

Abstract
In the mature brain, the neurotransmitter GABA can cause a postsynaptic hyperpolarization via activation of chloride permeant GABAA receptor channels. This hyperpolarizing response critically depends on chloride extrusion via the KCl-cotransporter KCC2 1. Its knockdown in mice impairs synaptic inhibition by changing the electrochemical potential for chloride and thus increases neuronal excitability 2,3. Two independent groups provide first evidence now, published in EMBO reports, that rare variants of KCC2 confer an increased risk of epilepsy in men 4,5.
Epilepsy is characterized by recurrent disturbed neuronal activity in the brain, which causes seizures. During seizures, affected patients experience abnormal behavior and sensations and/or loss of consciousness. With a prevalence of roughly 1% worldwide, epilepsy is among the most common neurological disorders. Epilepsy can either arise symptomatically as part of an underlying disease or as a disorder on its own. The latter is often referred to as idiopathic epilepsy and is thought to be mainly determined by genetic factors. Twin studies clearly show a higher concordance in monozygotic compared to dizygotic twins. According to epidemiological studies, the risk is increased two- to four-fold in first-degree relatives of individuals suffering from epilepsy. Approximately 2% of epilepsies are inherited as monogenic disorders, and several genes with a major effect have been identified. However, in most cases, epilepsy is sporadic without family history, and unraveling the genetic mechanisms in these patients is challenging.
“Two independent studies […] provide first evidence now that dysfunction of KCC2 may indeed predispose to epilepsy in humans.”
In a simplified model, epilepsy-causing defects render the neuronal network more excitable and hence the brain more susceptible to seizures. The affected proteins include voltage-gated Na+ and K+ channels and ligand-gated ion channels such as receptors for acetylcholine, glutamate, or the main inhibitory neurotransmitter GABA. GABAA receptor chloride channels open upon binding of GABA. Because of the electrochemical potential for chloride, this typically results in a hyperpolarizing chloride influx in the mature brain. If the intraneuronal chloride concentration is high, opening of GABAA receptors can cause a depolarizing chloride efflux and can generate an action potential. This excitatory action of GABA is thought to play an important role for the formation of the neuronal network during maturation of the brain 6. Early network activity is reduced upon disruption of the Na+-dependent KCl-cotransporter NKCC1 7, which uses the Na+ gradient to raise the intracellular chloride concentration. Only when the chloride concentration is lowered by expression of the Na+-independent KCl-cotransporter KCC2 1,8, encoded by SLC12A5, the GABA-mediated hyperpolarizing chloride current is established (Fig 1), which typically occurs during the first postnatal weeks in rodents. Mice with a total deletion of Slc12a5 display impaired GABAergic inhibition in the spinal cord and die immediately after birth because of a failure to breathe 2. Disruption of only the developmentally regulated KCC2 splice variant, however, which accounts for most of the KCC2 protein in adult brain, results in a less severe phenotype with epilepsy and lethality in the second postnatal week of life. The heterozygotes do not show overt epilepsy but a reduced seizure threshold in response to the pro-convulsive drug pentylenetetrazole 3.
Figure 1. In the brain, excitatory and inhibitory activities are finely tuned.
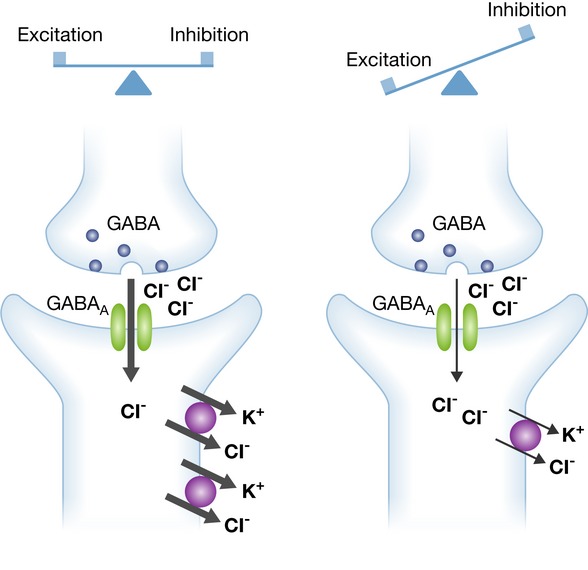
In a simplified model, mutations identified in epilepsy disturb this delicate balance and thereby increase the excitability of the neuronal network. GABA as the main inhibitory neurotransmitter acts via chloride permeant GABAA receptor channels. The hyperpolarizing chloride current upon activation of GABAA receptors critically depends on the electrochemical gradient for chloride, which is mainly established by the KCl-cotransporter KCC2 (left). Supporting this concept, KCC2 variants with impaired chloride extrusion impair synaptic inhibition and thus confer an increased risk for epilepsy (right).
“Both [KCC2] variants together clearly showed statistical association with IGE.”
Based on this information, mutations in SLC12A5 were predicted and have long been awaited in humans affected by epilepsy. Two independent studies published by EMBO reports provide first evidence now that dysfunction of KCC2 may indeed predispose to epilepsy in humans. In the first study, Puskarjov et al 4 report on a small Australian family with partial co-segregation of a rare C-terminal KCC2 variant (R952H) with febrile seizures. As this variant was unable to rescue normal spine maturation in KCC2 knockout neurons and resulted in reduced surface expression and impaired chloride extrusion, its functional characterization supports a contribution in the development of febrile seizures in the reported family. In the second study, Kahle et al 5 identify the same R952H variant as well as another C-terminal KCC2 variant, R1049C, in Canadian patients suffering from idiopathic generalized epilepsy (IGE). Although the number of the R952H alleles compared to a large control cohort was not significantly increased, the R1049C variant and in particular both variants together clearly showed statistical association with IGE. As far as parental DNA samples were available, however, parents of affected individuals carrying the variants were not affected. The functional characterization confirmed a decreased surface expression for R952H, while this was not the case for R1049C. Both variants displayed impaired chloride extrusion and entailed decreased phosphorylation of a functionally relevant KCC2 serine residue.
“The genetic contributions in most affected individuals are complex, and the relevant different risk alleles remain largely unclear.”
Both reports suggest that the KCC2 variants are involved in the pathogenesis of epilepsy. However, the associated risks to develop epilepsy appear to be quite small, because there was no clear co-segregation with the phenotype and both variants were also found in controls. What are the other genetic or environmental factors that are necessary to result in overt epilepsy and what are the factors that cause the different phenotypes? Although next-generation sequencing allows for a mostly comprehensive analysis of genetic variants and thus offers an unprecedented ability to identify rare variants, the genetic contributions in most affected individuals are complex and the relevant different risk alleles remain largely unclear. Exome sequencing failed to identify single rare variants of large effect in IGE, and it was proposed that gene-based approaches in large cohorts might be more successful for future studies on epilepsy predisposition 9. Because of the importance of ion channels for neuronal excitability, Klassen et al 10 sequenced 237 ion channel genes in 152 individuals with idiopathic epilepsy and 139 healthy controls. Channel variants predicted to be disease causing were found in both unaffected controls and patients. As such, the authors conclude that causality cannot be assigned to a particular variant in most cases, but rather depends on a complex individual interplay of different variants. Identifying these patterns of risk alleles within the large number of rare and potentially functional variants found in each human genome presents a considerable diagnostic challenge.
“Identifying these patterns of risk alleles […] presents a considerable diagnostic challenge.”
Conflict of interest
The author declares that he has no conflict of interest.
References
- Rivera C, Voipio J, Payne JA, et al. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Hübner CA, Stein V, Hermans-Borgmeyer I, et al. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Woo NS, Lu JM, England R, et al. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- Puskarjov M, Seja P, Heron SE, et al. EMBO Rep. 2014;15:723–729. doi: 10.1002/embr.201438749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Merner ND, Friedel P, et al. EMBO Rep. 2014;15:766–774. doi: 10.15252/embr.201438840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, et al. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Pfeffer CK, Stein V, Keating DJ, et al. J Neurosci. 2009;29:3419–3. doi: 10.1523/JNEUROSCI.1377-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, Hermans-Borgmeyer I, Jentsch TJ, et al. J Comp Neurol. 2004;468:57–64. doi: 10.1002/cne.10983. [DOI] [PubMed] [Google Scholar]
- Heinzen EL, Depondt C, Cavalleri GL, et al. Am J Hum Genet. 2012;91:293–302. doi: 10.1016/j.ajhg.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen T, Davis C, Goldman A, et al. Cell. 2011;145:1036–1048. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]