

Abstract
We have investigated in whole cells whether, at low oxygen concentrations ([O2]), endogenous nitric oxide (NO) modulates the redox state of the mitochondrial electron transport chain (ETC), and whether such an action has any signaling consequences. Using a polarographic-and-spectroscopic-coupled system, we monitored redox changes in the ETC cytochromes bH, cc1, and aa3 during cellular respiration. The rate of O2 consumption (VO2) remained constant until [O2] fell below 15 μM, whereas the onset of reduction of cytochromes aa3, part of the terminal ETC enzyme cytochrome c oxidase, occurred at ≈50 μM O2. Incubation of the cells with an inhibitor of NO synthase lowered significantly (P < 0.05) the [O2] at which reduction of the cytochromes occurred. We also measured intracellular superoxide (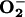 ) production at different [O2] and found there was no increase in
) production at different [O2] and found there was no increase in 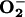 generation in control cells, or those treated with the NO synthase inhibitor, when incubated at 21% O2. However, after 30-min exposure of control cells to 3% O2, an increase in
generation in control cells, or those treated with the NO synthase inhibitor, when incubated at 21% O2. However, after 30-min exposure of control cells to 3% O2, an increase in 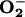 generation was observed, accompanied by translocation to the nucleus of the transcription factor NF-κB. Both of these responses were diminished by NO synthase inhibition. Our results suggest that endogenous NO, by enhancing the reduction of ETC cytochromes, contributes to a mechanism by which cells maintain their VO2 at low [O2]. This, in turn, favors the release of
generation was observed, accompanied by translocation to the nucleus of the transcription factor NF-κB. Both of these responses were diminished by NO synthase inhibition. Our results suggest that endogenous NO, by enhancing the reduction of ETC cytochromes, contributes to a mechanism by which cells maintain their VO2 at low [O2]. This, in turn, favors the release of 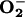 , which initiates the transcriptional activation of NF-κB as an early signaling stress response.
, which initiates the transcriptional activation of NF-κB as an early signaling stress response.
The role of nitric oxide (NO) as a signaling molecule involved in the physiology of the cardiovascular, pulmonary, and nervous systems has been clearly demonstrated (1). It is widely accepted that many of the signaling consequences of NO are mediated through activation of the biochemical target soluble guanylyl cyclase (2). In the last decade, however, another potential target has emerged, namely cytochrome c oxidase (CcO, complex IV), the mitochondrial enzyme responsible for reduction of O2 into water in the final stage of the electron transport chain (ETC). NO reversibly inhibits CcO by competing with O2 for the binuclear binding site (3–5). Moreover, activation of endothelial NO synthase (NOS) by bradykinin in respiring endothelial cells results in a decrease in the rate of O2 consumption (VO2), an effect that can be reversed by an inhibitor of NOS (6). The high affinity of CcO for NO suggests that the nanomolar concentrations of NO generated in tissues under basal conditions may play a role in the regulation of VO2 (7).
We have recently developed a polarographic-and-spectroscopic-coupled system based on visible light spectroscopy (VLS) to monitor changes in the redox states of the mitochondrial ETC cytochromes during cellular respiration (8). Using this system, we have confirmed previous studies showing an early reduction of cytochrome c at low O2 concentration [O2], without a change in VO2 (9). This phenomenon has been postulated to be part of a mechanism to maintain VO2 at decreasing [O2], although there is no consensus as to the factors that control this process (10, 11). Increased generation of mitochondrial reactive oxygen species (ROS) at low [O2] has previously been linked to the activation of various adaptive signaling pathways (12). However, at present, the mechanism responsible for the observed increase in ROS at low [O2] is not clear (see ref. 13 for review). One possibility is that NO, via its contribution to the early reduction of ETC cytochromes, favors the generation of ROS. We have therefore studied the effects of endogenous NO on the ETC redox state in endothelial and monocytic cells and the subsequent signaling consequences, specifically the release of ROS and the activation of NF-κB (14).
Materials and Methods
Reagents. Hepes, N-dodecyl-β-d-maltoside, cytochrome c (from bovine heart mitochondria), N,N,N′,N′ tetramethyl-p-phenylenediamine, sodium ascorbate, and dihydroethidium (DHE) were purchased from Sigma; (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate (DETA-NO) and NG-monomethyl-l-arginine monoacetate (l-NMMA) were purchased from Alexis (Nottingham, U.K.).
CcO Activity. Experiments with the purified enzyme (from bovine heart mitochondria) were carried out using the VLS system described previously (8). Briefly, the sample was placed in an air-tight respiration chamber (Rank Brothers, Cambridge, U.K.) maintained at 37°C, in which O2 and NO were measured polarographically, and emergent light from a broad-band tungsten-halogen source was detected in the visible region (490–650 nm). Intensity spectra were converted to changes in optical attenuation, to which the oxidized-minus-reduced absorption spectra of mitochondrial cytochromes were fitted using a linear least-squares algorithm. Changes in cytochrome redox states were thus monitored simultaneously with [O2] and VO2 every 0.5 seconds.
In the enzyme studies, 1 ml of 20 mM Hepes buffer containing 5 μl of Intralipid-10% (Pharmacia–Upjohn) at pH 6.8 was placed in the respiration chamber. Intralipid-10% is an i.v. nutrient often used in optical studies to simulate the turbidity of cells or tissue (15). Before closing the chamber, 0.1% dodecyl maltoside, 2 mM sodium ascorbate, 20 μM N,N,N′,N′ tetramethyl-p-phenylenediamine, and 1 μM cytochrome c were also added. Changes in VO2 and the absorption spectra of cytochromes aa3 (from CcO) and c were recorded after the addition of 100 nM CcO and monitored from the steady-state turnover until full reduction of the cytochromes, that is, when O2 in the sample was exhausted (defined as anoxia). The enzyme turnover number with these concentrations of enzyme and substrates was 15.2 ± 0.4 electrons·s-1 (n = 4).
The O2 dependence of CcO activity was characterized by the [O2] at which the VO2 decreased to half its maximal value during the steady-state, termed the P50. Values of the redox state of cytochromes aa3, denoted Y, were normalized to the total redox change from the steady-state (Y = 1) to the fully reduced form at anoxia (Y = 0). Cytochromes aa3 are not fully oxidized during turnover, therefore Y = 1 does not represent 100% oxidation of the cytochromes. Indeed, we have previously estimated cytochromes aa3 to be ≈31% reduced during steady-state respiration in human leukocytes (8). The C50 was defined as the [O2] at which Y = 0.5, i.e., when the cytochrome redox change from the steady state was 50% of the total.
Experiments with the NO donor DETA-NO were initiated 20 min after its incubation in reaction buffer (without enzyme), at which time there was a constant release of NO, as detected using the NO electrode. The concentration of released NO was determined by measuring the decrease in NO concentration after the addition of 5 μM oxyhemoglobin.
Cells. The murine monocytic cell line RAW 246.7 was purchased from American Type Culture Collection (Manassas, VA); human umbilical venous endothelial cells (HUVEC) were purchased from PromoCell (Heidelberg).
HUVEC were grown until passage 4 in phenol red-free endothelial cell growth medium EGM-1-PRF (PromoCell) in 175-cm2 flasks at 37°C in a 5% CO2/humidified air incubator. Cells were harvested by trypsinization and resuspended in EGM-1-PRF medium at a concentration of 2–4 × 107 cells per ml. The concentration in the medium of the NOS substrate l-arginine was 300 μM. Cell viability was >90%, as determined by the Trypan blue exclusion method.
RAW 246.7 cells were grown in RPMI medium 1640 supplemented with 25 mM Hepes, penicillin–streptomycin (100 units·ml-1 and 100 μg·ml-1), 2 mM glutamine, and 10% heat-inactivated New Zealand fetal bovine serum (Life Technologies, Paisley, U.K.). The cell suspension was maintained by growing cells in 500-ml stirring bottles in a 5% CO2/humidified air incubator. Cells were harvested by centrifugation at 400 × g for 5 min, washed, and resuspended in supplemented phenol redfree, l-arginine-free RPMI medium 1640 (to which 50 μM of l-arginine was added) at 4–6 × 107 cells per ml. Cells were used only when the viability, as measured by Trypan blue, was >95%.
Redox Changes in the ETC During Cellular Respiration. Cells were placed in the respiration chamber of the VLS system to monitor redox changes in cytochrome bH of complex bc1 (complex III, cytochrome c reductase), cytochromes aa3 of CcO, and their shared substrate cytochrome c (in combination with cytochrome c1 of complex bc1) during cellular respiration to anoxia. We have previously demonstrated the ability of our system to measure absolute concentration changes by characterizing the total light pathlength through the cells (8). In the case of RAW 246.7 cells, we determined the optical pathlength to be 2.4 ± 0.1 cm (n = 12) in the cell concentration range of interest, allowing the on-line measurement of absolute cytochrome concentration changes. In general, however, the redox states of the cytochromes were expressed in terms of the normalized change Y, as described above for the purified enzyme. This enabled a better comparison of the O2 dependence of cytochrome redox states in the two cell lines. As with the isolated enzyme, P50 and C50 are defined as the [O2] at which cellular VO2 is half-maximal and Y = 0.5, respectively.
The term reducing equivalents (RE) is used to describe components of the ETC in their reduced form. To provide an indication of the redox state of the ETC at any given [O2], we have defined RE as the normalized sum of all increases, relative to the steady-state, in reduced cytochromes, such that 0% represents the steady-state and 100% the total redox change on anoxia.
Intracellular Superoxide Production. RAW 246.7 cells at a concentration of 5 × 107 cells per ml were incubated for 1 h in the presence or absence of 750 μM l-NMMA at 21% O2 (i.e., atmospheric conditions) and 37°C. After this time, 10 μM DHE was added to the cell suspensions as a marker of 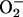 production; one-half of each group was maintained at 21% O2; the other half was transferred to an O2-controlled hypoxic chamber (Coy Laboratory Products; Ann Arbor, MI) maintained at 3% O2 and 37°C. Cells were agitated gently and aliquots of 5 × 106 cells were taken every 15 min and washed with PBS or with 3% O2-equilibrated PBS, respectively, to remove excess DHE. Cells were fixed for 10 seconds with 0.5 ml of 70% ethanol on ice and were then resuspended in 1 ml of normal PBS and analyzed immediately by flow cytometry (FACSCalibur, Becton Dickinson). Data were acquired and analyzed using cellquest software (Becton Dickinson). Results are expressed as the mean fluorescence intensity (MFI).
production; one-half of each group was maintained at 21% O2; the other half was transferred to an O2-controlled hypoxic chamber (Coy Laboratory Products; Ann Arbor, MI) maintained at 3% O2 and 37°C. Cells were agitated gently and aliquots of 5 × 106 cells were taken every 15 min and washed with PBS or with 3% O2-equilibrated PBS, respectively, to remove excess DHE. Cells were fixed for 10 seconds with 0.5 ml of 70% ethanol on ice and were then resuspended in 1 ml of normal PBS and analyzed immediately by flow cytometry (FACSCalibur, Becton Dickinson). Data were acquired and analyzed using cellquest software (Becton Dickinson). Results are expressed as the mean fluorescence intensity (MFI).
Activation of NF-κB. RAW 246.7 cells at a concentration of 5 × 107 cells per ml-1 were incubated for 30 min in the presence or absence of 750 μM l-NMMA at 21% O2 and 37°C. A sample from each group was kept in these conditions, and the rest was transferred to the hypoxic chamber maintained at 3% O2 and 37°C. Aliquots were taken every 15 min for determination of NF-κB translocation to the nucleus. The first step of nuclear extraction from the cells kept at 3% O2 was carried out inside the hypoxic chamber to avoid the effect of cellular reoxygenation. Aliquots of 1.25 × 107 cells per ml were washed in ice-cold O2-equilibrated PBS with phosphatase inhibitors, and nuclear extraction was carried out according to the manufacturer's instructions (nuclear extract kit, Active Motif Europe, Rixensart, Belgium). Protein content in the nuclear extract was determined to adjust the amount to 30 μg per well for each sample. Samples of 30 μg of protein were analyzed by Western blotting using a rabbit polyclonal antibody (Biomol Research Laboratories, Plymouth Meeting, PA) against NF-κB (p65 subunit) followed by an anti-rabbit horseradish peroxidase conjugate (Vector Laboratories). The protein band was detected by enhanced chemiluminescence (Amersham Pharmacia).
Measurement of Intercellular PO2. Measurements of the partial pressure of O2 (PO2) in a cell suspension open to the environment were carried out by using an O2 probe based on O2 quenching of fluorescence (OxyLite, Oxford Optronics, Oxford, U.K.). The sensor was used to measure the PO2 in the medium of cell suspensions exposed either to 21% (atmospheric) or 3% (hypoxic chamber) O2, in a shaking water bath maintained at 37°C. Measurements of PO2 were converted to [O2] in μM, taking into account the salinity and temperature of the medium and daily atmospheric and water vapor pressure (16).
Statistical Analysis. Means ± standard deviations were calculated for quantitative analysis of the results. Statistically significant differences (P < 0.05) between samples were determined using Student's paired t test.
Results
Effect of Exogenous NO on the Redox State of Purified CcO. Studies of the redox changes (Y) in purified CcO in turnover showed that increasing concentrations of NO led to a progressive increase in C50 (see Fig. 1), with an equivalent effect on P50. Table 1 summarizes these parameters for given concentrations of DETA-NO (the NO donor). NO also caused a progressive increase in the [O2] at which the reduction of cytochromes aa3 occurred; at the concentrations of NO used, the change in Y was linearly related to the decrease in VO2.
Fig. 1.

Effect of exogenous NO on the redox state of purified CcO. Normalized changes in the redox state of cytochromes aa3 from the steady state (Y = 1) to anoxia (Y = 0) against [O2] for 100 nM of purified CcO in turnover (solid black line) and the effect of exogenous NO administered as 50 μM (solid gray line) and 200 μM (dotted black line) DETA-NO.
Table 1. Effect of exogenous NO on purified CcO enzyme in turnover.
| DETA-NO, μM | NO, nM | C50, μM | P50, μM |
|---|---|---|---|
| 0 | 0 | 2.8 ± 0.3 | 2.8 ± 0.5 |
| 50 | 118 ± 19 | 5.3 ± 1.1 | 5.0 ± 0.7 |
| 200 | 470 ± 27 | 29.0 ± 10.0 | 30.5 ± 11.0 |
Values of [O2] for which VO2 and the change in the redox state of cytochromes aa3 are half-maximal (P50 and C50, respectively), obtained from purified CcO in turnover measured using the VLS system (n = 3). The concentration of NO released from the donor DETA-NO was determined by the decrease in NO measured by the NO electrode after the addition of 5 μM oxyhemoglobin (n = 3).
Effect of Endogenous NO on Cellular Respiration. The on-line time course of [O2] and VO2 was monitored in RAW 246.7 cells at a concentration of 5.3 ± 1.4 × 107 cells per ml (n = 12). The maximum VO2 during steady-state respiration was 56.2 ± 9.7 μM·min-1; this was maintained until [O2] decreased to ≈15 μM (see Fig. 2A). The corresponding changes in the redox states of cytochromes bH, cc1, and aa3 in terms of absolute concentration changes (i.e., corrected for the optical pathlength) were also monitored (Fig. 2B). An early reduction of cytochromes aa3 and cc1 occurred at an [O2], at which VO2 was still maximal (see Fig. 2A). This is also shown in Fig. 2C, in which VO2 and Y are plotted against [O2]. In contrast, the redox state of cytochrome bH was maintained in its steady-state until the [O2] was ≈10 μM.
Fig. 2.

Cellular respiration and changes in the mitochondrial cytochrome redox states monitored by VLS. RAW 246.7 cells were placed in a closed respiration chamber and allowed to consume O2 until anoxia. (A) On-line recording of changes in [O2] (solid black line) and VO2 (dotted gray line) against time using the VLS system. (B) On-line recording of changes in concentration of cytochromes bH (filled circles), cc1 (filled triangles), and aa3 (open squares) from the steady-state against time. Negative values indicate an increase in reduced and a decrease in oxidized cytochromes and vice versa. (C) Changes in VO2 (dotted gray line), normalized between 1 and 0, and Y, the normalized changes in cytochrome redox states for cytochromes bH (filled circles), cc1 (filled triangles), and aa3 (open squares), against [O2].
To determine whether basal concentrations of NO in intact cells modulate the O2 dependence of the redox state of cytochromes aa3, as observed in purified CcO, we compared cellular respiratory parameters in the presence and absence of the NOS inhibitor l-NMMA. Table 2 summarizes values of C50 and P50 in RAW 246.7 cells and HUVEC, with and without l-NMMA. The P50 was significantly (P < 0.05) decreased in both cell types by treatment with l-NMMA. Values of C50 were also significantly (P < 0.05) decreased in the presence of the NOS inhibitor.
Table 2. Effect of endogenous NO on cellular respiration and cytochrome redox states.
| Cell type | l-NMMA | [Cell] (107 cells per ml) | C50, μM | P50, μM |
|---|---|---|---|---|
| RAW 246.7 | - | 5.3 ± 1.0 | 8.3 ± 3.0 | 6.1 ± 2.7 |
| + | 5.2 ± 1.7 | 4.9 ± 2.6 | 2.9 ± 1.7 | |
| HUVEC | - | 2.9 ± 0.4 | 10.8 ± 5.6 | 8.1 ± 4.3 |
| + | 3.1 ± 0.7 | 5.6 ± 3.1 | 4.1 ± 2.8 |
Values of [O2] for which VO2 and the change in the redox state of cytochromes aa3 are half-maximal (P50 and C50, respectively), obtained from intact cells (RAW 246.7 and HUVEC) during respiration measured using the VLS system (n = 6) in the absence (-) and presence (+) of the NOS inhibitor l-NMMA.
Effect of Endogenous NO on Formation of RE in the ETC. Fig. 3 shows the increase in RE with decreasing [O2] in RAW 246.7 cells and HUVEC, respectively, in the absence and presence of l-NMMA. Above 50 μM O2, the redox state of the ETC cytochromes was independent of [O2], and there was no difference in RE between control and l-NMMA-treated cells. As [O2] decreased below 50 μM, the presence of basal concentrations of NO in control cells led to a more pronounced increase in RE at any given [O2] compared to l-NMMA-treated cells. For example, a 10% increase in RE from the steady-state occurred at an [O2] of 20.2 ± 6.2 and 22.6 ± 7.6 μM (n = 6) in control RAW 246.7 cells and HUVEC, respectively. This occurred at significantly (P < 0.05) lower [O2] in l-NMMA-treated cells, with values of 15.7 ± 5.5 and 15.0 ± 3.8 μM, respectively (Fig. 3). In both control and l-NMMA-treated cells, the VO2 was still maximal at the [O2] at which RE had increased by 10%. At the [O2] at which a 10% decrease in VO2 was observed (11.9 ± 2.3 μM in control RAW 246.7 cells and 13.6 ± 5.7 μM in control HUVEC), the increase in RE was 20.1 ± 4.6 and 21.9 ± 4.3%, respectively.
Fig. 3.

Effect of endogenous NO on the increase in RE in the ETC at low [O2]. Increases in RE in the ETC are represented by the normalized sum of changes in the cytochrome redox states. Dotted lines indicate a 10% increase in RE and the corresponding [O2] (significantly different, P < 0.05, with and without l-NMMA). Data are the mean of six experiments. (A) Changes in RE against [O2] for RAW 246.7 cells in the absence (filled circles) or presence (open circles) of 750 μM l-NMMA. (B) Changes in RE against [O2] for HUVEC in the absence (filled triangles) or presence (open triangles) of 500 μM l-NMMA. Broken lines show the [O2] at which there is a 10% increase in RE.
Effect of Endogenous NO on the Release of 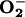 and Nuclear Migration of NF-κB. The production of
and Nuclear Migration of NF-κB. The production of 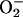 was studied in RAW 246.7 cells incubated at 21% O2 for 1 h in the presence or absence of l-NMMA. DHE was then added to the cells; one-half of each group was maintained at 21% O2 and the other half at 3% O2. [O2] values of 62 and 4 μM were measured in the medium of cells (≈4 × 107 cells per ml) incubated at 21% and 3% O2, respectively.
was studied in RAW 246.7 cells incubated at 21% O2 for 1 h in the presence or absence of l-NMMA. DHE was then added to the cells; one-half of each group was maintained at 21% O2 and the other half at 3% O2. [O2] values of 62 and 4 μM were measured in the medium of cells (≈4 × 107 cells per ml) incubated at 21% and 3% O2, respectively.
Fig. 4A shows that there was no significant difference in DHE fluorescence in cells incubated for 30 min at 21% O2 in the presence or absence of l-NMMA, with a MFI of 3.3 ± 0.7 and 3.0 ± 0.6 (n = 3) for control and l-NMMA-treated cells, respectively. When cells were incubated for 30 min at 3% O2 (see Fig. 4B), there was a significant (P < 0.05) increase in fluorescence in control but not in l-NMMA-treated cells (MFI of 7.2 ± 0.9 and 3.9 ± 1.3, respectively).
Fig. 4.

Effect of endogenous NO on the production of 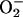 and nuclear migration of NF-κB at different [O2]. (A) Fluorescence, as measured by flow cytometry from intact cells incubated with DHE for 30 min, indicating intracellular
and nuclear migration of NF-κB at different [O2]. (A) Fluorescence, as measured by flow cytometry from intact cells incubated with DHE for 30 min, indicating intracellular  production in the presence (shaded) or absence (clear) of l-NMMA at 21% O2. (B) Fluorescence, as described in A, indicating intracellular
production in the presence (shaded) or absence (clear) of l-NMMA at 21% O2. (B) Fluorescence, as described in A, indicating intracellular 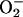 production in the presence (shaded) or absence (clear) of l-NMMA at 3% O2 (C) Western blot of nuclear extracts of cells incubated at 21 and 3% O2 detecting NF-κB (p65 subunit) in the absence (-) or presence (+) of l-NMMA at 30 and 60 min.
production in the presence (shaded) or absence (clear) of l-NMMA at 3% O2 (C) Western blot of nuclear extracts of cells incubated at 21 and 3% O2 detecting NF-κB (p65 subunit) in the absence (-) or presence (+) of l-NMMA at 30 and 60 min.
To study the downstream consequences of this increase in 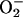 , we analyzed the time course of NF-κB migration to the nucleus in the same system. Fig. 4C shows the absence of NF-κB in the nuclear extracts of cells exposed to 21% O2, both with and without l-NMMA. When the cells were incubated in the hypoxic chamber at 3% O2, however, there was a large time-dependent increase of NF-κB in control cells that was diminished by treatment with l-NMMA.
, we analyzed the time course of NF-κB migration to the nucleus in the same system. Fig. 4C shows the absence of NF-κB in the nuclear extracts of cells exposed to 21% O2, both with and without l-NMMA. When the cells were incubated in the hypoxic chamber at 3% O2, however, there was a large time-dependent increase of NF-κB in control cells that was diminished by treatment with l-NMMA.
Discussion
The finding that NO inhibits CcO in a manner that is competitive with O2 and reversible led to the concept that NO might regulate mitochondrial VO2. Because the potency of NO increases as the [O2] decreases, it is likely that the low concentrations of NO found in tissues (17) may, under certain conditions, successfully compete with O2 for the binuclear binding site of CcO (4, 18). The nature of the interaction between NO and purified CcO or isolated mitochondria has now been extensively studied (for reviews, see refs. 19 and 20). However, a role for endogenous NO in the regulation of VO2 remains to be fully understood, and so far only a few studies have demonstrated that basal concentrations of endogenous NO are able to modulate respiration physiologically both in vitro and in vivo (6, 21, 22).
Using our recently developed VLS system, we have previously demonstrated in whole cells that exogenously applied NO causes a reversible inhibition of VO2, with a concomitant decrease in the redox state of cytochromes aa3 and consequently, the upstream cytochromes cc1 and bH (8). We have now used this system to study the effects of NO on the activity and redox behavior of purified CcO at a relatively low turnover (≈15 electrons·s-1) and have confirmed the well-known increase in the apparent Km for O2 (4). Furthermore, at concentrations at which NO inhibited VO2, we found a progressive increase in the [O2] at which the onset of reduction of cytochromes occurred.
In respiring cells, however, we observed an early reduction in cytochromes aa3 and cc1 relative to the decrease in VO2. We and others have previously reported this early reduction of cytochromes in cells (8, 9), and other authors have described it in vivo (23). Although the nature and biological significance of this phenomenon remain controversial (9, 24), one interesting suggestion is that the early reduction of mitochondrial cytochromes may be a manifestation of a mechanism by which cells maintain their respiratory rate at low [O2]. Our results with l-NMMA indicate that such a mechanism may be partly attributable to endogenous NO. This is supported by the observation that in yeast, in which the effects of NO are abolished by the scavenging action of a constitutive flavohemoglobin protein (25), no early reduction of the cytochromes occurs (24). Interestingly, we did not observe an early reduction in cytochrome bH. Although the explanation for this phenomenon is not clear, it is possible that it could be due in part to a difference in midpoint potentials of the cytochromes and/or could suggest that complex III is a site of 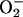 production.
production.
Mitochondrial 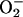 is generated by the interaction between O2 and RE, and thus the rate of its formation in any given situation will depend on the concentrations of these two reactants (13). Under atmospheric conditions, high concentrations of exogenous NO have been shown to produce
is generated by the interaction between O2 and RE, and thus the rate of its formation in any given situation will depend on the concentrations of these two reactants (13). Under atmospheric conditions, high concentrations of exogenous NO have been shown to produce 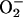 and H2O2 in submitochondrial particles, rat heart mitochondria (26), and Langendorf preparations of isolated hearts (27). Furthermore, we have previously shown that treatment of human lymphoid T cells with a proapoptotic stimulus (anti-Fas Ab) leads to an increase in
and H2O2 in submitochondrial particles, rat heart mitochondria (26), and Langendorf preparations of isolated hearts (27). Furthermore, we have previously shown that treatment of human lymphoid T cells with a proapoptotic stimulus (anti-Fas Ab) leads to an increase in 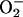 generation that depends on endogenous NO (28). All these experiments, however, were carried out at high [O2], unlike those found in vivo.
generation that depends on endogenous NO (28). All these experiments, however, were carried out at high [O2], unlike those found in vivo.
As the [O2] decreases, it is likely that there will be a point at which the increase in RE resulting from a more potent interaction between NO and CcO compensates for the decreased availability of O2, thus creating a favorable situation for the formation of 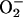 . Our results showing an increase in intracellular generation of
. Our results showing an increase in intracellular generation of 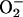 at low [O2] compared with atmospheric conditions confirm this and are in agreement with the reports of others (14, 29, 30). As expected, we found that
at low [O2] compared with atmospheric conditions confirm this and are in agreement with the reports of others (14, 29, 30). As expected, we found that 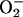 production was significantly diminished by treatment with l-NMMA.
production was significantly diminished by treatment with l-NMMA.
Mitochondrial production of ROS has been linked to the translocation of NF-κB to the nucleus as a hypoxic stress signaling response (14). Our experiments clearly show that NO is involved in this process, because the increase in mitochondrial RE, the generation of mitochondrial 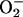 , and the activation of NF-κB were all closely correlated and could be attenuated by treatment with l-NMMA. Interestingly, this mechanism appears to be activated at [O2] well above those that are critical for the maintenance of steady-state cellular respiration and also at concentrations of NO below those at which this gas inhibits respiration. Thus the generation of
, and the activation of NF-κB were all closely correlated and could be attenuated by treatment with l-NMMA. Interestingly, this mechanism appears to be activated at [O2] well above those that are critical for the maintenance of steady-state cellular respiration and also at concentrations of NO below those at which this gas inhibits respiration. Thus the generation of 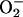 and the translocation of NF-κB might not, strictly speaking, be a cellular response to hypoxia. Although decreasing [O2] may be one of the triggers, any factors that enhance interaction between NO and CcO, leading to an increase in RE, could result in the activation of this signaling cascade, even at high [O2]. Our results thus provide a clearer understanding of the role of NO in this “early warning” system, in which a change in the mitochondrial ETC redox state triggers a nuclear mechanism for cellular defense.
and the translocation of NF-κB might not, strictly speaking, be a cellular response to hypoxia. Although decreasing [O2] may be one of the triggers, any factors that enhance interaction between NO and CcO, leading to an increase in RE, could result in the activation of this signaling cascade, even at high [O2]. Our results thus provide a clearer understanding of the role of NO in this “early warning” system, in which a change in the mitochondrial ETC redox state triggers a nuclear mechanism for cellular defense.
Acknowledgments
We acknowledge P. Rich (Department of Biology, University College London) for donating the purified enzyme, K. Ilic (Wolfson Institute for Biomedical Research, University College London) for technical support with cell culture, and A. Higgs for critical revision of the manuscript. M.Q. is the recipient of a short-term fellowship from the European Molecular Biology Organisation (Grant ASTF198-2003). S.M. is supported by a grant from the Medical Research Council.
Abbreviations: ETC, electron transport chain; NOS, NO synthase; CcO, cytochrome c oxidase; VO2, rate of O2 consumption; VLS, visible light spectroscopy; ROS, reactive oxygen species; DHE, dihydroethidium; HUVEC, human umbilical venous endothelial cells; RE, reducing equivalents; DETA-NO, (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate; l-NMMA, NG-monomethyl-l-arginine.
References
- 1.Moncada, S. & Higgs, A. (1993) N. Engl. J. Med. 329, 2002-2012. [DOI] [PubMed] [Google Scholar]
- 2.Bellamy, T. C. & Garthwaite, J. (2002) Br. J. Pharmacol. 136, 95-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cleeter, M. W., Cooper, J. M., Darley-Usmar, V. M., Moncada, S. & Schapira, A. H. (1994) FEBS Lett. 345, 50-54. [DOI] [PubMed] [Google Scholar]
- 4.Brown, G. C. & Cooper, C. E. (1994) FEBS Lett. 356, 295-298. [DOI] [PubMed] [Google Scholar]
- 5.Schweizer, M. & Richter, C. (1994) Biochem. Biophys. Res. Commun. 204, 169-175. [DOI] [PubMed] [Google Scholar]
- 6.Clementi, E. Brown, G. C., Foxwell, N. & Moncada, S. (1999) Proc. Natl. Acad. Sci. USA 96, 1559-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moncada, S. & Erusalimsky, J. D. (2002) Nat. Rev. Mol. Cell Biol. 3, 214-220. [DOI] [PubMed] [Google Scholar]
- 8.Hollis, V. S., Palacios-Callender, M., Springett, R. J., Delpy, D. T. & Moncada, S. (2003) Biophys. Biochim. Acta 1607, 191-202. [DOI] [PubMed] [Google Scholar]
- 9.Wilson, D. F., Erecinska, M., Drown, C. & Silver, I. A. (1979) Arch. Biochem. Biophys. 195, 485-493. [DOI] [PubMed] [Google Scholar]
- 10.Wilson, D. F. (1994) Med. Sci. Sports Exerc. 26, 37-43. [PubMed] [Google Scholar]
- 11.Korzeniewski, B. (2001) Biochim. Biophys. Acta 1504, 31-45. [DOI] [PubMed] [Google Scholar]
- 12.Chandel, N. S., Maltepe, E., Goldwasser, E., Mathieu, C. E., Simon, M. C. & Schumacker, P. T. (1998) Proc. Natl. Acad. Sci. USA 95, 11715-11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turrens, J. F. (2003) J. Physiol. 552, 335-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pearlstein, D. P., Ali, M. H., Mungai, P. T., Hynes, K. L., Gewertz, B. L. & Schumacker, P. T. (2002) Arterioscler. Thromb. Vasc. Biol. 22, 566-573. [DOI] [PubMed] [Google Scholar]
- 15.Van Staveren, H. G., Moes, C. J. M., van Marle, J., Prahl, S. A. & van Gemert, M. J. C. (1991) Appl. Opt. 30, 4507-4514. [DOI] [PubMed] [Google Scholar]
- 16.Hitchman, M. L. (1978) in Measurement of Dissolved Oxygen, eds. Elving, P. J. & Winefordner, J. D. (Wiley, New York), pp. 7-34.
- 17.Malinski, T., Taha, Z., Grunfeld, S., Patton, S., Katpurczak, M. & Tomboulian, P. (1993) Biochem. Biophys. Res. Commun. 193, 1076-1082. [DOI] [PubMed] [Google Scholar]
- 18.Torres, J. & Wilson, M. T. (1996) Methods Enzymol. 269, 3-11. [DOI] [PubMed] [Google Scholar]
- 19.Cooper, C. E. (2002) Trends Biochem. Sci. 27, 33-39. [DOI] [PubMed] [Google Scholar]
- 20.Sarti, P., Giuffre, A., Barone, M. C., Forte, E., Mastronicola, D. & Brunori, M. (2003) Free Radic. Biol. Med. 34, 509-520. [DOI] [PubMed] [Google Scholar]
- 21.Sarti, P., Lendaro, E., Ippoliti, R., Bellelli, A., Benedetti, P. A. & Brunori, M. (1999) FASEB J. 13, 191-197. [DOI] [PubMed] [Google Scholar]
- 22.Shen, W., Xu, X., Ochoa, M., Zhao, G., Wolin, M. S. & Hintze, T. H. (1994) Circ. Res. 75, 1086-1095. [DOI] [PubMed] [Google Scholar]
- 23.Stingele, R., Wagner, B., Kameneva, M. V., Williams, M. A., Wilson, D. A., Thakor, N. V., Traystman, R. J. & Hanley, D. F. (1996) Am. J. Physiol. 271, H579-H587. [DOI] [PubMed] [Google Scholar]
- 24.Chance, B. (1988) FEBS Lett. 226, 343-346. [DOI] [PubMed] [Google Scholar]
- 25.Liu, L., Zeng, M., Hausladen, A., Heitman, J. & Stamler, J. S. (2000) Proc. Natl. Acad. Sci. USA 97, 4672-4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poderoso, J. J., Carreras, M. C., Lisdero, C., Riobo, N., Schopfer, F. & Boveris, A. (1996) Arch. Biochem. Biophys. 328, 85-92. [DOI] [PubMed] [Google Scholar]
- 27.Poderoso, J. J., Peralta, J. G., Lisdero, C. L., Carreras, M. C., Radisic, M., Schopfer, F., Cadenas, E. & Boveris, A. (1998) Am. J. Physiol. 274, C112-C119. [DOI] [PubMed] [Google Scholar]
- 28.Beltran, B., Quintero, M., Garcia-Zaragoza, E., O'Connor, E., Esplugues, J. V. & Moncada, S. (2002) Proc. Natl. Acad. Sci. USA 99, 8892-8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schroedl, C., McClintock, D. S., Budinger, G. R. & Chandel, N. S. (2002) Am. J. Physiol. 283, L922-L931. [DOI] [PubMed] [Google Scholar]
- 30.Schafer, M., Schafer, C., Ewald, N., Piper, H. M. & Noll, T. (2003) Circ. Res. 92, 1010-1015. [DOI] [PubMed] [Google Scholar]