embMyo2F | TACGTGGTCTCGACTGCCCTAGGTTAGATCCTTAAG |
| embMyo2R | GACAAGGTCTCGCAGTTAACACGGTAACACCATG |
| ca353F | GAAACGTCTTGCTCGAGGCCGCGATTAAATTC |
| ca544R | GAGTAGGTCTCGGATCCTGCGAAGCGGAATTAATTC |
| MyoF | GATCCCATGGTTCTGTCTGAAGGTGAATGGC |
| MyoR | CCGGGGTACCTCAATGGTGATGGTGATGATGTCC |
| 24F | GTTGAAGCTGACGTCGCTAGGACATGGTCAGGACATCTTG |
| 24R | CAAGATGTCCTGACCATGTCCTAGCGACGTCAGCTTCAAC |
| 75F | GTGTTAACTGCCCTAGGTTAGATCCTTAAGAAAAAAGGG |
| 75R | CCCTTTTTTCTTAAGGATCTAACCTAGGGCAGTTAACAC |
| J17FPstl | CTTGCTGCAGGTGGCGCCCCATCAAAAAAATATTC |
| J17RXmal | ACTAACCCGGGACCAAGTTTACTCATATATAC |
Cloning and Expression of Synthetase Genes. Genomic DNA was prepared from a P. horikoshii lyophilized cell pellet obtained from American Type Culture Collection (ATCC 700860) by using the DNeasy kit (Qiagen, Valencia, CA). The gene for the P. horikoshii tRNA synthetase (PhKRS) was amplified by PCR with oligonucleotides vis15 and ca419R and subsequently digested with NcoI and EcoRI. For constitutive expression, the synthetase gene was ligated into the NcoI/EcoRI sites of pGLN and pKQ (21). These plasmids contain the pUC origin of replication, a polylinker, Myc and His-6 tags, and the rrnB terminator from pBAD/Myc-HisA (Invitrogen) with the E. coli glutaminyl-tRNA synthetase (glnRS) promoter of plasmid pHYQRS (22). Plasmid pGLN confers resistance to ampicillin, whereas pKQ confers resistance to kanamycin. Similarly, the gene for E. coli lysyl-tRNA synthetase (EcKRS) was amplified by PCR from the lysU locus of E. coli strain HB101 with oligonucleotides ca468F and ca468R, digested with BsaI, and ligated into the NcoI/EcoRI sites of pGLN and pKQ.
Complementation Analysis. E. coli strain PALΔSΔUTR(pMAKlysU) was obtained from P. Plateau of the Laboratoire de Biochimie, Ecole Polytechnique (Palaiseau, France). The temperature-sensitive plasmid pMAKlysU cannot replicate at 43°C and is unable to complement the lysU/lysS synthetase deletions present in the genome of PALΔSΔUTR. PALΔSΔUTR(pMAKlysU) cells were transformed with pGLN derivatives for expression of PhKRSΔ, EcKRS, or no synthetase and were rescued on LB-agar plates containing 50 μg/ml ampicillin at 30°C, a permissive temperature. Single colonies were then grown to saturation at 30°C in 2YT media supplemented with ampicillin, diluted, and spread on duplicate LB agar plates with no antibiotics at densities of 100 colony-forming units per plate. One plate was incubated at 43°C to assay complementation, whereas the second plate was incubated at 30°C as a positive control. GeneHogs cells harboring the pGLN synthetase-expressing plasmids were similarly plated at 30°C and 43°C as a positive control to eliminate the possibility of toxic effects due to synthetase expression.
Construction of tRNA Expression Plasmids. Genes for tRNAs were constructed by the overlap extension of synthetic oligonucleotides and inserted into the EcoRI and PstI sites of pACKO-A184TAG or pACKO-A184AGGA (21). These reporter plasmids are derived from pACYC184, which has the p15A origin of replication, a chloramphenicol resistance gene, and a strong constitutive lpp promoter controlling expression of the tRNA genes. Specifically, a gene for AKtRNACUA was constructed by using oligonucleotides ca400F/R and inserted into pACKO-A184TAG to obtain plasmid pAC-AKtRNACUA. Similarly, plasmid pAC-AKtRNAUCCU was obtained by extension of oligonucleotides ca502F and ca502R and insertion into plasmid pACKO-A184AGGA. The acceptor stem library from which AK514 was selected was constructed from oligonucleotides ca514F/R in pACKO-A184AGGA. The library contained 1 × 107 members, 150-fold more than the theoretical diversity.
The pAC-AKtRNAUCCU library was introduced into cells harboring plasmid pKQ-PhKRSΔ and subjected to two rounds of ampicillin selection at 200 μg/ml. The resulting tRNA plasmids were then separated from the synthetase plasmids by retransformation of GeneHogs cells. To identify orthogonal tRNAs, colonies were picked, grown to saturation, and spotted on plates containing various concentrations of ampicillin. Of the 384 colonies picked, 192 were unable to grow at 20 μg/ml ampicillin. These clones were pooled and then transformed with plasmid pKQ-PhKRSΔ. Transformants were subjected to two rounds of selection at 200 and 500 μg/ml ampicillin. tRNA-expressing plasmids were isolated again, and individual clones were characterized.
Construction of pKQ-PhKRSΔ. XL1-red E. coli cells (Stratagene) were transformed with plasmid pKQ-PhE444G (pKQ-PhKRS with an E444G mutation in the synthetase, resulting in lower toxicity) and plated on LB-agar plates supplemented with kanamycin. XL1-red cells have several genomic mutations that cause a high rate of mutagenesis in transformed plasmids. Approximately 100 colonies were scraped from the plate and serial-cultured twice in 2YT media supplemented with kanamycin. Because nontoxic mutants of pKQ-PhE444G have faster growth rates, serial culture of the cells should lead to the accumulation of these mutants. After two rounds of serial culture with 10,000-fold dilution at each step, plasmids were isolated from the cells and introduced into GeneHogs cells (Invitrogen) containing plasmid pAC-AKtRNACUA. These cells were plated on LB-agar plates containing kanamycin, chloramphenicol, and various concentrations of ampicillin. Plasmid pKQ-PhKep was identified as a nontoxic variant of pKQ-PhE444G from this population and was able to survive on LB-agar plates containing 1,000 μg/ml ampicillin in the presence of pAC-AKtRNACUA. The synthetase contained an insertion of 778 bp of unknown origin, a sequence homologous to insAcp1 from plasmid p1658/97, after residue S357. To confirm that this insertion yields a truncated PhKRS, the N-terminal peptide of PhKRS extending to residue S357 was amplified with vis15 and ca510R by using pKQ-PhKep as template, digested with NcoI/EcoRI, and inserted into plasmid pKQ to give pKQ-PhKRSΔ.
Determination of Suppression Efficiency. β-Lactamase minimum inhibitory concentrations (MIC) were used to quantify the suppression efficiency of individual clones. To determine these values, cells were spread at densities of 100 colony forming units per plate on a series of LB-agar plates supplemented with different ampicillin concentrations and the appropriate antibiotics for plasmid maintenance. The MIC was scored as the highest concentration of ampicillin at which growth was observed.
Construction of the PhKRSΔ Active-Site Library. The PhKRSΔ-derived library was constructed in plasmid pKQ by two rounds of enzymatic inverse PCR (23). First, position Y268 was randomized. Plasmid pKQ-PhKRSΔ was amplified by PCR with oligonucleotides ca480F/R, digested with BsmBI, recircularized by ligation, and introduced into cells by electroporation. This library was miniprepped and used as template for PCR with oligonucleotides ca501F and ca503R, digested with BsaI, ligated, and introduced into cells by electroporation to mutate site E41. The final two-site library contained 1 × 106 members and was shown to be free of sequence bias by sequencing.
Identification of Homoglutamine-Specific Synthetases. To screen variants of pKQ-PhKRSΔ, a T7 RNA polymerase/GFP reporter plasmid, pREP2-AKtRNACUA, was constructed in two steps from plasmid pAC-AKtRNACUA. First, plasmid pRYC-AKtRNACUA was constructed by combining the NcoI/PstI fragment containing AKtRNACUA with the NcoI/PstI fragment of pRYC-HLAA02, a homolog of plasmid pACM(D112TAG) (24). Second, plasmid pRYC-AKtRNACUA was digested with SacII and ClaI and the tRNA-bearing fragment was inserted into corresponding sites of pREP(2)YC-JYCUA (25). The PhKRSΔ-derived library was cotransformed with pREP2-AKtRNACUA, and cells were plated on GMML-agar plates supplemented with 1 mM hGln and kanamycin and tetracycline. Fluorescent colonies were picked, diluted in GMML media, and spread on two GMML-agar plates, one with homoglutamine and one without. Clones that were fluorescent in the presence of 1 mM hGln but white in its absence were selected for further characterization.
Expression and Characterization of Myoglobin Containing hGln. For single-site incorporation experiments, position Gly-24 of the sperm whale myoglobin was mutated to an AGGA codon by overlap PCR. In brief, the N-terminal and C-terminal segments of the myoglobin gene were first amplified separately from pBAD-JYAMB-4TAG (26) with primer sets MyoF/24R and 24F/MyoR, respectively. The two fragments were then gel-purified and used as templates to generate the full-length gene with primers MyoF and MyoR. The PCR product corresponding to the entire gene was then digested with NcoI and KpnI and inserted into pBAD/Myc-HisA (Invitrogen). The region containing araC, the arabinose promoter, and the myoglobin mutant gene was then amplified by PCR with primers ca385N and ca303C, digested with MscI and XmaI, and inserted into the EcoRV and XmaI sites of pAC-AK514 to obtain the final expression vector pMyo24-AK514. This plasmid contains a p15A origin of replication and genes for chloramphenicol acetyltransferase, AK514, and the myoglobin gene under the control of the arabinose promoter.
For two-site amino acid incorporation, plasmid pMyo24/75-AK514J17 was constructed by introducing a TAG codon at position Ala-75 of the myoglobin gene in pMyo24-AK514 by an overlap PCR strategy as before, with primers 75F and 75R instead of 24F/R to give pMyo24/75-AK514. The orthogonal tRNATyr, J17, together with its own lpp promoter was then inserted immediately downstream of AK514 as follows. The PstI site on the 3′ end of J17 in vector pAC-J17 (27) was removed by digestion with PstI, made blunt with Deep Vent polymerase (NEB), and circularized by ligation. This vector was used as a template for PCR with primers J17FPstI and J17RXmaI to obtain the J17 expression cassette, which was digested with PstI and XmaI and inserted into pMyo24/75-AK514 to give the final vector pMyo24/75-AK514J17. A plasmid containing the two synthetases hGlnRS and OMeTyrRS (1), pKQ-PhKRSΔOMeTyrRS, was constructed by inserting a PCR fragment [amplified from pBK-OMYRS (1) with oligonucleotides ca353F and ca544R and digested with BsaI and XhoI] containing OMeTyrRS along with its own glnRS promoter into the BamHI and XhoI sites of pKQ-PhKRSΔ.
GeneHogs cells harboring appropriate synthetase and myoglobin expression plasmids were grown at 37°C to OD600 = 0.5 in GMML media supplemented with 18 amino acids (lysine and tyrosine were deleted) at 0.4 mg/ml each, vitamins (riboflavin, niacinamide, pyridoxine monohydrochloride, and thiamine) at 1 μg/ml, and 1 mM hGln (Sigma). Cultures were induced with 0.02% arabinose and then grown to saturation at 37°C. Cells were lysed by sonication, and myoglobin was purified with the Qiagen QIAexpressionist kit and identified by Western blot analysis with anti-His antibodies (Invitrogen 46–0693 and Santa Cruz Biotechnology sc-2308). Bands were detected with ECF substrate (Amersham Pharmacia Biosciences no. RPN5785) and imaged with a Storm 860 PhosphoImager from Molecular Dynamics. Tryptic digestion of the protein and matrix-assisted laser desorption ionization-MS analysis of the tryptic fragments were performed at The Scripps Research Institute Proteomics Facility. Full-length protein was desalted by microbore reversed-phase HPLC (Michrom, Auburn, CA) and the purity and mass were assessed by electrospray ionization-ion trap MS (Bruker-Agilent, Palo Alto, CA) with mass accuracy within 1 amu.
Results and Discussion
An Orthogonal Lysyl-tRNA-Synthetase Pair. We initially focused on the lysyl synthetase/tRNA pair of the archaebacterium P. horikoshii (PhKRS) as a candidate orthogonal pair for the following reasons: (i) This pair is likely to be orthogonal because of the conservation of A73 in prokaryotes and G73 in archaea (28). (ii) Unlike the M. jannaschii tyrosyl synthetase, PhKRS is tolerant of substitutions in the anticodon loop of its cognate tRNA and, consequently, will likely charge both three- and four-base suppressor tRNAs (29). (iii) The crystal structure of PhKRS is available (29), facilitating changes in amino acid specificity. Unfortunately, PhKRS is toxic to E. coli when expressed constitutively under the glnRS promoter. However, serial culture of a pGLN plasmid containing PhKRS in the E. coli mutator strain XL1-red led to the isolation of a nontoxic variant, PhKRSΔ. This mutant is truncated after residue S357, resulting in deletion of the α-helix bundle-like domain and the α-helix cage domain, which are involved in anticodon recognition (29).
To determine whether this truncated synthetase is orthogonal in E. coli, it was necessary to verify that the mutant cannot charge E. coli tRNAs to any significant extent. Genes for PhKRSΔ and EcKRS under the constitutive glnRS promoter of the pGLN expression vector were used to transform temperature-sensitive E. coli cells PALΔSΔUTR(pMAKlysU) (30). The deletion strain PALΔSΔUTR is deficient in lysyl-tRNA synthetase because of lysS/lysU double mutations; pMAKlysU is a temperature-sensitive plasmid that encodes lysU and complements the lysS/lysU mutations. The inability of plasmid pMAKlysU to replicate at 43°C causes cell death at this temperature. We found that PhKRSΔ was unable to complement the lysS/lysU deletions in PALΔSΔUTR at 43°C, whereas EcKRS afforded normal growth. Thus, PhKRSΔ is unable to substitute for EcKRS and would likely compete poorly with endogenous E. coli synthetases for charging of E. coli tRNAs in vivo.
A cognate suppressor tRNA was then designed for PhKRSΔ based on sequence analysis of all archaeal tRNALys tRNAs from available genomic sequences (21). A consensus tRNA sequence generated from this analysis retains all the conserved recognition sites for the archaeal lysine synthetase, while eliminating unnecessary mismatched base pairs that can be deleterious to suppression efficiency (21). The anticodon loop sequence of the consensus tRNA was changed to CUCUAAA to afford an amber suppressor tRNA, AKtRNACUA (Fig. 1). This anticodon loop sequence was shown to be optimal for amber suppression in E. coli (31). The initial use of an amber suppressor (versus a frameshift suppressor) simplifies the subsequent characterization and evolution of the orthogonal PhKRSΔ/AKtRNA pair because the positive and negative selection schemes previously developed in this laboratory, which are based on amber suppression, can be used.
Fig. 1.

Construction of amber and four-base suppressor tRNAs. An amber suppressor tRNA was generated from sequence alignments of multiple archaeal tRNALys sequences. The orthogonal AGGA suppressor tRNA was isolated by selection from an acceptor stem library.
The gene for AKtRNACUA was synthesized and inserted into plasmid pACKO-A184TAG (21) to examine suppression efficiency and cross-reactivity of the orthogonal tRNA. This plasmid contains a gene for β-lactamase (bla) with a TAG codon at the permissive site A184. In the absence of an amber suppressor tRNA, translation affords a truncated inactive β-lactamase, and cells are sensitive to 5 μg/ml ampicillin. With AKtRNACUA alone, no increase in ampicillin resistance was observed, indicating that this tRNA is not charged by E. coli synthetases to any significant extent (it is orthogonal to E. coli synthetases). When coexpressed with PhKRSΔ, pAC-AKtRNACUA affords resistance to 1,000 μg/ml ampicillin, indicating that PhKRSΔ is expressed and active in E. coli, and that the aminoacylated AKtRNACUA is an efficient amber suppressor tRNA. These results demonstrate that the PhKRSΔ/AKtRNACUA pair is both functional and orthogonal in E. coli.
Generation of a tRNA That Decodes a Quadruplet Codon. Earlier experiments have explored the effects of codon/anticodon size and sequence on suppression efficiency in E. coli. In those studies, a tRNA library in which the anticodon loop of 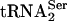 (positions 31–38) was replaced with all possible 8-bp sequences was examined for the suppression of all possible four-base codon sequences substituted at position S70 of β-lactamase (13). Efficient pairs of suppressor and four-base codon were identified, including a mutant tRNA with an anticodon loop sequence of CUUCCUAG that efficiently suppressed an AGGA codon at position S70 of β-lactamase (14). In this case, suppression of AGGA is competing with the rare codon AGG, which may contribute to the efficiency and lack of toxicity of the suppressor tRNA.
(positions 31–38) was replaced with all possible 8-bp sequences was examined for the suppression of all possible four-base codon sequences substituted at position S70 of β-lactamase (13). Efficient pairs of suppressor and four-base codon were identified, including a mutant tRNA with an anticodon loop sequence of CUUCCUAG that efficiently suppressed an AGGA codon at position S70 of β-lactamase (14). In this case, suppression of AGGA is competing with the rare codon AGG, which may contribute to the efficiency and lack of toxicity of the suppressor tRNA.
Based on these results, a four-base AGGA suppressor tRNA was constructed from AKtRNACUA by mutating the anticodon loop of AKtRNACUA to CUUCCUAA and expressing the corresponding gene in plasmid pACKO-A184AGGA. This plasmid contains an AGGA codon at position A184 in β-lactamase resulting in abortive translation and resistance to only 5 μg/ml ampicillin. Unfortunately, the frameshift suppressor AK-tRNAUCCU was no longer orthogonal to E. coli synthetases; cells containing this tRNA survive to 200 μg/ml ampicillin both in the absence and presence of PhKRSΔ, indicating some degree of suppression of the frameshift mutation.
To identify orthogonal AKtRNAUCCU variants, a library was constructed in which the last 4 bp of the acceptor stem, positions 1–4 and 69–72, were simultaneously randomized. Because many aminoacyl-tRNA synthetases rely on these positions and the discriminator base for recognition of their cognate tRNAs (32), it should be possible to find a combination of bases that preserves the interaction with PhKRSΔ while eliminating the background interactions with endogenous E. coli synthetases (21). These tRNAs were coexpressed with PhKRSΔ and the A184AGGA mutant β-lactamase, and cells were subjected to two rounds of ampicillin selection at 200 μg/ml, resulting in a pool of active AGGA suppressor tRNAs. Plasmids containing the mutant tRNAs were then isolated and 384 individual clones were screened for sensitivity to ampicillin in the absence of PhKRSΔ. Of these, the most efficient AGGA suppressor affords resistance to 700 μg/ml ampicillin in the presence of PhKRSΔ, but resistance to only 5 μg/ml in its absence. This tRNA, AK514, contains multiple acceptor stem substitutions and a serendipitous A37C substitution in the anticodon loop (Fig. 1). Because PhKRSΔ lacks an anticodon-binding domain, it likely does not interact with A37. However, many other aminoacyl-tRNA synthetases use the 37 position for positive recognition. Therefore, the A37 to C substitution may serve to improve discrimination against AK514 by noncognate synthetases.
Evolution of a hGln-Specific Synthetase. To incorporate an unnatural amino acid in response to the quadruplet codon AGGA, it was next necessary to alter the specificity of the cognate synthetase PhKRSΔ to recognize an unnatural amino acid, but not lysine or another endogenous amino acid. hGln was chosen as an initial target to test the degree of translational fidelity one can achieve with this orthogonal pair because hGln is similar in size to lysine and has both hydrogen bond-donating and -accepting properties (Fig. 2A). Examination of the PhKRS crystal structure reveals that only two residues specifically recognize the ε-amino group of lysine, Glu-41 and Tyr-268. Both the glutamate carboxylate group and the tyrosine hydroxyl group interact with the ε-amino group of bound lysine (Fig. 2B). Based on the crystal structure, and the similarity between hGln and Lys, a two-site library of PhKRSΔ mutants was constructed in which Glu-41 and Tyr-268 were simultaneously randomized.
Fig. 2.

(A) Structure of homoglutamine. (B) Schematic illustration of the PhKRS active site; residues Glu-41 and Tyr-268 make specific contacts with the lysine substrate (29). These two residues were simultaneously randomized.
To screen for hGln-specific synthetases, a GFP reporter plasmid, pREP2-AKtRNACUA, was used, which encodes the gene for the amber suppressor AKtRNACUA, a T7 RNA polymerase gene (T7RNAP) with two TAG codons at permissive positions M1 and Q107, and GFPuv under the control of a T7 promoter (25). Again, by using the amber suppressor rather than AK514, well characterized selection schemes developed for the M. jannaschii orthogonal pair could be used to evolve the specificity of PhKRSΔ. Moreover, because PhKRSΔ does not bind the anticodon loop of the suppressor tRNA, any hGln-specific synthetase should also function with AK514. When cotransformed with pREP2-AKtRNACUA and Ph-KRSΔ, cells are fluorescent, indicating that the amber codons in T7RNAP are sufficiently suppressed to produce GFPuv. In the absence of the synthetase, cells are white, allowing active synthetases to be identified by fluorescence. Cells were cotransformed with pREP2-AKtRNACUA and the library was then spread on GMML plates containing 1 mM hGln. Individual green colonies were isolated and grown with and without the unnatural amino acid to identify clones whose fluorescence depended on the presence of hGln. Of 15 colonies screened, five showed higher fluorescence on plates containing hGln. Four of the five clones had a Tyr-268 to Ser substitution; Ser-268 may form a hydrogen bond with either the carbonyl or the amide group of the hGln side chain. The synthetase with the highest activity for hGln versus Lys, hGlnRS, has Ile-41 and Ser-268 mutations. Ile-41 may form hydrophobic interactions with the carbon chain of hGln and discriminate against any charged substrates such as Lys.
To show that the evolved synthetase can insert homoglutamine in response to a quadruplet codon, a Gly-24 → AGGA mutant myoglobin gene was expressed by using the orthogonal hGlnRS and the AK514 suppressor (Gly-24 is a permissive site). On expression of the mutant myoglobin gene in the absence of hGlnRS, no detectable protein was produced, which is consistent with the previous experiment, showing that PhKRSΔ and AK514 are an orthogonal pair (Fig. 3A). When coexpressed with hGlnRS, 2 mg/liter myoglobin was isolated. In comparison, 11 mg/liter of protein was produced under similar conditions from the wild-type myoglobin gene. Matrix-assisted laser desorption ionization–time- of-flight analysis of tryptic fragments of the purified protein revealed a peptide of mass 1,676.85 Da, consistent with the predicted mass of 1,676.88 Da. No evidence of peptides containing lysine or glutamine at position 24 was observed (Fig. 4A).
Fig. 3.

Expression of mutant myoglobin by AGGA suppression. (A) Protein was produced from the myoglobin gene with an AGGA codon at position G24 (Myo24AGGA) only in the presence of all three components, AK514 tRNA, hGlnRS (an hGln-specific variant of PhKRSΔ), and hGln (lanes 2–5). Expression of myoglobin by amber suppression at position 75 (Myo75TAG) was only possible with JYRS and its cognate amber tRNA, demonstrating the mutual orthogonality of the lysyl and tyrosyl pairs (lanes 6–9). (B) Protein was expressed from a myoglobin gene containing AGGA at position 24 and TAG at position 75 (Myo24AGGA/75TAG) in the presence of both the lysyl and tyrosyl pairs. hGln was incorporated by AGGA suppression at position 24 and OMeTyr was incorporated by amber suppression at position 75 in a single polypeptide by using hGlnRS and OMeTyrRS. Protein was visualized with an anti-His6 antibody.
Fig. 4.

(A) Matrix-assisted laser desorption/ionization–time-of-flight analysis of tryptic fragments containing hGln (Upper) or Lys (Lower). (B) Electrospray MS analysis of full-length myoglobin containing hGln at position 24 and OMeTyr at position 75.
The doubling time of cells incorporating hGln is twice that of GeneHogs. The same reduction in growth rate was observed in both the presence and absence of hGln, suggesting that this is attributed to synthetase and tRNA expression rather than AGGA suppression. Therefore, cross-reactivity of the AGGA suppressor with rare AGG codons does not limit the practical application of frameshift suppression.
Simultaneous Incorporation of Two Unnatural Amino Acids. We next investigated the possibility of using two distinct orthogonal tRNA-synthetase pairs that decode two different codons to simultaneously incorporate two unnatural amino acids into a single polypeptide chain. The two candidate pairs are the PhKRSΔ/AK514 pair from P. horikoshii and a previously generated orthogonal tRNA/tyrosyl-tRNA synthetase (JYRS/J17) pair from M. jannaschii (1). The obvious requirement is that these two pairs have to be mutually orthogonal: JYRS should not charge the four-base suppressor tRNA, AK514, and PhKRSΔ should not charge the amber suppressor tRNA, J17. To demonstrate this lack of cross-reactivity, wild-type JYRS was coexpressed with the Gly-24 → AGGA mutant myoglobin gene and AK514. No full-length protein was detected (Fig. 3A), showing that JYRS is unable to charge AK514. Conversely, we attempted to express myoglobin from plasmid pBAD/JYAMB-75TAG, which contains a myoglobin gene with a TAG codon at the permissive site 75 (4). Coexpression of this plasmid with JYRS affords 4 mg/liter of myoglobin, but no protein is observed when JYRS is replaced with PhKRSΔ. Therefore, PhKRSΔ is unable to charge the amber suppressor J17. As such, these two synthetase/tRNA pairs are mutually orthogonal.
To simultaneously incorporate two unnatural amino acids into myoglobin, genes for AK514 and J17 were combined into a single plasmid expressing a mutant myoglobin gene with Gly-24→AGGA and Ala-75→TAG mutations. An OMeTyr-specific synthetase, OMeTyrRS [derived from JYRS (1)] and hGlnRS were encoded in a second plasmid, with each synthetase gene under its own glnRS promoter. When cells were cotransformed with both plasmids and grown in the presence of both unnatural amino acids, 1.7 mg/liter of myoglobin was produced (Fig. 3B). Although the yield is about 6-fold less than wild-type myoglobin, it is sufficient to make useful quantities of protein. Additional mutations to either tRNA may lead to improved dual-site suppression efficiency. No protein was produced when either of the two unnatural amino acids was excluded. Electrospray MS analysis of the full-length protein revealed a mass of 18,546.40 Da (SD 0.11), consistent with the predicted mass of 18,546.60 Da for myoglobin containing both unnatural amino acids, compared with the calculated mass of 18,518.3 Da for myoglobin with G24K and A75Y substitutions (Fig. 4B).
Conclusion
In summary, we have shown that a quadruplet codon can be used to site-specifically incorporate an unnatural amino acid into proteins in E. coli. It should be possible to identify lysyl-tRNA synthetase variants that permit the incorporation of additional unnatural amino acids in E. coli by using a larger synthetase library with additional active-site residues randomized simultaneously. Other quadruplet codons that can be efficiently suppressed in E. coli (e.g., CCCU or CUAG) should allow additional amino acids to be genetically encoded. Furthermore, the mutual orthogonality of the P. horikoshii-derived lysyl-tRNA synthetase/tRNA pair and the pair derived from the M. jannaschii tyrosyl-tRNA synthetase/tRNA allows the simultaneous incorporation of two unnatural amino acids into a single protein. This will be useful for a range of biophysical studies including NMR and fluorescent energy-transfer experiments. Finally, we have begun to explore the possibility of constructing an E. coli genome with limited codon degeneracy, thereby making more triplet codons available for additional amino acids.
Acknowledgments
We thank Peter Rudberg and Badry Bursulaya for assistance in the analysis of the PhKRS active site. J.C.A. is a National Science Foundation Predoctoral Fellow. N.W. is supported by a Canadian Institutes of Health Research fellowship. S.W.S. is supported in part by a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund. Support was provided by Department of Energy Grant DE-FG03-00ER45812. This is manuscript 16151-CH of The Scripps Research Institute.
Abbreviations: hGln, l-homoglutamine; OMeTyr, O-methyl-l-tyrosine; glnRS, glutaminyl-tRNA synthetase; PhKRS, Pyrococcus horikoshii tRNA synthetase; EcKRS, Escherichia coli lysyl-tRNA synthetase.
References
- 1.Wang, L., Brock, A., Herberich, B. & Schultz, P. G. (2001) Science 292, 498-500. [DOI] [PubMed] [Google Scholar]
- 2.Wang, L., Zhang, Z., Brock, A. & Schultz, P. G. (2003) Proc. Natl. Acad. Sci. USA 100, 56-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chin, J. W., Martin, A. B., King, D. S., Wang, L. & Schultz, P. G. (2002) Proc. Natl. Acad. Sci. USA 99, 11020-11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehl, R. A., Anderson, J. C., Santoro, S. W., Wang, L., Martin, A. B., King, D. S., Horn, D. M. & Schultz, P. G. (2003) J. Am. Chem. Soc. 125, 935-939. [DOI] [PubMed] [Google Scholar]
- 5.Zhang, Z., Gildersleeve, J., Yang, Y. Y., Xu, R., Loo, J. A., Uryu, S., Wong, C. H. & Schultz, P. G. (2004) Science 303, 371-373. [DOI] [PubMed] [Google Scholar]
- 6.Chin, J. W., Cropp, T. A., Anderson, J. C., Mukherji, M., Zhang, Z. & Schultz, P. G. (2003) Science 301, 964-967. [DOI] [PubMed] [Google Scholar]
- 7.Chin, J. W. & Schultz, P. G. (2002) Chembiochem 3, 1135-1137. [DOI] [PubMed] [Google Scholar]
- 8.Zhang, Z., Wang, L., Brock, A. & Schultz, P. G. (2002) Angew Chem. Int. Ed. Engl. 41, 2840-2842. [DOI] [PubMed] [Google Scholar]
- 9.Alfonta, L., Zhang, Z., Uryu, S., Loo, J. A. & Schultz, P. G. (2003) J. Am. Chem. Soc. 125, 14662-14663. [DOI] [PubMed] [Google Scholar]
- 10.Curran, J. F. & Yarus, M. (1987) Science 238, 1545-1550. [DOI] [PubMed] [Google Scholar]
- 11.Bossi, L. & Roth, J. R. (1981) Cell 25, 489-496. [DOI] [PubMed] [Google Scholar]
- 12.O'Connor, M. (2002) Nucleic Acids Res. 30, 1985-1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Magliery, T. J., Anderson, J. C. & Schultz, P. G. (2001) J. Mol. Biol. 307, 755-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson, J. C., Magliery, T. J. & Schultz, P. G. (2002) Chem. Biol. 9, 237-244. [DOI] [PubMed] [Google Scholar]
- 15.Hohsaka, T., Ashizuka, Y., Murakami, H. & Sisido, M. (1996) J. Am. Chem. Soc. 118, 9778-9779. [Google Scholar]
- 16.Murakami, H., Hohsaka, T., Ashizuka, Y. & Sisido, M. (1998) J. Am. Chem. Soc. 120, 7520-7529. [Google Scholar]
- 17.Hohsaka, T. & Sisido, M. (2002) Curr. Opin. Chem. Biol. 6, 809-815. [DOI] [PubMed] [Google Scholar]
- 18.Hohsaka, T., Ashizuka, Y., Murakami, H. & Sisido, M. (2001) Nucleic Acids Res. 29, 3646-3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hohsaka, T., Ashizuka, Y. & Sisido, M. (1999) Nucleic Acids Symp. Ser. 42, 79-80. [DOI] [PubMed] [Google Scholar]
- 20.Taki, M., Hohsaka, T., Murakami, H., Taira, K. & Sisido, M. (2002) J. Am. Chem. Soc. 124, 14586-14590. [DOI] [PubMed] [Google Scholar]
- 21.Anderson, J. C. & Schultz, P. G. (2003) Biochemistry 42, 9598-9608. [DOI] [PubMed] [Google Scholar]
- 22.Liu, D. R. & Schultz, P. G. (1999) Proc. Natl. Acad. Sci. USA 96, 4780-4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stemmer, W. P. & Morris, S. K. (1992) BioTechniques 13, 214-220. [PubMed] [Google Scholar]
- 24.Pastrnak, M., Magliery, T. J. & Schultz, P. G. (2000) Helv. Chim. Acta 83, 2277-2286. [Google Scholar]
- 25.Santoro, S. W., Wang, L., Herberich, B., King, D. S. & Schultz, P. G. (2002) Nat. Biotechnol. 20, 1044-1048. [DOI] [PubMed] [Google Scholar]
- 26.Chin, J. W., Santoro, S. W., Martin, A. B., King, D. S., Wang, L. & Schultz, P. G. (2002) J. Am. Chem. Soc. 124, 9026-9027. [DOI] [PubMed] [Google Scholar]
- 27.Wang, L., Magliery, T. J., Liu, D. R. & Schultz, P. G. (2000) J. Am. Chem. Soc. 122, 5010-5011. [Google Scholar]
- 28.Ibba, M., Losey, H. C., Kawarabayasi, Y., Kikuchi, H., Bunjun, S. & Soll, D. (1999) Proc. Natl. Acad. Sci. USA 96, 418-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terada, T., Nureki, O., Ishitani, R., Ambrogelly, A., Ibba, M., Soll, D. & Yokoyama, S. (2002) Nat. Struct. Biol. 9, 257-262. [DOI] [PubMed] [Google Scholar]
- 30.Chen, J., Brevet, A., Lapadat-Tapolsky, M., Blanquet, S. & Plateau, P. (1994) J. Bacteriol. 176, 2699-2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hou, Y. M., Schimmel, P. & Houweling, P. L. (1992) Biochemistry 31, 4157-4160. [DOI] [PubMed] [Google Scholar]
- 32.Martinis, S. A. & Schimmel, P. (1995) tRNA: Structure, Biosynthesis, and Function (Am. Soc. Microbiol., Washington, DC).