

Abstract
In this work, we investigate the dynamic motions of fatty acid binding protein 4 (FABP4) in the absence and presence of a ligand by explicitly solvated all-atom molecular dynamics simulations. The dynamics of one ligand-free FABP4 and four ligand-bound FABP4s is compared via multiple 1.2 μs simulations. In our simulations, the protein interconverts between the open and closed states. Ligand-free FABP4 prefers the closed state, whereas ligand binding induces a conformational transition to the open state. Coupled with opening and closing of FABP4, the ligand adopts distinct binding modes, which are identified and compared with crystal structures. The concerted dynamics of protein and ligand suggests that there may exist multiple FABP4–ligand binding conformations. Thus, this work provides details about how ligand binding affects the conformational preference of FABP4 and how ligand binding is coupled with a conformational change of FABP4 at an atomic level.
Fatty acid binding protein (FABP), a family of proteins that reversibly bind with fatty acids and other lipids with high affinity, is indispensable for intracellular transport, storage, and metabolism of lipids.1 They also play a central role in lipid-mediated biological processes and metabolic and immune response pathways.2 In spite of diverse sequence similarity (22–73%), the whole FABP family shares a very similar tertiary structure consisting of 10 antiparallel β-strands organized into two nearly orthogonal β-sheet and two α-helices linking the first two β-strands (Figure 1A).3 The binding site for ligands is buried in an interior cavity and surrounded by β-strands.
Figure 1.

Crystal structures of FABP4. (A) Solvated FABP4. Sodium (Na+) is shown as a purple sphere. Chlorine (Cl–) is shown as a green sphere. Waters are shown as lines, and oxygen is red. (B) Closed conformation of FABP4 (PDB entry 2QM9). (C) Open conformation of FABP4 (PDB entry 3HK1). Carbon atoms of the ligands are white, carbon atoms of the protein are green, oxygen is red, nitrogen is blue, and sulfur is yellow.
Among nine FABP family members, FABP4 (also known as adipocyte FABP, AFABP, or aP2) has been recognized as a potential target for the treatment of type 2 diabetes, atherosclerosis, and ovarian cancer.4,5 FABP4 delivers hydrophobic ligands from the cytoplasm to the nucleus, and directly channels them to peroxisome proliferator activated receptor gamma (PPARγ), thereby regulating its transcriptional activity.6,7 Thus, opening and closing of the binding cavity are vital for the shuttling function of FABP4, which controls access and egress of ligands and water molecules. On the basis of crystallography and mutation studies, it has been proposed that a ligand enters and exits the binding cavity through the portal region consisting of helix αII and loops between βC−βD and βE−βF (Figure 1).8−13 The kinetics study of fatty acid binding to different FABPs indicates that the rate-limiting step in the binding process is the entry or release of ligand through the portal.14 Phe57, located at the mouth of the portal, has long been recognized as the key residue that serves as the gate keeper in FABP4 opening/closing. In other FABPs, the residue homologous to Phe57 of FABP4 also plays the same role.15 Although FABP4 can bind a variety of compounds with high affinity,16 only some of them can activate the nuclear translocation of FABP4.6 Comparison of different FABP4–ligand crystal structures suggests that FABP4 activation coincides with closure of the portal region.17
Previously, molecular dynamics (MD) simulations have been performed to investigate the structure and dynamics of FABP4 and other FABPs.18−25 However, the conformational transition between the open and closed forms has not been investigated, which is directly related with ligand binding and activity. In addition, such conformational changes need to be considered in the development of potent FABP4 inhibitors. Dynamic information about conformational changes is hardly obtained from static structures. Additionally, incorporation of protein dynamics may be helpful in computer-aided discovery of novel FABP4 inhibitors. To explicitly account for protein flexibility in virtual screening, multiple protein conformations have been employed for improving docking accuracy,26−28 which has been reviewed.29 The performance of protein structures extracted from MD simulations in virtual screening has been assessed.30 Moreover, a recent study indicates that sampling multiple binding modes of the ligand is required to correctly predict binding affinities.31 Therefore, sampling binding-relevant conformations by MD simulations is helpful for accurately calculating binding affinities and developing potent FABP4 inhibitors, which may be used in therapy of associated disorders such as diabetes and cancers.
MD simulations on the microsecond time scale have been recently employed for studies of protein–ligand interactions.32−36 Long MD simulations that directly describe the dynamics of protein–ligand complexes are expected to provide mechanistic insight with atomic detail.37 Here, we present multiple 1.2 μs MD simulations of FABP4 in the absence and presence of a ligand. Dynamics of five FABP4 structures, one apo form and four holo forms with distinct ligands, is compared. For each structure, two independent 1.2 μs all-atom MD simulations with explicit solvent have been carried out, and, in total, 12 μs MD trajectories are analyzed. Backbone dynamics of FABP4 in its apo and holo forms are examined and compared with experimental results. The effect of ligand binding on the opening and closing of FABP4 is investigated. In our simulations, opening and closing of the portal region are repeatedly observed, and ligand binding induces a population redistribution of FABP4 conformations. We also find that the opening/closing events are coupled with movement of the ligand. The coupling effect of protein dynamics and ligand dynamics is consistent with recent crystallography and NMR studies,38−40 which indicate that different protein conformations prefer distinct binding orientations of the ligand. To our knowledge, this is the first report of the concerted dynamics of the protein and its ligand through MD simulations. Thus, our work provides details about how ligand binding affects the conformational preference of the protein and how ligand binding is coupled with the conformational change of the protein at an atomic level.
Materials and Methods
FABP4 and Ligands
The PDB entries used in this study are 1ALB (apo-FABP4),411ADL (FABP4–ACD),422ANS (FABP4–ANS),432QM9 (FABP4–TGZ),17 and 3HK1 (FABP4–AOB).441ALB is the apo form of FABP4 and adopts the closed conformation. 1ADL is the complex form of FABP4 and arachidonic acid (ACD). 2ANS is the complex form of FABP4 and 2-anilino-8-naphthalenesulfonate (ANS). 2QM9 is the bound state of FABP4 and troglitazone (TGZ). 3HK1 is the bound state of FABP4 and 4-((2-(methoxycarbonyl)-5-(2-thienyl)-3-thienyl)amino)-4-oxo-2-butenoic acid (AOB). The four ligands (Figure 2) were extracted from the PDB files and geometry-optimized using Jaguar v7.9 with the B3LYP functional and the 6-31G* basis set. An electrostatic potential (ESP) for each ligand was generated by Jaguar. The atomic RESP charges were then determined by fitting with the RESP procedure implemented in Antechamber.
Figure 2.

Chemical structures of four FABP4 ligands: ACD, arachidonic acid (PDB entry 1ADL); ANS, 2-anilino-8-naphthalenesulfonate (PDB entry 2ANS); TGZ, troglitazone (PDB entry 2QM9); and AOB, 4-((2-(methoxycarbonyl)-5-(2-thienyl)-3-thienyl)amino)-4-oxo-2-butenoic acid (PDB entry 3HK1).
Molecular Dynamics Simulations
All MD simulations were carried out with Amber11.45 The equations of motion were solved with the leapfrog integration algorithm with a time step of 2 fs. The lengths of all bonds involving hydrogen atoms were kept constrained with the SHAKE algorithm. The particle mesh Ewald (PME) method was applied for treating long-range electrostatic interactions. Periodic boundary conditions were used in all simulations. A random seed was generated based on the current date and time for every run to assign initial velocities.
The protein was modeled using the Amber ff03 force field,46 and the ligands were modeled using the general Amber force field (GAFF).47 The starting structure was explicitly solvated in a rectangular box of TIP3P water molecules with a minimal distance of 10 Å from the protein/complex to the edges of the box. Chloride ions were added to neutralize uncompensated charges, and further salt (NaCl) was added to represent 0.15 M ionic concentration and thus to mimic the physiological environment. After the whole system was set up, a series of energy minimizations and equilibrations was performed. First, the water molecules, hydrogen atoms, and salt ions were subjected to 3000 steps of steepest descent minimization followed by 12 000 steps of conjugate gradient minimization, whereas other heavy atoms were constrained with the harmonic force of 2 kcal mol–1 Å–2. Next, the whole system was energy-minimized with 20 000 steps of the L-BFGS algorithm without any harmonic restraint. Then, coupled to a Langevin thermostat, the system was heated from 10 K to 300 K by increments of 100 K in 20 ps and continued to run for 40 ps at 300 K at constant volume. Finally, the system was equilibrated for 200 ps in the NPT ensemble with the Langevin thermostat and isotropic position scaling, at 300 K and 1 bar. The production run for each protein/complex was carried out for 1.2 μs in the NVT ensemble with the Langevin thermostat at 300 K using the parallel CUDA version of PMEMD on 2 GPUs. The trajectories were sampled at a time interval of 10 ps. All simulations were performed on our Linux cluster with 2 GPUs and 12 CPUs on each node. The analysis of trajectories was performed using PTRAJ. Time evolution of system energy and temperature during MD simulations is plotted in the Supporting Information (Figure S1).
Free Energy Surface (FES)
The calculation of the FES is given by Boltzmann weighting the free energy values
where W0 is the minimum of the FES, P is the probability distribution, kB is the Boltzmann constant, and T is the temperature. For 1D FES, r is the distance between the center of mass (COM) of Thr29 and Phe57, describing the opening and closing of FABP4. The distribution function is calculated with the histogram analysis method HIST implemented in R v3.0. The bin width is set to 0.1 Å using the Freedman–Diaconis algorithm. The optimal bin width is determined on the basis of Figure S2. For 2D FES, r = (r1, r2). r1 is the distance between the COM of Thr29 and Phe57. r2 is the distance between the COM of Cys117 and the ligand, describing the movement of the ligand in the binding cavity. The 2D distribution function is computed using 2D histogram analysis method HIST2D implemented in the package of GPLOTS with a bin area of 0.3 × 0.3 Å2. The effect of bin area on FES is shown in Figures S3–S6. For each FABP4 structure, the aggregate simulation time is 2.4 μs, and 240 000 snapshot structures are included in the FES calculation. The convergence of 1D free energy surfaces is presented in the Supporting Information (Figure S7), which is calculated over the distance of Thr29–Phe57/Cys117–ligand at an interval of 200 ns of simulation time. The error on the calculation of free energy (1D and 2D FES) is estimated by the variation observed in the last 200 ns MD simulations. On the basis of the analysis in Figures S8 and S9, the error is estimated to be 0.6 kcal mol–1.
Results and Discussion
Structures of FABP4 and Ligands
Currently, more than 30 crystal structures of FABP4 have been deposited in Protein Data Bank. Among these structures, two distinct FABP4 forms are available, in which Phe57, located at the entrance of the binding cavity, adopts completely different conformations. In the closed form (Figure 1B), Phe57 points inwardly and blocks access to the cavity inside. In the open form (Figure 1C), Phe57 projects outwardly away from the cavity and exposes the ligand to the solvent.
To investigate the effect of ligand binding on the conformational preference of the protein, four FABP4–ligand complexes with distinct compounds have been selected (Figure 2). Among these ligands, ACD is a fatty acid and has a Kd value of 4.4 μM.42 The other three are different small-molecule compounds. ANS and TGZ have a similar Kd value in the nanomolar range (31.5 and 17.0 nM, respectively).17 The experimental Kd value of AOB is not available, and the Ki value is 670 nM.44 Two ligands (ACD and AOB) are bound to the open form of FABP4, and the other two (ANS and TGZ) are bound with the closed form in the crystal structures. It has been reported that ACD cannot induce FABP4 nuclear accumulation, whereas ANS and TGZ can.17 It is unknown if AOB can activate FABP4 nuclear translocation.
FABP4 Backbone Dynamics
FABP4 dynamics in the absence and presence of a ligand is evaluated by calculating the root-mean-squared deviation (RMSD) of backbone atoms (Figure 3A and Table 1). The average RMSD fluctuates between 1.7 and 2.7 Å in all simulations (Table 1). Examination of RMSD plots (Figure 3A) shows that ligand-free FABP4 varies widely during 1.2 μs simulations. The standard deviations in two trajectories of apo-FABP4 are a little higher than those of the others (Table 1). The one-sample t-test results, calculated by the T.TEST procedure implemented in R v3.0, show that the difference in the standard deviations with and without ligand is statistically significant (t = −7.90 and p-value = 0.004). After ligand binding, the standard deviation decreases 0.34 in average (95% confidence interval: 0.28–0.55). By contrast, RMSD plots for ligand-bound FABP4s (Figure 3A) are very stable during the simulation, such as for FABP4–ACD, or become very steady after a few hundred nanoseconds, such as for FABP4–TGZ. The simulation results are consistent with the experimental finding that ligand binding reduces backbone flexibility of FABPs.48−50 Moreover, we examined the atomic fluctuation of each residue by calculating the root-mean-squared fluctuation (RMSF) of backbone atoms (Figure 3B and Table 2). For ligand-free FABP4, the portal region is subject to great fluctuation (Figure 3B) owing to the flexibility of helix αII (residues 27–34), the βC−βD loop (residues 55–58), and the βE−βF loop (residues 74–78). The average RMSF of the three parts is around 3.0 Å (Table 2). This agrees well with experimental results demonstrating that the portal region is highly flexible in apo-FABPs.51−53 After ligand binding, RMSF values of helix αII, the βC−βD loop, and the βE−βF loop are all reduced (Table 2), suggesting that ligand binding stabilizes the portal region in the complex forms. Among them, the βC−βD loop slightly decreases in average RMSF values, whereas helix αII and the βE−βF loop have significant decreases (Table 2). Therefore, our results indicate that ligand binding stabilizes the backbone conformation of FABP4, largely due to restraining the mobility of the portal region, especially helix αII and the βE−βF loop.
Figure 3.

Dynamic properties of five FABP4 structures in 1.2 μs MD simulations. For each structure, two MD trajectories are presented. The upper row is denoted as trajectory 1, and the lower row is trajectory 2, in accordance with Tables 1 and 2. (A) Time evolution of backbone RMSD with respect to the crystal structure. RMSD values of sampled structures are shown in gray. The smoothed line, computed with LOESS implemented in R v3.0, is shown in black. (B) Backbone RMSF per residue. RMSF values are calculated using PTRAJ implemented in Amber 11. All backbone heavy atoms in a residue are considered.
Table 1. Mean Values of Backbone RMSD of FABP4s in MD Simulations with Standard Deviation.
| backbone
RMSD (Å) |
|||||
|---|---|---|---|---|---|
| apo-FABP4 | FABP4–ACD | FABP4–ANS | FABP4–TGZ | FABP4–AOB | |
| trajectory 1 | 2.5 ± 0.7 | 1.9 ± 0.3 | 2.5 ± 0.5 | 2.1 ± 0.4 | 1.7 ± 0.3 |
| trajectory 2 | 2.4 ± 0.8 | 2.3 ± 0.3 | 1.8 ± 0.4 | 2.7 ± 0.6 | 2.7 ± 0.5 |
Table 2. Mean Values of Backbone RMSF of the Portal Region in MD Simulations with Standard Deviationa.
| |
backbone
RMSF (Å) |
|||||
|---|---|---|---|---|---|---|
| apo-FABP4 | FABP4–ACD | FABP4–ANS | FABP4–TGZ | FABP4–AOB | ||
| trajectory 1 | Helix αII | 3.1 ± 0.6 | 1.8 ± 0.2 | 1.7 ± 0.2 | 2.3 ± 0.2 | 1.5 ± 0.2 |
| βC−βD loop | 3.0 ± 0.5 | 2.4 ± 0.3 | 1.5 ± 0.2 | 2.8 ± 0.5 | 2.6 ± 0.6 | |
| βE−βF loop | 3.7 ± 0.6 | 1.7 ± 0.2 | 2.3 ± 0.3 | 2.9 ± 0.3 | 1.9 ± 0.2 | |
| trajectory 2 | Helix αII | 3.2 ± 0.8 | 1.5 ± 0.2 | 1.7 ± 0.2 | 1.7 ± 0.2 | 1.8 ± 0.2 |
| βC−βD loop | 2.5 ± 0.3 | 2.3 ± 0.4 | 2.0 ± 0.3 | 2.7 ± 0.1 | 1.9 ± 0.2 | |
| βE−βF loop | 2.9 ± 0.3 | 1.8 ± 0.3 | 2.2 ± 0.2 | 1.8 ± 0.3 | 2.0 ± 0.2 | |
The RMSF values for helix αII, βC−βD loop, and βE−βF loop are computed with residues 27–34, 55–58, and 74–78, respectively.
Opening/Closing of FABP4
Opening/closing of the portal is controlled by relative movements of helix αII and loops between βC−βD and βE−βF. It can be well-described by the distance between the COM of Thr29 and Phe57, sitting on opposite sides of the cavity mouth. Thr29 is located in helix αII, and Phe57 is located in loop βC−βD (Figure 1B,C). In the closed form, Thr29 and Phe57 are directly in contact, and the distance between them is about 7 Å. In the open form, Phe57 points outwardly away from Thr29, and the distance between them is about 10 Å.
Time-dependence plots of the Thr29–Phe57 distance (Figure 4A) show that the protein interconverts repeatedly between the open and closed forms in all MD simulations, suggesting that FABP4 is in a dynamic equilibrium and undergoes rapid fluctuation with or without a ligand. On calculated 1D free energy surfaces (Figure 4B), a handful of energy wells are observable. For apo-FABP4, the deepest basin appears at 7.7 Å, suggesting that the closed form is preferred. A shallow basin positioned at 10.3 Å corresponds to the open form. For FABP4–ACD, one deep basin is observable at 10.3 Å with an energy barrier of 1.0 kcal mol–1 between the open and closed forms, indicating that the open form is the most populated ensemble (Figure 5). For the other three complexes (FABP4–ANS, FABP4–TGZ, and FABP4–AOB), two deep basins, corresponding to the open and closed forms, are observable, indicating that both of them are thermodynamically stable. The curves in Figure 4B are plotted with a bin width of 0.1 Å. FES curves with various bin sizes are shown in Figure S2. We find that the bin width has little effect on the 1D FES when it is between 0.1 and 0.5 Å. The energy difference between the basins is less than 1.0 kcal mol–1, suggesting that the transition between the open and closed forms may occur easily with little energy cost.
Figure 4.

Opening and closing of FABP4 in MD simulations. (A) Time evolution of the distance between Thr29 and Phe57. The distance is shown in gray. Two black lines denote distances of 7 and 10 Å, respectively. Fluctuation of Thr29–Phe57 distance in all 10 MD simulations is shown. (B) One-dimensional free energy surface. For each structure, the free energy surface is computed by combining two MD trajectories. The free energy values are calculated using the histogram analysis method implemented in R v3.0 with a bin width of 0.1 Å. The opening/closing of FABP4 is measured by the distance between the center of mass of Thr29 and Phe57. On the basis of crystal structures, the distance between them is about 10 Å in the open form, whereas in the closed form, the distance is about 7 Å.
Figure 5.
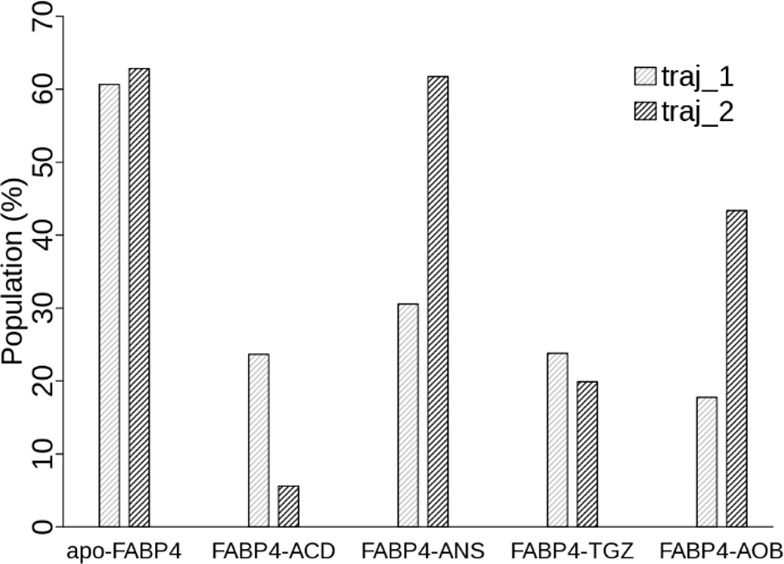
Population of the closed state in the absence and presence of a ligand. Population of the closed state is obtained by integration of the population distribution along the Thr29–Phe57 distance coordinate between 0 and 8.5 Å.
To investigate the effect of ligand binding on population distribution of FABP4 different states, we define the closed state to be all conformations with a Thr29–Phe57 distance less than 8.5 Å. Population of the closed state in each simulation (Figure 5) is computed by integration of the population distribution over the Thr29–Phe57 distance. For apo-FABP4, the average population of the closed state is 0.62. After ligand binding, the average population decreases to 0.15 (FABP4–ACD), 0.46 (FABP4–ANS), 0.22 (FABP4–TGZ), and 0.31 (FABP4–AOB). The one-sample t-test results, calculated with the T.TEST procedure implemented in R v3.0, show that the population difference before and after ligand binding is statistically significant (t = −5.01 and p-value = 0.015). After ligand binding, the population of the closed conformation decreases 0.34 in average (95% confidence interval: 0.07–0.50). Our results indicate that FABP4 shifts from the closed state to the open state upon ligand binding. Two mechanisms, induced fit and conformational selection, have been proposed to describe the conformational transition in biomolecular recognition.54 Our results support the hybrid view that the open state is already reachable in ligand-free FABP4 as a minor event and that ligand binding induces a population redistribution between the open and closed states.
Concerted Dynamics of FABP4 and Ligands
To monitor dynamics of FABP4 and ligands, we have identified a two-dimensional order parameter (r1, r2): r1 describes the distance between the COM of Thr29 and Phe57, as illustrated in the previous section; r2 describes the distance between the COM of Cys117 and the ligand, which measures the movement of ligands within FABP4. Cys117 is located at the bottom of the FABP4 binding pocket and is in direct contact with ligands in the crystal structures (Figure 1B,C). Therefore, the distance between Cys117 and the ligand indicates how far the ligand moves toward the portal.
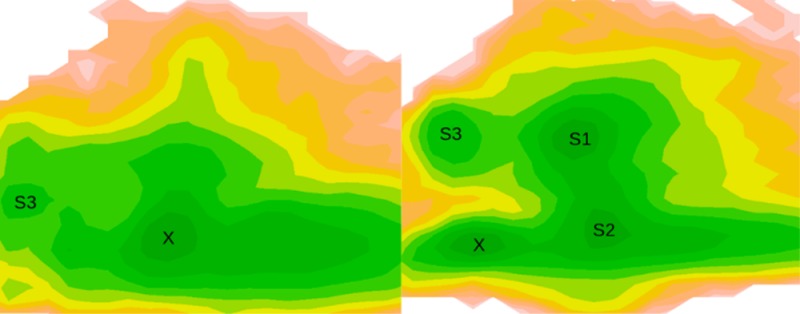
With the order parameters, two-dimensional FESs of FABP4–ligand complexes were calculated. On the surfaces, we found a handful of well-defined basins, suggesting multiple binding modes between the protein and ligands (Figure 6). Snapshot structures in each energy well were collected and compared. One representative structure for each basin was selected with the script of average_structure implemented in Maestro v9.3. The representative structures were then compared with crystal structures. In Figure 6, the populated ensemble X corresponds to the crystal structure. The heavy-atom RMSD of ligand in X with respect to the crystal structure is 2.0, 1.3, 0.7, and 1.0 Å, respectively, for FABP4–ACD, FABP4–ANS, FABP4–TGZ, and FABP4–AOB after alignment of protein structures (Figure 7A), indicating the successful reproduction of the binding mode that was experimentally determined.
Figure 6.

Two-dimensional free energy surfaces of four FABP4–ligand complexes. For each structure, two MD simulations are employed in calculation of the free energy surface. The free energy values are computed using 2D histogram analysis method HIST2D implemented in the package of GPLOTS with a bin area of 0.3 × 0.3 Å2. X corresponds to the X-ray crystal structure. S1 and S2 denote the two most populated basins, and S3 denotes a less populated basin.
Figure 7.

Representative structures of populated ensembles on 2D free energy surfaces. (A) Superposition of ligands in the crystal structure and state X in Figure 6. Carbons in the crystal structure are white, and carbons in X are green. (B) Representative structure of S3 for the FABP4–ACD complex. In S3, FABP4 adopts the closed form and ACD stretches out of the binding cavity from the aperture enclosed by Val80 and Trp97. (C) Representative structures of S1 and S2 for FABP4–ANS. (D) Representative structures of S1 and S2 for FABP4–TGZ. (E) Representative structures of S1 and S2 for FABP4–AOB. Carbons of ligands are green, carbons of residues are cyan, oxygen is red, nitrogen is blue, and sulfur is yellow.
Moreover, other binding modes are observable, which have not been solved experimentally. For the FABP4–ACD complex, a shallow basin is located at S3 (Figure 6). Unlike in the crystal structure, in S3 FABP4 adopts the closed conformation and ACD is near the portal. As shown in Figure 7B, ACD stretches out of the binding cavity from the orifice enclosed by loops βE−βF and βG−βH. It does not interact with Arg106 and Cys117, which are conserved in the crystal structures, but makes contacts with Phe57, Val80 on strand βF, and Trp97 on loop βG−βH. For the FABP4–ANS complex, two most populated ensembles are observable: S1 is in the closed state and S2 is in the open state (Figures 6 and 7C). In S1, the sulfonate group of ANS moves close to strands βC/βD, and the interaction with the triad of Arg106/Arg126/Tyr128 is lost. In S2, ANS makes a more hydrophobic interaction with helix αII, and the sulfonate moiety is exposed to water because of the opening of the portal. For the FABP4–TGZ complex, two most populated basins (S1 and S2) are available (Figures 6 and 7D). Both of them adopt the open conformation. In S1, TGZ protrudes from the binding cavity through the aperture formed by rotation of Phe57. TGZ does not interact with Arg106, Cys117, or Tyr128, but it is in contact with Phe57, even if Phe57 points outwardly. In contrast, S2 displays a similar binding mode with that from the crystal structure, and TGZ is in contact with Arg106, C117, R126, and Y128, not Phe57. For the FABP4–AOB complex, two most populated ensembles are observable with distinct protein conformations (Figures 6 and 7E). S1 adopts the closed conformation, and AOB moves close to helix αII. In S2, AOB sticks out of the binding cavity through the open portal and does not interact with Cys117.
Coupled with opening and closing of FABP4, the ligand may exist in two different binding modes: close to the bottom of the cavity or close to the portal. When the ligand stays close to the bottom, it forms polar contacts with Arg106 and Arg126 and extensive hydrophobic contacts with the binding cavity including Phe16, Cys117, and Tyr128. When the ligand moves close to the portal, most of the above contacts are lost except for Phe16. The interaction between the ligand and Phe16 is highly conserved and appears in all populated conformations. In contrast, the interaction between the ligand and Arg106/Cys117 completely disappears when the ligand is close to the portal. As the gate keeper, Phe57 is always in contact with the ligand when FABP4 is closed, and this interaction can also be observed in FABP4–TGZ (S1) and FABP4–AOB (S2) complexes when the ligand protrudes from the open portal. Our results are well-supported by experimental data. Mutation of R126L/Y128F does not affect the binding of oleic acid to FABP4, although the interaction between ligands and the reactive triad of Arg106/Arg126/Tyr128 is highly conserved in crystal structures, suggesting that there may exist an alternative binding mode.10,55 In the study of the FABP4–ANS reaction, two ligand-dependent relaxation times are available from the stopped-flow data, suggesting that ANS may have two binding sites.43
Interestingly, a high energy barrier (about 3.5 kcal mol–1) is observable between state X and S3 for FABP4–TGZ (Figure 6). The energetically favorable pathway for the FABP4–TGZ transition (X → S2 → S1 → S3) is coupled with FABP4 opening and closing. This suggests that the transition between different states is energetically unfavorable when FABP4 is closed. In contrast, for FABP4–ANS and FABP4–AOB, the energy barrier is about 1.0 kcal mol–1 between different states. This difference may be caused by the size of ligands. The volume of ANS, TGZ, and AOB, calculated within Maestro v9.3, is 230.0, 343.0, and 241.0 Å3, respectively. TGZ is much bigger than the other two ligands. Although FABP4 has a large binding cavity, there may be not enough room left for TGZ to move freely when the protein is closed.
An Alternative Opening Site
Although significant evidence supports the hypothesis that ligands enter and exit the binding cavity of FABP4 through the portal region, a computer simulation21 suggests that there may exist another entrance near the N-terminus. In our simulations, a new opening site is observable besides the portal region. Opening of the cavity allows direct access of the ligand to the solvent, providing a possible channel for the ligand to leave the internal cavity. In MD simulations of FABP4–ACD, an opening between loops βE−βF and βG−βH is observed in the less populated ensemble S3. As shown in Figure 7B, helix αII and loops between βC−βD and βE−βF move together, and the portal is fully closed. This movement leaves a gap enclosed by Val80 on strand βF and Trp97 on the βG−βH loop. The carboxyl group of ACD sticks out of the binding cavity through this orifice, suggesting another possible route for release of ligands. However, this opening is not observed in simulations of the other three complexes. On the contrary, TGZ and AOB both protrude from the binding cavity through the portal (S1 in Figure 7D and S2 in Figure 7E). The aperture between loops βE−βF and βG−βH is measured by the distance between Val80 and Trp97. In the open state, the distance between Val80 and Trp97 is about 6 Å. It is sufficient for water molecules to pass through, but it may not be for bulky ligands such as TGZ and AOB.
Conclusions
In this work, we compared dynamic properties of ligand-free and ligand-bound FABP4s. Although the open and closed conformation was observed for all FABP4s in our simulations, the population of the closed form decreased after ligand binding, suggesting that ligand binding stabilizes the open state. Furthermore, we investigated the concerted dynamics of FABP4–ligand complexes and found that, coupled with opening and closing of FABP4, the ligand could adopt distinct binding modes. Although our findings have not been directly verified in experiments, a series of experimental data suggests that there may exist different binding orientations.
Mounting evidence31,38−40 suggests that the protein and ligand may adopt multiple binding modes due to the inherent flexibility of the protein and ligand. A simple inspection of FABP4 ligands shows that three ligands, ANS, TGZ, and AOB, who have a Kd or Ki value in the nanomolar range, adopt more binding modes than does ACD, which has a Kd value in the micromolar range. It is still not clear how these different binding conformations make contributions to binding affinities. Understanding of the interaction between FABP4 and inhibitors will be helpful in developing potent FABP4 inhibitors for cancer therapy.
Glossary
Abbreviations
- ACD
arachidonic acid
- ANS
2-anilino-8-naphthalenesulfonate
- TGZ
troglitazone
- AOB
4-((2-(methoxycarbonyl)-5-(2-thienyl)-3-thienyl)amino)-4-oxo-2-butenoic acid
- FABP4
fatty acid binding protein 4
Supporting Information Available
Additional information on the MD simulations and the convergence of free energy calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ Y.L. and X.L. contributed equally to this work. Y.L. and X.L. performed the experiments and analyzed the data. Y.L., X.L., and Z.D. wrote the manuscript.
This work was supported by The Hormel Foundation and National Institutes of Health grant nos. CA172457, CA166011, and R37 CA081064.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zimmerman A. W.; Veerkamp J. H. (2002) New insights into the structure and function of fatty acid-binding proteins. Cell. Mol. Life Sci. 59, 1096–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M.; Hotamisligil G. S. (2008) Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discovery 7, 489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch J.; McDermott L. (2009) Structural and functional analysis of fatty acid-binding proteins. J. Lipid Res. 50, S126–S131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M.; Tuncman G.; Gorgun C. Z.; Makowski L.; Atsumi G.; Vaillancourt E.; Kono K.; Babaev V. R.; Fazio S.; Linton M. F.; Sulsky R.; Robl J. A.; Parker R. A.; Hotamisligil G. S. (2007) Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature 447, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieman K. M.; Kenny H. A.; Penicka C. V.; Ladanyi A.; Buell-Gutbrod R.; Zillhardt M. R.; Romero I. L.; Carey M. S.; Mills G. B.; Hotamisligil G. S.; Yamada S. D.; Peter M. E.; Gwin K.; Lengyel E. (2011) Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 17, 1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan N. S.; Shaw N. S.; Vinckenbosch N.; Liu P.; Yasmin R.; Desvergne B.; Wahli W.; Noy N. (2002) Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol. Cell. Biol. 22, 5114–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers S. D.; Nedrow K. L.; Gillilan R. E.; Noy N. (2007) Continuous nucleocytoplasmic shuttling underlies transcriptional activation of PPAR gamma by FABP4. Biochemistry 46, 6744–6752. [DOI] [PubMed] [Google Scholar]
- Sacchettini J. C.; Scapin G.; Gopaul D.; Gordon J. I. (1992) Refinement of the structure of Escherichia coli-derived rat intestinal fatty acid binding protein with bound oleate to 1.75-Å resolution. Correlation with the structures of the apoprotein and the protein with bound palmitate. J. Biol. Chem. 267, 23534–23545. [PubMed] [Google Scholar]
- Xu Z. H.; Bernlohr D. A.; Banaszak L. J. (1993) The adipocyte lipid-binding protein at 1.6-Å resolution. Crystal structures of the apoprotein and with bound saturated and unsaturated fatty acids. J. Biol. Chem. 268, 7874–7884. [PubMed] [Google Scholar]
- Ory J.; Kane C. D.; Simpson M. A.; Banaszak L. J.; Bernlohr D. A. (1997) Biochemical and crystallographic analyses of a portal mutant of the adipocyte lipid-binding protein. J. Biol. Chem. 272, 9793–9801. [DOI] [PubMed] [Google Scholar]
- Simpson M. A.; Bernlohr D. A. (1998) Analysis of a series of phenylalanine 57 mutants of the adipocyte lipid-binding protein. Biochemistry 37, 10980–10986. [DOI] [PubMed] [Google Scholar]
- Richieri G. V.; Ogata R. T.; Kleinfeld A. M. (1999) Fatty acid interactions with native and mutant fatty acid binding proteins. Mol. Cell. Biochem. 192, 77–85. [PubMed] [Google Scholar]
- Jenkins A. E.; Hockenberry J. A.; Nguyen T.; Bernlohr D. A. (2002) Testing of the portal hypothesis: analysis of a V32G, F57G, K58G mutant of the fatty acid binding protein of the murine adipocyte. Biochemistry 41, 2022–2027. [DOI] [PubMed] [Google Scholar]
- Ogata R. T. (1996) Kinetics of fatty acid interactions with fatty acid binding proteins from adipocyte, heart, and intestine. J. Biol. Chem. 271, 11291–11300. [DOI] [PubMed] [Google Scholar]
- Hanhoff T.; Lücke C.; Spener F. (2002) Insights into binding of fatty acids by fatty acid binding proteins. Mol. Cell. Biochem. 239, 45–54. [PubMed] [Google Scholar]
- Richieri G. V.; Ogata R. T.; Zimmerman A. W.; Veerkamp J. H.; Kleinfeld A. M. (2000) Fatty acid binding proteins from different tissues show distinct patterns of fatty acid interactions. Biochemistry 39, 7197–7204. [DOI] [PubMed] [Google Scholar]
- Gillilan R. E.; Ayers S. D.; Noy N. (2007) Structural basis for activation of fatty acid-binding protein 4. J. Mol. Biol. 372, 1246–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich M. R.; Evans J. S. (1996) Molecular dynamics simulations of adipocyte lipid-binding protein: effect of electrostatics and acyl chain unsaturation. Biochemistry 35, 1506–1515. [DOI] [PubMed] [Google Scholar]
- Woolf T. B. (1998) Simulations of fatty acid-binding proteins suggest sites important for function. I. Stearic acid. Biophys. J. 74, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf T. B.; Tychko M. (1998) Simulations of fatty acid-binding proteins. II. Sites for discrimination of monounsaturated ligands. Biophys. J. 74, 694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman R.; Nachliel E.; Gutman M. (2005) Molecular dynamics simulations of the adipocyte lipid binding protein reveal a novel entry site for the ligand. Biochemistry 44, 4275–4283. [DOI] [PubMed] [Google Scholar]
- Friedman R.; Nachliel E.; Gutman M. (2006) Fatty acid binding proteins: same structure but different binding mechanisms? Molecular dynamics simulations of intestinal fatty acid binding protein. Biophys. J. 90, 1535–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsfadia Y.; Friedman R.; Kadmon J.; Selzer A.; Nachliel E.; Gutman M. (2007) Molecular dynamics simulations of palmitate entry into the hydrophobic pocket of the fatty acid binding protein. FEBS Lett. 581, 1243–1247. [DOI] [PubMed] [Google Scholar]
- Levin L. B.-A.; Nachliel E.; Gutman M.; Tsfadia Y. (2009) Molecular dynamics study of the interaction between fatty acid binding proteins with palmitate mini-micelles. Mol. Cell. Biochem. 326, 29–33. [DOI] [PubMed] [Google Scholar]
- Long D.; Mu Y.; Yang D. (2009) Molecular dynamics simulation of ligand dissociation from liver fatty acid binding protein. PLoS One 4, e6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Kim D. J.; Ma W. Y.; Lubet R. A.; Bode A. M.; Dong Z. G. (2011) Discovery of novel checkpoint kinase 1 inhibitors by virtual screening based on multiple crystal structures. J. Chem. Inf. Model. 51, 2904–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahasenan K. V.; Li C. L. (2012) Novel inhibitor discovery through virtual screening against multiple protein conformations generated via ligand-directed modeling: a maternal embryonic leucine zipper kinase example. J. Chem. Inf. Model. 52, 1345–1355. [DOI] [PubMed] [Google Scholar]
- Rueda M.; Totrov M.; Abagyan R. (2012) ALiBERO: evolving a team of complementary pocket conformations rather than a single leader. J. Chem. Inf. Model. 52, 2705–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuriev E.; Ramsland P. A. (2013) Latest developments in molecular docking: 2010–2011 in review. J. Mol. Recognit. 26, 215–239. [DOI] [PubMed] [Google Scholar]
- Nichols S. E.; Baron R.; Ivetac A.; McCammon J. A. (2011) Predictive power of molecular dynamics receptor structures in virtual screening. J. Chem. Inf. Model. 51, 1439–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. L.; Deng Y. Q.; Knight J. L.; Wu Y. J.; Kim B.; Sherman W.; Shelley J. C.; Lin T.; Abel R. (2013) Modeling local structural rearrangements using FEP/REST: application to relative binding affinity predictions of CDK2 inhibitors. J. Chem. Theory Comput. 9, 1282–1293. [DOI] [PubMed] [Google Scholar]
- Du Y.; Yang H. Y.; Xu Y. C.; Cang X. H.; Luo C.; Mao Y. Y.; Wang Y. Y.; Qin G. R.; Luo X. M.; Jiang H. L. (2012) Conformational transition and energy landscape of ErbB4 activated by neuregulin1 beta: one microsecond molecular dynamics simulations. J. Am. Chem. Soc. 134, 6720–6731. [DOI] [PubMed] [Google Scholar]
- Lee J. Y.; Lyman E. (2012) Agonist dynamics and conformational selection during microsecond simulations of the A2A adenosine receptor. Biophys. J. 102, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai A.; Delarue P.; Senet P. (2013) Conformational dynamics of full-length inducible human Hsp70 derived from microsecond molecular dynamics simulations in explicit solvent. J. Biomol. Struct. Dyn. 31, 1111–1126. [DOI] [PubMed] [Google Scholar]
- Yuan S. G.; Wu R. L.; Latek D.; Trzaskowski B.; Filipek S. (2013) Lipid receptor S1P1 activation scheme concluded from microsecond all-atom molecular dynamics simulations. PLoS Comput. Biol. 9, e1003261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe J. I.; El-Nahal W. G.; Shirts M. R. (2014) Investigating the mutation resistance of nonnucleoside inhibitors of HIV-RT using multiple microsecond atomistic simulations. Proteins 82, 130–144. [DOI] [PubMed] [Google Scholar]
- Klepeis J. L.; Lindorff-Larsen K.; Dror R. O.; Shaw D. E. (2009) Long-timescale molecular dynamics simulations of protein structure and function. Curr. Opin. Struct. Biol. 19, 120–127. [DOI] [PubMed] [Google Scholar]
- Bruning J. B.; Parent A. A.; Gil G.; Zhao M.; Nowak J.; Pace M. C.; Smith C. L.; Afonine P. V.; Adams P. D.; Katzenellenbogen J. A.; Nettles K. W. (2010) Coupling of receptor conformation and ligand orientation determine graded activity. Nat. Chem. Biol. 6, 837–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T. S.; Chalmers M. J.; Novick S.; Kuruvilla D. S.; Chang M. R.; Kamenecka T. M.; Rance M.; Johnson B. A.; Burris T. P.; Griffin P. R.; Kojetin D. J. (2012) Ligand and receptor dynamics contribute to the mechanism of graded PPAR gamma agonism. Structure 20, 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S.; Nwachukwu J. C.; Parent A. A.; Cavett V.; Nowak J.; Hughes T. S.; Kojetin D. J.; Katzenellenbogen J. A.; Nettles K. W. (2013) Ligand-binding dynamics rewire cellular signaling via estrogen receptor-alpha. Nat. Chem. Biol. 9, 326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z. H.; Bernlohr D. A.; Banaszak L. J. (1992) Crystal structure of recombinant murine adipocyte lipid-binding protein. Biochemistry 31, 3484–3492. [DOI] [PubMed] [Google Scholar]
- Lalonde J. M.; Levenson M. A.; Roe J. J.; Bernlohr D. A.; Banaszak L. J. (1994) Adipocyte lipid-binding protein complexed with arachidonic-acid. Titration calorimetry and X-ray crystallographic studies. J. Biol. Chem. 269, 25339–25347. [DOI] [PubMed] [Google Scholar]
- Ory J. J.; Banaszak L. J. (1999) Studies of the ligand binding reaction of adipocyte lipid binding protein using the fluorescent probe 1,8-anilinonaphthalene-8-sulfonate. Biophys. J. 77, 1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzel A. V.; Hellberg K.; Reynolds J. M.; Kruse A. C.; Juhlmann B. E.; Smith A. J.; Sanders M. A.; Ohlendorf D. H.; Suttles J.; Bernlohr D. A. (2009) Identification and characterization of a small molecule inhibitor of fatty acid binding proteins. J. Med. Chem. 52, 6024–6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Cheatham T. E.; Darden T.; Gohlke H.; Luo R.; Merz K. M.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y.; Wu C.; Chowdhury S.; Lee M. C.; Xiong G. M.; Zhang W.; Yang R.; Cieplak P.; Luo R.; Lee T.; Caldwell J.; Wang J. M.; Kollman P. (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012. [DOI] [PubMed] [Google Scholar]
- Wang J. M.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Hodsdon M. E.; Cistola D. P. (1997) Ligand binding alters the backbone mobility of intestinal fatty acid-binding protein as monitored by 15N NMR relaxation and 1H exchange. Biochemistry 36, 2278–2290. [DOI] [PubMed] [Google Scholar]
- Lu J. Y.; Lin C. L.; Tang C. G.; Ponder J. W.; Kao J. L. F.; Cistola D. P.; Li E. (2000) Binding of retinol induces changes in rat cellular retinol-binding protein II conformation and backbone dynamics. J. Mol. Biol. 300, 619–632. [DOI] [PubMed] [Google Scholar]
- Franzoni L.; Lucke C.; Perez C.; Cavazzini D.; Rademacher M.; Ludwig C.; Spisni A.; Rossi G. L.; Ruterjans H. (2002) Structure and backbone dynamics of apo- and holo-cellular retinol-binding protein in solution. J. Biol. Chem. 277, 21983–21997. [DOI] [PubMed] [Google Scholar]
- Constantine K. L.; Friedrichs M. S.; Wittekind M.; Jamil H.; Chu C. H.; Parker R. A.; Goldfarb V.; Mueller L.; Farmer B. T. (1998) Backbone and side chain dynamics of uncomplexed human adipocyte and muscle fatty acid-binding proteins. Biochemistry 37, 7965–7980. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Gonzalez L. H.; Ludwig C.; Hohoff C.; Rademacher M.; Hanhoff T.; Ruterjans H.; Spener F.; Lucke C. (2002) Solution structure and backbone dynamics of human epidermal-type fatty acid-binding protein (E-FABP). Biochem. J. 364, 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Yang X.; Wang H.; Estephan R.; Francis F.; Kodukula S.; Storch J.; Stark R. E. (2007) Solution-state molecular structure of apo and oleate-liganded liver fatty acid-binding protein. Biochemistry 46, 12543–12556. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; Nussinov R.; Wright P. E. (2009) The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha R. S.; Kane C. D.; Xu Z. H.; Banaszak L. J.; Bernlohr D. A. (1993) Modulation of ligand-binding affinity of the adipocyte lipid-binding protein by selective mutation. Analysis in vitro and in situ. J. Biol. Chem. 268, 7885–7892. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.