

Abstract
The TRP (Transient Receptor Potential) superfamily of cation channels is remarkable in that it displays greater diversity in activation mechanisms and selectivities than any other group of ion channels. The domain organizations of some TRP proteins are also unusual, as they consist of linked channel and enzyme domains. A unifying theme in this group is that TRP proteins play critical roles in sensory physiology, which include contributions to vision, taste, olfaction, hearing, touch, and thermo- and osmosensation. In addition, TRP channels enable individual cells to sense changes in their local environment. Many TRP channels are activated by a variety of different stimuli and function as signal integrators. The TRP superfamily is divided into seven subfamilies: the five group 1 TRPs (TRPC, TRPV, TRPM, TRPN, and TRPA) and two group 2 subfamilies (TRPP and TRPML). TRP channels are important for human health as mutations in at least four TRP channels underlie disease.
Keywords: calcium, diacylglycerol, kidney disease, mucolipidosis type IV, phospholipase C, sensory transduction
INTRODUCTION
Members of the TRP (Transient Receptor Potential) superfamily of channels share the common features of six transmembrane segments, varying degrees of sequence homology, and permeability to cations. Despite these similarities, TRP channels are highly unusual among the known families of ion channels in that they display an impressive diversity of cation selectivities and specific activation mechanisms. Perhaps most amazing is that a single TRP channel can be activated through seemingly disparate mechanisms. In many cases, TRPs can be considered as multiple signal integrators, as the response to one input is modified by another. Nevertheless, a common thread among the superfamily is that TRP channels play critical roles in the responses to all major classes of external stimuli, including light, sound, chemicals, temperature, and touch. TRP channels also imbue individual cells with the ability to sense changes in the local environment, such as alterations in osmolarity.
TRP Classification and Phylogenetic Distribution
TRP channels are expressed and function in a great variety of multicellular organisms, including worms, fruit flies, zebrafish, mice, and humans. The TRP superfamily is broadly divided into groups 1 and 2, which are themselves divided into seven subfamilies (Figure 1) (1). An eighth subfamily, TRPY, consists of yeast TRPs, which are distantly related to the group 1 and group 2 TRPs (2, 3). The existence of TRPs in yeast indicates that the origin of TRP channels predates the emergence of metazoan organisms.
Figure 1.
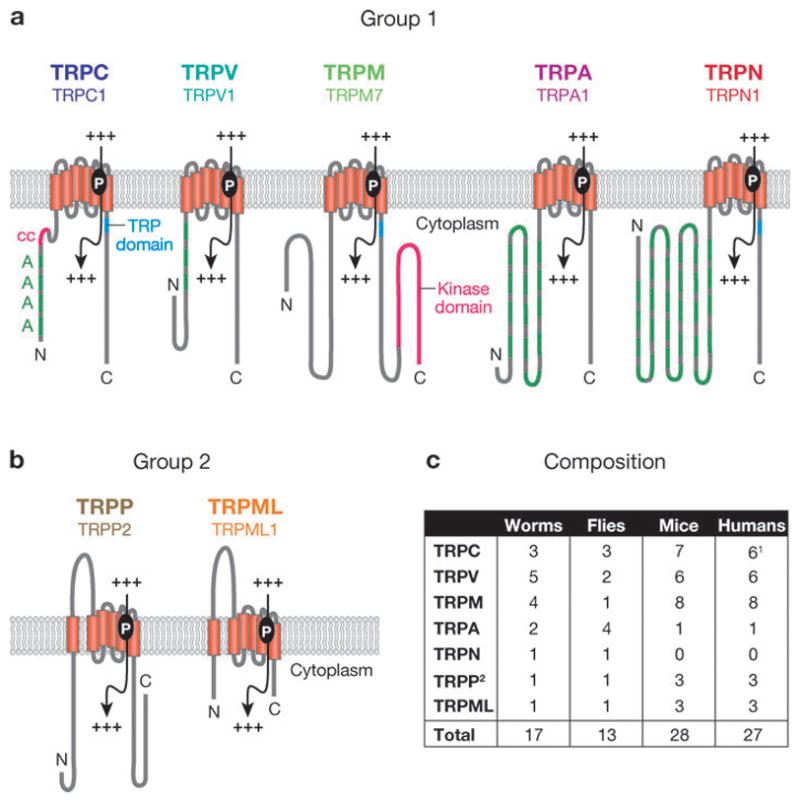
The TRP superfamily. (a) Single members from each of the five group 1 subfamilies. (b) Single members from each of the two group 2 subfamilies. The following domains are indicated: A, ankyrin repeats; cc, coiled-coil domain; protein kinase domain; TRP domain. Also shown are transmembrane segments (vertical rectangles) and pore loop (P), allowing the passage of cations (+++). (c) Composition of the TRP superfamily in worms, flies, mice, and humans. 1Human TRPC2 is a pseudogene and is not counted. 2TRPP1-like proteins are not counted. Modified and reprinted with permission from Reference 1.
The separation of the group 1 and 2 TRPs is based on sequence and topological differences. The group 1 TRPs consist of five subfamilies, which bear the strongest sequence homology to the founding member of the superfamily, Drosophila TRP (4). Those channels most related to Drosophila TRP are referred to as the classical or TRPC channels. The TRPV, TRPM, TRPA, and TRPN subfamilies are so named based on the original name of the initially described member of each subfamily. Depending on the organism, the group 1 TRP family consists of between 11 and 22 members (Figure 1c). The TRPN proteins are not found in mammals, although they are expressed in some vertebrates, such as zebrafish.
The group 1 TRPs include several notable sequence elements and domains. The largest region of sequence homology spans the six transmembrane segments, including the pore loop situated between the fifth and sixth transmembrane segments (Figure 1a). TRPC, TRPM, and TRPN channels also contain a TRP domain, which follows the sixth transmembrane segment (Figures 1 and 2). The most conserved portions of the TRP domain are the TRP boxes 1 and 2 (Figure 2). With the exception of the TRPM channels, the TRPs in group 1 have multiple N-terminal ankyrin repeats. Three TRPM proteins are so-called chanzymes (5), with C-terminal enzyme domains.
Figure 2.

The TRP domain. Highly conserved region of 23–25 amino acids C-terminal to the transmembrane domains in TRPCs, TRPNs, and TRPMs. TRP box 1 is invariant in TRPCs, and variations of TRP box 1 are also present in TRPNs and TRPMs. The TRP box 2 is a proline-rich region. Sequences are from mammals, except zTRPN1 is from zebrafish; cNOMPC is from Caenorhabditis elegans; and TRP, TRPL, TRPγ, and dTRPM are from Drosophila.
The two subfamilies TRPP and TRPML comprise the group 2 TRPs, which are distantly related to the group 1 TRPs. The TRPP and TRPML proteins share sequence homology over the transmembrane segments and contain a large loop separating the first two transmembrane domains (Figure 1b). The TRPP subfamily may be the most ancient, as members of this subfamily extend from yeast to mammals (6). The founding TRPP and TRPML proteins were discovered as gene products mutated in autosomal dominant polycystic kidney disease (ADPKD) and mucolipidosis type IV (MLIV), respectively (7–10). Mutations in two group 1 TRP proteins also underlie human diseases. Autosomal dominant segmental glomerulosclerosis results from disruption of TRPC6 (11, 12), and mutations in TRPM6 cause hypomagnesemia and hypocalcemia (13, 14). Thus TRP channels are also of relevance to human health and disease.
ACTIVATION AND REGULATION OF CHANNEL ACTIVITY
The mechanism(s) through which a given TRP is activated and regulated cannot be predicted reliably on the basis of its subfamily assignment. For example, thermally activated TRPs belong to members of the TRPV, TRPM, and TRPA subfamilies, while regulated exocytosis stimulates cation influx via several TRPC, TRPV, and TRPM channels. However, in some cases there are similarities among subfamily members, as all TRPC channels are activated through pathways coupled to stimulation of phospholipase C (PLC). The recurring theme is that most TRP channels are activated through a diversity of mechanisms. Remarkably, there are TRP channels, such as TRPV1, which respond to stimuli ranging from heat to botanical compounds, proinflammatory agents, and exocytosis.
Invertebrate TRPCs
The gene encoding the first member of the TRP superfamily, Drosophila TRP (4), is required for maintaining the light response (15). The Drosophila phototransduction cascade is initiated when light strikes the G-protein-coupled receptor rhodopsin. This leads to stimulation of a heterotrimeric G protein, activation of PLC, and subsequent cation influx into the photoreceptor cells (15). Mutations in the trp locus result in a transient light response, characterized by a tenfold decrease in Ca2+ entry (16). These results indicate that Drosophila TRP is either a Ca2+ permeable cation channel or a regulatory protein required for channel activation. The predicted TRP topology, consisting of multiple transmembrane segments (4), combined with heterologous expression studies (17, 18), indicated that TRP was a new type of Ca2+-permeable cation channel.
The presence of a transient light response in the absence of functional TRP indicates a role for additional channels. Indeed, there are two other TRPC channels expressed in flies, TRPL (TRP-like) and TRPγ (19, 20), and the light response is eliminated in flies with mutations in both trp and trpl (21). Although TRP is the major source for Ca2+ entry in the fly eye (16, 21), TRPL has a role in maintaining a sustained response during prolonged illuminations (22). Whereas TRP channels are relatively selective for Ca2+, TRPL and TRPγ are nonselective cation channels (23).
A key question concerns the mechanisms through which stimulation of PLC leads to the activation of TRP and TRPL in vivo. Stimulation of PLC cleaves phosphoinositide-4,5-bisphosphate (PIP2), leading to the production of diacylglycerol (DAG) and the soluble second messenger inositol-1,4,5-trisphosphate (IP3). There are at least three models for the mechanism by which TRP and TRPL may be activated. According to one model, IP3 diffuses rapidly through the cytoplasm and binds to Ca2+-permeable IP3 receptors present in the endoplasmic reticulum (ER) membrane. This leads to Ca2+ release and depletion of the ER Ca2+ stores, which in turn activates TRP and TRPL. In fact, Drosophila TRP and several mammalian TRPCs are activated through such a store-operated mechanism in heterologous expression systems (1). However, neither TRP nor TRPL is activated by either thapsigargin, a specific inhibitor of the ER Ca2+ ATPase, or IP3-mediated store depletion in native photoreceptor cells, providing evidence against a role of ER Ca2+ store depletion on channel activation in vivo (24, 25). Furthermore, mutation of the lone Drosophila homolog of the mammalian IP3 receptor has no effect on channel activation or the photoresponse (26).
A second model suggests that PLC regulates Drosophila TRPC activity by reducing the concentration of inhibitory PIP2. PIP2 has been reported to inhibit the activity of exogenously expressed TRPL channels in inside-out patches (27). Therefore, PLC activity could lead to channel activation by alleviating the inhibitory effects of PIP2. Although PIP2 may be inhibitory, depletion of PIP2 may not be sufficient for channel activation in vivo.
It appears that TRP channel activation is mediated in vivo by DAG or its metabolites, polyunsaturated fatty acids (PUFAs). PUFAs activate TRP and TRPL channels in dissected photoreceptor cells (28), and DAG/PUFAs activate TRPL channels expressed exogenously in insect cells (27, 28). Furthermore, disruption of the eye enriched DAG kinase, which phosphorylates DAG to phosphatidic acid (PA), causes constitutive activity of TRP and TRPL (29). Although PUFA levels were not measured in the DAG-kinase mutants, diversion of DAG to PUFAs instead of PA could elevate PUFA levels, thereby increasing channel activity (29). Further evidence for a role of DAG in TRP activation in vivo is that flies with mutations in the PA phosphatase (PAP), which converts PA back to DAG, have small light responses (30), whereas overexpression of PAP increases channel activity (31).
The activity of TRPL is also downregulated by its light-dependent shuttling out of the phototransducing portion of the photoreceptor cells, the rhabdomeres, into the extrarhabdomeral cell bodies (32). A Caenorhabditis elegans TRPC channel, TRP-3, which functions in fertilization, undergoes a developmentally regulated translocation (33). In spermatids, TRP-3 is situated in intracellular vesicles; however, during sperm activation, the channel translocates to the plasma membrane. This translocation is correlated with an increase in TRP-3 activity.
Mammalian TRPCs
The first mammalian homologs of Drosophila TRPs identified were referred to as TRPC1, TRPC2, and TRPC3 (34, 35). Since then, seven mammalian TRPC proteins (TRPC1–7) have been described (36) (Figure 3, Table 1). However, humans express only six because human TRPC2 is a pseudogene (34). The seven mammalian homologs share ≥30% amino acid identity with each other and with the Drosophila TRPCs over the N-terminal 750–900 amino acids. Based on amino acid similarities, the mammalian TRPCs fall into four subsets: TRPC1, TRPC2, TRPC3/6/7, and TRPC4/5. Whereas some TRPCs (such as TRPC1) are broadly expressed (34, 35), others are enriched in the nervous system (Table 1).
Figure 3.

Phylogenetic tree showing the relatedness of the TRP proteins. The dendrogram of vertebrate TRPs includes mostly human TRPs, except for mouse TRPC2 (cartoon of a mouse) and zebrafish TRPN1 (cartoon of a zebrafish). White text and cartoons highlight the TRP proteins from worms and flies. One C. elegans and one Drosophila member of each subfamily are included.
Table 1.
Properties of vertebrate TRPC and TRPV proteins
| Gene name | Chromosomal localization | Selectivity PCa:PNa | Modulation of activity | Highest expression |
|---|---|---|---|---|
| TRPC subfamily | ||||
| TRPC1 | 3q22–q24 | Nonselective | Store depletion, conformational coupling, mechanical stretchb | Heart, brain, testis, ovary, liver, spleen |
| TRPC2a | 7, 50.0 cM | 2.7 | Diacylglycerol (DAG) | VNO, testis |
| TRPC3 | 4q27 | 1.6 | Store depletion, conformational coupling, DAG, exocytosis | Brain |
| TRPC4 | 13q13.1–q13.2 | 7 | Store depletion (?), exocytosis | Brain, endothelia, adrenal gland, retina, testis |
| TRPC5 | Xq23 | 9.5 | Store depletion (?), sphingosine-1-phosphate, exocytosis | Brain |
| TRPC6 | 11q21–q22 | 5 | Conformational coupling, DAG, PIP3 | Lung, brain, placenta, ovary |
| TRPC7 | 5q31.2 | 1.9c, 5d | Store depletion, DAG | Eye, heart, lung |
| TRPV subfamily | ||||
| TRPV1 | 17p13.3 | 3.8 (heat), 9.6 (vanilloids) | Heat (43°C), vanilloids, anandamide, camphor, piperine (black pepper), allicin (garlic), ethanol, nicotine, proinflammatory cytokines, protons, PIP2, phosphorylation exocytosis | TG, DRG, neurons, urinary bladder, testis |
| TRPV2 | 17p11.2 | 3 | Heat (52°C), osmotic cell swelling, exocytosis | DRG, spinal cord, brain, spleen, intestine |
| TRPV3 | 17p13.3 | 2.6 | Warm (33–39°C); PUFAs; menthol; compounds from oregano, cloves, and thymes | TG, DRG, spinal cord, brain, keratinocytes, tongue |
| TRPV4 | 12q24.1 | 6 | Warm (27–34°C), osmotic cell swelling, 5′6′-EET, exocytosis | DRG, kidney, lung, spleen, testis, heart, keratinocytes, heart, liver, endothelia |
| TRPV5 | 7q35 | >100 | Low intracellular Ca2+, hyperpolarization, exocytosis | Kidney, intestine, pancreas, placenta |
| TRPV6 | 7q33–q34 | >100 | Store depletion, exocytosis | Small intestine, pancreas, placenta |
Chromosomal localizations apply to human genes except TRPC2, which is a pseudogene in humans. Therefore, the chromosomal localization of mouse TRPC2 is provided.
Abbreviations: Question mark depicts controversial results; DRG, dorsal root ganglia; PIP2, phosphoinositide-4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PUFAs, polyunsaturated fatty acids; TG, trigeminal ganglia; VNO, vomeronasal organ.
Spontaneous current.
ATP-enhanced current.
All mammalian TRPCs require PLC for activation (1, and references therein). However, there is variability in their selectivities for Ca2+ versus other cations and in the mechanisms coupling PLC activity to channel stimulation. Several TRPC proteins may be activated by depletion of Ca2+ from internal stores, brought about by application of either IP3 or thapsigargin (listed in Reference 37) (Table 1). However, the dependence of exogenously expressed TRPC-channel activity on store depletion is controversial (1 and 37, and references therein).
One hypothesis regarding the relationship between the status of the Ca2+ stores and TRPC channel activity is that there is direct conformational coupling between TRPCs and ER Ca2+ release channels. Such a coupling mechanism would require close apposition between the ER and plasma membranes. Indeed, separation of the two membranes by pharmacological reorganization of the actin cytoskeleton into a cortical ring prevents the activation of exogenously expressed TRPC3 channels (38). However, modifying the actin cytoskeleton may also decrease channel activity by leading to their internalization (39). Although requirements for direct interactions between TRPCs and ER Ca2+ release channels (both IP3 and ryanodine receptors) have been described (1 and 37, and references therein), TRPC channels are completely functional in DT40 avian B-lymphocytes in which all three IP3-receptor homologs are knocked out (40–42). However, a role for conformational coupling cannot be excluded because TRPC channel activation could be coupled to ryanodine receptors in the absence of functional IP3 receptors.
Several mammalian TRPCs are coupled to PLC activation owing to a requirement for DAG. When expressed exogenously, some TRPC channels are potently stimulated by DAG and its analogs (43) (Table 1). Supporting a role for DAG in vivo, native TRPC2 channels, which are expressed in the mouse vomeronasal organ (VNO) (44), are activated exclusively by DAG, and not by IP3 receptors, Ca2+ stores, or the PUFA arachidonic acid (45). Furthermore, elimination of native TRPC6/7 channels in cultured cells prevents the development of DAG-dependent conductances (46, 47). At least one TRPC channel, TRPC1, is mechanically gated as it is activated by the development of tension across the lipid bilayer in reconstituted liposomes (48) in the absence of proteins.
The discrepancies with regard to the activation mechanisms of TRPC channels underscore the complications in arriving at conclusions based exclusively on heterologous expression studies. A native TRPC3-mediated conductance in primary pontine neurons from neonatal rat brains is activated through a pathway initiated by the neurotrophin brain-derived neutotrophic factor (BDNF) and the TrkB receptor (49). This native conductance (IBDNF ) depends on PLC stimulation, an elevation of cytosolic Ca2+, and functional IP3 receptors (49). Surprisingly, IBDNF is not gated by DAG (49), raising the question as to why TRPC3 is activated by DAG when overexpressed in tissue culture cells. DAG is also required for protein kinase C (PKC)-mediated inactivation of TRPCs (42), and DAG activates native TRPC7 in DT40 cells only after PKC inhibition (50). Therefore, discrepancies in the observed contributions of DAG to TRPC activation may reflect distinct contributions of PKC-mediated inhibition in different cells. In addition, as described above, there is strong evidence that some native TRPC conductances are activated by DAG (45).
Agonist-mediated exocytosis of TRPCs also modulates the activity of the channels (1) (Table 1). Incorporation of TRPC4 and TRPC5 into the plasma membrane is promoted by stimulation of the epidermal growth factor (EGF) receptor. Activation of the EGF receptor augments insertion of TRPC4 through tyrosine phosphorylation of TRPC4 by Src, which in turn increases interaction with a scaffold protein, the Na+/H+ exchanger regulatory factor (NHERF) (51). EGF-induced translocation of TRPC5 occurs through a separate mechanism, involving phosphatidylinositide-3-kinase, the Rho GTPase Rac1, and phosphatidylinositol 4-phosphate 5-kinase (52). In the case of TRPC3, agonist-induced exocytosis involves direct interactions with the v-SNARE protein, vesicle-associated membrane protein 2 (53). Regulated translocation of TRPCs from internal vesicles raises the possibility that some TRPCs are constitutively active and contribute to cation influx by insertion into the plasma membrane (1).
TRPV
TRPV proteins (Figure 3) share ~25% homology with the TRPC channels over a region spanning transmembrane domains 5 and 6 (1). The C. elegans OSM-9 and rat TRPV1 were the first TRPVs identified (54). The biophysical features of the OSM-9-dependent current have not been described, although PUFAs such as arachidonic acid induce both Ca2+ transients and characteristic avoidance behaviors in the ASH neurons, which express OSM-9 and function in osmosensation, mechanosensation, and chemosensation (55). This arachidonic acid-induced Ca2+ rise is not observed in osm-9 mutants. Mutants with defects in PUFA biosynthesis show similar behavioral phenotypes, further suggesting that OSM-9 may be activated by PUFAs (55).
The first mammalian TRPV was identified by expression cloning in a search for channels activated by the inflammatory vanilloid compound capsaicin, which gives spicy foods their characteristic hot taste (56) (Figure 3, Table 1). In addition, TRPV1 is activated by heat (≥43°C) (56) and many other chemicals, which include an endocannabinoid, anandamide (57); the topical analgesic, camphor (58); and the pungent compounds present in black pepper (piperine) (59) and garlic (allicin) (60). TRPV1-mediated cation influx initiated by the application of noxious chemicals or heat is further enhanced by low pH (56). In fact, pH ≤ 5.9, characteristic of proalgesic tissue acidosis associated with injury, induces a shift in the thermal activation threshold of TRPV1 so that it can be activated at room temperature (1). Shifts in the activation threshold of TRPV1 and potentiation of capsaicin-mediated responses are also brought about by ethanol (61), nicotine (62), and proinflammatory cytokines (63). Furthermore, TRPV1 is potentiated by a decrease in PIP2 levels and PKC-mediated phosphorylation of the channel, both following PLC activation induced by proalgesic agents such as bradykinin and nerve growth factor (1). Therefore, TRPV1 is a multiple signal integrator capable of transducing signals evoked by several noxious stimuli (Table 1).
Activation by heat is also a feature of TRPV2, TRPV3, and TRPV4. TRPV2 is activated by noxious heat (≥52°C) but not by capsaicin or changes in pH (64). TRPV1 and TRPV2 are expressed in distinct dorsal root ganglia neurons of different sizes (64), maintaining the fidelity of signaling in response to different temperatures. TRPV3 and TRPV4 are activated at warm temperatures in the ranges of 33–39°C and 27–34°C, respectively (Table 1) (1).
TRPV2–4 are also polymodally activated and are capable of integrating various stimuli. TRPV2 function is upregulated by phosphatidylinositol-3-kinase activation (65) and hypotonicity-induced cell swelling (66). TRPV3 is activated in cultured cells by PLC stimulation (67); by application of PUFAs such as arachidonic acid (68); by menthol (69); and by compounds present in spices such as oregano, cloves, and thymes (67) (Table 1).
In addition to warm temperatures, TRPV4 is activated by extracellular hypotonicity (1). The hypotonicity-induced activation of TRPV4 requires its interaction with the water channel aquaporin 5 (70). Chemicals such as anandamide and arachidonic acid activate TRPV4 via a mechanism involving cytochrome P450-dependent formation of epoxyeicosatrienoic acids (71). The different modes through which TRPV4 can be activated do not occur through the same mechanism because a point mutation in the third transmembrane domain inhibits activation by heat but not by cell swelling or arachidonic acid (72) (Table 1).
Consistent with the theme that a change in osmolarity is an evolutionarily conserved mode for TRPV activation, the worm TRPVs (OSM-9 and OCR-2) are required for osmosensation, although it is unclear if they are activated directly or indirectly by changes in osmolarity. Nevertheless, both Drosophila TRPV proteins, Nanchung and Inactive, are activated by hypoosmolarity when expressed in a heterologous expression system (73, 74).
TRPV5 and TRPV6 are distinct from other TRPV channels in that they are not activated by heat and are the most Ca2+ selective (PCa:PNa > 100) of the mammalian TRPs (1) (Table 1). TRPV6 has been reported to have properties similar to a store-operated conductance known as Ca2+-release-activated Ca2+ current or CRAC (75). Although the channels exhibit high Ca2+ selectivity, they are permeable to monovalent cations in the absence of extracellular Ca2+ (75). However, some biophysical properties of TRPV6 and CRAC have clear distinctions (76), and the involvement of Ca2+ stores in TRPV6 activation remains controversial.
Several TRPV channel activities are regulated by insertion or retention in the plasma membrane (Table 1). TRPV1 interacts with members of the SNARE-dependent exocytic pathway, and these interactions promote PKC-mediated translocation of TRPV1 to the plasma membrane (77). Furthermore, proalgesic factors such as nerve growth factor promote surface insertion of TRPV1 via Src kinase-mediated tyrosine phosphorylation of the channel (78). TRPV2 activity may also be regulated by channel insertion into the plasma membrane from internal vesicles, induced by the application of insulin-like growth factor-I in cultured cells (1). Glycosylation in the pore loop (79) and interactions with proteins called PACSINs (involved in synaptic vesicle trafficking) (80) regulate the surface expression of TRPV4. Insertion of TRPV5 and TRPV6 into the plasma membrane regulates their activity as well (81). Retention of TRPV5 in the plasma membrane is promoted by the β-glucuronidase Klotho through hydrolysis of extracellular sugar residues on the channel (82).
TRPM
The mammalian TRPM subfamily consists of eight members (Figure 3), which share ~20% amino acid identities to TRPC channels over the C-terminal five transmembrane domains (1). Similar to TRPCs, TRPM proteins have a TRP domain C-terminal to the transmembrane segments (Figure 2). The total amino acid lengths (~1000–2000) and sequences of the C-terminal regions of these proteins vary considerably. Based on similarities in amino acid sequences, these proteins fall into subsets consisting of TRPM1/3, TRPM4/5, and TRPM6/7 (Figure 3). TRPM2 and TRPM8 are not placed in any subset, although they are most closely related to each other. TRPM2 and TRPM6/7 are unusual in that they are chanzymes, with C-terminal enzyme domains.
The first mammalian TRPM identified, TRPM1, was initially named melastatin, as its expression levels correlated inversely with the metastatic potential in some melanomic cell lines (83). In addition to the full-length clone (TRPM1-L), a short variant of TRPM1 (TRPM1-S), which is devoid of transmembrane domains, interacts with TRPM1-L and inhibits its translocation to the plasma membrane (84) (Table 2).
Table 2.
Properties of vertebrate TRPM, TRPA, and TRPN proteins
| Gene name | Chromosomal localizationa | Selectivity PCa:PNa | Modulation of activity | Highest expression |
|---|---|---|---|---|
| TRPM subfamily | ||||
| TRPM1 | 15q13–q14 | Nonselective | Translocation (?)b | Brain, melanosomes |
| TRPM2 | 21q22.3 | ~0.3 | ADP-ribose, cADP-ribose, pyrimidine nucleotides, arachidonic acid, NAD, H2O2, Ca2+ | Brain, bone marrow, spleen |
| TRPM3 | 9q21.11 | 1.6 | Osmotic cell swelling, store depletion (?) | Kidney, brain, pituitary |
| TRPM4 | 19q13.33 | Monovalent cation selective | Ca2+, voltage modulated, PIP2 | Prostate, colon, heart, kidney, testis |
| TRPM5 | 11p15.5 | Monovalent cation selective | Ca2+, voltage modulated, PIP2, heat (15–35°C) | Intestine, liver, lung, taste cells |
| TRPM6 | 9q21.13 | Divalent cation selective (Mg2+ and Ca2+) | Mg2+ inhibited, translocation | Kidney, small intestines |
| TRPM7 | 15q21 | Divalent cation selective (Mg2+ and Ca2+) | Mg2+ inhibited, ATP, protons, phosphorylation, PIP2 | Kidney, heart, pituitary, bone, adipose |
| TRPM8 | 2q37.2 | 3.3 | Cool (23–28°C), menthol, icilin, pH modulated, PIP2 | DRG, TG, prostate, liver |
| TRPA and TRPN | ||||
| TRPA1 | 8q13 | 0.8 | Cold (17°C) (?), icilin, isothiocyanates (mustard oil, horseradish, and wasabi), allicin (garlic), cinnamaldehyde (cinnamon oil), acrolein (tear gas), cannabinoids, bradykinin, DAG, PUFAs, mechanically gated (?) | DRG, hair cells, ovary, spleen, testis |
| zTRPN | – | ? | Mechanically gated (?) | Ear, eye |
All chromosomal localizations are of human proteins except for the zebrafish TRPN1 (zTRPN1) because TRPN1 proteins are absent in humans.
Abbreviations: Question mark depicts controversial results; DAG, diacylglycerol; DRG, dorsal root ganglia; PIP2, phosphoinositide-4,5-bisphosphate; PUFAs, polyunsaturated fatty acids; TG, trigeminal ganglia.
TRPM3 forms a constitutively active Ca2+ and Mn2+ permeable channel when expressed exogenously in cultured cells (85). The spontaneous TRPM3 currents are enhanced by hypotonic extracellular solutions and cell swelling (85) (Table 2). Alternatively, TRPM3 might be activated by store depletion (86) or sphingolipids (87). TRPM3 also has multiple splice variants with distinct ion selectivities (88).
TRPM4 and TRPM5 are unusual among the TRP superfamily in that they are voltage-modulated, Ca2+-activated, monovalent cation selective channels (VCAMs) (89–93) (Table 2). The monovalent selectivity, which sets these channels apart from other TRP channels, is dictated by a short acidic stretch of six amino acids in its pore loop (94). TRPM4 is expressed as two splice variants (84, 90), one of which (TRPM4b) is a VCAM (89, 93), whereas the other (TRPM4a) displays little discernible activity (84, 89, 90). The voltage dependence of TRPM4b and TRPM5 ensures the modulation of both activation and inactivation by changes in membrane potentials (91–93). The lack of Ca2+ permeability, Ca2+ activation, and voltage modulation suggests that the activities of VCAMs could be affected by the activity of voltage-gated Ca2+ channels. In turn, VCAM activation would decrease the membrane potential and the driving force for Ca2+ through other channels (90).
TRPM4b and TRPM5, similar to many other TRP channels, are modulated by PIP2, with PIP2 reversing the Ca2+-dependent desensitization of the channels and thereby increasing channel activity (91, 95, 96) (Table 2). TRPM5 is also temperature sensitive and activated by heat in the range of 15–35°C (97) (Table 2). Thus TRPM5 is another example of a TRP channel that integrates thermal input with other modes of activation.
The TRPM6 and TRPM7 channels have unusual architectures as they are chanzymes with C-terminal atypical protein kinase domains (13, 14, 98, 99). TRPM7 is a divalent-specific cation channel, which is permeable to virtually all physiological divalent cations including Mg2+ and trace metals such as Ni2+ (100). It is a Mg2+-inhibited channel, and because it fluxes Mg2+ (99), inhibition by Mg2+ provides an effective mode of feedback regulation. The TRPM6 chanzyme is also permeable to trace metals, Mg2+, and Ca2+ and is inhibited by intracellular Mg2+ (101) (Table 2).
TRPM7 activity is regulated by pH, ATP, lipids, and translocation. TRPM7 is upregulated tenfold following a decrease in extracellular pH to 4.0 from the physiological pH of 7.4 (102). Thus a portion of the TRPM7 pool may be situated in an acidic environment, such as acidic vesicles. ATP significantly increases TRPM7 currents (98); however, the basis for this effect is controversial. TRPM7 activity may be enhanced by ATP, forming Mg· ATP, which decreases intracellular free Mg2+ (99) (Table 2). Alternatively, the requirement for ATP might reflect consumption of ATP for PIP2 generation because PIP2 depletion causes a decrease in TRPM7 activity (103). Relatively rapid plasma membrane insertion of TRPM7 (<2 min) is induced by physiologically relevant fluid flow, leading to an increase in activity (104). TRPM7 is expressed in a vascular smooth muscle cell line (104) and could be activated by blood flow in areas in which the endothelium lining blood vessels is breached.
The role of the kinase domain of TRPM7 is controversial. According to one study, mutations in the ATP-binding region of the kinase domain lead to markedly decreased channel function (98). Subsequently, it was reported that mutations in the kinase domain do not prevent channel activation, although they change the sensitivity of the channel to Mg2+ inhibition (105). Nucleotide-mediated inhibition of TRPM7 is dependent on the nucleotide-binding site in the kinase domain, which works in concert with a Mg2+-binding site on TRPM7 outside of the kinase domain (106). The kinase domain also plays a role in trafficking or assembly of the channel (107).
TRPM2 is a chanzyme with a C-terminal ADP-ribose pyrophosphatase domain (NUDT9 homology domain) (108, 109). TRPM2 forms a Ca2+ permeable nonspecific cation channel that is activated by intracellular ADP-ribose, pyrimidine nucleotides, and NAD (108, 109) (Table 2). This channel forms a cellular redox sensor and is activated by peroxide or other agents that produce reactive oxygen and nitrogen species (110) (Table 2). This activation of TRPM2 by oxidative and nitrosative stress requires an intact ADP-ribose binding cleft in the NUDT9 domain of TRPM2 (111). Furthermore, cADP-ribose, arachidonic acid, and intracellular Ca2+ also positively modulate TRPM2 function (110, 112, 113) (Table 2). A shorter variant of TRPM2, which is missing the last four transmembrane domains and C terminus, acts as a dominant negative inhibitor of TRPM2 (114).
TRPM8 is a thermally regulated channel activated by moderately cool temperatures (<23–28°C) and by compounds that evoke a sensation of coolness, such as menthol, eucalyptol, and icilin (115, 116) (Table 2). Activation by cold and menthol can be separated because several mutations having a profound effect on activation by menthol have only minimal effects on activation by cool temperatures (117). These data indicate that the domains involved in activation by menthol and thermal input are distinct. In further support of this conclusion, modulation of TRPM8 activity by pH has differential effects on activation by icilin and cold versus menthol (118). The mechanism for thermal activation of TRPV1 and TRPM8 by hot and cool temperatures, respectively, appears to be similar (119, 120). These channels are voltage dependent, and their respective activation temperatures as well as ligands lead to shifts in their voltage thresholds toward more physiological membrane potentials (119, 120).
TRPM8 activity is also regulated by PIP2 (121) (Table 2), as depletion of PIP2 reduces activity induced by menthol or thermal cool. PIP2-mediated regulation of TRPM8 is reduced by mutations in basic residues in the TRP domain (122). Although these mutations affect the PIP2-modulated responses to both menthol and cool (122), they lie close or adjacent to mutations identified in the screen for residues that specifically function in the menthol response (117).
TRPA and TRPN
The first TRPA protein, human TRPA1, was identified in a screen for genes downregulated following oncogenic transformation of fibroblasts (123). TRPA1 is the only TRPA protein present in humans and other mammals (1) and was previously called ANKTM1 because the protein consists of many N-terminal ankyrin repeats. There are two and four TRPA proteins encoded in the C. elegans and Drosophila genomes, respectively (Figure 1c).
TRPA1 is chemically activated by the psychoactive component in marijuana, environmental irritants, and pungent compounds. These include ingredients present in wasabi, horseradish, and mustard oils (isothiocyanates); garlic (allicin); cinnamon oil (cinnamaldehyde); marijuana (tetrahydrocannabinol); and tear gas (acrolein) (60, 124–126). TRPA1 can also be activated by stimulation of the PLC pathway by bradykinin and by the subsequently produced metabolites DAG and PUFAs (125) (Table 2).
There is controversy as to whether TRPA1 is a thermally or mechanically gated channel. When exogenously expressed in cultured cells, TRPA1 has been reported as a noxious cold (<17°C) activated channel (125, 127) (Table 2). However, the cold activation of TRPA1 in both cultured cells (124) and TRPA1-deficient mice is controversial (126, 128). TRPA1 channels from mouse and zebrafish have been suggested to be mechanically gated channels, and the multiple ankyrin repeats of TRPA1 may form a gating spring capable of transducing mechanical force and thereby facilitating channel opening (129, 130). However, analyses of TRPA1-deficient mice described below mitigate this possibility (126, 128).
Despite the controversy concerning TRPA1, three Drosophila TRPAs (dTRPA1, Pyrexia, and Painless) function in thermosensation. Although TRPA1 may be a cold thermosensor, Drosophila TRPA1 and Pyrexia are activated in vitro by warm (≥24–27°C) and noxious (≥40°C) heat, respectively (131–133). Painless has not been functionally expressed in vitro but is predicted to be gated by noxious heat (≥38°C) and the isothiocyanates present in wasabi (134, 135).
The TRPN subfamily is so named based on the founding member, Drosophila NOMPC (136). Single TRPN members are present in worms, flies, and zebrafish, whereas mammals do not encode any TRPN homologs. These proteins have 29 ankyrin repeats situated in tandem N-terminal to the transmembrane domains. Based on the functional analyses described below, they are likely to be mechanically gated, possibly involving a gating spring comprised of the ankyrin repeats. Presently, there is a lack of functional information concerning TRPN’s ability to form channels.
TRPP
TRPP2 (polycystin-2 or PKD2) is the archetypal TRPP channel and was discovered as a protein disrupted in ADPKD (10). TRPP2 and other group 2 TRPs have very low sequence similarity with members of the group 1 branch of the TRP superfamily. The group 2 TRPs are also distinguished from the group 1 TRPs by a large loop between transmembrane domains one and two (Figure 1b). Nevertheless, TRPP2 and the related mammalian proteins, referred to as TRPP3 (polycystin-L, PKD2L1) and TRPP5 (PKD2L2), have six transmembrane segments (1). Invertebrate TRPPs have been described in C. elegans (137), Drosophila (AMO) (138, 139), and the sea urchin (suPC2) (140). TRPPs might comprise the most primitive TRP subfamily, as microbial homologs of TRPP2 are found in yeast (6).
The transmembrane segments in TRPP2 have sequence homology with 6 of the 11 C-terminal transmembrane domains of another protein disrupted in ADPKD, referred to as TRPP1 (polycystin-1 or PKD1) (141). This protein, which includes a ~2500-amino acid extracellular domain, is also referred to as TRPP1, based on its sequence relatedness to TRPP2. TRPP2 binds to TRPP1, and this association induces the translocation of TRPP2 to the plasma membrane where it serves as a Ca2+ permeable nonselective cation channel (1) (Table 3). Translocation of TRPP2 to the primary cilia, where it is localized in many cells, requires a stretch of 15 amino acids on the N terminus of TRPP2 and is independent of TRPP1 (142). Chaperone-like factors can also induce translocation of TRPP2 to the plasma membrane from intracellular pools (143, 144). The TRPP1/TRPP2 complex may contribute to TRPP2 function at the plasma membrane because anti-TRPP1 antibodies can induce structural rearrangements leading to TRPP2 activation (145). Furthermore, the addition of a C-terminal fragment of TRPP1 enhances the channel activity of TRPP2 (146).
Table 3.
Properties of vertebrate TRPP and TRPML proteins
| Gene name | Chromosomal localizationa | Selectivity PCa:PNa | Modulation of activity | Highest expression |
|---|---|---|---|---|
| TRPP2 | 4q21–q23 | Nonspecific | Ca2+, translocation, TRPP1, EGF, PIP2, fluid flow, actin cytoskeletonb | Widely expressed, kidney |
| TRPP3 | 10q24 | 4.3 | Ca2+ | Kidney, heart |
| TRPP5 | 5q31 | – | – | Testis, heart |
| TRPML1 | 19p13.2–p13.3 | Monovalent cation selective (?) | pH, Ca2+, proteolytic cleavage | Brain, heart, skeletal muscle |
| TRPML2 | 1p22 | – | – | – |
| TRPML3 | 1p22.3 | – | – | Cochlear hair cellsc |
All chromosomal localizations are of human proteins.
Abbreviations: Question mark depicts controversial results; EGF, epidermal growth factor; PIP2, phosphoinositide-4,5-bisphosphate.
Localization performed in the mouse.
Activation of TRPP2 may occur through mechanical gating or via a transduction pathway initiated by growth factors. The presence of TRPP proteins on both motile and primary cilia could render them capable of sensing fluid flow, osmolarity, and mechanical stretch. Accordingly, TRPP1 and TRPP2 have been implicated in flow-induced Ca2+ elevation in cultured kidney cells (147) (Table 3). Furthermore, TRPP2-deficient embryos do not display the normal flow-induced intracellular Ca2+ elevation at nodal cilia (148). TRPP2 increases EGF-induced, PLC-dependent Ca2+ influx in cultured kidney epithelial cells (149). EGF activates TRPP2-dependent Ca2+ influx by lifting PIP2-induced inhibition of TRPP2 (149). Reconstitution of TRPP2 in lipid bilayers and exogenous expression in cultured cells lead to the development of a Ca2+ permeable, nonselective cation channel with multiple conductance states that are inhibited by Ca2+ and the diuretic amiloride (150) (Table 3). The channel activity of TRPP2 in lipid bilayers is enhanced by its interaction with α-actinin, implicating a role for the actin cytoskeleton in the modulation of TRPP2 (151).
In addition to functioning as a cation influx channel, TRPP2 is also localized in the ER membrane where it is proposed to serve as a new type of Ca2+ release channel (152). The C. elegans homolog of TRPP2 also appears to be a Ca2+ release channel and modulates IP3- and ryanodine-mediated Ca2+ release from intracellular stores (153). Localization of mammalian TRPP2 to the ER is mediated by the phosphofurin acidic cluster sorting protein-2 (PACS-2) interacting with an acidic cluster in the C terminus of TRPP2 (154). Another study reports that TRPP2 impacts Ca2+ release by interacting with the IP3 receptor, thereby modulating IP3-mediated Ca2+ release (155). TRPP3 has been reported to be a voltage-modulated, Ca2+-activated nonselective cation channel (156). TRPP3 is also permeable to monovalent organic cations (157).
TRPML
TRPML1 (mucolipin-1 or MCOLN1) is the founding member of the TRPML subfamily (7–9). Mutations in this protein are responsible for the lysosomal storage disorder mucolipidosis IV, which is characterized by severe neurodegeneration. In addition to TRPML1, mammals encode two other closely related proteins referred to as TRPML2 and TRPML3 (Figure 3) (1). TRPML1 and TRPML2 have lysosomal targeting signals and reside in the lysosomal membrane (158–160). TRPML3 does not have such a targeting signal and resides predominantly in the ER membrane when expressed in cultured cells (158). The C. elegans and Drosophila genomes each encode one TRPML protein (Figure 1c).
TRPML1 is the only member of this subfamily shown to be a cation channel. When expressed in Xenopus oocytes, TRPML1 translocates to the plasma membrane upon elevations in intracellular Ca2+ and is reported to form a nonselective Ca2+ channel (161) (Table 3). Another study found that TRPML1 has multiple conductance states whose function is inhibited by a decrease in pH (162). However, the Ca2+ permeability and pH regulation of TRPML1 are controversial. TRPML1 has been reported to be a monovalent cation permeable channel (160) that fluxes protons, thereby providing a proton leak pathway, regulating intralysosomal pH (163). Indeed, the absence of TRPML1 leads to the acidification of the lysosomal lumen (163). Cleavage of TRPML1 by the lysosomal protease cathepsin B has been reported to inactivate the channel (160) (Table 3). Since cathepsin B is highly pH sensitive, this provides an elegant model for pH-mediated feedback regulation of TRPML function.
HETEROMULTIMERIZATION AMONG TRP PROTEINS
A feature shared by many members of the TRP superfamily of proteins is that closely related TRP channels form heteromultimers. Heteromultimerization is an effective mode for modulating the function, subcellular localization, and biophysical properties of the interacting channels. In the Drosophila eye, TRP is tenfold more abundant than TRPL or TRPγ and is present primarily as a homomultimer. However, TRPL appears to form obligatory heteromultimers, resulting in TRP/TRPL as well as TRPL/TRPγ heteromultimers in vivo and when expressed exogenously in cultured cells (18, 20). Such heteromultimerization provides a mechanism for regulating the channel conductance as the biophysical properties of the heteromultimers are distinct from those of the homomultimers (18, 20).
Mammalian TRPC proteins also form heteromultimers. Heteromultimeric interactions tend to be favored between members that are closely related to each other (164, 165), although this is not a strict rule. TRPC1 is most related to TRPC4/TRPC5, and TRPC1 and TRPC5 have overlapping distribution in the hippocampus and form heteromultimeric channels when coexpressed in cultured cells (166). Interactions between TRPC1 and TRPC5 lead to the generation of novel conductances with biophysical properties distinct from those of TRPC1 and TRPC5 homomultimers (166). Furthermore, biochemical analyses have identified TRPC1, TRPC4, and TRPC5 heteromultimers in rat embryonic brains, which form channels with novel conductances when expressed in vitro (167).
TRPC1 can coassemble with members of the TRPC3/6/7 subfamily as well (18, 167, 168). Native TRPC3 and TRPC6 interact with TRPC1 in the rat embryonic brain (167). Furthermore, a TRPC3-associated conductance in cultured cells requires coexpression of TRPC1 (167, 168). Evidence exists for multiple combinations of interactions between TRPC1, TRPC3/6, and TRPC4/5 (167). Such combinations between different TRP proteins could generate an incredible diversity of channels with an array of distinct biophysical properties and biological functions.
Members of other TRP subfamilies also form heteromultimers. The C. elegans TRPV proteins OSM-9 and OCR-2 may interact in vivo, as each is required for the localization of the other in the sensory cilia (169). Similar interactions might also exist between the Drosophila TRPV proteins, Nanchung and Inactive, as elimination of one of the two proteins in vivo results in instability of the other protein (73). TRPV5 and TRPV6 form heteromultimers (81). TRPM6 and TRPM7 also coassemble, and this interaction is required for the generation of a novel conductance larger than that of either homomultimer and also for the insertion of TRPM6 into the plasma membrane (170). Heteromultimerization-induced trafficking of TRPM6 to the plasma membrane is of particular significance because mutations that disrupt the interactions between TRPM6 and TRPM7 prevent TRPM6 from reaching the plasma membrane, leading to hypomagnesemia and hypocalcemia (170).
Heteromultimeric interactions among group 2 TRPs can also effect subcellular distributions of the channels. The C terminus of TRPP1 interacts with the C terminus of TRPP2, and as mentioned above, this interaction drives the translocation of TRPP2 from intracellular compartments to the plasma membrane (1). The other group 2 TRP channel, TRPML1, interacts strongly with the closely related protein TRPML3 and induces the translocation of the latter to the lysosomes, which appears to be the ultimate destination for the TRPML proteins (158).
SUPRAMOLECULAR COMPLEXES
Several TRP proteins associate with scaffold proteins, which link the channels to large macromolecular assemblies. A key function of such complexes is to control the subcellular distribution of the associated TRP channels.
The Drosophila Signalplex
The first evidence for a TRP-containing signaling complex (signalplex) was demonstrated in Drosophila photoreceptor cells (23). The molecular scaffold, which nucleates the complex, is Inactivation No After Potential D (INAD), a protein consisting largely of five PDZ protein-protein interaction modules. Homomeric interactions between individual INAD molecules further increase INAD’s capacity to simultaneously bind multiple target proteins in the signalplex (23). The signalplex includes two types of proteins: core and noncore. The core complex consists of four proteins—INAD, TRP, PLC (NORPA), and PKC—which are present at similar concentrations and are most likely constitutively bound to INAD (23). Additional noncore INAD binding proteins are TRPL, rhodopsin, calmodulin, myosin III (NINAC), and the immunophilin FKBP59 (23). These noncore proteins are not present in stoichiometric concentrations with INAD and may interact transiently with INAD.
A primary function of the signalplex is to retain the proteins that comprise the core complex in the light-sensitive microvillar structures of the photoreceptor cells, the rhabdomeres (15). Components of the phototransduction cascade are enriched in the rhabdomeres, and mutations in the gene encoding INAD disrupt the rhabdomere localization of TRP, PLC, and PKC (23). The interaction with INAD is required for the retention rather than the targeting of these proteins to the signalplex (171, 172). Furthermore, TRP, PLC, and PKC are unstable when they are not retained in the signalplex owing to a lack of interactions with INAD (23). This has important physiological consequences because altered stoichiometries of these signaling molecules reduce the overall amplitude and affect the termination kinetics of the photoresponse.
There is a reciprocal requirement between INAD and TRP for retention in the rhabdomeres. In addition to the dependence on INAD for the localization of TRP, the retention of INAD in the rhabdomeres requires binding to TRP. Thus TRP has an additional role as an anchoring protein (171, 172).
It might appear intuitive that the primary function of nucleating the key components of the phototransduction cascade in the signalplex is to facilitate fast signaling. This is a particularly attractive hypothesis because cation influx takes place within milliseconds of initiation of the phototransduction cascade. However, a direct association between TRP and INAD is not required for rapid activation because elimination of the TRP-INAD interaction by mutating the INAD-binding site in TRP does not affect the photoresponse (171). The mutated TRP could still interact with the signalplex indirectly via interactions with other INAD binding proteins. Nevertheless, the signalplex does play a signaling role because disruption of the myosin III (NINAC)/INAD interaction causes a profound delay in response termination. This defect results from signaling because noncore INAD binding proteins, such as the myosin III (NINAC), do not depend on INAD for normal stability or localization in the rhabdomeres (23).
Mammalian TRP-Containing Signaling Complexes
Mammalian TRP channels also appear to be assembled into macromolecular complexes. Native TRPC3 in rat pontine neurons associates in vivo with the BDNF receptor TrkB (49). Other members of the TRPC3/TrkB complex have not been defined. TRPC1 localizes to cholesterol-enriched lipid raft domains, caveolae, where it appears to associate with the IP3 receptor and Gαq (173). Disruption of caveolae by cholesterol depletion prevents Ca2+ influx (173). Although this is only indirect evidence for the involvement of a macromolecular complex with TRPC1 for Ca2+ entry, it suggests that disruption of specific membrane domains such as lipid rafts, which may harbor TRP-containing macromolecular complexes, prevents Ca2+ entry.
Mammalian TRPC4 may associate with a macromolecular complex that includes the double-PDZ-domain-containing protein NHERF (or EBP50) (1). Interaction between TRPC4 and NHERF is required for the translocation of the channel to the plasma membrane (1). Furthermore, NHERF can multimerize, allowing it to cluster additional proteins. Known NHERF binding proteins include PLC and the G protein–coupled β2-adrenergic receptor (1).
In addition to PDZ-domain-containing proteins, components involved in TRP signaling also interact with different isoforms of another scaffold protein referred to as Homer. TRPC1 binds to Homer in the brain and localizes it to a complex that contains IP3 receptors and group 1 metabotropic glutamate receptors (mGluR1) (174). The significance of such a complex tethered by Homer is underscored by the involvement of native TRPC1 channels in the development of mGluR1-induced conductances in Purkinje neurons (175). Furthermore, Homer 1b exists in a complex with TRPC3 and IP3 receptors and mediates both TRPC3 trafficking and gating by IP3 receptors (176).
FUNCTIONS OF TRPs
The TRP superfamily of channels participates in a diversity of functions in both excitable and nonexcitable cells. However, a recurring theme is that TRP channels have critical roles in sensory physiology. These include sensory roles in the broadest sense. TRP channels are not only essential in allowing animals to sense the outside world, but also allow individual cells to sense their local environment.
Sensing the Outside World
TRP channels play critical roles in sensory modalities, such as touch, hearing, taste, olfaction, vision, and thermal sensation, in animals ranging from worms to flies, mice, and humans.
Chemosensation
Genetic analyses of the C. elegans osm-9 gene, which encodes one of the two archetypal TRPV proteins, provided the first evidence that TRP channels participate in chemosensation (54). Mutations in osm-9 (as well as in another TRPV gene, ocr-2) cause defects in the response to odorants (54, 169). Given that both osm-9 and ocr-2 are expressed in neurons that sense odorants, it is likely they are involved in olfactory transduction. In addition, the OSM-9 and OCR-2 proteins are required for multiple sensory modalities, including nociception and osmosensation (see below).
Several mammalian TRP channels are required for chemosensation, one of which (TRPM5) is a taste transduction channel (177, 178). TRPM5 is broadly required for the taste modality as the responses to sweet, bitter, and amino acids are greatly reduced, if not eliminated, owing to knockout of the mouse TRPM5 gene (97, 178).
TRPC2 functions in the pheromone response, which in mammals is mediated through the VNO. TRPC2 is expressed in the VNO (44), and TRPC2-deficient male mice display intriguing behavioral phenotypes (179, 180). These include a failure to show aggression toward intruding males and a propensity to mate with either males or females. It is noteworthy that TRPC2 is a pseudogene in humans (34), and the VNO is generally thought to be a vestigial organ. In addition to TRPM5 and TRPC2, thermoTRPs contribute to the perception of chemical stimuli and, therefore, function in chemosensation (see below).
Thermosensation and nociception
Multiple mammalian TRP channels are activated by temperature changes and account for a large proportion of the temperature range to which mammals respond. As described above, TRPV1 and TRPV2 are sensors for uncomfortably warm (>43°C) (56) and very hot (>52°C) (64) temperatures, respectively, whereas TRPV3 (>30–39°C) (181–183) and TRPV4 (~25–34°C) (184, 185) contribute to the perception of moderate temperatures. TRPM8 appears to function in our perception of cool temperatures (115, 116), and TRPA1 may be a cold sensor (127, 128); however, this latter conclusion is controversial (124, 126). Although the thermoTRPs are expressed in neurons such as those in the dorsal root or trigeminal ganglia, which are known to function in thermosensation, TRPV3 and TRPV4 are also expressed in skin keratinocytes (181, 184). These channels appear to be heat activated in these skin cells (181, 186, 187), although the mechanism through which temperature stimulation of keratinocytes contributes to thermosensation remains a matter of speculation (reviewed in 188). One possibility is that the activated keratinocytes release a factor, such as ATP, which activates an ATP-gated channel in the sensory neurons that innervate the skin (188).
The requirements for TRPV1, TRPV3, and TRPV4 in thermosensation have been confirmed in knockout mice. TRPV1-deficient mice do not display a response to noxious heat until the temperature is increased to ~55°C (189, 190). Mice missing TRPV3 exhibit a thermal preference defect in a temperature range consistent with the biophysical analyses. When given a choice between 35°C and room temperature, wild-type animals have a strong bias to occupy the 35°C zone, whereas the TRPV3-knockout mice display only a slight preference for the higher temperature (191). TRPV4−/− mutant mice also show a variety of behavioral phenotypes, including an inability to discriminate between 30 and 34°C (192, 193).
There are two reports describing TRPA1-deficient mice, and whether these mice are defective in the cold-temperature detection remains controversial. According to one study, TRPA1−/− mice do not show impairment in temperature sensitivity (126), whereas a second study reports a defect in paw withdrawal from a cold (0°C) surface (128). It is unclear whether these differences result from slightly different behavioral paradigms, genetic backgrounds, or the production of truncated versions of TRPA1, which in turn could affect other channels.
Despite the controversy concerning the thermosensory role of TRPA1, all three Drosophila TRPs known to function in thermosensation are TRPA proteins. The Drosophila homolog of the mammalian TRPA1, dTRPA1, has a role in thermotaxis. When given a choice between two temperature zones, 27–31°C and 35–41°C, wild-type larvae choose the cooler zone. Reduction of dTRPA1 expression, using small interfering RNAs (siRNAs), eliminates this preference (133). Whereas mammalian TRPA1 may be a cold sensor (127), dTRPA1 is necessary for detecting warm temperatures (132, 133). Painless, the first invertebrate thermoTRP identified, is required for the avoidance response in larvae to noxious heat (>39–41°C) (134). Another TRPA protein, Pyrexia, is necessary for flies to withstand several minutes of exposure to warm temperatures, such as 40°C, without becoming paralyzed (131).
ThermoTRPs are also required for the response to chemical stimuli
A shared feature of mammalian thermoTRPs is that they respond to chemical stimuli. This property was initially found for TRPV1, providing a molecular explanation for the long-known observation that certain compounds, such as capsaicin, elicit the same sensation as thermal heat (56). In addition to displaying impaired responses to heat and vanilloid compounds, TRPV1−/− mice are deficient in their reaction to protons and display minimal thermal hyperalgesia (189, 190), which is enhanced by proalgesic agents produced at the site of injury. Thus TRPV1 is a molecular integrator for noxious thermal and chemical stimuli. Although a role for TRPA1 as a cold sensor is still a matter of debate, there is agreement that TRPA1−/− animals are deficient in their response to chemical irritants, such as mustard oils (126, 128). The Drosophila TRPA Painless is also required for the avoidance of isothiocyanate, which is the pungent compound in wasabi and mustard oils (135).
Thermal and chemical stimuli both affect TRPM5 activity (97, 177, 178), and this convergence has fascinating effects on the response to sweet tastants. Stimulation of the chorda tympani nerve with sugars is normally increased by temperatures over the range of 15–35°C. However, the residual sugar response in TRPM5−/− mice is not augmented by temperature (97). Thus TRPM5 appears to be required for the enhanced perception of sweetness at higher temperatures.
Mechanosensation
At least one thermoTRP functions in mechanosensation. TRPV4 is activated in vitro by hypotonicity (194), and TRPV4−/− mice display defects in osmotic regulation (195). The role of TRPVs in osmosensation is conserved, as the C. elegans TRPVs OSM-9 and OCR-2 are required for the avoidance of high osmolarity and mechanical stimuli, in addition to functioning in chemosensation. Rat TRPV4 restores normal avoidance to hypoosmotic and mechanical stimuli in osm-9 mutant worms, when it is expressed in those neurons (ASH) that normally express osm-9 and function in these modalities (196). A conundrum is that TRPV4 is activated in vitro by hypo- rather than hypertonicity. A role for TRPs in osmosensation also occurs in yeast, as TRPY proteins function in the release of Ca2+ from vacuoles in response to hyperosmotic conditions (2, 3).
The identity of an auditory transduction channel in mammals is not resolved. Such a channel is predicted to be expressed at the tip of the stereocilia in hair cells of the inner ear, where mechanical force mediated through the gating spring is thought to open the channels. TRPA1 was a candidate auditory transduction channel as it is situated at the tips of the stereocilia, and the reduction of TRPA1 expression in zebrafish and in the mouse using morpholinos and siRNAs, respectively, reduced sound-evoked potentials (129). However, the auditory response is normal in TRPA1−/− mice (126, 128). It has been suggested that TRPA1 may be a hair-cell transduction channel only during early development because the morpholino and siRNA analyses were performed in early stage zebrafish and embryonic mice, whereas the response in the knockout animals was analyzed in postnatal mice (128).
Two mammalian TRP channels function in the auditory response; however, based on the expression pattern of the channels and/or the phenotypes of the mutant animals, neither appears to be a transduction channel. Mice with severe mutations in TRPML3 exhibit a variety of phenotypes, including progressive disorganization of the inner and outer hair cell bundles (197). TRPML3 is expressed in vesicles throughout the hair-cell bodies, leading to the speculation that it functions in vesicular or intracellular ion homeostasis in hair cells (197). The auditory response is normal in young TRPV4−/− mice; however, in older mutant mice, there is severe auditory impairment (198). TRPN1 is suggested to be a mechanotransduction channel in the zebrafish because TRPN1 RNA is expressed in hair cells, and the reduction in TRPN1 expression in larvae using morpholinos reduces sound-evoked potentials (199). In the frog the TRPN1 protein does not appear to be the transduction channel because it is not situated in the stereocilia but instead in a motile cilium in the hair cells (200).
The founding TRPN protein, Drosophila NOMPC (136), and two Drosophila TRPV proteins (Nanchung and Inactive) are required for auditory sensitivity (73, 74, 201). In wild-type animals, amplification of auditory signals is nonlinear. Small signals are more greatly amplified through mechanical feedback vibrations in the mechanosensory cells in the antenna. However, nompC mutant flies show linear amplification at all levels of sound stimuli (201). The converse is observed in the nanchung and inactive mutants, both of which exhibit profound defects in hearing (73, 74) as a result of excessive amplification at the lower sound intensities (201). Since the mechanotransduction channel is expected to be required for feedback and amplification, it has been proposed that NOMPC is an auditory transduction channel (201). There must be at least one additional such channel, given that the nompC mutation does not abolish the auditory response.
The proposal that Drosophila NOMPC protein is an auditory transduction channel is consistent with the initial report that it is a mechanotransduction channel required for touch (136). The nompC gene is expressed in mechanosensory organs, and the mechanoreceptor currents generated in wild-type animals in response to deflection of the sensory bristles are absent in nompC flies. The mutant animals show behavioral phenotypes—they are uncoordinated and insensitive to a light touch. The Drosophila trpA gene painless is dispensable for the normal response to a light touch but functions in the avoidance response to noxious mechanical stimuli (134). The worm NOMPC homolog TRP-4 is a stretch-sensitive channel, and mutations in trp-4 cause locomotor defects (202).
Phototransduction
Given that phototransduction in mammalian rods and cones operates through closure of a cGMP-gated channel, it is curious that Drosophila phototransduction utilizes TRP channels. Recent studies suggest that TRP channels may function in a small subset of mammalian retinal ganglion cells, which are photosensitive (pRGCs) and contribute to circadian rhythm (reviewed in 203). These pRGCs utilize a phototransduction cascade that bears intriguing similarities to Drosophila phototransduction. The cascade initiates with light activation of an opsin, melanopsin, and culminates with depolarization of the cells. Heterologous expression studies indicate that light activation of melanopsin operates through a PLC and TRPC3 (204–206); however, it is unclear which TRP channel is coupled to the light-activated cascade in the pRGCs. Nevertheless, the cascades functioning in pRGCs and Drosophila photoreceptor cells appear to have a common origin.
Sensing the Local Cellular Environment
In addition to sensing stimuli from the external environment, TRP channels enable cells to respond to cues in their local environment.
TRPC channels in growth cone guidance, neurite outgrowth, and synaptic activity
TRP channels play roles during neurite outgrowth and growth cone guidance in the developing nervous system. Among the known guidance cues are netrins and BDNF, which exert either attractive or repulsive effects on growth cones through the induction of Ca2+ influx. TRPC3 was a candidate for regulation of growth cone guidance by BDNF because it is activated in the developing mammalian brain by BDNF (49). In support of this possibility, knockdown of TRPC3 expression with siRNAs inhibits BDNF-induced turning of growth cones in cultured cerebellar granule cells (207). Similarly, reduction in expression of the Xenopus TRPC1 using morpholinos interferes with growth cone turning in response to either netrin-1 or BDNF (208, 209). Another TRP channel, TRPC5, operates in neurite extension, as exposure of hippocampal neurons to EGF promotes the plasma membrane insertion of TRPC5, which inhibits neurite outgrowth (210).
TRPC channels function postsynaptically in response to the activation of metabotropic receptors. Activation of metabotropic serotonin receptors in the dendrites of thalamic interneurons leads to release of the neurotransmitter GABA (γ-aminobutyric acid) from the dendrites and local GABAergic inhibition. In neurons obtained from TRPC4−/− knockout animals, the dendritic release of GABA is greatly reduced in thalamic interneurons (211). In contrast to this inhibitory role, TRPC1 appears to underlie an excitatory postsynaptic current, following activation of metabotropic glutamate receptors in Purkinje cells, because manipulations that interfere with TRPC1 activity reduce the excitatory postsynaptic current (175).
Vascular tone
Influx of Ca2+ into vascular endothelial cells causes the release of vasoactive agents, which in turn induces Ca2+ influx in smooth muscle cells and affects vascular tone and blood pressure. Many TRP channels are expressed in the endothelium and have been implicated in endothelial function, although the strongest genetic evidence has been provided for TRPC4. Mice lacking TRPC4 are deficient for an IP3-induced Ca2+ influx current and display greatly reduced vasorelaxation (212). Elimination of TRPC6 increases vascular smooth muscle contractility and blood pressure (213). Paradoxically, there is an increase in basal cation influx. This latter effect results from increased expression of TRPC3, which in the absence of TRPC6 shows constitutive activity. These latter results are consistent with the evidence that TRPC6 and TRPC3 form heteromultimers (43) and suggest that TRPC3 is constitutively active in the absence of TRPC6. This result is reminiscent of the suppression of constitutive activity of Drosophila TRPL, by interactions with TRP (18).
Secretion of fluids and hormones
TRP channels play important roles in the secretion of fluids, electrolytes, and hormones in vertebrates and invertebrates. In addition to functioning during phototransduction, TRPL is required for Ca2+-mediated fluid transport in a Drosophila epithelium (214). In mammals, TRPC1 may play a role in fluid secretion as it is expressed in human salivary gland cells and its level of expression is correlated with Ca2+ entry and fluid secretion in these cells (215).
Two TRPM channels are implicated in insulin secretion. TRPM4 is expressed in an insulin pancreatic β-cell line, and expression of a dominant negative form of this channel reduces glucose-induced insulin secretion (216). TRPM2 is also expressed in insulin-producing pancreatic β-cells, and TRPM2 siRNAs decrease insulin secretion (217). The activation of TRPM2 in these cells is proposed to require both cyclic ADP–ribose and 37°C because the channel is poorly activated by cyclic ADP–ribose at 25°C. Therefore, TRPM2 is an additional example of chemical and thermal inputs converging on a TRP channel.
Kidney, urinary bladder, and bone function
At least one TRP is required for urinary bladder function as mutations in TRPV1 result in an increase in urinary bladder capacity and inefficient voiding (218). Urothelial cells from the TRPV1−/− animals show a defect in stretch-induced ATP release, which may underlie the bladder phenotype because purinergic signaling has been linked to bladder function. It is not known whether TRPV1 is a mechanosensitive channel or whether it is responding indirectly to membrane stretch.
Definitive evidence that TRP channels are required for renal function in humans is that mutations in TRP channels underlie kidney diseases (see the following section below). Elimination of mouse TRPV5 causes the animals to excrete high levels of Ca2+ into the urine, owing to a defect in Ca2+ reabsorption along the kidney’s distal convolution, which is the site of TRPV5 expression (219). TRPV5 is also expressed in osteoclasts, and TRPV5−/− animals show a defect in transcellular Ca2+ transport during osteoclastic bone resorption (220). The critical role for TRPV5 in these processes is consistent with the observation that TRPV5 and the highly related TRPV6 are the most Ca2+-selective channels among the mammalian TRPs (1).
In the zebrafish, loss-of-function mutations in TRPM7 result in dwarfism, skeletal abnormalities, kidney stone formation, melanophore development, and touch-response defects (221). In addition, there is mineralization in the mesonephric tubules and changes in ossification. TRPM7 is permeable to Mg2+ and Ca2+ (99), and supplementation with either Mg2+ or Ca2+ partially rescues the melanophore-development phenotype, whereas supplementation with Ca2+ partially restores the touch responsiveness. In certain cell types, TRPM7 is essential for viability, as deletion of TRPM7 in the chicken DT-40 cell line results in cell lethality (99). This phenotype results from a defect in Mg2+ homeostasis, as the addition of Mg2+ to the media rescues the lethality (105).
TRP CHANNELS AND DISEASE
Four diseases have been shown to be caused by mutations in TRP channels, and preliminary data indicate that disruptions in TRP channels underlie many additional diseases. Uncontrolled activity of TRP channels may also have pathological consequences and cause neurodegeneration due to oxidative stress.
Hypomagnesemia and Hypocalcemia Owing to Mutations in TRPM6
TRPM proteins are critical for Mg2+ uptake in organisms ranging from worms to humans. TRPM6 is expressed in the intestinal brush border and in the renal convoluted tubule, where it is required for Mg2+ absorption (101). Of particular significance, mutations in the TRPM6 chanzyme result in hypomagnesemia with secondary hypocalcemia (13, 14).
A role for TRPM proteins in Mg2+ homeostasis is evolutionarily conserved. Two of the four trpm genes in worms are expressed in the intestines, and the inability of gon-2; gtl-1 mutants to reach adulthood is suppressed by Mg2+ supplementation (222). Furthermore, the gon-2; gtl-1 animals display increased tolerance to Ni2+ toxicity (222), reminiscent of the findings that TRPM7 is permeable to Ni2+ and other trace metals (99).
TRPC6, Podocyte Function, and Kidney Disease
Mutations in TRPC6 cause one type of kidney disorder, focal and segmental glomerulosclerosis (FSGS), which is characterized by a breach in the capillary/kidney permeability barrier and progression to end-stage renal disease (11, 12). A critical structure contributing to the filtration system in the kidney glomerulus is the series of interdigitated foot processes extending from the podocytes and surrounding the capillaries. The slit diaphragm, situated between the foot processes, forms a multiprotein barrier that allows the passage of small molecules from the capillaries to the glomerulus. Disruption of this permeability barrier, due to mutation of TRPC6, can lead to proteinuria and end-stage renal disease. TRPC6 has been proposed to be an essential channel in the slit diaphragm because it is localized in the foot processes, near the slit diaphragm, and directly interacts with two proteins, nephron and podocin, known to be situated in these regions. FSGS patients also display hypertension, which is a clinical manifestation consistent with higher blood pressure in the TRPC6-knockout mice (213). Whether older TRPC6−/− mice display kidney defects remains to be reported. The mechanism of TRPC6 activation in the podocytes is also unresolved; given the mechanosensitivity of these cells, TRPC6 may be mechanically gated in podocytes.
TRPPs and Autosomal Dominant Polycystic Kidney Disease
ADPKD is the most common disease worldwide of monogenic origin and results from mutations either in the channel TRPP2 or the associated protein TRPP1 (reviewed in 223). The hallmark of this disorder is renal cysts and kidney failure, although this is a systemic disease, which can affect many tissues. The patients are heterozygous for the initial mutation in either TRPP1 or TRPP2, and the disease results from a somatic mutation in the second copy of the gene.
The normal functions of TRPP proteins remain poorly understood; however, a key insight is that the TRPP2 and TRPP1 tend to be localized in axonemal structures, such as monocilia (224). These cilia are small appendages that extend from most cells and are capable of sensing shear stress during fluid flow. TRPP2 is also expressed in the motile node cell monocilia (148, 225), which is critical in establishing left-right asymmetry during embryogenesis by generating fluid flow. Cells from TRPP2−/− embryos exhibit laterality defects and do not display the Ca2+ rise in the node cilia, which is induced in wild-type embryos upon fluid flow over the cilia (148, 225). Thus TRPP2 may function as a mechanosensor, which detects the shear force resulting from fluid flow (147).
Specialized axonemal structures related to monocilia include mechanosensors, the outer segments of mammalian photoreceptor cells, and sperm. In support of an evolutionarily conserved role for TRPPs in axonemal structures, worm TRPPs are concentrated in sensory cilia and function in male sensory behavior (137). The Drosophila TRPP2 AMO is localized to the sperm tail (139) and is essential for entry in the female storage organs (138, 139).
TRPML1 and Mucolipidosis Type IV
MLIV is a severe neurodegenerative disorder, which arises from mutations in TRPML1 (7–9). The clinical manifestations typically become evident by the time children are two to three years old of age and include neurodegeneration, mental retardation, achlorhydria, and retinal degeneration. MLIV is primarily a lysosomal storage disorder, and as is typical of other lysosomal storage disorders, cells from patients with MLIV contain many large vesicles and accumulate lysosomal storage components, such as phospholipids, mucopolysaccharides, and gangliosides (226). There does not appear to be a defect in lysosomal enzymes, and the specific role for TRPML1 is not known. TRPML1 may be required for lysosomal reformation/biogenesis (227), and this model is supported by analyses of worms harboring a mutation in cup-5, the C. elegans homolog of TRPML1 (228).
Oxidative Stress, Constitutively Active TRPs, and Neuronal Cell Death
Hypoxia can lead to excessive Ca2+ influx and cell death owing to Ca2+ overload. Such a phenomenon is of particular importance in the brain, where oxidative stress leads to neuronal cell death following stroke and certain chronic neurodegenerative diseases. There is accumulating evidence that TRP channels contribute significantly to neuronal cell death under anoxic conditions, although the first demonstration that oxidative stress causes constitutive activity of a TRP was obtained for TRPC3 in aortic endothelial cells (229). Drosophila TRP and TRPL are activated in vivo following application of mitochondrial uncouplers (230), and mutations that cause constitutive activation of TRP result in massive photoreceptor cell death (231). Evidence that this death indeed results from Ca2+ overload is that the retinal degeneration is suppressed by overexpression of the Na+/Ca2+ exchanger (232).
Two mediators of hypoxia-induced neuronal cell death in the mammalian brain appear to be TRPM2 and TRPM7. Cells heterologously expressing TRPM2 display an increased susceptibility to cell death following exposure to reactive oxygen species (110). Moreover, inhibition of native TRPM2 expression in a rat insulinoma cell line and in cultured cortical neurons reduces H2O2-induced cell death (110, 233). Expression of a naturally occurring truncated form of TRPM2 decreased the sensitivity to cell death owing to oxidative stress (114). In a related model for neuronal cell death induced by oxygen glucose deprivation, suppression of native TRPM7 expression inhibited cell death in cortical neuronal cultures (234).
The concept that TRP channels are activated under hypoxic conditions does not apply only to pathological conditions, such as during ischemia and in some chronic neurological diseases, but also most likely reflects a physiological role for TRP channels. In the worm, O2 levels affect multiple behaviors, and two TRPV channels (OSM-9 and OCR-2) are required for the avoidance of high O2 (235). The effects of O2 on OSM-9 or OCR-2 activity are not known.
FUTURE PERSPECTIVES
Based on an array of highly suggestive studies that are not reviewed here owing to space limitations, it is likely that many more diseases will be shown to result from mutations or changes in the activities of TRP channels. These include cardiovascular diseases, sensory deficits, abnormal sensitivities to pain, gastrointestinal diseases, additional renal and neurodegenerative diseases, certain cancers, asthma, and even psychiatric diseases. Consistent with this last possibility is the association of alterations in TRPM2 with bipolar disorder (236, 237). Finally, TRP channels are exciting targets for drug development. Inhibitors of TRP channels hold the potential for the control of pain, asthma, and neurodegenerative diseases, which are associated with increased activities of TRP channels.
Acknowledgments
Work in the laboratory of C.M. is supported by the NEI, NIDCD, and the March of Dimes.
Glossary
- TRP
Transient Receptor Potential
- PLC
phospholipase C
- TRPL
TRP-like
- PIP2
phosphoinositide-4,5-bisphosphate
- DAG
diacylglycerol
- IP3
inositol-1,4,5-trisphosphate
- PUFA
polyunsaturated fatty acid
- PKC
protein kinase C
- INAD
inactivation no after potential D
LITERATURE CITED
- 1.Montell C. Sci STKE. 2005;2005:re3. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- 2.Palmer CP, Zhou XL, Lin J, Loukin SH, Kung C, Saimi Y. Proc Natl Acad Sci USA. 2001;98:7801–5. doi: 10.1073/pnas.141036198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Denis V, Cyert MS. J Cell Biol. 2002;156:29–34. doi: 10.1083/jcb.200111004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montell C, Rubin GM. Neuron. 1989;2:1313–23. doi: 10.1016/0896-6273(89)90069-x. [DOI] [PubMed] [Google Scholar]
- 5.Montell C, Birnbaumer L, Flockerzi V. Cell. 2002;108:595–98. doi: 10.1016/s0092-8674(02)00670-0. [DOI] [PubMed] [Google Scholar]
- 6.Palmer CP, Aydar E, Djamgoz MB. Biochem J. 2005;387:211–19. doi: 10.1042/BJ20041710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun M, Goldin E, Stahl S, Falardeau JL, Kennedy JC, et al. Hum Mol Genet. 2000;9:2471–78. doi: 10.1093/hmg/9.17.2471. [DOI] [PubMed] [Google Scholar]
- 8.Bassi MT, Manzoni M, Monti E, Pizzo MT, Ballabio A, Borsani G. Am J Hum Genet. 2000;67:1110–20. doi: 10.1016/s0002-9297(07)62941-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bargal R, Avidan N, Ben-Asher E, Olender Z, Zeigler M, et al. Nat Genet. 2000;26:118–23. doi: 10.1038/79095. [DOI] [PubMed] [Google Scholar]
- 10.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, et al. Science. 1996;272:1339–42. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 11.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, et al. Science. 2005;308:1801–4. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 12.Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, et al. Nat Genet. 2005;37:739–44. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walder RY, Landau D, Meyer P, Shalev H, Tsolia M, et al. Nat Genet. 2002;31:171–74. doi: 10.1038/ng901. [DOI] [PubMed] [Google Scholar]
- 14.Schlingmann KP, Weber S, Peters M, Nejsum LN, Vitzthum H, et al. Nat Genet. 2002;31:166–70. doi: 10.1038/ng889. [DOI] [PubMed] [Google Scholar]
- 15.Montell C. Annu Rev Cell Dev Biol. 1999;15:231–68. doi: 10.1146/annurev.cellbio.15.1.231. [DOI] [PubMed] [Google Scholar]
- 16.Hardie RC, Minke B. Neuron. 1992;8:643–51. doi: 10.1016/0896-6273(92)90086-s. [DOI] [PubMed] [Google Scholar]
- 17.Vaca L, Sinkins WG, Hu Y, Kunze DL, Schilling WP. Am J Physiol Cell Physiol. 1994;266:C1501–5. doi: 10.1152/ajpcell.1994.267.5.C1501. [DOI] [PubMed] [Google Scholar]
- 18.Xu XZ, Li HS, Guggino WB, Montell C. Cell. 1997;89:1155–64. doi: 10.1016/s0092-8674(00)80302-5. [DOI] [PubMed] [Google Scholar]
- 19.Phillips AM, Bull A, Kelly LE. Neuron. 1992;8:631–42. doi: 10.1016/0896-6273(92)90085-r. [DOI] [PubMed] [Google Scholar]
- 20.Xu XZ, Chien F, Butler A, Salkoff L, Montell C. Neuron. 2000;26:647–57. doi: 10.1016/s0896-6273(00)81201-5. [DOI] [PubMed] [Google Scholar]
- 21.Niemeyer BA, Suzuki E, Scott K, Jalink K, Zuker CS. Cell. 1996;85:651–59. doi: 10.1016/s0092-8674(00)81232-5. [DOI] [PubMed] [Google Scholar]
- 22.Leung HT, Geng C, Pak WL. J Neurosci. 2000;20:6797–803. doi: 10.1523/JNEUROSCI.20-18-06797.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montell C. J Physiol. 2005;567.1:45–51. doi: 10.1113/jphysiol.2005.092551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hardie RC, Raghu P. Cell Calcium. 1998;24:153–63. doi: 10.1016/s0143-4160(98)90125-7. [DOI] [PubMed] [Google Scholar]
- 25.Ranganathan R, Bacskai BJ, Tsein RY, Zuker CS. Neuron. 1994;13:837–48. doi: 10.1016/0896-6273(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 26.Acharya JK, Jalink K, Hardy RW, Hartenstein V, Zuker CS. Neuron. 1997;18:881–87. doi: 10.1016/s0896-6273(00)80328-1. [DOI] [PubMed] [Google Scholar]
- 27.Estacion M, Sinkins WG, Schilling WP. J Physiol. 2001;530:1–19. doi: 10.1111/j.1469-7793.2001.0001m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chyb S, Raghu P, Hardie RC. Nature. 1999;397:255–59. doi: 10.1038/16703. [DOI] [PubMed] [Google Scholar]
- 29.Raghu P, Usher K, Jonas S, Chyb S, Polyanovsky A, Hardie RC. Neuron. 2000;26:169–79. doi: 10.1016/s0896-6273(00)81147-2. [DOI] [PubMed] [Google Scholar]
- 30.Kwon Y, Montell C. Curr Biol. 2006;16:723–29. doi: 10.1016/j.cub.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Murillas I, Pettitt T, Macdonald E, Okkenhaug H, Georgiev P, et al. Neuron. 2006;49:533–46. doi: 10.1016/j.neuron.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 32.Bähner M, Frechter S, Da Silva N, Minke B, Paulsen R, Huber A. Neuron. 2002;34:83–93. doi: 10.1016/s0896-6273(02)00630-x. [DOI] [PubMed] [Google Scholar]
- 33.Xu XZ, Sternberg PW. Cell. 2003;114:285–97. doi: 10.1016/s0092-8674(03)00565-8. [DOI] [PubMed] [Google Scholar]
- 34.Wes PD, Chevesich J, Jeromin A, Rosenberg C, Stetten G, Montell C. Proc Natl Acad Sci USA. 1995;92:9652–56. doi: 10.1073/pnas.92.21.9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu X, Chu PB, Peyton M, Birnbaumer L. FEBS Lett. 1995;373:193–98. doi: 10.1016/0014-5793(95)01038-g. [DOI] [PubMed] [Google Scholar]
- 36.Montell C, Birnbaumer L, Flockerzi V, Bindels RJ, Bruford EA, et al. Mol Cell. 2002;9:229–31. doi: 10.1016/s1097-2765(02)00448-3. [DOI] [PubMed] [Google Scholar]
- 37.Venkatachalam K, van Rossum DB, Patterson RL, Ma HT, Gill DL. Nat Cell Biol. 2002;4:E263–72. doi: 10.1038/ncb1102-e263. [DOI] [PubMed] [Google Scholar]
- 38.Ma HT, Patterson RL, van Rossum DB, Birnbaumer L, Mikoshiba K, Gill DL. Science. 2000;287:1647–51. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- 39.Lockwich T, Singh BB, Liu XB, Ambudkar IS. J Biol Chem. 2001;276:42401–8. doi: 10.1074/jbc.M106956200. [DOI] [PubMed] [Google Scholar]
- 40.Vazquez G, Lievremont JP, Bird GS, Putney JW., Jr Proc Natl Acad Sci USA. 2001;98:11777–82. doi: 10.1073/pnas.201238198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venkatachalam K, Ma HT, Ford DL, Gill DL. J Biol Chem. 2001;276:33980–85. doi: 10.1074/jbc.C100321200. [DOI] [PubMed] [Google Scholar]
- 42.Venkatachalam K, Zheng F, Gill DL. J Biol Chem. 2003;278:29031–40. doi: 10.1074/jbc.M302751200. [DOI] [PubMed] [Google Scholar]
- 43.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Nature. 1999;397:259–63. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 44.Liman ER, Corey DP, Dulac C. Proc Natl Acad Sci USA. 1999;96:5791–96. doi: 10.1073/pnas.96.10.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lucas P, Ukhanov K, Leinders-Zufall T, Zufall F. Neuron. 2003;40:551–61. doi: 10.1016/s0896-6273(03)00675-5. [DOI] [PubMed] [Google Scholar]
- 46.Lievremont JP, Numaga T, Vazquez G, Lemonnier L, Hara Y, et al. J Biol Chem. 2005;280:35346–51. doi: 10.1074/jbc.M507606200. [DOI] [PubMed] [Google Scholar]
- 47.Soboloff J, Spassova M, Xu W, He LP, Cuesta N, Gill DL. J Biol Chem. 2005;280:39786–94. doi: 10.1074/jbc.M506064200. [DOI] [PubMed] [Google Scholar]
- 48.Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP. Nat Cell Biol. 2005;7:179–85. doi: 10.1038/ncb1218. [DOI] [PubMed] [Google Scholar]
- 49.Li HS, Xu XZ, Montell C. Neuron. 1999;24:261–73. doi: 10.1016/s0896-6273(00)80838-7. [DOI] [PubMed] [Google Scholar]
- 50.Vazquez G, Bird GS, Mori Y, Putney JW., Jr J Biol Chem. 2006;281:25250–58. doi: 10.1074/jbc.M604994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Odell AF, Scott JL, van Helden DF. J Biol Chem. 2005;280:37974–87. doi: 10.1074/jbc.M503646200. [DOI] [PubMed] [Google Scholar]
- 52.Bezzerides V, Ramsey S, Greka A, Clapham DE. Nat Cell Biol. 2004;6:709–20. doi: 10.1038/ncb1150. [DOI] [PubMed] [Google Scholar]
- 53.Singh BB, Lockwich TP, Bandyopadhyay BC, Liu X, Bollimuntha S, et al. Mol Cell. 2004;15:635–46. doi: 10.1016/j.molcel.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 54.Colbert HA, Smith TL, Bargmann CI. J Neurosci. 1997;17:8259–69. doi: 10.1523/JNEUROSCI.17-21-08259.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kahn-Kirby AH, Dantzker JL, Apicella AJ, Schafer WR, Browse J, et al. Cell. 2004;119:889–900. doi: 10.1016/j.cell.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 56.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. Nature. 1997;389:816–24. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 57.Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, et al. Nature. 1999;400:452–57. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]
- 58.Xu H, Blair NT, Clapham DE. J Neurosci. 2005;25:8924–37. doi: 10.1523/JNEUROSCI.2574-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McNamara FN, Randall A, Gunthorpe MJ. Br J Pharmacol. 2005;144:781–90. doi: 10.1038/sj.bjp.0706040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Macpherson LJ, Geierstanger BH, Viswanath V, Bandell M, Eid SR, et al. Curr Biol. 2005;15:929–34. doi: 10.1016/j.cub.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 61.Trevisani M, Smart D, Gunthorpe MJ, Tognetto M, Barbieri M, et al. Nat Neurosci. 2002;5:546–51. doi: 10.1038/nn0602-852. [DOI] [PubMed] [Google Scholar]
- 62.Liu L, Zhu W, Zhang ZS, Yang T, Grant A, et al. J Neurophysiol. 2004;91:1482–91. doi: 10.1152/jn.00922.2003. [DOI] [PubMed] [Google Scholar]
- 63.Zhang N, Inan S, Cowan A, Sun R, Wang JM, et al. Proc Natl Acad Sci USA. 2005;102:4536–41. doi: 10.1073/pnas.0406030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D. Nature. 1999;398:436–41. doi: 10.1038/18906. [DOI] [PubMed] [Google Scholar]
- 65.Penna A, Juvin V, Chemin J, Compan V, Monet M, Rassendren FA. Cell Calcium. 2006;39:495–507. doi: 10.1016/j.ceca.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 66.Muraki K, Iwata Y, Katanosaka Y, Ito T, Ohya S, et al. Circ Res. 2003;93:829–38. doi: 10.1161/01.RES.0000097263.10220.0C. [DOI] [PubMed] [Google Scholar]
- 67.Xu H, Delling M, Jun JC, Clapham DE. Nat Neurosci. 2006;9:628–35. doi: 10.1038/nn1692. [DOI] [PubMed] [Google Scholar]
- 68.Hu HZ, Xiao R, Wang C, Gao N, Colton CK, et al. J Cell Physiol. 2006;208:201–12. doi: 10.1002/jcp.20648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Macpherson LJ, Hwang SW, Miyamoto T, Dubin AE, Patapoutian A, Story GM. Mol Cell Neurosci. 2006;32:335–43. doi: 10.1016/j.mcn.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 70.Liu X, Bandyopadhyay B, Nakamoto T, Singh B, Liedtke W, et al. J Biol Chem. 2006;281:15485–95. doi: 10.1074/jbc.M600549200. [DOI] [PubMed] [Google Scholar]
- 71.Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Nature. 2003;424:434–38. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- 72.Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Proc Natl Acad Sci USA. 2004;101:396–401. doi: 10.1073/pnas.0303329101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gong ZF, Son WS, Chung YD, Kim JW, Shin DW, et al. J Neurosci. 2004;24:9059–66. doi: 10.1523/JNEUROSCI.1645-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim J, Chung YD, Park DY, Choi SK, Shin DW, et al. Nature. 2003;424:81–84. doi: 10.1038/nature01733. [DOI] [PubMed] [Google Scholar]
- 75.Yue LX, Peng JB, Hediger MA, Clapham DE. Nature. 2001;410:705–9. doi: 10.1038/35070596. [DOI] [PubMed] [Google Scholar]
- 76.Voets T, Prenen J, Fleig A, Vennekens R, Watanabe H, et al. J Biol Chem. 2001;276:47767–70. doi: 10.1074/jbc.C100607200. [DOI] [PubMed] [Google Scholar]
- 77.Morenilla-Palao C, Planells-Cases R, Garcia-Sanz N, Ferrer-Montiel A. J Biol Chem. 2004;279:25665–72. doi: 10.1074/jbc.M311515200. [DOI] [PubMed] [Google Scholar]
- 78.Zhang XM, Huang JH, McNaughton PA. EMBO J. 2005;24:4211–23. doi: 10.1038/sj.emboj.7600893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu HS, Fu Y, Tian W, Cohen DM. Am J Physiol Renal Physiol. 2006;290:F1103–9. doi: 10.1152/ajprenal.00245.2005. [DOI] [PubMed] [Google Scholar]
- 80.Cuajungco MP, Grimm C, Oshima K, D’Hoedt D, Nilius B, et al. J Biol Chem. 2006;281:18753–62. doi: 10.1074/jbc.M602452200. [DOI] [PubMed] [Google Scholar]
- 81.Hoenderop JG, Voets T, Hoefs S, Weidema F, Prenen J, et al. EMBO J. 2003;22:776–85. doi: 10.1093/emboj/cdg080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG. Science. 2005;310:490–93. doi: 10.1126/science.1114245. [DOI] [PubMed] [Google Scholar]
- 83.Duncan LM, Deeds J, Hunter J, Shao J, Holmgren LM, et al. Cancer Res. 1998;58:1515–20. [PubMed] [Google Scholar]
- 84.Xu XZ, Moebius F, Gill DL, Montell C. Proc Natl Acad Sci USA. 2001;98:10692–97. doi: 10.1073/pnas.191360198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grimm C, Kraft R, Sauerbruch S, Schultz G, Harteneck C. J Biol Chem. 2003;278:21493–501. doi: 10.1074/jbc.M300945200. [DOI] [PubMed] [Google Scholar]
- 86.Lee N, Chen J, Sun L, Wu SJ, Gray KR, et al. J Biol Chem. 2003;278:20890–97. doi: 10.1074/jbc.M211232200. [DOI] [PubMed] [Google Scholar]
- 87.Grimm C, Kraft R, Schultz G, Harteneck C. Mol Pharmacol. 2005;67:798–805. doi: 10.1124/mol.104.006734. [DOI] [PubMed] [Google Scholar]
- 88.Oberwinkler J, Lis A, Giehl KM, Flockerzi V, Philipp SE. J Biol Chem. 2005;280:22540–48. doi: 10.1074/jbc.M503092200. [DOI] [PubMed] [Google Scholar]
- 89.Hofmann T, Chubanov V, Gudermann T, Montell C. Curr Biol. 2003;13:1153–58. doi: 10.1016/s0960-9822(03)00431-7. [DOI] [PubMed] [Google Scholar]
- 90.Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R, Kinet JP. Cell. 2002;109:397–407. doi: 10.1016/s0092-8674(02)00719-5. [DOI] [PubMed] [Google Scholar]
- 91.Liu D, Liman ER. Proc Natl Acad Sci USA. 2003;100:15160–65. doi: 10.1073/pnas.2334159100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Prawitt D, Monteilh-Zoller MK, Brixel L, Spangenberg C, Zabel B, et al. Proc Natl Acad Sci USA. 2003;100:15166–71. doi: 10.1073/pnas.2334624100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nilius B, Prenen J, Droogmans G, Voets T, Vennekens R, et al. J Biol Chem. 2003;278:30813–20. doi: 10.1074/jbc.M305127200. [DOI] [PubMed] [Google Scholar]
- 94.Nilius B, Prenen J, Janssens A, Owsianik G, Wang C, et al. J Biol Chem. 2005;280:22899–906. doi: 10.1074/jbc.M501686200. [DOI] [PubMed] [Google Scholar]
- 95.Zhang Z, Okawa H, Wang Y, Liman ER. J Biol Chem. 2005;280:39185–92. doi: 10.1074/jbc.M506965200. [DOI] [PubMed] [Google Scholar]
- 96.Nilius B, Mahieu F, Prenen J, Janssens A, Owsianik G, et al. EMBO J. 2006;25:467–78. doi: 10.1038/sj.emboj.7600963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Talavera K, Yasumatsu K, Voets T, Droogmans G, Shigemura N, et al. Nature. 2005;438:1022–25. doi: 10.1038/nature04248. [DOI] [PubMed] [Google Scholar]
- 98.Runnels LW, Yue L, Clapham DE. Science. 2001;291:1043–47. doi: 10.1126/science.1058519. [DOI] [PubMed] [Google Scholar]
- 99.Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, et al. Nature. 2001;411:590–95. doi: 10.1038/35079092. [DOI] [PubMed] [Google Scholar]
- 100.Monteilh-Zoller MK, Hermosura MC, Nadler MJ, Scharenberg AM, Penner R, Fleig A. J Gen Physiol. 2003;121:49–60. doi: 10.1085/jgp.20028740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Voets T, Nilius B, Hoefs S, van der Kemp AW, Droogmans G, et al. J Biol Chem. 2004;279:19–25. doi: 10.1074/jbc.M311201200. [DOI] [PubMed] [Google Scholar]
- 102.Jiang J, Li M, Yue L. J Gen Physiol. 2005;126:137–50. doi: 10.1085/jgp.200409185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Runnels LW, Yue L, Clapham DE. Nat Cell Biol. 2002;4:329–36. doi: 10.1038/ncb781. [DOI] [PubMed] [Google Scholar]
- 104.Oancea E, Wolfe JT, Clapham DE. Circ Res. 2006;98:245–53. doi: 10.1161/01.RES.0000200179.29375.cc. [DOI] [PubMed] [Google Scholar]
- 105.Schmitz C, Perraud AL, Johnson CO, Inabe K, Smith MK, et al. Cell. 2003;114:191–200. doi: 10.1016/s0092-8674(03)00556-7. [DOI] [PubMed] [Google Scholar]
- 106.Demeuse P, Penner R, Fleig A. J Gen Physiol. 2006;127:421–34. doi: 10.1085/jgp.200509410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Matsushita M, Kozak JA, Shimizu Y, McLachlin DT, Yamaguchi H, et al. J Biol Chem. 2005;280:20793–803. doi: 10.1074/jbc.M413671200. [DOI] [PubMed] [Google Scholar]
- 108.Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, et al. Nature. 2001;411:595–99. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- 109.Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, et al. Science. 2001;293:1327–30. doi: 10.1126/science.1062473. [DOI] [PubMed] [Google Scholar]
- 110.Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, et al. Mol Cell. 2002;9:163–73. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- 111.Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, et al. J Biol Chem. 2005;280:6138–48. doi: 10.1074/jbc.M411446200. [DOI] [PubMed] [Google Scholar]
- 112.Kolisek M, Beck A, Fleig A, Penner R. Mol Cell. 2005;18:61–69. doi: 10.1016/j.molcel.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 113.McHugh D, Flemming R, Xu SZ, Perraud AL, Beech DJ. J Biol Chem. 2003;278:11002–6. doi: 10.1074/jbc.M210810200. [DOI] [PubMed] [Google Scholar]
- 114.Zhang W, Chu X, Tong Q, Cheung JY, Conrad K, et al. J Biol Chem. 2003;278:16222–29. doi: 10.1074/jbc.M300298200. [DOI] [PubMed] [Google Scholar]
- 115.McKemy DD, Neuhausser WM, Julius D. Nature. 2002;416:52–58. doi: 10.1038/nature719. [DOI] [PubMed] [Google Scholar]
- 116.Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, et al. Cell. 2002;108:705–15. doi: 10.1016/s0092-8674(02)00652-9. [DOI] [PubMed] [Google Scholar]
- 117.Bandell M, Dubin AE, Petrus MJ, Orth A, Mathur J, et al. Nat Neurosci. 2006;9:493–500. doi: 10.1038/nn1665. [DOI] [PubMed] [Google Scholar]
- 118.Andersson DA, Chase HW, Bevan S. J Neurosci. 2004;24:5364–69. doi: 10.1523/JNEUROSCI.0890-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Voets T, Droogmans G, Wissenbach U, Janssens A, Flockerzi V, Nilius B. Nature. 2004;430:748–54. doi: 10.1038/nature02732. [DOI] [PubMed] [Google Scholar]
- 120.Brauchi S, Orio P, Latorre R. Proc Natl Acad Sci USA. 2004;101:15494–99. doi: 10.1073/pnas.0406773101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu B, Qin F. J Neurosci. 2005;25:1674–81. doi: 10.1523/JNEUROSCI.3632-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rohacs T, Lopes CM, Michailidis I, Logothetis DE. Nat Neurosci. 2005;8:626–34. doi: 10.1038/nn1451. [DOI] [PubMed] [Google Scholar]
- 123.Jaquemar D, Schenker T, Trueb B. J Biol Chem. 1999;274:7325–33. doi: 10.1074/jbc.274.11.7325. [DOI] [PubMed] [Google Scholar]
- 124.Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, et al. Nature. 2004;427:260–65. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- 125.Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, et al. Neuron. 2004;41:849–57. doi: 10.1016/s0896-6273(04)00150-3. [DOI] [PubMed] [Google Scholar]
- 126.Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, et al. Cell. 2006;124:1269–82. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 127.Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, et al. Cell. 2003;112:819–29. doi: 10.1016/s0092-8674(03)00158-2. [DOI] [PubMed] [Google Scholar]
- 128.Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, et al. Neuron. 2006;50:277–89. doi: 10.1016/j.neuron.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 129.Corey DP, Garcia-Anoveros J, Holt JR, Kwan KY, Lin SY, et al. Nature. 2004;432:723–30. doi: 10.1038/nature03066. [DOI] [PubMed] [Google Scholar]
- 130.Howard J, Bechstedt S. Curr Biol. 2004;14:R224–26. doi: 10.1016/j.cub.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 131.Youngseok Lee, Yong Lee, Jaejung Lee, Bang S, Hyun S, et al. Nat Genet. 2005;37:305–10. doi: 10.1038/ng1513. [DOI] [PubMed] [Google Scholar]
- 132.Viswanath V, Story GM, Peier AM, Petrus MJ, Lee VM, et al. Nature. 2003;423:822–23. doi: 10.1038/423822a. [DOI] [PubMed] [Google Scholar]
- 133.Rosenzweig M, Brennan KM, Tayler TD, Phelps PO, Patapoutian A, Garrity PA. Genes Dev. 2005;19:419–24. doi: 10.1101/gad.1278205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tracey WDJ, Wilson RI, Laurent G, Benzer S. Cell. 2003;113:261–73. doi: 10.1016/s0092-8674(03)00272-1. [DOI] [PubMed] [Google Scholar]
- 135.Al-Anzi B, Tracey WDJ, Benzer S. Curr Biol. 2006;16:1034–40. doi: 10.1016/j.cub.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 136.Walker RG, Willingham AT, Zuker CS. Science. 2000;287:2229–34. doi: 10.1126/science.287.5461.2229. [DOI] [PubMed] [Google Scholar]
- 137.Barr MM, DeModena J, Braun D, Nguyen CQ, Hall DH, Sternberg PW. Curr Biol. 2001;11:1341–46. doi: 10.1016/s0960-9822(01)00423-7. [DOI] [PubMed] [Google Scholar]
- 138.Gao Z, Ruden DM, Lu X. Curr Biol. 2003;13:2175–78. doi: 10.1016/j.cub.2003.11.053. [DOI] [PubMed] [Google Scholar]
- 139.Watnick TJ, Jin Y, Matunis E, Kernan MJ, Montell C. Curr Biol. 2003;13:2179–84. doi: 10.1016/j.cub.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 140.Neill AT, Moy GW, Vacquier VD. Mol Reprod Dev. 2004;67:472–77. doi: 10.1002/mrd.20033. [DOI] [PubMed] [Google Scholar]
- 141.IPKD Consortium. Cell. 1995;81:289–98. doi: 10.1016/0092-8674(95)90339-9. [DOI] [PubMed] [Google Scholar]
- 142.Geng L, Okuhara D, Yu Z, Tian X, Cai Y, et al. J Cell Sci. 2006;119:1383–95. doi: 10.1242/jcs.02818. [DOI] [PubMed] [Google Scholar]
- 143.Vassilev PM, Guo L, Chen XZ, Segal Y, Peng JB, et al. Biochem Biophys Res Commun. 2001;282:341–50. doi: 10.1006/bbrc.2001.4554. [DOI] [PubMed] [Google Scholar]
- 144.Luo Y, Vassilev PM, Li X, Kawanabe Y, Zhou J. Mol Cell Biol. 2003;23:2600–7. doi: 10.1128/MCB.23.7.2600-2607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Delmas P, Nauli SM, Li X, Coste B, Osorio N, et al. FASEB J. 2004;18:740–42. doi: 10.1096/fj.03-0319fje. [DOI] [PubMed] [Google Scholar]
- 146.Xu GM, Gonzalez-Perrett S, Essafi M, Timpanaro GA, Montalbetti N, et al. J Biol Chem. 2003;278:1457–62. doi: 10.1074/jbc.M209996200. [DOI] [PubMed] [Google Scholar]
- 147.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, et al. Nat Genet. 2003;33:129–37. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 148.McGrath J, Somlo S, Makova S, Tian X, Brueckner M. Cell. 2003;114:61–73. doi: 10.1016/s0092-8674(03)00511-7. [DOI] [PubMed] [Google Scholar]
- 149.Ma R, Li WP, Rundle D, Kong J, Akbarali HI, Tsiokas L. Mol Cell Biol. 2005;25:8285–98. doi: 10.1128/MCB.25.18.8285-8298.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gonzalez-Perrett S, Kim K, Ibarra C, Damiano AE, Zotta E, et al. Proc Natl Acad Sci USA. 2001;98:1182–87. doi: 10.1073/pnas.98.3.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li Q, Montalbetti N, Shen PY, Dai XQ, Cheeseman CI, et al. Hum Mol Genet. 2005;14:1587–603. doi: 10.1093/hmg/ddi167. [DOI] [PubMed] [Google Scholar]
- 152.Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, et al. Nat Cell Biol. 2002;4:191–97. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- 153.Koulen P, Duncan RS, Liu J, Cohen NE, Yannazzo JA, et al. Cell Calcium. 2005;37:593–601. doi: 10.1016/j.ceca.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 154.Köttgen M, Benzing T, Simmen T, Tauber R, Buchholz B, et al. EMBO J. 2005;24:705–16. doi: 10.1038/sj.emboj.7600566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li Y, Wright JM, Qian F, Germino GG, Guggino WB. J Biol Chem. 2005;280:41298–306. doi: 10.1074/jbc.M510082200. [DOI] [PubMed] [Google Scholar]
- 156.Liu Y, Li Q, Tan M, Zhang YY, Karpinski E, et al. FEBS Lett. 2002;525:71–76. doi: 10.1016/s0014-5793(02)03071-5. [DOI] [PubMed] [Google Scholar]
- 157.Dai XQ, Karpinski E, Chen XZ. Biochim Biophys Acta. 2006;1758:197–2. doi: 10.1016/j.bbamem.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 158.Venkatachalam K, Hofmann T, Montell C. J Biol Chem. 2006;281:17517–27. doi: 10.1074/jbc.M600807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Manzoni M, Monti E, Bresciani R, Bozzato A, Barlati S, et al. FEBS Lett. 2004;567:219–24. doi: 10.1016/j.febslet.2004.04.080. [DOI] [PubMed] [Google Scholar]
- 160.Kiselyov K, Chen J, Rbaibi Y, Oberdick D, Tjon-Kon-Sang S, et al. J Biol Chem. 2005;280:43218–23. doi: 10.1074/jbc.M508210200. [DOI] [PubMed] [Google Scholar]
- 161.LaPlante JM, Falardeau J, Sun M, Kanazirska M, Brown EM, et al. FEBS Lett. 2002;532:183–87. doi: 10.1016/s0014-5793(02)03670-0. [DOI] [PubMed] [Google Scholar]
- 162.Raychowdhury MK, Gonzalez-Perrett S, Montalbetti N, Timpanaro GA, Chasan B, et al. Hum Mol Genet. 2004;13:617–27. doi: 10.1093/hmg/ddh067. [DOI] [PubMed] [Google Scholar]
- 163.Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, Bashllari E, Bisceglia J, et al. J Biol Chem. 2006;281:7294–301. doi: 10.1074/jbc.M508211200. [DOI] [PubMed] [Google Scholar]
- 164.Goel M, Sinkins WG, Schilling WP. J Biol Chem. 2002;277:48303–10. doi: 10.1074/jbc.M207882200. [DOI] [PubMed] [Google Scholar]
- 165.Hofmann T, Schaefer M, Schultz G, Gudermann T. Proc Natl Acad Sci USA. 2002;99:7461–66. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. Neuron. 2001;29:645–55. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 167.Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. J Biol Chem. 2003;278:39014–19. doi: 10.1074/jbc.M306705200. [DOI] [PubMed] [Google Scholar]
- 168.Lintschinger B, Balzer-Geldsetzer M, Baskaran T, Graier WF, Romanin C, et al. J Biol Chem. 2000;275:27799–805. doi: 10.1074/jbc.M002705200. [DOI] [PubMed] [Google Scholar]
- 169.Tobin D, Madsen D, Kahn-Kirby A, Peckol E, Moulder G, et al. Neuron. 2002;35:307–18. doi: 10.1016/s0896-6273(02)00757-2. [DOI] [PubMed] [Google Scholar]
- 170.Chubanov V, Waldegger S, Schnitzler MM, Vitzthum H, Sassen MC, et al. Proc Natl Acad Sci USA. 2004;101:2894–99. doi: 10.1073/pnas.0305252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li HS, Montell C. J Cell Biol. 2000;150:1411–22. doi: 10.1083/jcb.150.6.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tsunoda S, Sun Y, Suzuki E, Zuker C. J Neurosci. 2001;21:150–58. doi: 10.1523/JNEUROSCI.21-01-00150.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lockwich TP, Liu X, Singh BB, Jadlowiec J, Weiland S, Ambudkar IS. J Biol Chem. 2000;275:11934–42. doi: 10.1074/jbc.275.16.11934. [DOI] [PubMed] [Google Scholar]
- 174.Yuan JP, Kiselyov K, Shin DM, Chen J, Shcheynikov N, et al. Cell. 2003;114:777–89. doi: 10.1016/s0092-8674(03)00716-5. [DOI] [PubMed] [Google Scholar]
- 175.Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ. Nature. 2003;426:285–91. doi: 10.1038/nature02162. [DOI] [PubMed] [Google Scholar]
- 176.Kim JY, Zeng W, Kiselyov K, Yuan JP, Dehoff MH, et al. J Biol Chem. 2006;281:32540–49. doi: 10.1074/jbc.M602496200. [DOI] [PubMed] [Google Scholar]
- 177.Pérez CA, Huang L, Rong M, Kozak JA, Preuss AK, et al. Nat Neurosci. 2002;5:1169–76. doi: 10.1038/nn952. [DOI] [PubMed] [Google Scholar]
- 178.Zhang Y, Hoon MA, Chandrashekar J, Mueller KL, Cook B, et al. Cell. 2003;112:293–301. doi: 10.1016/s0092-8674(03)00071-0. [DOI] [PubMed] [Google Scholar]
- 179.Stowers L, Holy TE, Meister M, Dulac C, Koentges G. Science. 2002;295:1493–500. doi: 10.1126/science.1069259. [DOI] [PubMed] [Google Scholar]
- 180.Leypold BG, Yu CR, Leinders-Zufall T, Kim MM, Zufall F, Axel R. Proc Natl Acad Sci USA. 2002;99:6376–81. doi: 10.1073/pnas.082127599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Peier AM, Reeve AJ, Andersson DA, Moqrich A, Earley TJ, et al. Science. 2002;296:2046–49. doi: 10.1126/science.1073140. [DOI] [PubMed] [Google Scholar]
- 182.Smith GD, Gunthorpe MJ, Kelsell RE, Hayes PD, Reilly P, et al. Nature. 2002;418:186–90. doi: 10.1038/nature00894. [DOI] [PubMed] [Google Scholar]
- 183.Xu H, Ramsey IS, Kotecha SA, Moran MM, Chong JA, et al. Nature. 2002;418:181–86. doi: 10.1038/nature00882. [DOI] [PubMed] [Google Scholar]
- 184.Güler AD, Lee HS, Iida T, Shimizu I, Tominaga M, Caterina M. J Neurosci. 2002;22:6408–14. doi: 10.1523/JNEUROSCI.22-15-06408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Watanabe H, Vriens J, Suh SH, Benham CD, Droogmans G, Nilius B. J Biol Chem. 2002;277:47044–51. doi: 10.1074/jbc.M208277200. [DOI] [PubMed] [Google Scholar]
- 186.Chung MK, Lee H, Caterina MJ. J Biol Chem. 2003;278:32037–46. doi: 10.1074/jbc.M303251200. [DOI] [PubMed] [Google Scholar]
- 187.Chung MK, Lee H, Mizuno A, Suzuki M, Caterina MJ. J Biol Chem. 2004;279:21569–75. doi: 10.1074/jbc.M401872200. [DOI] [PubMed] [Google Scholar]
- 188.Lee H, Caterina MJ. Pflüg Arch. 2005;451:160–67. doi: 10.1007/s00424-005-1438-y. [DOI] [PubMed] [Google Scholar]
- 189.Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, et al. Science. 2000;288:306–13. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- 190.Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, et al. Nature. 2000;405:183–87. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- 191.Moqrich A, Hwang SW, Earley TJ, Petrus MJ, Murray AN, et al. Science. 2005;307:1468–72. doi: 10.1126/science.1108609. [DOI] [PubMed] [Google Scholar]
- 192.Todaka H, Taniguchi J, Satoh J, Mizuno A, Suzuki M. J Biol Chem. 2004;279:35133–38. doi: 10.1074/jbc.M406260200. [DOI] [PubMed] [Google Scholar]
- 193.Lee HS, Iida T, Mizuno A, Suzuki M, Caterina MJ. J Neurosci. 2005;25:1304–10. doi: 10.1523/JNEUROSCI.4745.04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, et al. Cell. 2000;103:525–35. doi: 10.1016/s0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liedtke W, Friedman JM. Proc Natl Acad Sci USA. 2003;100:13698–703. doi: 10.1073/pnas.1735416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Liedtke W, Tobin DM, Bargmann CI, Friedman JM. Proc Natl Acad Sci USA. 2003;100(Suppl 2):14531–36. doi: 10.1073/pnas.2235619100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Di Palma F, Belyantseva IA, Kim HJ, Vogt TF, Kachar B, Noben-Trauth K. Proc Natl Acad Sci USA. 2002;99:14994–99. doi: 10.1073/pnas.222425399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tabuchi K, Suzuki M, Mizuno A, Hara A. Neurosci Lett. 2005;382:304–8. doi: 10.1016/j.neulet.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 199.Sidi S, Friedrich RW, Nicolson T. Science. 2003;301:96–99. doi: 10.1126/science.1084370. [DOI] [PubMed] [Google Scholar]
- 200.Shin JB, Adams D, Paukert M, Siba M, Sidi S, et al. Proc Natl Acad Sci USA. 2005;102:12572–77. doi: 10.1073/pnas.0502403102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gopfert MC, Albert JT, Nadrowski B, Kamikouchi A. Nat Neurosci. 2006;9:999–1000. doi: 10.1038/nn1735. [DOI] [PubMed] [Google Scholar]
- 202.Li W, Feng Z, Sternberg PW, Xu XZ. Nature. 2006;440:684–87. doi: 10.1038/nature04538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Foster RG. Nature. 2005;433:698–99. doi: 10.1038/433698a. [DOI] [PubMed] [Google Scholar]
- 204.Panda S, Nayak SK, Campo B, Walker JR, Hogenesch JB, Jegla T. Science. 2005;307:600–4. doi: 10.1126/science.1105121. [DOI] [PubMed] [Google Scholar]
- 205.Qiu X, Kumbalasiri T, Carlson SM, Wong KY, Krishna V, et al. Nature. 2005;433:745–49. doi: 10.1038/nature03345. [DOI] [PubMed] [Google Scholar]
- 206.Isoldi MC, Rollag MD, Castrucci AM, Provencio I. Proc Natl Acad Sci USA. 2005;102:1217–21. doi: 10.1073/pnas.0409252102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Li Y, Jia YC, Cui K, Li N, Zheng ZY, et al. Nature. 2005;434:894–98. doi: 10.1038/nature03477. [DOI] [PubMed] [Google Scholar]
- 208.Wang GX, Poo MM. Nature. 2005;434:898–904. doi: 10.1038/nature03478. [DOI] [PubMed] [Google Scholar]
- 209.Shim S, Goh EL, Ge S, Sailor K, Yuan JP, et al. Nat Neurosci. 2005;8:730–35. doi: 10.1038/nn1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Greka A, Navarro B, Oancea E, Duggan A, Clapham DE. Nat Neurosci. 2003;6:837–45. doi: 10.1038/nn1092. [DOI] [PubMed] [Google Scholar]
- 211.Munsch T, Freichel M, Flockerzi V, Pape HC. Proc Natl Acad Sci USA. 2003;100:16065–70. doi: 10.1073/pnas.2535311100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Freichel M, Suh SH, Pfeifer A, Schweig U, Trost C, et al. Nat Cell Biol. 2001;3:121–27. doi: 10.1038/35055019. [DOI] [PubMed] [Google Scholar]
- 213.Dietrich A, Mederos YSM, Gollasch M, Gross V, Storch U, et al. Mol Cell Biol. 2005;25:6980–89. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.MacPherson MR, Pollock VP, Kean L, Southall TD, Giannakou ME, et al. Genetics. 2005;169:1541–52. doi: 10.1534/genetics.104.035139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Singh BB, Zheng C, Liu X, Lockwich T, Liao D, et al. FASEB J. 2001;15:1652–54. doi: 10.1096/fj.00-0749fje. [DOI] [PubMed] [Google Scholar]
- 216.Cheng H, Beck A, Launay P, Gross SA, Stokes AJ, et al. Cell Calcium. 2007;41:51–61. doi: 10.1016/j.ceca.2006.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Togashi K, Hara Y, Tominaga T, Higashi T, Konishi Y, et al. EMBO J. 2006;25:1804–15. doi: 10.1038/sj.emboj.7601083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Birder LA, Nakamura Y, Kiss S, Nealen ML, Barrick S, et al. Nat Neurosci. 2002;5:856–60. doi: 10.1038/nn902. [DOI] [PubMed] [Google Scholar]
- 219.Hoenderop JG, van Leeuwen JP, van der Eerden BC, Kersten FF, van der Kemp AW, et al. J Clin Invest. 2003;112:1906–14. doi: 10.1172/JCI19826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.van der Eerden BC, Hoenderop JG, de Vries TJ, Schoenmaker T, Buurman CJ, et al. Proc Natl Acad Sci USA. 2005;102:17507–12. doi: 10.1073/pnas.0505789102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Elizondo MR, Arduini BL, Paulsen J, MacDonald EL, Sabel JL, et al. Curr Biol. 2005;15:667–71. doi: 10.1016/j.cub.2005.02.050. [DOI] [PubMed] [Google Scholar]
- 222.Teramoto T, Lambie EJ, Iwasaki K. Cell Metab. 2005;1:343–54. doi: 10.1016/j.cmet.2005.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sutters M, Germino GG. J Lab Clin Med. 2003;141:91–101. doi: 10.1067/mlc.2003.13. [DOI] [PubMed] [Google Scholar]
- 224.Cantiello HF. Trends Mol Med. 2003;9:234–36. doi: 10.1016/s1471-4914(03)00073-x. [DOI] [PubMed] [Google Scholar]
- 225.Pennekamp P, Karcher C, Fischer A, Schweickert A, Skryabin B, et al. Curr Biol. 2002;12:938–43. doi: 10.1016/s0960-9822(02)00869-2. [DOI] [PubMed] [Google Scholar]
- 226.Bach G. Pflüg Arch. 2005;451:313–17. doi: 10.1007/s00424-004-1361-7. [DOI] [PubMed] [Google Scholar]
- 227.Piper RC, Luzio JP. Trends Cell Biol. 2004;14:471–73. doi: 10.1016/j.tcb.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 228.Fares H, Greenwald I. Nat Genet. 2001;28:64–68. doi: 10.1038/ng0501-64. [DOI] [PubMed] [Google Scholar]
- 229.Balzer M, Lintschinger B, Groschner K. Cardiovasc Res. 1999;42:543–49. doi: 10.1016/s0008-6363(99)00025-5. [DOI] [PubMed] [Google Scholar]
- 230.Agam K, von Campenhausen M, Levy S, Ben-Ami HC, Cook B, et al. J Neurosci. 2000;20:5748–55. doi: 10.1523/JNEUROSCI.20-15-05748.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Yoon J, Ben-Ami HC, Hong YS, Park S, Strong LL, et al. J Neurosci. 2000;20:649–59. doi: 10.1523/JNEUROSCI.20-02-00649.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Wang T, Xu H, Oberwinkler J, Gu Y, Hardie RC, Montell C. Neuron. 2005;45:367–78. doi: 10.1016/j.neuron.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 233.Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, et al. J Pharmacol Sci. 2006;101:66–76. doi: 10.1254/jphs.fp0060128. [DOI] [PubMed] [Google Scholar]
- 234.Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, et al. Cell. 2003;115:863–77. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- 235.Rogers C, Persson A, Cheung B, de Bono M. Curr Biol. 2006;16:649–59. doi: 10.1016/j.cub.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 236.Xu C, Macciardi F, Li PP, Yoon IS, Cooke RG, et al. Am J Med Genet B. 2006;141:36–43. doi: 10.1002/ajmg.b.30239. [DOI] [PubMed] [Google Scholar]
- 237.McQuillin A, Bass NJ, Kalsi G, Lawrence J, Puri V, et al. Mol Psychiatry. 2006;11:134–42. doi: 10.1038/sj.mp.4001759. [DOI] [PubMed] [Google Scholar]