

Abstract
Myeloperoxidase (MPO), the phagocyte hemoprotein involved in neutrophil host defense and consuming nitric oxide (•NO), induces the nitration of extracellular matrix proteins and tissue remodeling subsequent to its transcytosis across the endothelial barrier. We addressed the role of an interaction of MPO with albumin as a requirement for MPO transport across the endothelium. Matrix-assisted laser desorption/ionization MS analysis of 80- and 60-kDa proteins purified from human lung tissue [with a human serum albumin (HSA)-affinity column] identified these albumin-binding proteins as MPO and MPO-heavy chain. A peptide corresponding to the MPO-heavy chain residues 425–454 demonstrated high-affinity binding to HSA. Replacement of the positively charged residues, R and K with G, prevented the binding of HSA to the peptide. We observed that albumin increased the binding of 125I-MPO to lung microvascular endothelial cells by 2-fold and the rate of transendothelial flux of 125I-MPO in cultured monolayers and intact vessels. Disruption of caveolae with cyclodextrin prevented the albumin-induced increase in transendothelial flux of 125I-MPO. We also observed by confocal imaging that albumin induced the rapid internalization of MPO and its colocalization with albumin-labeled vesicles. MPO colocalized with the caveolae markers cholera toxin subunit B and caveolin 1 in the endocytosed vesicles. Thus, transcytosis of MPO by caveolae induced by its charge-dependent interaction with albumin is an important means of delivering MPO to the subendothelial space. Albumin-mediated transport of MPO may thereby regulate NO bioavailability and formation of NO-derived oxidants in the vessel wall.
Myeloperoxidase (MPO), expressed in polymorphonuclear neutrophils (PMNs), plays an important role in innate immunity (1) and regulation of the generation of nitric oxide (NO)-derived oxidants (2–7). MPO exists as a 150-kDa tetramer composed of two glycosylated 59- to 64-kDa heavy chain and two nonglycosylated 14-kDa light chains (8). MPO is released from activated PMNs into the phagosome and extracellular space, where the enzyme utilizes H2O2 from the respiratory burst to catalyze the formation of hypochlorous acid (HOCl) and variety of other oxidizing species (1). MPO activity measurement is used as an index of inflammation, because the MPO levels are increased in inflammatory tissue largely secondary to PMN extravasation (9–11). MPO is also present in human atherosclerotic lesions (12), and plasma MPO activity may be an excellent marker of acute coronary syndromes (13–15). The MPO-induced catalysis of low-density lipoprotein oxidation converts low-density lipoprotein into its high-uptake form and thus contributes to the formation of foam cells and cholesterol deposition in the vessel wall (16, 17).
MPO activates the formation of protein nitrotyrosine (NO2Tyr), a characteristic of acute and chronic inflammation (2–7). The presence of NO2Tyr in proteins is considered as an index of peroxynitrite (ONOO-) production, the product of the reaction of •NO and superoxide anion (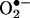 ) (18, 19). MPO induces NO2Tyr formation by oxidation of nitrite (
) (18, 19). MPO induces NO2Tyr formation by oxidation of nitrite (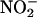 ) to nitrogen dioxide (
) to nitrogen dioxide (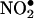 ) (7, 20, 21). MPO was shown to colocalize with NO2Tyr in the subendothelium of coronary and hepatic vessels and in the alveolar epithelial compartment of patients with sickle cell disease or acute rejection of lung transplants (21, 22). The subendothelium of pulmonary vessels from MPO-/- mice challenged with i.p. zymosan also showed significantly less NO2Tyr formation than wild-type mice (20, 22), indicating that MPO is essential for protein tyrosine nitration during vascular inflammation. For MPO to induce protein tyrosine nitration in the subendothelium, it needs to be transported across the endothelial barrier. Studies showed that MPO was transported by transcytosis after binding to the negatively charged endothelial cell surface (22). The MPO was subsequently localized with fibronectin in the extracellular matrix and induced tissue remodeling by nitration of matrix proteins (22). Although the transcytosis of MPO plays an important role in its deposition in the subendothelium, the mechanisms of MPO transport have not been defined.
) (7, 20, 21). MPO was shown to colocalize with NO2Tyr in the subendothelium of coronary and hepatic vessels and in the alveolar epithelial compartment of patients with sickle cell disease or acute rejection of lung transplants (21, 22). The subendothelium of pulmonary vessels from MPO-/- mice challenged with i.p. zymosan also showed significantly less NO2Tyr formation than wild-type mice (20, 22), indicating that MPO is essential for protein tyrosine nitration during vascular inflammation. For MPO to induce protein tyrosine nitration in the subendothelium, it needs to be transported across the endothelial barrier. Studies showed that MPO was transported by transcytosis after binding to the negatively charged endothelial cell surface (22). The MPO was subsequently localized with fibronectin in the extracellular matrix and induced tissue remodeling by nitration of matrix proteins (22). Although the transcytosis of MPO plays an important role in its deposition in the subendothelium, the mechanisms of MPO transport have not been defined.
Albumin is the predominant plasma protein responsible for maintaining the transendothelial oncotic pressure gradient and regulating the transport of fatty acids, steroids, thyroxine, and amino acids (23). Albumin transport by caveolae is a key determinant of transcellular endothelial permeability (24–27). The binding of albumin to albumin-binding proteins (ABPs) localized in caveolae was essential for the transcellular permeability of albumin (24–27). In the present study, we have isolated 80- and 60-kDa ABPs from human lung tissue by using a HSA-affinity column and identified these proteins as MPO by matrix-assisted laser desorption/ionization (MALDI) MS (28). The MPO-heavy chain (HC) sequence (409–454) was homologous with the HSA-docking sequence identified in certain bacterial proteins that interact with HSA (29). We observed that albumin interaction with MPO was essential in inducing the transcytosis of MPO by caveolae.
Materials and Methods
Materials. MPO purified from human PMNs and polyclonal MPO antibody (Ab) were purchased from Calbiochem. HSA and BSA were from Sigma. HSA-affinity Sepharose was prepared by coupling HSA with CNBr-activated Sepharose 4B from Pharmacia. Endothelial cell growth medium was from Invitrogen and FBS was from HyClone. Endothelial cell growth supplement was from BD Biosciences (Bedford, MA). Bovine lung microvessel endothelial cells (BLMVECs) and rat lung microvessel endothelial cells were from Vec Technologies (Rensselaer, NY). Peptides were synthesized as C-terminal amide (bioWORLD, Dublin, OH). Their purity and amino acid sequence were determined by HPLC and MS, respectively. Peptides used in this study were 98% pure. BSA-Alexa 594, cholera toxin subunit B (CTB)-Alexa 488, and secondary antibodies were from Molecular Probes.
Protein Purification and MALDI-MS. Human lung tissue was obtained from University of Illinois Hospital according to approved Institutional Review Board protocol. The tissue (180 g) was washed, minced, and homogenized with 20 volumes of buffer A (20 mM Hepes/Tris/0.15 M NaCl/0.1 mM phenylmethylsufonylfluoride/30 μM benzamidine, pH 7.4). The homogenate was centrifuged at 1,000 × g for 10 min, and the supernatant was centrifuged at 100,000 × g for 60 min. The pellet obtained was solubilized by using 2.5% sodium cholate and 4 M urea (30). The solubilized extract was concentrated by ethanol precipitation and reextracted with Triton X-100 (30). The Triton X-100 concentration in the extract was adjusted to 0.2% and applied on HSA-Sepharose column (1 × 10 cm) preequilibrated with buffer B (10 mM Tris·HCl/0.5 mM EDTA, pH 7.4, containing 0.1 mM PMSF, 30 μM benzamidine, and 0.2% Triton X-100). The column was washed, and bound proteins were eluted with 20 ml of 0.1 M citrate buffer (pH 4.0) containing 0.2% Triton X-100. The proteins eluted were separated on SDS/PAGE, stained with Coomassie Brilliant Blue R-250 (CBBR-250; Fig. 1A). The protein bands excised were digested with trypsin and peptides were analyzed by using MALDI-MS at Yale University Biotechnology Resource Laboratory (New Haven, CT). The peptide masses were compared with known sequences by using profound search analysis on the OWL database. In addition, EMBL/nonredundant database search was used to identify the amino acid sequences. Using these methods, we identified the 80- and 60-kDa proteins as MPO and MPO-HC, respectively (GenBank accession no. P05164).
Fig. 1.

Identification of MPO as an ABP. (A) Purification of proteins by using HSA-affinity column. Proteins eluted from HSA-affinity column were subjected to SDS/PAGE and stained with CBBR-250. Lane 1, human lung homogenate proteins; lane 2, proteins eluted from HSA-affinity column. (B) Identification of proteins by MALDI-MS. Schematics illustrations of the identified MPO sequences are shown (GenBank accession no. P05164). The mass of 17 tryptic peptides to mass of amino acids 295–303, 367–375, 395–406, 405–422, 441–448, 460–474, 472–482, 488–500, 498–512, 512–530, 536–549, 559–572, 578–591, 590–604, 691–702, 701–715, and 714–726 were obtained from 60-kDa proteins (MPO-HC). In the 80-kDa protein, the mass of amino acid sequences obtained were the same as the 60-kDa proteins, except for two additional masses of amino acids 173–193 and 218–228. LC, light chain. (C) MPO sequence homology with the HSA-binding domain bacterial proteins. (D) Binding of MPO peptides with 125I-HSA. Peptides were immobilized on nitrocellulose membranes at the indicated concentrations. The binding of 125I-HSA to the peptides was determined as described in Materials and Methods.(E) Effects of soluble MPO-WT and MPO-MT peptides on the binding of 125I-HSA to immobilized MPO-WT peptide. MPO-WT peptide (10 nmol) was spotted on the membranes (15 × 15 mm). Nonspecific binding was blocked and then incubated with 1 ml of 125I-HSA (0.3 μM) for 2 h at 22°C in the presence of varying concentrations of either WT peptide or MT peptide. Other details were described in Materials and Methods. The experiment was repeated three times in triplicate. *, the difference from control (in the absence of soluble peptide), P < 0.05; **, P < 0.001.
125I-Labeling of Proteins. HSA and MPO were labeled with 125I by using IODO-GEN reagent from Pierce.
125I-HSA Binding. 125I-HSA binding to the MPO peptides was determined by dot-blot analysis. Peptides were immobilized on nitrocellulose membranes, and nonspecific binding was blocked with bovine γ-globulin (2 mg/ml in PBS) for 1 h at 22°C. Membranes were washed two times with PBS and incubated with 125I-labeled HSA (0.3 μM) for 2 h at 22°C. Membranes were then washed four times with PBS containing 0.05% Tween 20 (PBST), and autoradiography was performed. In some experiments, the membranes were directly counted to determine the membrane-associated radioactivity.
Endothelial Cell Cultures. BLMVECs were grown in OPTI-MEM I supplemented with 15% FBS and endothelial cell growth supplement (15 μg/ml). Rat lung microvessel endothelial cells were grown in DMEM supplemented with 5% FBS.
125I-MPO Binding. BLMVECs grown to confluence in six-well culture plates were washed two times and incubated with 5 mM Hepes/Hanks' balanced salt solution (HBSS, pH 7.4; buffer C) overnight at 37°C. Cells were then washed two times, and the binding assay was initiated by adding 1 ml of 125I-MPO (10 nM) in buffer C containing bovine γ-globulin or BSA. Incubation was continued at 4°C for 1 h. Binding was terminated by washing three times with ice-cold buffer C, and the radioactivity associated with the BLMVEC monolayer was determined (30).
Transendothelial 125I-MPO Permeability. Transendothelial permeability of 125I-MPO in BLMVEC monolayers was determined by using Transwell filter units (Corning) (24, 27). BLMVEC monolayers were washed and incubated for 2 h with buffer C before experiments. Both luminal and abluminal chambers contained 5 mg/ml either bovine γ-globulin or BSA in buffer C at volumes of 0.5 and 1.5 ml, respectively. Tracer 125I-MPO (1 × 107 cpm) was added in the upper compartment, and 0.05-ml samples were collected from the lower compartment at 15-min intervals for 90 min for determination of transendothelial 125I-MPO permeability (24, 27).
125I-MPO Permeability in Intact Lung Vessels. Pulmonary vascular 125I-MPO permeability × surface area (PS) product, a measure of vascular permeability, was determined in the rat lung as described (26). In brief, Krebs-perfused lung preparations received 125I-MPO (100,000 cpm/ml) for 10 min followed by a 6-min washout with Krebs solution and a 3-min washout with 0.1 M acetate buffer (pH 4.5) at 12°C to remove any vascular surface bound 125I-MPO.
Immunostaining. Cellular localization of MPO, albumin, CTB (a caveolae marker), and caveolin-1 was determined by confocal microscopy (25, 27). BLMVECs grown to confluence on glass coverslips were incubated in serum-free medium (buffer C) for 12 h at 37°C. MPO uptake in BLMVECs was determined in the presence or absence of albumin or CTB. After incubation, the cells were washed three times with buffer C, fixed with 4% paraformaldehyde in HBSS, and blocked for 30 min in HBSS containing 5% goat serum and 0.1% Triton X-100. Primary Ab labeling was performed overnight at 4°C in HBSS containing 5% goat serum. Coverslips were washed three times and incubated with the appropriate secondary Ab (diluted 1:500) for 1 h. Cells were washed and mounted; images were acquired with the Zeiss LSM 510 confocal microscope. The experiments were repeated three times.
Statistical Analysis. Comparisons were made by using the two-tailed Student t test. Values were considered significant at P < 0.05.
Results
MPO Interacts with HSA. Using HSA-affinity column, we isolated 80- and 60-kDa proteins from human lung tissue, and by MALDI-MS, we identified these proteins as MPO (80 kDa) and MPO-HC (60 kDa) (Fig. 1 A and B). We compared the MPO sequence with the known HSA-binding protein sequences. The MPO-HC (residues 409–454) showed high homology with the HSA-binding domain of the M12 protein from Streptococcus pyogenes (29) and other bacterial HSA-binding proteins (29) (Fig. 1C). Positively charged amino acids R and K were enriched in the HSA-binding domain of bacterial proteins (Fig. 1C). Similar positively charged residues were also present in MPO-HC between residues 421 and 460 (Fig. 1C).
Because MPO is cationic (1, 8), we addressed the possibility that it interacts by means of charge with negatively charged albumin (pI = 4.7). Peptides were synthesized peptides from MPO-HC sequence 425–454, which exhibits the albumin-docking homology with bacterial protein HSA-binding domain sequence. 125I-HSA binding to the MPO peptides was determined (see Materials and Methods). MPO-HC sequence 425–454 [RLATELKSLNPRWDGERLYQEARKIVGAMV (MPO-WT peptide)] showed high-affinity binding to HSA (Fig. 1D and Table 1). The EARKIV motif showed no binding to HSA (Fig. 1D and Table 1). To address the role of the positive charges, we synthesized peptides by replacing K and R with G [GLATELGSLNPGWDGEGLYQEAGGIVGAMV (MPO-MT peptide)] and measured HSA binding. MPO-MT peptide failed to bind to 125I-HSA (Fig. 1D and Table 1). Thus, it appears that the length and charge on this MPO sequence are essential for binding to albumin. The binding characteristics of BSA or rat serum albumin to the MPO-WT peptide were similar to those of HSA (data not shown). To address the specificity of binding of MPO peptides to albumin, we immobilized WT peptide on nitrocellulose membrane and determined the binding of 125I-HSA in the presence of various concentrations of WT or MT peptide. Increasing concentrations of WT peptide prevented the binding of 125I-HSA to the immobilized WT peptide (Fig. 1E). However, MT peptide had no effect on 125I-HSA binding to WT peptide (Fig. 1E). The binding affinity (Kd) of MPO-WT peptide to HSA (determined by using nonlinear regression analysis) was determined to be 20 ± 1.5 μM.
Table 1. Binding of synthetic MPO peptides to 125I-HSA.
| MPO peptide, nmol/filter | 125I-HSA bound, pmol/filter |
|---|---|
| RLATELKSLNPRWDGERLYQEARKIVGAMV | |
| 1.0 | 0.31 ± 0.003 |
| 2.5 | 1.45 ± 0.04 |
| 5.0 | 2.93 ± 0.07 |
| 10.0 | 6.55 ± 0.23 |
| GLATELGSLNPGWDGEGLYQEAGGIVGAMV | |
| 1.0 | 0.01 ± 0.003 |
| 2.5 | 0.11 ± 0.035 |
| 5.0 | 0.14 ± 0.07 |
| 10.0 | 0.19 ± 0.045 |
| EARKIV | |
| 1.0 | 0.05 ± 0.02 |
| 2.5 | 0.06 ± 0.025 |
| 5.0 | 0.08 ± 0.04 |
| 10.0 | 0.11 ± 0.06 |
Peptides were immobilized on nitrocellulose membranes and then incubated with 1 ml of 0.3 μM 125I-HSA in PBS for 2 h. Other details were described in Materials and Methods. Nonspecific binding was determined by incubating membranes with 125I-HSA in the absence of any peptides. Results are shown as mean ± SEM for three separate experiments made in triplicate.
MPO Is Present in Cultured Endothelial Cells Grown in Serum. Because endothelial cells are typically grown in serum-containing medium to mimic physiological conditions, we determined whether cultured endothelial cells are coated with MPO. Cell surface staining of anti-MPO Ab and MPO-HC was evident in both rat lung microvessel endothelial cells and BLMVECs (data not shown).
Albumin Promotes Binding of MPO to Endothelial Cell Surface. To address the role of albumin in inducing the binding of MPO, we measured the binding of 125I-MPO to BLMVECs in the presence of albumin. Increasing the concentration (1–50 mg/ml) of BSA in the binding buffer increased the binding of 125I-MPO to the endothelial cell surface (Fig. 2A). In contrast, the presence of bovine γ-globulin (5 mg/ml) had no effect on 125I-MPO binding.
Fig. 2.

(A) Albumin increases the binding of 125I-MPO to endothelial cell surface. The binding of 125I-MPO to BLMVECs was measured as described in Materials and Methods. Results are shown as mean ± SEM of three separate experiments made in a triplicate-binding assay. *, difference from control, P < 0.05; **, P < 0.001, different from control (i.e., binding was determined in the presence of 5 mg/ml γ-globulin). The presence or absence of 5 mg/ml γ-globulin in the binding buffer did not affect the binding of 125I-MPO to endothelial cells. (B) Effects of albumin on the time course of transendothelial transport of MPO. BLMVECs grown on microporous Transwell filters were used to determine transendothelial transport of MPO as described in Materials and Methods. To study the methyl-β-CD effect, luminal chamber was incubated with 5.0 mM CD for 20 min before measuring 125I-MPO permeability. The results are shown as mean ± SEM of four separate experiments made in triplicate. *, difference from control (γ-globulin) group or CD-treated group, P < 0.001. (C) Albumin increases transendothelial 125I-MPO permeability. Experiment was made as described in B. The transendothelial 125I-MPO clearance rate was calculated as described (24, 27). *, difference from control or CD-treated group, P < 0.001. The results are shown as mean ± SEM of four separate experiments made in triplicate. (D) Pulmonary vascular permeability of 125I-MPO depends on the albumin concentration. Lung preparations were perfused at 37°C with Krebs-albumin solutions at the indicated concentrations; MPO PS (μl·min-1·g-1 of wet lung) was determined by perfusing 125I-MPO for 10 min and removing the vascular tracer with Krebs for 6 min and removing the bound tracer with cold (12°C) acetate buffer (pH 4.5) for 3 min. 125I-MPO PS increased 2-fold on increasing [BSA] from 0.05 to 0.4 g/100 ml. The results are shown as mean ± SEM of four separate experiments made in triplicate.
Albumin Induces Transendothelial Transport of 125I-MPO. We next measured the effects of albumin on the transendothelial transport of 125I-MPO. The appearance of 125I-MPO in the abluminal chamber was measured after adding the tracer in the luminal chamber. Transendothelial 125I-MPO permeability increased >2-fold in the presence of albumin, as compared with γ-globulin (Fig. 2B). Addition of the cholesterol-binding agent methyl-β-cyclodextrin (CD; 5.0 mM for 20 min) prevented the transport of 125I-MPO in the presence of albumin (Fig. 2C).
Albumin Induces Pulmonary Vascular Permeability of MPO. To assess vessel wall MPO permeability, we addressed the role of albumin in inducing MPO transport in lung microvessels by using the isolated-perfused rat lung model (see Materials and Methods). Pulmonary vascular 125I-MPO permeability as determined by 125I-MPO PS depended on the albumin concentration (Fig. 2D). Albumin concentrations of 0.05 and 0.1 g/100 ml produced 2- to 3-fold increases in the 125I-MPO PS. However, MPO PS did not increase further at higher BSA concentrations (0.25 and 0.4 g/100 ml), indicating a saturable effect of albumin on MPO transport.
Albumin-Induced Transcellular MPO Transport Occurs by Means of Caveolae. Because albumin is transported across endothelial barrier by a caveolae-dependent pathway (24–27), we addressed the possible role of caveolae as the carriers responsible for MPO transport. BLMVECs were incubated with medium containing either 25 nM purified MPO or 25 nM purified MPO plus 1 mg/ml BSA and Alexa 594-BSA tracer for 30 min at 37°C (see Materials and Methods). High-resolution confocal images (<1.0-μm optical thickness; pinhole set to achieve 1 Airy unit) were obtained. In the absence of albumin, MPO staining was observed only on the cell surface (Fig. 3A Left), whereas in the presence of albumin, MPO staining was markedly colocalized with albumin-containing vesicles (Fig. 3A Right). To identify the MPO-labeled caveolae, we carried out an MPO-uptake study in BLMVECs in the presence of the caveolae marker, Alexa 488-CTB (green), and BSA (1 mg/ml). After 30 min of incubation at 37°C, cells were washed, fixed, permeabilized, and stained with anti-MPO-Ab and secondary Ab (red). Fig. 3B shows that the internalized MPO colocalized with CTB-labeled vesicles, indicating that caveolae are the primary carriers of MPO transport. To address whether MPO and CTB were internalized together in caveolae in the presence of albumin, z-section images (0.4 μm) of immunostained MPO and CTB-Alexa 488 uptake were acquired. As shown in the x–y orthogonal view of the z stack of images in Fig. 3C, MPO was colocalized with CTB on incubation with albumin (Left), but MPO staining did not colocalize with CTB-positive vesicles in the absence of albumin (Right). The y–z single line and projection images show distinctly that MPO is internalized in caveolae. In another experiment to address whether caveolae are required for MPO transport, we pretreated BLMVECs with 5.0 mM CD for 20 min at 37°C and measured albumin-induced MPO uptake. CD treatment prevented the albumin-induced MPO uptake (Fig. 3D).
Fig. 3.

(A) Albumin induces the endocytosis of MPO in endothelial cells. BLMVECs grown on glass coverslips were incubated with buffer C for 12 h. Cells were then incubated with buffer C containing 25 nM MPO alone or in combination with Alexa 594 BSA (50 μg/ml) and unlabeled BSA (1 mg/ml) for 30 min at 37°C. Cells were washed three times, fixed with 4% paraformaldehyde in HBSS for 30 min at 22°C, and blocked with 5% goat serum in HBBS containing 0.1% Triton X-100 (blocking buffer) for 30 min at 22°C. After washing, the cells were incubated with anti-MPO Ab diluted (1:1,000) in blocking buffer at 4°C overnight. After washing two times, cells were incubated with Alexa 488-labeled (green) secondary Ab in blocking buffer for 60 min at 4°C. Confocal images were obtained as described in Materials and Methods. In the absence of BSA, anti-MPO Ab staining (green) was observed at the cell surface (Left). In the presence of BSA, anti-MPO Ab staining (green) was seen in vesicles containing the Alexa 594 BSA (red). Note the marked colocalization of MPO (green) and Alexa 594 BSA (red) in the merged image (yellow, Right). (B) Albumin induces the colocalization of MPO with CTB in endothelial cells. BLMVECs were grown as described in A. Cells were incubated with Alexa 488 CTB; (20 μg/ml), MPO (25 nM), and BSA (1 mg/ml) in buffer C for 30 min at 37°C. After this, cells were washed, fixed, permeabilized, and stained with anti-MPO Ab and Alexa A594-labeled secondary antibody. Cells were visualized by confocal microscopy after mounting. Confocal images of CTB (green, Left) and anti-MPO Ab staining (red, Center) in vesicles are shown. Overlay of MPO staining and Alexa 488 CTB uptake shows marked colocalization in the merged image (Right). (C) Vesicular staining pattern and colocalization of the albumin–MPO complex with CTB. High-resolution z stacks of images combined with orthogonal and projection view image displays were used to colocalize MPO internalized in the presence of albumin with the endocytosed CTB (see B). Note the marked merging in vesicles in the y–z projection image (Left) and single-frame (x–y) or single-line (y–z and x–z) images (Left Center) observed in the presence of albumin compared to the green CTB-positive/MPO-negative vesicles observed in the absence albumin (Right Center and Right). (D) Cyclodextrin prevents albumin-induced endocytosis of MPO. BLMVECs were grown as described in A. Cells were incubated with 5 mM CD in buffer C for 20 min at 37°C. Cells were then incubated with 25 nM MPO plus BSA (1 mg/ml) in buffer C for 30 min at 37°C. After this incubation, cells were stained with anti-MPO Ab as described above. Confocal images were obtained at the midplane of the cell to visualize the internalized vesicles. CD treatment prevented the endocytosis of MPO in BLMVECs. (E) Internalized MPO colocalizes with caveolin 1 in endothelial cells. BLMVECs were grown and incubated with serum-free medium as described in A. Cells were incubated with 25 nM MPO in the presence and absence of BSA (1 mg/ml) for 30 min at 37°C. The cells were then fixed and stained with rabbit anti-MPO Ab and anti-caveolin-1 mAb (1 μg/ml) overnight at 4°C. After washing, the cells were incubated with goat anti-rabbit Alexa 594- or anti-mouse Alexa 488-labeled secondary Ab for 1 h at 22°C. Confocal images were acquired by using identical settings. Note the increase in MPO uptake in the presence of albumin and the colocalization of the internalized MPO with caveolin 1.
We also determined whether the internalized MPO colocalized with caveolin 1, a specific protein marker of caveolae. BLMVECs were incubated at 37°C for 30 min in medium containing 25 nM MPO in the presence BSA (5 mg/ml),and the cells were stained with anti-MPO Ab and anti-caveolin 1 mAb. MPO staining was only seen to colocalize with caveolin 1 in the presence of albumin (Fig. 3E).
Discussion
In the present study, we purified MPO from human lung tissue by using HSA-affinity column and demonstrated the specific interactions of MPO with HSA. We observed that the positive charge on MPO-HC residues 425–454 was required for the MPO binding to HSA. Because albumin is transported across the endothelial barrier by caveolae-derived vesicles after binding of albumin to cell surface ABPs such as gp60 (24–27), we addressed whether MPO binding with albumin was also required for the transport of MPO. We observed that MPO interaction with albumin induced the transcytosis of MPO by caveolae in endothelial cells.
Serum albumin, a monomeric protein containing 17 disulfide bridges, consists of three homologous domains (I–III) (23). Albumin is an anionic protein at a pH of 7.4 with net charges for domains I, II, and III of -9, -8, and +2, respectively, for human albumin (23). To address whether MPO has a specific albumin-binding sequence, we compared MPO sequence with known HSA-binding sequences. Protein alignment revealed that MPO-HC (molecular mass, 60 kDa) sequence (409–454) has a high homology with the HSA-docking sequence identified in bacterial proteins (29). We showed that HSA binds with high affinity to the MPO-HC peptide sequence 425–454 (MPO-WT peptide). HSA–MPO-binding ability was abolished by altering the charge on this sequence by substituting G for R and K. The albumin-binding domain in bacterial proteins showed binding only to HSA and failed to interact with BSA (29). However, we observed that both BSA and rat serum albumin bound to the MPO-WT-peptide sequence as effectively as HSA (data not shown). It is possible that the sequence ELKSLNPRWDGE in MPO-WT peptide (Fig. 1C) contributed to the binding of the multiple albumin forms (29). The EARKIV motif is conserved in MPO-HC and bacterial proteins containing the albumin-binding domain (29). To address the importance of this motif in HSA binding, we performed 125I-HSA binding to the EARKIV peptide. HSA did not bind to the EARKIV, suggesting that the flanking sequence is critical for the MPO interaction with albumin.
Albumin with plasma concentration ranging from 400 to 675 μM constitutes >60% of plasma protein in human blood (23). Albumin at very low concentrations (≈0.05 g/100 ml) maintains the endothelial barrier integrity by interactions with cell surface and extracellular matrix components (23). Studies have shown that caveolae-mediated vesicular transport of albumin is the primary mode of transendothelial albumin permeability (24–27). The ABPs localized in caveolae play an important role in the mechanism of transcellular transport of albumin in endothelial cells (24–27) and intact microvessels (26). Signaling by Src tyrosine kinase phosphorylation is critical in the mechanism of transcellular transport of albumin and fluid-phase solutes carried with albumin in vesicles (24).
The release of MPO from PMNs catalyzes the conversion of H2O2 to HOCl, a potent antibacterial agent (1). MPO transport across the endothelial cell barrier and its accumulation in the subendothelium is crucial in oxidative events because MPO is an enzymatic source of NO-derived oxidants and nitrotyrosine formation (2–7). Recent studies showed that MPO levels in plasma provide a useful measure of the severity of acute coronary syndromes (12, 13). MPO levels in the plasma increased (ranging from pM to nM) in patients with different inflammatory pathologies (13–15). Because MPO and MPO peptides interact with albumin, we tested the postulate that this interaction is important in the mechanism of MPO transport across the endothelial barrier. We observed that 125I-MPO binding to the endothelial cell surface increased in the presence of albumin, and albumin induced the transendothelial transport of MPO. MPO transport resulting from its interaction with albumin depended on the albumin concentration and was saturable. These findings are consistent with the transport of albumin occurring through a nonhydraulic transcellular pathway that requires albumin binding to endothelial cell surface ABPs such as gp60 (24–27). Thus, our results indicate that the albumin–MPO interaction induces MPO transport by a transcellular pathway dependent on albumin binding to endothelia cells. Because plasma albumin concentration is 2,000- to 5,000-fold greater than MPO, the generation of MPO and its interaction with plasma albumin is likely to be the dominant means of MPO transport across the endothelial barrier.
Caveolae are the non-clathrin-coated pits in endothelial cells responsible for transcytosis (31). To address the role of caveolae in the albumin-induced MPO transport, we disturbed the organization of caveolae by treating endothelial cells with methyl-β-CD and measured 125I-MPO permeability. CD prevented the albumin-induced increase in transendothelial 125I-MPO permeability. Further, we observed that, in the absence of albumin, MPO localized at the cell surface; however, in the presence of albumin, MPO was rapidly internalized and colocalized with the albumin-containing vesicles. We also showed that in the presence of albumin, the internalized MPO colocalized with CTB, indicating that caveolae-mediated endocytosis of albumin induces the uptake and transport of MPO. We observed that MPO and caveolin 1 [the structural protein of caveolae (31)] colocalized in endothelial cells after incubation with albumin; however, little MPO was colocalized with caveolin 1 in the absence of albumin. Thus, our results suggest a previously unreported model of MPO transport across the endothelial barrier (Fig. 4). Albumin interaction with the ABPs such as gp60 localized in caveolae induces vesicle trafficking across the endothelium (24–27). Because MPO interacts with albumin by a specific binding domain, albumin enables the transcytosis of MPO by caveolae. The accumulation of MPO in the subendothelial space, as regulated by specific MPO interaction with albumin, may promote extracellular matrix remodeling by generating NO-derived reactive species and nitrotyrosine formation and thereby interfere with endothelial integrity.
Fig. 4.

Albumin-induced transcytosis of MPO in endothelial cells. Binding of positively charged domain of MPO to albumin in plasma promotes MPO–albumin interaction and binding to endothelial plasmalemma in caveolae by ABP (e.g., gp60). Albumin plays an essential role in the mechanism of MPO transcytosis.
Acknowledgments
This work was supported by National Institutes of Health Grants P01HL60678 and GM58531.
Abbreviations: MPO, myeloperoxidase; HC, heavy chain; HSA, human serum albumin; MALDI, matrix-assisted laser desorption/ionization; BLMVEC, bovine lung microvascular endothelial cell; ABP, albumin-binding protein; HBSS, Hanks' balanced salt solution; PMN, polymorphonuclear neutrophil; CTB, cholera toxin subunit B; CD, cyclodextrin; PS, permeability × surface area.
References
- 1.Winterbourn, C. C., Vissers, M. C. & Kettle, A. J. (2000) Curr. Opin. Hematol. 7, 53-58. [DOI] [PubMed] [Google Scholar]
- 2.Abu-Soud, H. M. & Hazen, S. L. (2000) J. Biol. Chem. 275, 37524-37532. [DOI] [PubMed] [Google Scholar]
- 3.Leeuwenburgh, C., Hardy, M. M., Hazen, S. L., Wagner, P., Oh-ishi, S., Steinbrecher, U. P. & Heinecke, J. W. (1997) J. Biol. Chem. 272, 1433-1436. [DOI] [PubMed] [Google Scholar]
- 4.Shishehbor, M. H., Aviles, R. J., Brennan, M. L., Fu, X., Goormastic, M., Pearce, M. S., Gokce, N., Keaney, J. F., Jr., Penn, M. S., Sprecher, D. L., et al. (2003) J. Am. Med. Assoc. 289, 1675-1680. [DOI] [PubMed] [Google Scholar]
- 5.Lamb, N. J., Gutteridge, J. M., Baker, C., Evans, T. W. & Quinlan, G. J. (1999) Crit. Care Med. 27, 1738-1744. [DOI] [PubMed] [Google Scholar]
- 6.Van Der Vliet, A., Nguyen, M. N., Shigenaga, M. K., Eiserich, J. P., Marelich, G. P. & Cross, C. E. (2000) Am. J. Physiol. 279, L537-L546. [DOI] [PubMed] [Google Scholar]
- 7.Brennan, M. L., Wu, W., Fu, X., Shen, Z., Song, W., Frost, H., Vadseth, C., Narine, L., Lenkiewicz, E., Borchers, M. T., et al. (2002) J. Biol. Chem. 277, 17415-17427. [DOI] [PubMed] [Google Scholar]
- 8.Anderson, E., Hellman, L., Gullberg, U. & Olssson, I. (1998) J. Biol. Chem. 273, 4747-4753. [DOI] [PubMed] [Google Scholar]
- 9.Tate, RM. & Repine, J. E. (1983) Am. Rev. Respir. Dis. 128, 552-559. [DOI] [PubMed] [Google Scholar]
- 10.Harlan, J. M. (1987) Acta Med. Scand. Suppl. 715, 123-129. [DOI] [PubMed] [Google Scholar]
- 11.Malik, A. B. & Lo, S. K. (1996) Pharmacol. Rev. 48, 213-229. [PubMed] [Google Scholar]
- 12.Daugherty, A., Dunn, J. L., Rateri, D. L. & Heinecke, J. W. (1994) J. Clin. Invest. 94, 437-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brennan, M. L., Penn, M. S., Lente, F. V., Nambi, V., Shishehbor, M. H., Aviles, R. J., Goormastic, M., Pepoy, M. L., McErlean, E. S., Topol, E. J., et al. (2003) N. Engl. J. Med. 349, 1595-1604. [DOI] [PubMed] [Google Scholar]
- 14.Baldus, S., Heeschen, C., Meinertz, T., Zeiher, A. M., Eiserich, J. P., Munzel, T., Simoons, M. L. & Hamm, C. W., on behalf of the CAPTURE Investigators (2003) Circulation 108, 1440-1445. [DOI] [PubMed] [Google Scholar]
- 15.Zhang, R., Brennan, M. L., Fu, X., Aviles, R. J., Pearce, G. L., Penn, M. S., Topol, E. J., Sprecher, D. L. & Hazen, S. L. (2001) J. Am. Med. Assoc. 286, 2136-2142. [DOI] [PubMed] [Google Scholar]
- 16.Heller, J. I., Crowley, J. R., Hazen, S. L., Salvay, D. M., Wagner, P., Pennathur, S. & Heinecker, J. W. (2000) J. Biol. Chem. 275, 9957-9962. [DOI] [PubMed] [Google Scholar]
- 17.Podrez, E. A., Schmitt, D., Hoff, H. F. & Hazen, S. L. (1999) J. Clin. Invest. 103, 1547-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beckman, J. S. & Koppenol, W. H. (1996) Am. J. Physiol. 271, C1424-C1437. [DOI] [PubMed] [Google Scholar]
- 19.Radi, R., Denicola, A. & Freeman, B. A. (1999) Methods Enzymol. 301, 353-367. [DOI] [PubMed] [Google Scholar]
- 20.Eiserich, J. P., Baldus, S., Brennan, M. L., Ma, W., Zhang, C., Tousson, A., Castro, L., Lusis, A. J., Nauseef, W. M., White, C. R. & Freeman B. A. (2002) Science 296, 2391-2394. [DOI] [PubMed] [Google Scholar]
- 21.Baldus, S., Eiserich, J. P., Brennan, M. L., Jackson, R. M., Alexander, C. B. & Freeman, B. A. (2002) Free Radical Biol. Med. 33, 1010-1019. [DOI] [PubMed] [Google Scholar]
- 22.Baldus, S., Eiserich, J. P., Mani, A., Casro, L., Figueroa, M., Chumley, P., Ma, W., Tousson, A., White, C. R., Bullard, D. C., et al. (2001) J. Clin. Invest. 108, 1759-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peters, T. (1996) All About Albumin: Biochemistry, Genetics, and Medical Applications (Academic, San Diego), pp. 1-413.
- 24.Tiruppathi, C., Song, W., Bergenfeldt, M., Sass, P. & Malik, A. B. (1997) J. Biol. Chem. 272, 25968-25975. [DOI] [PubMed] [Google Scholar]
- 25.Minshall, R. D., Tiruppathi, C., Vogel, S. M., Niles, W. D., Gilchrist, A., Hamm, H. E. & Malik, A. B. (2000) J. Cell Biol. 150, 1057-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vogel, S. M., Minshall, R. D., Pilipovic, M., Tiruppathi, C. & Malik, A. B. (2001) Am. J. Physiol. 281, L1512-L1522. [DOI] [PubMed] [Google Scholar]
- 27.John, T. A., Vogel, S. M., Tiruppathi, C., Malik, A. B. & Minshall, R. D. (2003) Am. J. Physiol. 284, L187-L196. [DOI] [PubMed] [Google Scholar]
- 28.Karas, M. & Hillenkamp, F. (1988) Anal. Chem. 60, 2299-2301. [DOI] [PubMed] [Google Scholar]
- 29.Retnoningrum, D. S. & Cleary, P. P. (1994) Infect. Immun. 62, 2387-2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tiruppathi, C., Finnegan, A. & Malik, A. B. (1996) Proc. Natl. Acad. Sci. USA 93, 250-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carver, L. A. & Schnitzer, J. E. (2003) Nat. Rev. Cancer. 3, 571-581. [DOI] [PubMed] [Google Scholar]