

Abstract
Density functional tight binding (DFTB), which is ∼100–1000 times faster than full density functional theory (DFT), has been used to simulate the structure and properties of protic ionic liquid (IL) ions, clusters of ions and the bulk liquid. Proton affinities for a wide range of IL cations and anions determined using DFTB generally reproduce G3B3 values to within 5–10 kcal/mol. The structures and thermodynamic stabilities of n-alkyl ammonium nitrate clusters (up to 450 quantum chemical atoms) predicted with DFTB are in excellent agreement with those determined using DFT. The IL bulk structure simulated using DFTB with periodic boundary conditions is in excellent agreement with published neutron diffraction data.
1. Introduction
Ionic liquids (ILs) are liquids that are comprised entirely of ions, differentiated from typical ionic salts by having melting points below 100 °C.1 IL melting points are low because electrostatic interactions between component ions are weaker and crystal lattice packing is hindered. This is typically achieved by making at least one of the ions large, unsymmetrical and organic. The physicochemical properties of ILs can be tuned through a judicious choice of ions.2−5 This “flexibility” has driven wide-ranging research into their use as solvents in “green” chemistry,1,6 energy,7 electrochemical applications,8,9 pharmaceuticals,10−12 and lubricants.13−15
Protic ILs are formed by proton transfer from a Brønsted acid and a Brønsted base. While molecular liquids like water are structurally homogeneous, many ILs are nanostructured in the bulk16−24 and at interfaces.14,25−27 Nanostructure in protic ILs is usually spongelike.3,28−30 IL nanostructure is driven by the amphiphilicity of (usually) the cation, with short-range forces driving the formation of longer-range self-assembled structures. IL nanostructure is a consequence of the solvophobic effect;31 strong electrostatic attractions between IL charged groups lead to the assembly of charged (polar) domains. Cation alkyl chains are solvophobically excluded from these charged regions and cluster together to form uncharged (apolar) domains.16,32 Many IL properties can be correlated with nanostructure, and the short-range interactions within it, such as hydrogen bonding.33 To fully exploit IL nanostructure for control of physicochemical properties an atomistic understanding of the interactions between IL ions is required. While this cannot currently be achieved using experiments, it can be using classical molecular dynamics,34ab initio molecular dynamics (AIMD),35 as well as static quantum chemical calculations.36,37
IL simulations generally fall into two categories that balance accuracy and system size. Accurate quantum chemical simulations yield a detailed picture of IL structure at the atomic scale, but are limited in large-scale applications by their computational expense. This means that fundamental aspects of real IL systems, such as self-assembled nanostructure,38−45 are not captured. One possible approach to circumventing this problem is the use of continuum solvation models,46 but this method is complicated by the heterogeneous liquid nanostructure. Noncontinuum solvation methods based on the reference interaction site model (RISM)47,48 are a possible alternative, but an accurate description of the solvating “bulk” region is still required; this becomes particularly problematic in the case of conformationally flexible ions. Conversely, large-scale model IL systems consisting of hundreds of ion pairs can be studied using classical potentials,3,35,49−54 though such potentials often lead to reduced accuracy and are invariably IL-specific. If molecular dynamics (MD) is employed, the time scale of the simulation is another parameter limiting accuracy, and this often necessitates smaller systems.
Density functional tight binding (DFTB) is a fast quantum chemical method with great potential for IL simulations. Density functional tight binding has accuracy comparable to density functional theory (DFT), but is 100–1000 times faster. Potentially, therefore, DFTB enables both molecular and nanoscale structures to be captured simultaneously, with quantum chemical accuracy. However, the performance of DFTB in predicting structure and properties of protic ILs has not yet been assessed.
In this work, we compare proton affinities (PAs) computed with DFTB against DFT and electron correlated ab initio calculations, and DFTB-predicted bulk structure against neutron-diffraction data.3 Protic ILs are synthesized by proton transfer from a Brønsted acid to base. Therefore, PAs of component ions provide the best elementary test of the accuracy of a method. We demonstrate that DFTB predicts accurate PA values for a wide range of prototypical IL cations and anions (see Figures 1 and 2),55 along with the bulk IL structure. These results indicate that DFTB is an ideal method for efficiently and accurately predicting properties and structure for protic ILs in general, over both molecular-scale and nanoscale domains.
Figure 1.

Protic IL cations considered in this work: (1–3) primary, secondary, tertiary n-alkyl amines, (4) ethanolammonium, (5,6) mono, 1,2-dimethyl imidizolium (MIm, 1,2-DMIm), (7) 1,1,3,3-tetramethylguanidine, and (8) caprolactam.
Figure 2.

Protic IL anions considered in this work: (9) dimethylamide, (10) dicyanamide (dca), (11) tricyanomethanide (tcm), (12) acetate, (13) methoxy, (14) isobutane, and (15) nitrate.
2. Computational Details
2.1. Density Functional Tight Binding
DFTB is an extended Hückel-like method, parametrized with DFT (see refs (56 and 57) for recent reviews), and consists of electronic and repulsive terms. The DFTB method assumes the Foulkes-Haydock ansatz; the electron density ρ is treated as a reference density ρ0 plus a small perturbation (i.e., ρ = ρ0 + Δρ), where ρ0 is computed using DFT. The exchange-correlation potential can thus be expanded in a Taylor series around some reference density ρ0. The DFTB energy can be written as
| 1 |
where the first and second terms are the electronic and repulsive terms, respectively. The third term accounts for charge transfer between atoms A and B (γAB is a distance-dependent function of the chemical hardness of A and B, and Δq is the difference between an atom’s charge and its charge arising from the reference density ρ0), and the fourth term accounts for how each atom’s contribution to the reference density ρ0 relaxes in the presence of every other atom (ΓAB describes how the chemical hardness (γAB) changes as a function of the chemical environment due to atoms A and B). The DFTB3 method is the most recent extension to the original DFTB method pioneered by Fraunheim and Seifert,58,59 and includes all terms up to and including third-order.60 The inclusion of third-order effects in the exchange-correlation potential expansion enables an accurate treatment of hydrogen bonding60,61 and so is particularly important in the context of ILs. In this work, we will consider both the self-consistent charge DFTB method (i.e., the second order expansion in eq 1, DFTB2) and full DFTB3.
All DFTB calculations presented in this work were performed with the DFTB+ program.62 As a demonstration of DFTB’s transferability, we rely on the mio-0-163 and 3ob-1-1 sets,64 both of which are already available, and do not develop any new “purpose-built” parameters in this work. Many previous investigations have demonstrated the importance of dispersion forces in IL structure and properties.65−67 Therefore, all DFTB calculations include dispersion forces calculated using a Slater–Kirkwood polarizable atom model;68 however, dispersion is anticipated to be significant only for large cluster and bulk calculations.
2.2. Proton Affinities
For each of the ions in Figures 1 and 2, PAs calculated with DFTB2 and DFTB3 were compared with those obtained using DFT and second-order Møller–Plesset perturbation theory (MP2). Density functional theory (DFT) and MP2 calculations were performed in conjunction with the 6-31G(d) and 6-311G(2d,2p) Pople basis sets, in order to assess basis set effects. Addition of diffuse functions (+ and ++) to these basis sets was also investigated. For DFT calculations, the PBE,69 B3LYP,70,71 and M06-2X72 DFT functionals were employed. The PBE functional was included because of its use in the parametrization of the electronic term in eq 1, B3LYP was chosen as a hybrid functional and also because of its popularity, and M06-2X was included because, in several previous ILs, it accurately reproduces properties and structure.54,73
The proton affinity is defined as the negative enthalpy for the reaction A– + H+ → AH in the gas phase. The PA is thus computed via
| 2 |
For all DFTB2 and DFTB3 calculations, EH+ values of 141.90 and 151.04 kcal/mol, respectively, were employed.60 For all DFT and MP2 calculations, PAs are based solely on the electronic potential energy and zero-point energy corrections and excluded thermal contributions. DFT, MP2, and DFTB PAs are compared with those determined using the G3B3 method.74−76 All PA calculations reported here (except DFTB) were performed with Gaussian09.77
2.3. Bulk and Cluster Nanostructure
The Kick3 stochastic generation algorithm54,78 was used to determine low-energy structures of ethyl-, propyl-, and butyl-ammonium nitrate (EAN, PAN, and BAN) clusters and the bulk liquid. The power of the Kick3 approach, when it is combined with DFTB3, is that it can be used to quickly and reliably elucidate complex molecular structure in conformationally flexible systems. Clusters consisting of 2–10, 15, and 20 ion pairs were generated, and all ethyl, propyl, and butyl chains were given full conformational flexibility. Between 1000 and 2000 starting structures were generated for cluster sizes of 2–10 ion pairs, whereas, for the larger clusters (15 or 20 ion pairs), 5000 starting structures were generated. This typically afforded ca. 500 “unique” structures for each system. Uniqueness is defined using energetic and geometric criteria. If the DFTB3 energy of two structures differed by less than 1 mEh, their geometries were compared by computing the centroid of each ion and then computing the root mean square deviation (RMSD) of cation–cation, anion–anion, and cation–anion intercentroid distances. A relatively loose threshold of 1.0 Å is applied to each RMSD, since it is expected that the potential energy surface (PES) will be comparatively flat, particularly for larger clusters. The energetically higher of the two structures is discarded as a duplicate only if all three geometric criteria fall below this threshold.
Each starting structure was optimized using DFTB3-D in conjunction with the mio-0–1 parameter set. Hubbard derivatives were the standard values published by Gaus et al.60 To ensure that DFTB3-D provides reliable thermodynamic trends, single-point energies of each unique structure were calculated using M06-2X/6-311G(d,p) at the optimized DFTB3-D geometry. For smaller clusters (fewer than 6 ion pairs), DFTB3-D structures were reoptimized with M06-2X/6-311G(d,p). Harmonic vibrational frequencies were also calculated with M06-2X/6-311G(d,p) to ensure they correspond to a local minimum on the PES. This is consistent with the approach employed in our recent investigation of imidazolium-based IL clusters.
For the bulk structure of EAN, PAN, and BAN, this Kick3 prescreening process was essentially repeated in the presence of cubic periodic boundary conditions. Cubic supercells of 1.58 nm × 1.58 nm × 1.58 nm (i.e., 4 nm3) were filled with 24 (EAN) or 20 (PAN, BAN) ion pairs, yielding densities of 1.077 g/cm3 (EAN), 1.013 g/cm3 (PAN), and 1.129 g/cm3 (BAN). These values correspond approximately to experimental densities.55,79,80 The absence of interatomic correlations at distances larger than 0.9 nm in previously published neutron diffraction data indicates that the box used here is sufficiently large to capture IL nanostructure.3,53 Following the Kick3 optimization of each bulk liquid (performed at 0 K), molecular dynamics (MD) with a time step of 1 fs was used to calculate partial gij(r) distribution functions for each liquid. An NVT ensemble at 298 K was enforced via a Nosé–Hoover chain thermostat (chain length = 3, coupling strength = 500 cm–1).81,82 Each liquid was first equilibrated for 50 ps. Partial gij(r) distribution functions were then sampled from a subsequent 50 ps period of simulation. All periodic calculations used 1 × 1 × 1 Monckhorst-Pack sampling (i.e., at the Γ-point only). Partial gij(r) distribution functions for EAN and PAN determined using DFTB3-D are compared to experimental data.3
3. Results and Discussion
3.1. Proton Affinities in Protic Ionic Liquids
The most elementary test of any first-principles method in the present context is the prediction of PA; a method incapable of yielding accurate PAs is unlikely to predict accurately larger-scale structures in protic ILs. PAs for cations 1–8 (Figure 1) calculated using DFTB2 and DFTB3 are presented in Table 1. The cation 1 PA value in Table 1 refers to n = 0 (i.e., methylammonium cation); supplementary calculations show that PAs are essentially the same for n = 1–3 (i.e., EA+, PA+, and BA+).
Table 1. Deviations in Computed Proton Affinities from G3B3 Values, for Protic IL Cations 1–8a.
| Cation
Proton Affinity (kcal/mol) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 (n = 0) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | MADb | max dev | |
| 6-31G(d) | ||||||||||
| PBE | 12.8 | 11.2 | 9.7 | –0.4 | 11.3 | 11.3 | 13.4 | 14.1 | 10.4 | 14.1 |
| B3LYP | 12.7 | 11.5 | 10.2 | –0.5 | 12.0 | 12.0 | 13.7 | 15.6 | 10.9 | 15.6 |
| M06-2X | 10.4 | 9.0 | 7.8 | –3.0 | 8.7 | 8.5 | –0.3 | 10.2 | 6.5 | 10.4 |
| MP2 | 13.8 | 13.6 | 13.4 | 0.5 | 14.7 | 14.6 | 15.7 | 17.7 | 13.0 | 17.7 |
| 6-31+G(d) | ||||||||||
| PBE | 9.2 | 8.1 | 6.9 | –4.8 | 7.3 | 7.3 | 9.2 | 9.5 | 6.6 | 9.5 |
| B3LYP | 8.3 | 7.7 | 6.9 | –5.7 | 7.1 | 7.1 | 8.7 | 9.9 | 6.2 | 9.9 |
| M06-2X | 6.9 | 6.1 | 5.2 | –7.1 | 4.9 | 4.6 | 6.0 | 5.6 | 4.0 | 6.9 |
| MP2 | 11.5 | 11.6 | 11.5 | –2.3 | 11.5 | 11.5 | 12.8 | 13.9 | 10.3 | 13.9 |
| 6-31++G(d) | ||||||||||
| PBE | 9.2 | 8.2 | 7.0 | –4.8 | 7.4 | 7.4 | 9.2 | 9.5 | 6.6 | 9.5 |
| B3LYP | 8.3 | 7.7 | 6.9 | –5.7 | 7.1 | 7.1 | 8.6 | 9.9 | 6.2 | 9.9 |
| M06-2X | 6.9 | 6.2 | 5.3 | –7.1 | 4.9 | 4.7 | 6.0 | 5.6 | 4.1 | 6.9 |
| MP2 | 11.6 | 11.8 | 11.7 | –2.3 | 11.6 | 11.6 | 12.8 | 13.9 | 10.3 | 13.9 |
| 6-311G(2d,2p) | ||||||||||
| PBE | 12.4 | 11.4 | 10.5 | –0.8 | 11.8 | 11.8 | 13.2 | 14.1 | 10.5 | 14.1 |
| B3LYP | 11.6 | 11.1 | 10.5 | –1.7 | 11.8 | 11.7 | 12.9 | 14.8 | 10.3 | 14.8 |
| M06-2X | 9.5 | 9.2 | 8.8 | –3.9 | 8.6 | 7.8 | 9.6 | 9.6 | 7.4 | 9.6 |
| MP2 | 15.8 | 16.1 | 16.6 | 2.5 | 16.4 | 16.5 | 17.2 | 19.0 | 15.0 | 19.0 |
| 6-311++G(2d,2p) | ||||||||||
| PBE | 9.7 | 9.4 | 8.9 | –3.8 | 9.6 | 9.7 | 11.0 | 11.1 | 8.2 | 11.1 |
| B3LYP | 8.5 | 8.7 | 8.6 | –5.1 | 9.3 | 9.3 | 10.4 | 11.4 | 7.7 | 11.4 |
| M06-2X | 7.0 | 7.3 | 7.3 | –6.6 | 6.5 | 6.5 | 7.6 | 6.9 | 5.3 | 7.6 |
| MP2 | 13.8 | 14.5 | 15.2 | 0.2 | 14.4 | 14.6 | 15.4 | 16.5 | 13.1 | 16.5 |
| DFTB2/mio-0-1 | –7.5 | –8.6 | –9.0 | –19.3 | 5.9 | 4.7 | 4.9 | 0.5 | 2.7 | 9.0 |
| DFTB3/mio-0-1 | –4.2 | –3.0 | –1.9 | –16.4 | 13.1 | 12.0 | 8.8 | 6.2 | 4.1 | 13.1 |
| DFTB3/3ob-1-1 | –1.9 | 1.1 | 3.9 | –13.8 | 16.0 | 14.8 | 11.1 | 9.8 | 5.6 | 16.0 |
| G3B3 | 214.2 | 221.5 | 226.1 | 227.8 | 229.3 | 234.2 | 245.5 | 214.4 | ||
Energies are given as Emethod – EG3B3.
Mean absolute deviation.
Each cation modeled has an acidic nitrogen, and therefore poses a challenge to any minimal basis set or semi-empirical method,83 because of the well-known nitrogen hybridization issue (which leads to large deprotonation errors). This is complicated further by the presence of both sp2- and sp3-hybridized nitrogen atoms in our test set. Table 1 reveals that the mean absolute deviations (MADs) of DFTB-predicted PAs from those determined using G3B3 are within ca. 5 kcal/mol. G3B3 values74 deviate from experimental values84 by, at most, 2 kcal/mol for these species. Based on previous reports,60,85 this level of accuracy is acceptable. DFTB3 is more accurate for cations 1–3, which have sp3-hybridized nitrogens (i.e., the alkyl ammonium cations), while DFTB2 provides greater accuracy for the other cations which have sp2-hybridized nitrogens.
For sp3 cations 1–3, DFTB3 is most accurate when the 3ob-1-1 parameter set is used, with maximum errors of 3.9 kcal/mol. This is surprising, because the 3ob-1-1 parameters have previously been shown to be inaccurate in modeling the sp3 nitrogen PAs,60 which prompted the development of the NHmod parameter set. However, in this study, the NHmod parameters produced deviations ca. 10 kcal/mol higher than those obtained using 3ob-1-1 for sp3 cations 1–3. The underlying cause of this is a topic of further study. However, we note that for cation 4, NHmod parameters substantially reduce the deviation in PA obtained using 3ob-1-1, from −13.8 kcal/mol to 5.0 kcal/mol. For the sp2 cations 5–8, deviations between DFTB3 and G3B3 data are as large as 16 kcal/mol. The most significant deviations occur for the MIm and 1,2-DMIm cations; we attribute this to DFTB’s treatment of charge delocalization in the imidazole ring (similar behavior for DFT has been noted previously for IL anions43).
Table 2 presents PAs for the protic IL anions presented in Figure 2. For anions with acidic oxygen (12, 13, 15), DFTB2-predicted PAs are within ca. 6 kcal/mol of the G3B3 values. PAs are generally reduced by 1–2 kcal/mol using DFTB3 in conjunction with mio-0-1 parameters, but when the 3ob-1-1 parameters are used PAs are underestimated for anions 12 and 14. The charge of the dca and tcm anions (anions 10, 11) is highly localized because of the cyano ligands. For isobutene (anion 14), DFTB2 underestimates the G3B3 PA by −18.1 kcal/mol, but third-order corrections increase accuracy substantially to within −12.8 kcal/mol. In general, however, DFTB PAs deviate from G3B3 values by as much as 25 kcal/mol, since DFTB cannot accurately treat localized charges.61 DFT suffers this shortcoming also, albeit to a lesser extent;43 deviations from G3B3 PA values are as large as 16 kcal/mol. The smallest deviations for DFT for these anions (<1 kcal/mol) occur with the smallest basis set (6-31G(d)), and this is most likely due to a fortuitous cancellation of errors. Adding a set of diffuse functions (i.e., 6-31+G(d)) increases the MAD by ca. 7 kcal/mol; however, the addition of further diffuse functions to hydrogen atoms (i.e., 6-31++G(d)) provides no further improvement. Similarly, adding diffuse functions to the larger 6-311G(2d,2p) basis set increases DFT MADs by ca. 4 kcal/mol. Conversely, for MP2, adding diffuse functions to the 6-31G(d) and 6-311G(2d,2p) basis sets decreases MAD values to 4.0 kcal/mol (6-31++G(d)) and 0.5 kcal/mol (6-311++G(2d,2p)), respectively.
Table 2. Deviations in Computed Proton Affinities from G3B3 Values, for Protic IL Anionsa.
| Anion
Proton Affinity (kcal/mol) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 9 | 10 | 11 | 12 | 13 | 14 | 15 | MADb | max dev | |
| 6-31G(d) | |||||||||
| PBE | –0.6 | 0.5 | –0.4 | 4.5 | 2.5 | 4.4 | 10.3 | 3.3 | 10.3 |
| B3LYP | –0.4 | 2.0 | 1.7 | 5.0 | 2.7 | 4.9 | 11.3 | 4.0 | 11.3 |
| M06-2X | –2.3 | 0.5 | 0.6 | 3.2 | 2.5 | 2.1 | 7.5 | 2.7 | 7.5 |
| MP2 | 7.7 | 4.1 | 2.6 | 5.5 | 7.8 | 13.4 | 1.9 | 6.1 | 13.4 |
| 6-31+G(d) | |||||||||
| PBE | –13.5 | –8.2 | –8.3 | –10.4 | –13.5 | –13.4 | –4.9 | 10.3 | 13.5 |
| B3LYP | –15.1 | –8.6 | –8.1 | –12.0 | –15.3 | –15.1 | –6.4 | 11.5 | 15.3 |
| M06-2X | –14.7 | –7.8 | –7.0 | –10.7 | –13.2 | –14.9 | –6.6 | 10.7 | 14.9 |
| MP2 | –2.6 | –2.7 | –3.1 | –5.2 | –5.5 | –0.6 | –7.8 | 3.9 | 7.8 |
| 6-31++G(d) | |||||||||
| PBE | –14.6 | –8.2 | –8.3 | –10.4 | –14.1 | –15.2 | –4.8 | 10.8 | 15.2 |
| B3LYP | –16.4 | –8.5 | –8.0 | –12.1 | –15.9 | –17.1 | –6.3 | 12.0 | 17.1 |
| M06-2X | –15.3 | –7.8 | –6.9 | –10.7 | –13.5 | –16.2 | –6.6 | 11.0 | 16.2 |
| MP2 | –2.9 | –2.7 | –3.1 | –5.2 | –5.6 | –1.0 | –7.7 | 4.0 | 7.7 |
| 6-311G(2d,2p) | |||||||||
| PBE | –4.4 | 0.1 | –1.9 | 4.1 | 1.3 | –5.2 | 14.3 | 4.5 | 14.3 |
| B3LYP | –5.0 | 0.7 | –1.0 | 3.3 | 0.4 | –5.6 | 14.1 | 4.3 | 14.1 |
| M06-2X | –5.3 | –0.7 | –1.6 | 2.5 | 1.7 | –6.9 | 10.6 | 4.2 | 10.6 |
| MP2 | 6.4 | 4.9 | 3.3 | 8.3 | 9.3 | 7.6 | 9.1 | 7.0 | 9.3 |
| 6-311++G(2d,2p) | |||||||||
| PBE | –12.7 | –6.0 | –7.1 | –5.8 | –9.9 | –15.1 | 1.4 | 8.3 | 15.1 |
| B3LYP | –14.1 | –6.3 | –7.0 | –7.6 | –11.7 | –16.6 | –0.3 | 9.1 | 16.6 |
| M06-2X | –12.7 | –6.6 | –6.5 | –6.6 | –9.1 | –15.3 | –1.2 | 8.3 | 15.3 |
| MP2 | –0.8 | –0.5 | –1.2 | 0.4 | –0.6 | –0.1 | 0.0 | 0.5 | 0.8 |
| DFTB2/mio-0-1 | –9.0 | 13.1 | 20.0 | 5.1 | –3.2 | –18.1 | 2.1 | 7.5 | 18.1 |
| DFTB3/mio-0-1 | –6.2 | 13.7 | 24.6 | 2.2 | –1.8 | –12.8 | –3.2 | 5.3 | 12.8 |
| DFTB3/3ob-1-1 | –4.4 | 14.3 | 24.9 | –3.4 | –5.6 | –9.2 | –6.1 | 5.8 | 9.2 |
| G3B3 | 412.4 | 316.6 | 308.3 | 361.4 | 400.4 | 435.0 | 329.2 | ||
Energies are given as (Emethod – EG3B3).
Mean absolute deviation.
These results show that comparable, if not greater, accuracy can be obtained systematically with DFTB than can be obtained using the PBE, B3LYP, and M06-2X DFT functionals with either small (6-31G(d)/6-31+G(d)) or large (6-311G(2d,2p)) basis sets. For anions 9–15, DFT calculations with 6-31G(d) and 6-311G(2d,2p) basis sets both produce deviations from G3B3 data that are <5 kcal/mol (however, for the small basis set, this is most likely through a fortuitous cancellation of errors, as noted above). For the cations (1–8), DFT and MP2 with all basis sets typically overestimate PA values, by up to ca. 20 kcal/mol. However, using the 6-31G(d) basis set with a single diffuse function improves accuracy by ∼5 kcal/mol. To improve this accuracy further, thermal/vibrational corrections, and corrections for basis set superposition error (particularly in the case of modest basis sets), are required. Even in small IL clusters, such corrections are computationally intractable. Thus, DFTB represents an efficient and accurate alternative for predicting PAs for both IL anions and cations.
3.2. Structure and Binding in EAN, PAN, and BAN Clusters
The performance of DFTB3 in larger systems is assessed using the n-alkyl ammonium nitrate ILs EAN, PAN, and BAN. These ILs have been chosen, as the bulk structures are known from both neutron diffraction and MD simulations.3,53 In order to establish that DFTB accurately reproduces the IL nanostructure, the ion arrangements determined by DFTB for clusters will be validated against DFT calculations. Once the cluster structure is established as correct, periodic boundary conditions will be employed to model the bulk liquid, and the structure will be compared to that determined experimentally by comparison of probability distribution functions.
The structure of DFTB ion pairs is first assessed. Previous DFT studies86 of n-alkyl ammonium nitrate ILs suggested that, for isolated ion pairs in the gas phase, an ammonium proton is transferred to the NO3– anion to form the amine and the acid. This is determined by comparison of the N–H and O–H bond lengths. Figure 3a shows the optimized N–H and O–H bond lengths for EAN, PAN, and BAN ion pairs computed using the M06-2X DFT functional and DFTB3. As per previous studies, both methods predict that the proton resides on the NO3– anion, and the O–H bond lengths are in good quantitative agreement, with deviations of <0.02 Å. There is a larger difference between the predicted N–H bond distances, ca. 0.3 Å. However, the distance between the N and H atoms (>1.5 Å) suggests that a bond is not present, accounting for the large variation in “bond” distance. The length of this hydrogen bridge using DFT is sensitive to the choice of functional, but not the basis set. For example, supplementary B3LYP/6-311G(d,p) calculations yield an N–H bond length ca. 0.07 Å larger than the M06-2X/6-311G(d,p) value. MP2/6-311G(d,p) increases this bond length by an additional 0.01 Å. Conversely, this bond length does not change using M06-2X with either a smaller basis set (6-31G(d)) or a larger basis set (aug-cc-pVTZ). Reoptimizing the DFTB/mio-0-1 structure using 3ob-1-1 parameters yields an N–H distance of 1.70 Å. This is a marked improvement over mio-0-1 data, as anticipated,64 and is in close agreement with B3LYP and MP2 results. This tendency for proton transfer is eliminated for larger clusters (2 or more ion pairs) (c.f. Figure 3), which is consistent with previous results.86 Increased competition between a greater number of hydrogen bonding donor and acceptor sites in larger clusters decreases the acidity of the ammonium group protons, preventing formation of the neutral species.
Figure 3.

DFTB3 equilibrium geometries of (a) EAN, (b) PAN, and (c) BAN monomers. N–H and O–H bond lengths (Å) are shown for DFTB3 (top number) and M06-2X/6-311G(d,p) (bottom number). (d) The most energetically stable EAN dimer structure and (e) an EAN dimer structure 25 kcal/mol higher in energy; both are shown from the top (left) and side (right) views. Structures shown are those optimized with DFTB3.
With structural accuracy established, the energy of the structures is now compared for cluster sizes between 2 and 15 ion pairs. Figure 4a–c plots the DFT and DFTB cluster energies against each other for cluster sizes of 2, 6, and 15 ion pairs for the unique structures identified using the Kick3 search for EAN. Corresponding data for PAN and BAN is presented in the Supporting Information. The closer these data fall to the line of unit gradient, the better the agreement between methods. For the three cluster sizes for EAN, PAN, and BAN, the agreement between models is excellent, except for the highest energy structures (energies >100 kcal/mol), which are not of chemical relevance. The agreement between DFTB3 and M06-2X is corroborated by the cluster binding energy, shown in Figure 4d. The cluster binding energy (BE) for n ion pairs is defined as
| 3 |
where Ecluster is the energy of the relaxed cluster, and Eanion and Ecation are the energies of the relaxed isolated anion and cation structures. For M06-2X/6-311G(d,p), Ecluster is the energy at the optimized DFTB3 structure.
Figure 4.
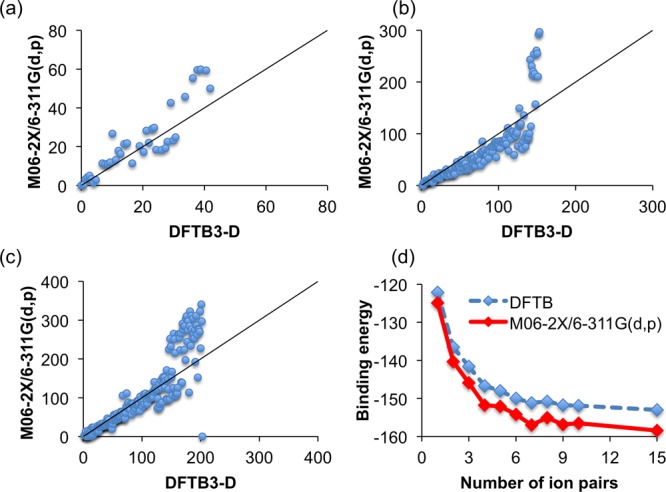
Comparison of relative energies (in kcal/mol) for (a) 2 (39 unique structures), (b) 6 (152 unique structures), and (c) 15 EAN ion-pair clusters (204 unique structures), computed using DFTB3 and M06-2X/6-311G(d,p). DFTB3 energies are fully optimized, M06-2X/6-311G(d,p) are single-point energies. (d) Binding energies (kcal/mol per ion pair) for EAN clusters, as a function of cluster size, computed using DFTB3 and M06-2X/6-311G(d,p). Corresponding data for PAN and BAN are included in the Supporting Information (Figure S1).
Figure 4d shows the BE per ion pair for EAN clusters of between 2 and 15 ion pairs. Corresponding data for PAN and BAN are presented in the Supporting Information (Figure S1). As the number of ion pairs increases, the BE and the form of the data obtained using DFT and DFTB is similar, but the BE for DFTB is ∼10 kcal/mol higher for all cluster sizes. The BE is almost constant for both methods when the cluster size is 7 ion pairs or more, thus providing a confident estimate of the bulk liquid BE per ion pair. Equivalent behavior was observed in our previous work considering imidazolium nitrate-based ILs.54 We note that two sources of error are present in these DFT values. Firstly, the values are calculated from single-point energies at the DFTB geometries and thus represent an upper bound to the value that would be obtained were the clusters to be fully optimized using DFT. Secondly, the modest basis set employed is likely to result in some degree of basis set superposition error, which is an error that, by construction, does not affect DFTB.
The binding energies in PAN and BAN also converge to values between −150 and −160 kcal/mol. Since increasing the cation alkyl chain length does not measurably influence the BE, this suggests that the BE is primarily determined by electrostatic interactions between the ammonium and nitrate charge groups. This is consistent with the predicted structures for EAN, PAN, and BAN clusters,54,86 where the cluster core is enriched in charged groups with n-alkyl chains expelled to an outer shell. The tendency for alkyl chains to be solvophobically expelled from between charged groups toward is evident even in the smallest clusters (c.f. Figures 3d and 3e).
3.3. Bulk Structure in EAN, PAN, and BAN
To model the bulk liquid structure rather than a cluster, a 1.58 nm × 1.58 nm × 1.58 nm (i.e., 4 nm3) supercell was filled with 24 ion pairs (EAN), or 20 ion pairs (PAN, BAN) to approximate the EAN, PAN, and BAN liquid densities.55 Periodic boundary conditions were employed to allow ions to move in the same way as they would in the bulk fluid, and the supercell volume and temperature were fixed via an NVT ensemble. The accuracy of the bulk structures obtained from DFTB is assessed by comparison of partial gij(r) distribution functions with published experimental (EAN and PAN)3 and simulation (BAN) data (c.f. Figures 5 and 6, respectively). The labeling scheme used to identify different atoms is shown in these figures. These DFTB3 simulations employ the mio-0-1 parameter set, and we note here that this should generally be avoided (3ob-1-1 parameters should be used instead). However, at least with respect to bulk structure in EAN, these parameter sets result in essentially identical bulk structures (see Figure S2a in the Supporting Information). All gij(r) functions for EAN are in near-perfect agreement, except for the Na–Na correlation, which loses the pronounced double-peak structure evident in Figure 5. Near perfect agreement between DFTB2-D/mio-0-1 and DFTB3-D/mio-0-1 bulk structure is also observed (Figure S2b in the Supporting Information).
Figure 5.

Comparison of partial gij(r) distribution functions for EAN bulk (left) and PAN bulk (right), computed with DFTB3-D/MD at 298 K (solid lines) and obtained via neutron diffraction3 (dotted lines). The inset shows the labeling scheme. The inset shows the C/N/O labeling scheme for PAN. The same scheme is used for EAN; note that the C3 carbon and associated H atoms are absent in EA+.
Figure 6.
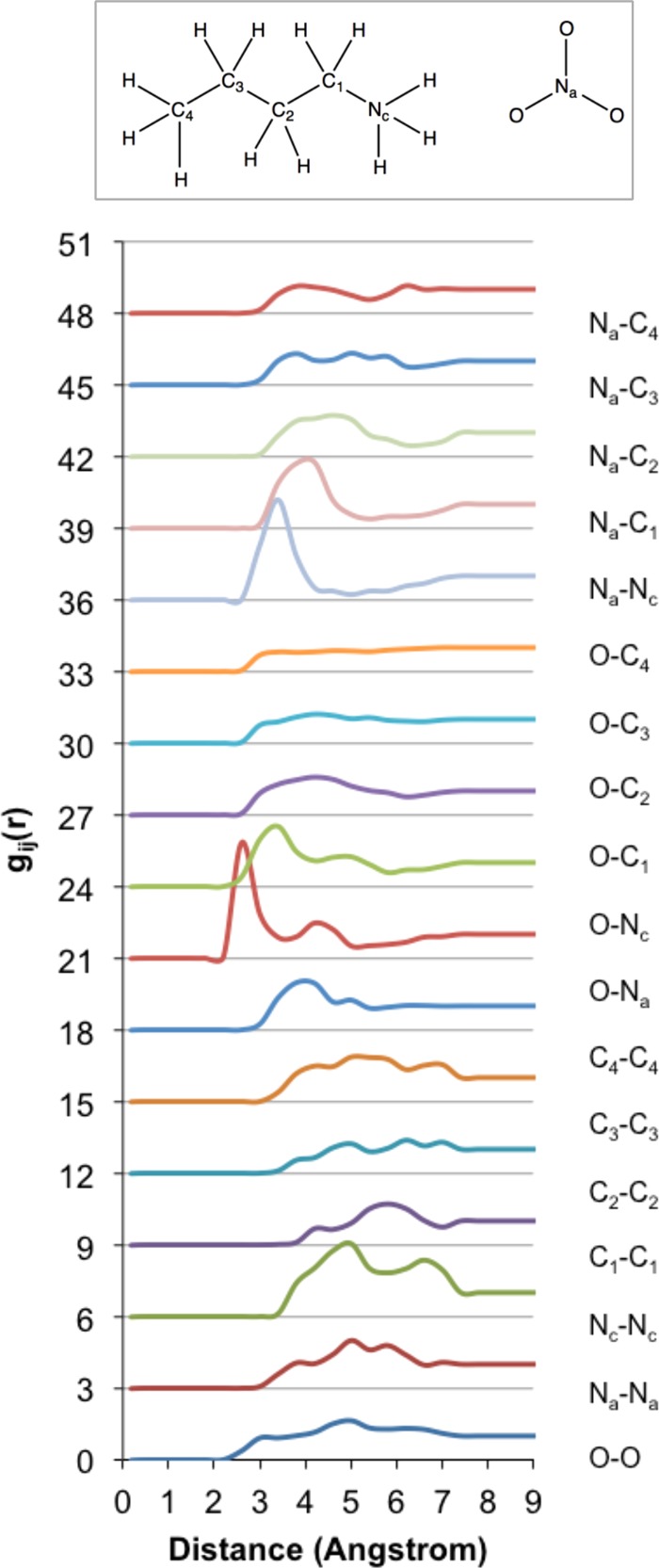
Partial gij(r) distribution functions for BAN predicted using DFTB3/MD at 298 K. The inset shows the C/N/O labeling scheme.
For EAN and PAN, agreement between the gij(r) distribution functions determined with DFTB3 and neutron diffraction is excellent; any deviations are primarily a consequence of DFTB overbinding effects,60 which tend to shift gij(r) peaks to shorter distances (usually by ∼0.5 Å). Overbinding here contracts covalent bonds in each ion, resulting in a more localized charge distribution and therefore a stronger electrostatic interaction between ions, causing the observed contraction in gij(r) peaks. The fact that simulated and experimental gij(r) distribution functions are in such good agreement here also indicates that the simulation box size is sufficient. For instance, if the box size was too small, or the simulated density too high, spurious gij(r) peaks not observed experimentally would be obtained, since ions would be forced into unphysical arrangements in the bulk liquid. This is not the case, as can be seen from Figure 5. The agreement between the gij(r) distribution functions from experiment and simulation reveals that DFTB3 reproduces the protic IL bulk sponge nanostructure, with segregated polar and apolar domains in the liquid. As such, the best way to assess the accuracy of the simulation is to examine the arrangements of charged and uncharged groups separately.
The accuracy of charge group arrangements can be assessed by examination of the Na–Nc, Nc–Nc, and Na– Nagij(r) distribution functions. The primary peak position and height in the Na–Ncgij(r) distribution function is almost perfectly reproduced by DFTB3. For the Na–Nagij(r), the agreement between the model and the experiment for the peak at short distances is good; however, in the simulation, the second peak is shifted to shorter distances. Similarly, for the Nc–Ncgij(r) distribution function peaks, both peaks are shifted to shorter distances.
The correctness of the cation alkyl chain arrangements in the nonpolar domains is determined by comparison of the arrangements C1–C1 and C2–C2gij(r) distribution function from DFTB3 and the experiment. The C1–C1gij(r) distribution function is in almost exact agreement with the experiment, but for the terminal C2gij(r) (C3 for PAN), the prominent correlation observed experimentally (3.4 Å) is underestimated by DFTB3. This is also a consequence of the overbinding effect. When charge groups associate more closely (even slightly), the nanostructure “contracts”, such that the space available for alkyl chain packing is reduced. Alkyl chains interdigitate to compensate, leading to the primary peak intensity being reduced in the C2–C2gij(r) with a concomitant increase in the intensity of the second peak.
Song et al.53 have recently reported on the bulk heterogeneity within primary n-alkyl ammonium nitrate ILs, including BAN, on the basis of X-ray diffraction and classical MD simulations. The prominent features in the simulated BAN gij(r) distribution functions are a sharp peak in the Na–Nc correlation function at 3.18 Å, broad peaks in the O–C4 and Na–C4 correlation functions at 3.7 and 3.99 Å, respectively, and a C4–C4 peak at 3.99 Å. The Nc–O correlation function features a double peak, with the first solvation shell located at 3.99 Å. The DFTB3 partial gij(r) distribution functions for BAN are presented in Figure 6, and agree well with those reported by Song et al.53 In consideration of the overbinding effect, peaks are generally shifted to shorter distances.
Experimental trends between EAN, PAN, and BAN regarding the extent of aggregation within both charged and uncharged domains are also reproduced here, using DFTB3. As the cation alkyl chain length is increased, the intensity of the correlation between alkyl chain terminal carbons increases. This is consistent with better segregation between charged and uncharged domains, attributed to stronger solvophobic interactions.
4. Conclusions
We have presented a systematic assessment of the density functional tight binding (DFTB) method for protic ionic liquids (ILs). DFTB is an exciting, but as yet underutilized, alternative method for simulating IL structure and IL-related phenomena. However, it is a quantum chemical method; its tight binding formulation enables computational speeds ca. 100–1000 times faster than conventional density functional theory (DFT) (depending on the system in question, and the number of terms included in the expansion of the exchange-correlation potential). A single DFTB3 energy and gradient calculation87 on a 15-ion-pair EAN cluster, for example, requires only 37 s. The scaling of DFTB3-D in the current context is shown explicitly in Figure S3 in the Supporting Information for EAN, which shows a near-perfect logarithmic relationship between cluster size and computation time. It is emphasized here that DFTB is also transferable—all calculations presented here have been performed with already available and general-purpose parameters. We note here that, despite recently reported DFTB3 parameters for S and P88 and DFTB2 parameters for halogens,89 third-order parameters (specifically the derivative of the Hubbard parameter, U) for halogens have not been reported to our knowledge. The application of DFTB3 to aprotic ILs including common anions, such as NTf2–, BF4–, and PF6– (for example), is currently not possible.
For ammonium nitrate ILs, DFTB is capable of predicting (1) the PAs of isolated gas-phase ions with accuracy often exceeding that achieved with both DFT and MP2 using extended basis sets, (2) the structure in IL clusters (with respect to DFT), and (3) the bulk structure (with respect to neutron diffraction data).3 As such, DFTB, and particularly DFTB3, is an efficient and accurate method for studying properties and structure in protic ILs.
Acknowledgments
This research was undertaken with the assistance of resources provided at the NCI National Facility Systems at The Australian National University and INTERSECT Systems, through the National Computational Merit Allocation Scheme, supported by the Australian Government. This work was supported by Australian Research Council Discovery Projects. R.A. acknowledges the Australian Research Council for a Future Fellowship. R.S. acknowledges financial support from the University of Newcastle.
Supporting Information Available
Comparison of DFTB and M06-2X/6-311G(d,p) binding energies for PAN and BAN clusters, up to 15 ion pairs. Comparison of radial distribution functions for EAN using DFTB2-D/mio-0-1, DFTB3-D/mio-0-1 and DFTB3-D/3ob-0-1. Computational scaling of DFTB3-D/mio-0-1 for ionic liquid clusters. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Seddon K. R. Ionic Liquids for Clean Technology. J. Chem. Technol. Biotechnol. 1997, 68, 351–356. [Google Scholar]
- Hayes R.; El Abedin S. Z.; Atkin R. Pronounced Structure in Confined Aprotic Room-Temperature Ionic Liquids. J. Phys. Chem. B 2009, 113, 7049–7052. [DOI] [PubMed] [Google Scholar]
- Hayes R.; Imberti S.; Warr G. G.; Atkin R. Pronounced sponge-like nanostructure in propylammonium nitrate. Phys. Chem. Chem. Phys. 2011, 13, 13544–13551. [DOI] [PubMed] [Google Scholar]
- Greaves T. L.; Drummond C. J. Ionic liquids as amphiphile self-assembly media. Chem. Soc. Rev. 2008, 37, 1709–1726. [DOI] [PubMed] [Google Scholar]
- Greaves T. L.; Drummond C. J. Protic Ionic Liquids: Properties and Applications. Chem. Rev. 2008, 108, 206–237. [DOI] [PubMed] [Google Scholar]
- Rodgers R. D.; Seddon K. R. Ionic Liquids—Solvents of the Future?. Science 2003, 302, 792. [DOI] [PubMed] [Google Scholar]
- Matic A.; Scrosati B. Ionic liquids for energy applications. MRS Bull. 2013, 38, 533–537. [Google Scholar]
- Chen I.-W. P.; Cottinet P.-J.; Tsai S.-Y.; Foster B.; Liang R.; Wang B.; Zhang C. Improved performance of carbon nanotube buckypaper and ionic-liquid-in-Nafion actuators for rapid response and high durability in the open air. Sens. Actuators B 2012, 171–172, 515–521. [Google Scholar]
- Terasawa N.; Ono N.; Mukai K.; Koga T.; Higashi N.; Asaka K. High performance polymer actuators based on multi-walled carbon nanotubes that surpass the performance of those containing single-walled carbon nanotubes: Effects of ionic liquid and composition. Sens. Actuators B 2012, 163, 20–28. [Google Scholar]
- Coleman D.; Spulak M.; Garcia M. T.; Gathergood N. Antimicrobial toxicity studies of ionic liquids leading to a “hit” MRSA selective antibacterial imidazolium salt. Green Chem. 2012, 14, 1350–1356. [Google Scholar]
- Ferraz R.; Branco L. C.; Marrucho I. M.; Araujo J. M. M.; Rebelo L. P. N.; da Ponte M. N.; Prudencio C.; Noronha J. P.; Petrovski Z. Development of novel ionic liquids based on ampicillin. Med. Chem. Commun. 2012, 3, 494–497. [Google Scholar]
- Lovejoy K. S.; Corley C. A.; Cope E. K.; Valentine M. C.; Leid J. G.; Purdy G. M.; Wilkes J. S.; Koppisch A. T.; Del Sesto R. E. Utilization of Metal Halide Species Ambiguity to Develop Amorphous, Stabilized Pharmaceutical Agents As Ionic Liquids. Cryst. Growth Des. 2012, 12, 5357–5364. [Google Scholar]
- Sweeney J.; Hausen F.; Hayes R.; Webber G. B.; Endres F.; Rutland M. W.; Bennewitz R.; Atkin R. Control of Nanoscale Friction on Gold in an Ionic Liquid by a Potential-Dependent Ionic Lubricant Layer. Phys. Rev. Lett. 2012, 109, 155502. [DOI] [PubMed] [Google Scholar]
- Li H.; Endres F.; Atkin R. Effect of alkyl chain length and anion species on the interfacial nanostructure of ionic liquids at the Au(111)–ionic liquid interface as a function of potential. Phys. Chem. Chem. Phys. 2013, 15, 14624–14633. [DOI] [PubMed] [Google Scholar]
- Perkin S.; Albrecht T.; Klein J. Layering and shear properties of an ionic liquid, 1-ethyl-3-methylimidazolium ethylsulfate, confined to nano-films between mica surfaces. Phys. Chem. Chem. Phys. 2010, 12, 1243–1247. [DOI] [PubMed] [Google Scholar]
- Atkin R.; Warr G. G. The Smallest Amphiphiles: Nanostructure in Protic Room-Temperature Ionic Liquids with Short Alkyl Groups. J. Phys. Chem. B 2008, 112, 4164–4166. [DOI] [PubMed] [Google Scholar]
- Hardacre C.; Holbrey J. D.; McMath J. S. E.; Bowron D. T.; Soper A. K. Structure of molten 1,3-dimethylimidazolium chloride using neutron diffraction. J. Chem. Phys. 2003, 118, 273. [Google Scholar]
- Hardacre C.; Holbrey J. D.; Mullan C. L.; Nieuwenhuyzen M.; Youngs T. G. A.; Bowron D. T. Liquid Structure of the Ionic Liquid, 1-Methyl-4-cyanopyridinium Bis{(trifluoromethyl)sulfonyl}imide Determined from Neutron Scattering and Molecular Dynamics Simulations. J. Phys. Chem. B 2008, 112, 8049–8056. [DOI] [PubMed] [Google Scholar]
- Hardacre C.; Holbrey J. D.; Nieuwenhuyzen M.; Youngs T. G. A. Structure and Solvation in Ionic Liquids. Acc. Chem. Res. 2007, 40, 1146. [DOI] [PubMed] [Google Scholar]
- Triolo A.; Russina O.; Bleif H.-J.; Di Cola E. Nanoscale Segregation in Room Temperature Ionic Liquids. J. Phys. Chem. B 2007, 111, 4641–4644. [DOI] [PubMed] [Google Scholar]
- Triolo A.; Russina O.; Fazio B.; Triolo R.; Di Cola E. Morphology of 1-alkyl-3-methylimidazolium hexafluorophosphate room temperature ionic liquids. Chem. Phys. Lett. 2008, 457, 362–365. [Google Scholar]
- Wang Y.; Jiang W.; Yan T.; Voth G. A. Understanding Ionic Liquids through Atomistic and Coarse-Grained Molecular Dynamics Simulations. Acc. Chem. Res. 2007, 40, 1193–1199. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Voth G. A. Unique Spatial Heterogeneity in Ionic Liquids. J. Am. Chem. Soc. 2005, 127, 12192. [DOI] [PubMed] [Google Scholar]
- Canongia Lopes J. N. A.; Pádua A. A. H. Nanostructural Organization in Ionic Liquids. J. Phys. Chem. B 2006, 110, 3330–3335. [DOI] [PubMed] [Google Scholar]
- Niga P.; Wakeham D.; Nelson A.; Warr G. G.; Rutland M.; Atkin R. Structure of the Ethylammonium Nitrate Surface: An X-ray Reflectivity and Vibrational Sum Frequency Spectroscopy Study. Langmuir 2010, 26, 8282–8288. [DOI] [PubMed] [Google Scholar]
- Segura J. J.; Elbourne A.; Wanless E. J.; Warr G. G.; Voitchovsky K.; Atkin R. Adsorbed and near surface structure of ionic liquids at a solid interface. Phys. Chem. Chem. Phys. 2013, 15, 3320–3328. [DOI] [PubMed] [Google Scholar]
- Elbourne A.; Sweeney J.; Webber G. B.; Wanless E. J.; Warr G. G.; Rutland M. W.; Atkin R. Adsorbed and near-surface structure of ionic liquids determines nanoscale friction. Chem. Commun. 2013, 49, 6797–6799. [DOI] [PubMed] [Google Scholar]
- Greaves T. L.; Kennedy D. F.; Kirby N.; Drummond C. J., Nanostructure changes in protic ionic liquids (PILs) through adding solutes and mixing PILs. Phys. Chem. Chem. Phys. 2011, 13, 13501–13509. [DOI] [PubMed] [Google Scholar]
- Greaves T. L.; Kennedy D. F.; Mudie S. T.; Drummond C. J. Diversity Observed in the Nanostructure of Protic Ionic Liquids. J. Phys. Chem. B 2010, 114, 10022–10031. [DOI] [PubMed] [Google Scholar]
- Greaves T. L.; Kennedy D. F.; Weerawardena A.; Tse N. M. K.; Kirby N.; Drummond C. J.. Nanostructured Protic Ionic Liquids Retain Nanoscale Features in Aqueous Solution While Precursor Bronsted Acids and Bases Exhibit Different Behavior. J. Phys. Chem. B 115, 2055–2066. [DOI] [PubMed] [Google Scholar]
- Ray A. Solvophobic Interactions and Micelle Formation in Structure Forming Nonaqueous Solvents. Nature 1971, 231, 313–315. [DOI] [PubMed] [Google Scholar]
- Hayes R.; Imberti S.; Warr G. G.; Atkin R. Amphiphilicity determines nanostructure in protic ionic liquids. Phys. Chem. Chem. Phys. 2011, 13, 3237–3247. [DOI] [PubMed] [Google Scholar]
- Hayes R.; Imberti S.; Warr G. G.; Atkin R. The Nature of Hydrogen Bonding in Protic Ionic Liquids. Angew. Chem., Int. Ed. 2013, 52, 4623–4627. [DOI] [PubMed] [Google Scholar]
- Morrow T. I.; Maginn E. J. Molecular Dynamics Study of the Ionic Liquid 1-n-Butyl-3-methylimidazolium Hexafluorophosphate. J. Phys. Chem. B 2002, 106, 12807–12813. [Google Scholar]
- Del Pópolo M. G.; Lynden-Bell R. M.; Kohanoff J. Ab Initio Molecular Dynamics Simulation of a Room Temperature Ionic Liquid. J. Phys. Chem. B 2005, 109, 5895–5902. [DOI] [PubMed] [Google Scholar]
- Izgorodina E. I.; Rigby J.; MacFarlane D. R. Large-scale ab initio calculations of archetypical ionic liquids. Chem. Commun. 2012, 48, 1493–1495. [DOI] [PubMed] [Google Scholar]
- Matthews R. P.; Welton T.; Hunt P. A. Competitive π interactions and hydrogen bonding within imidazolium ionic liquids. Phys. Chem. Chem. Phys. 2014, 16, 3238–3253. [DOI] [PubMed] [Google Scholar]
- Dong K.; Song Y.; Liu X.; Cheng W.; Yao X.; Zhang S. Understanding Structures and Hydrogen Bonds of Ionic Liquids at the Electronic Level. J. Phys. Chem. B 2012, 116, 1007–1017. [DOI] [PubMed] [Google Scholar]
- Fraser K. J.; Izgorodina E. I.; Forsyth M.; Scott J. L.; MacFarlane D. R. Liquids intermediate between “molecular” and “ionic” liquids: Liquid Ion Pairs?. Chem. Commun. 2007, 37, 3817–3819. [DOI] [PubMed] [Google Scholar]
- Hunt P. A.; Gould I. R. Structural Characterization of the 1-Butyl-3-methylimidazolium Chloride Ion Pair Using ab Initio Methods. J. Phys. Chem. A 2006, 110, 2269–2282. [DOI] [PubMed] [Google Scholar]
- Hunt P. A.; Gould I. R.; Kirchner B. The Structure of Imidazolium-Based Ionic Liquids: Insights From Ion-Pair Interactions. Aust. J. Chem. 2007, 60, 9–14. [Google Scholar]
- Hunt P. A.; Kirchner B.; Welton T. Characterising the Electronic Structure of Ionic Liquids: An Examination of the 1-Butyl-3-Methylimidazolium Chloride Ion Pair. Chem. Eur. J. 2006, 12, 6762–6775. [DOI] [PubMed] [Google Scholar]
- Izgorodina E. I.; Forsyth M.; MacFarlane D. R. Towards a Better Understanding of ’Delocalized Charge’ in Ionic Liquid Anions. Aust. J. Chem. 2007, 60, 15–20. [Google Scholar]
- Schmidt J.; Krekeler C.; Dommert F.; Zhao Y.; Berger R.; Site L. D.; Holm C. Ionic Charge Reduction and Atomic Partial Charges from First-Principles Calculations of 1,3-Dimethylimidazolium Chloride. J. Phys. Chem. B 2010, 114, 6150–6155. [DOI] [PubMed] [Google Scholar]
- Tsuzuki S.; Tokuda H.; Mikami M. Theoretical analysis of the hydrogen bond of imidazolium C2-H with anions. Phys. Chem. Chem. Phys. 2007, 9, 4780–4784. [DOI] [PubMed] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [DOI] [PubMed] [Google Scholar]
- Ten-no S.; Hirata F.; Kato S. A hybrid approach for the solvent effect on the electronic structure of a solute based on the RISM and Hartree-Fock equations. Chem. Phys. Lett. 1993, 214, 391–396. [Google Scholar]
- Ten-no S.; Hirata F.; Kato S. Reference interaction site model self-consistent field study for solvation effect on carbonyl compounds in aqueous solution. J. Chem. Phys. 1994, 100, 7443–7453. [Google Scholar]
- Bühl M.; Chaumont A.; Schurhammer R.; Wipff G. Ab Initio Molecular Dynamics of Liquid 1,3-Dimethylimidazolium Chloride. J. Phys. Chem. B 2005, 109, 18591–18599. [DOI] [PubMed] [Google Scholar]
- Gabl S.; Schröder C.; Steinhauser O. Computational studies of ionic liquids: Size does matter and time too. J. Chem. Phys. 2012, 137, 094501. [DOI] [PubMed] [Google Scholar]
- Kohagen M.; Brehm M.; Lingscheid Y.; Giernoth R.; Sangoro J.; Kremer F.; Naumov S.; Iacob C.; Kärger J.; Valiullin R.; Kirchner B. How Hydrogen Bonds Influence the Mobility of Imidazolium-Based Ionic Liquids. A Combined Theoretical and Experimental Study of 1-n-Butyl-3-methylimidazolium Bromide. J. Phys. Chem. B 2011, 115, 15280–15288. [DOI] [PubMed] [Google Scholar]
- Skarmoutsos I.; Dellis D.; Matthews R. P.; Welton T.; Hunt P. A. Hydrogen Bonding in 1-Butyl- and 1-Ethyl-3-methylimidazolium Chloride Ionic Liquids. J. Phys. Chem. B 2012, 116, 4921–4933. [DOI] [PubMed] [Google Scholar]
- Song X.; Hamano H.; Minofar B.; Kanzaki R.; Fujii K.; Kameda Y.; Kohara S.; Watanabe M.; Ishiguro S.-i.; Umebayashi Y. Structural Heterogeneity and Unique Distorted Hydrogen Bonding in Primary Ammonium Nitrate Ionic Liquids Studied by High-Energy X-ray Diffraction Experiments and MD Simulations. J. Phys. Chem. B 2012, 116, 2801–2813. [DOI] [PubMed] [Google Scholar]
- Addicoat M. A.; Fukuoka S.; Page A. J.; Irle S. Stochastic structure determination for conformationally flexible heterogenous molecular clusters. Application to ionic liquids. J. Comput. Chem. 2013, 34, 2591–2600. [DOI] [PubMed] [Google Scholar]
- Greaves T. L.; Drummond C. J. Protic Ionic Liquids: Properties and Applications. Chem. Rev. 2007, 108, 206–237. [DOI] [PubMed] [Google Scholar]
- Oliveira A. F.; Seifert G.; Heine T.; Duarte H. A. Density-functional based tight-binding: an approximate DFT method. J. Braz. Chem. Soc. 2009, 20, 1193–1205. [Google Scholar]
- Elstner M.; Seifert G.. Density functional tight binding. Philos. Trans. R. Soc., A 2014, 372, 20120483. [DOI] [PubMed] [Google Scholar]
- Porezag D.; Frauenheim T.; Köhler T.; Seifert G.; Kaschner R. Construction of tight-binding-like potentials on the basis of density-functional theory: Application to carbon. Phys. Rev. B 1995, 51, 12947–12957. [DOI] [PubMed] [Google Scholar]
- Seifert G.; Porezag D.; Frauenheim T. Calculations of molecules, clusters, and solids with a simplified LCAO-DFT-LDA scheme. Int. J. Quantum Chem. 1996, 58, 185–192. [Google Scholar]
- Gaus M.; Cui Q.; Elstner M. DFTB3: Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB). J. Chem. Theory. Comput. 2011, 7, 931–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Yu H.; York D.; Cui Q.; Elstner M. Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method: Third-Order Expansion of the Density Functional Theory Total Energy and Introduction of a Modified Effective Coulomb Interaction. J. Phys. Chem. A 2007, 111, 10861–10873. [DOI] [PubMed] [Google Scholar]
- Aradi B.; Hourahine B.; Frauenheim T. DFTB+, a Sparse Matrix-Based Implementation of the DFTB Method. J. Phys. Chem. A 2007, 111, 5678–5684. [DOI] [PubMed] [Google Scholar]
- Elstner M.; Porezag D.; Jungnickel G.; Elsner J.; Haugk M.; Frauenheim T.; Suhai S.; Seifert G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 1998, 58, 7260–7268. [Google Scholar]
- Gaus M.; Goez A.; Elstner M. Parametrization and Benchmark of DFTB3 for Organic Molecules. J. Chem. Theory Comput. 2012, 9, 338–354. [DOI] [PubMed] [Google Scholar]
- Izgorodina E. I.; Golze D.; Maganti R.; Armel V.; Taige M.; Schubert T. J. S.; MacFarlane D. R. Importance of dispersion forces for prediction of thermodynamic and transport properties of some common ionic liquids. Phys. Chem. Chem. Phys. 2014, 16, 7209–7221. [DOI] [PubMed] [Google Scholar]
- Zahn S.; Kirchner B. Validation of Dispersion-Corrected Density Functional Theory Approaches for Ionic Liquid Systems. J. Phys. Chem. A 2008, 112, 8430–8435. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Hujo W.; Kirchner B. Performance of dispersion-corrected density functional theory for the interactions in ionic liquids. Phys. Chem. Chem. Phys. 2012, 14, 4875–4883. [DOI] [PubMed] [Google Scholar]
- Elstner M.; Hobza P.; Frauenheim T.; Suhai S. a.; Kaxiras E. Hydrogen bonding and stacking interactions of nucleic acid base pairs: A density-functional-theory based treatment. J. Chem. Phys. 2001, 114, 5149–5155. [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [DOI] [PubMed] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Chen S.; Vijayaraghavan R.; MacFarlane D. R.; Izgorodina E. I. Ab Initio Prediction of Proton NMR Chemical Shifts in Imidazolium Ionic Liquids. J. Phys. Chem. B 2013, 117, 3186–3197. [DOI] [PubMed] [Google Scholar]
- Baboul A. G.; Curtiss L. A.; Redfern P. C.; Raghavachari K. Gaussian-3 theory using density functional geometries and zero-point energies. J. Chem. Phys. 1999, 110, 7650–7657. [Google Scholar]
- Montgomery J. A.; Frisch M. J.; Ochterski J. W.; Petersson G. A. A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J. Chem. Phys. 1999, 110, 2822–2827. [Google Scholar]
- Montgomery J. A.; Frisch M. J.; Ochterski J. W.; Petersson G. A. A complete basis set model chemistry. VII. Use of the minimum population localization method. J. Chem. Phys. 2000, 112, 6532–6542. [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Rev. D01, Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Kick3 is freely available at http://maddicoat.github.io/Kick3/.
- Capelo S. B.; Méndez-Morales T.; Carrete J.; López Lago E.; Vila J.; Cabeza O.; Rodríguez J. R.; Turmine M.; Varela L. M. Effect of Temperature and Cationic Chain Length on the Physical Properties of Ammonium Nitrate-Based Protic Ionic Liquids. J. Phys. Chem. B 2012, 116, 11302–11312. [DOI] [PubMed] [Google Scholar]
- We note here that it is currently not possible to optimize PBC lattice vectors and employ Slater–Kirkwood dispersion simultaneously in DFTB+.
- Martyna G. J.; Klein M. L.; Tuckerman M. Nosé–Hoover chains: The Canonical Ensemble via Continuous Dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar]
- Nose S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar]
- Winget P.; Selçuki C.; Horn A. H. C.; Martin B.; Clark T. Towards a “next generation” neglect of diatomic differential overlap based semiempirical molecular orbital technique. Theor. Chem. Acc. 2003, 110, 254. [Google Scholar]
- Hunter E. P. L.; Lias S. G. Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update. J. Phys. Chem. Ref. Data 1998, 27, 413–656. [Google Scholar]
- Bondar A.; Fischer S.; Smith J. C.; Elstner M.; Suhai S. Key Role of Electrostatic Interactions in Bacteriorhodopsin Proton Transfer. J. Am. Chem. Soc. 2004, 126, 14668. [DOI] [PubMed] [Google Scholar]
- Bodo E.; Mangialardo S.; Ramondo F.; Ceccacci F.; Postorino P. Unravelling the Structure of Protic Ionic Liquids with Theoretical and Experimental Methods: Ethyl-, Propyl- and Butylammonium Nitrate Explored by Raman Spectroscopy and DFT Calculations. J. Phys. Chem. B 2012, 116, 13878–13888. [DOI] [PubMed] [Google Scholar]
- Using an Intel Hexacore Xeon E5-2667 2.9 GHz CPU with 2GB RAM.
- Gaus M.; Lu X.; Elstner M.; Cui Q. Parameterization of DFTB3/3OB for Sulfur and Phosphorus for Chemical and Biological Applications. J. Chem. Theory Comput. 2014, 10, 1518–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubař T.; Bodrog Z.; Gaus M.; Köhler C.; Aradi B.; Frauenheim T.; Elstner M. Parametrization of the SCC-DFTB Method for Halogens. J. Chem. Theory Comput. 2013, 9, 2939–2949. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.