

Abstract
A20 (also known as TNFAIP3) is a deubiquitinating enzyme (DUB) that ensures optimal immune responses in cells stimulated by cytokines, such as TNF and IL-1, or pathogen components, such as lipopolysaccharide. Deletion of A20 in mice results in multi-organ inflammation and death within 2 weeks 1. The anti-inflammatory functions of A20 have been attributed to its ability to negatively regulate NF-κB signaling 2. The picture that has emerged over the last decade is that A20 attenuates NF-κB signaling by removing polyubiquitin chains from specific NF-κB signaling proteins. A study published in this issue of EMBO reports by Sankar Ghosh and colleagues 3 now shows that A20 knockin mice expressing a catalytically inactive A20 mutant that can no longer remove ubiquitin are normal and do not have an inflammatory phenotype. These results challenge the notion that A20 exerts its NF-κB inhibitory and anti-inflammatory function by acting as a DUB.
Polyubiquitination, the posttranslational modification by which a polyubiquitin chain is attached to a protein, has important roles in many cellular processes and physiological conditions, including innate and adaptive immunity. Formation of different ubiquitin linkages yields structurally diverse polyubiquitin signals, which can mark the modified protein for proteasomal degradation, alter subcellular localization, change activity, or modulate its interaction with other proteins. Defects in polyubiquitination have been linked with several diseases, including autoimmunity and cancer. Understanding how a cell controls the ubiquitination status of specific signaling proteins may thus guide the development of novel therapeutic strategies. Ubiquitination is catalyzed by a three-enzyme cascade involving an E1-ubiquitin-activating enzyme, E2-ubiquitin-conjugating enzymes, and E3 ubiquitin ligases and is reversed by specific DUBs. The NF-κB signaling pathway is key in driving immune responses, cell proliferation, and cell survival and is tightly controlled by multiple specific E2/E3 enzymes and DUBs. In this context, A20 has been described as a key molecule that controls the duration and intensity of proinflammatory NF-κB signaling 2. Mice deficient in A20 show enhanced NF-κB responses and die prematurely due to multi-organ inflammation 1, whereas cell type-specific deletion of the mouse A20 gene is associated with specific autoimmune responses 2. In humans, mutations in the A20 locus predispose to several autoimmune diseases and certain types of B cell lymphoma 2.
A20 belongs to the ovarian tumor (OTU) family of cysteine protease DUBs and has an N-terminal OTU domain and seven C-terminal zinc-finger (ZF) motifs (Fig 1). A20 can cleave unanchored K11-, K48-, and K63-linked polyubiquitin chains in vitro, but in cells only K63-linked polyubiquitin chains attached to NF-κB signaling proteins have been shown to be substrates 4. The C-terminal zinc fingers of A20 are involved in interaction with several proteins and mediate E3 ubiquitin ligase activity, leading to K48-linked polyubiquitination of RIP1 and E2 enzymes, hereby inducing their proteasomal degradation 4,5. This dual ubiquitin-editing activity of A20 is generally believed to be responsible for its NF-κB inhibitory and anti-inflammatory function. Overexpression of A20 mutants lacking the catalytic Cys103 residue or the whole DUB domain has been described to attenuate NF-κB signaling through non-catalytic mechanisms 6,7. However, as massive overexpression of mutant proteins may change the stoichiometry ratios between interacting proteins, such studies should be interpreted with care.
Figure 1. Domain structure of A20 and associated activities.
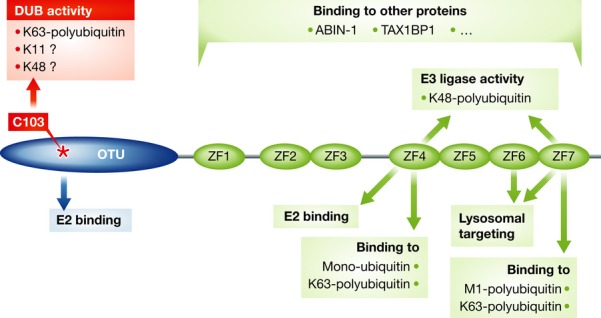
A20 has an N-terminal ovarian tumor (OTU) domain that mediates DUB activity. C103, which is critical for DUB activity, is indicated by an asterisk. The C-terminal region contains seven ZF motifs that bind multiple proteins implicated in NF-κB signaling. In addition, ZF4 and ZF7 bind distinct mono- and polyubiquitin species and have been suggested to contribute to the negative regulation of NF-κB signaling through multiple mechanisms.
“…whereas mice lacking A20 die prematurely […] animals expressing A20-C103A are viable and do not show any signs of inflammation.”
To elucidate the role of A20 DUB activity under physiological conditions, De et al genetically engineered mice to express a DUB-inactive mutant A20 (A20-C103A) 3. Surprisingly, whereas mice lacking A20 die prematurely 1, animals expressing A20-C103A are viable and do not show any signs of inflammation. A20 knockout mice have increased numbers of myeloid lineage cells in the bone marrow and spleen 1. In contrast, the differentiation and maintenance of myeloid, lymphoid, and erythroid lineages is normal in A20-C103A mice. De et al also investigated the response of A20-C103A mice to lipopolysaccharide (LPS)-induced shock. Although A20 is known to be important for restricting MyD88-mediated signaling in vivo, A20-C103A responded as wild-type mice to LPS-induced shock. Consistent with these findings, macrophages and dendritic cells from A20-C103A mice and wild-type mice expressed similar amounts of proinflammatory cytokines and surface activation markers and had similar activation of NF-κB, in response to LPS or TNF stimulation in vitro. Intriguingly, although inhibition of TNF-induced NF-κB activation by A20 has been proposed to involve RIP1 deubiquitination 4, De et al found roughly equal amounts of ubiquitinated RIP1 in macrophages of wild-type and A20-C103A mice after TNF stimulation, illustrating that the DUB activity of A20 does not substantially affect RIP1 ubiquitination. In addition to NF-κB signaling, A20 has been shown to inhibit TNF-induced MAP kinase activation and cell death 2. Again, De and collaborators could not find any difference in these responses in macrophages derived from wild-type and A20-C103A mice.
How can we reconcile the normal NF-κB response and phenotype of A20-C103A mice with A20’s ability to modulate ubiquitin-dependent NF-κB signaling and the severe inflammatory phenotype of mice lacking A20? One possible explanation is that the DUB activity of A20 is functionally redundant with another DUB for the regulation of NF-κB signaling. Cylindromatosis (CYLD) would be a prime candidate, as it is co-expressed with A20 in several cell types, shares its preference for K63-polyubiquitin chains, and has largely overlapping substrates (such as RIP1 and TRAF6). However, CYLD-deficient mice have a much milder phenotype than A20 knockout mice, indicating that at least some of the activities of A20 are not functionally redundant with CYLD. In this regard, some of A20’s C-terminal ZF motifs have been shown to be required for its NF-κB inhibitory function (Fig 1). ZF4 was shown to support E3 ligase activity and bind K63-polyubiquitin, which recruits A20 to specific NF-κB signaling proteins (such as RIP1 and E2 enzymes) 4,8. However, knockin mice harboring an A20 mutant with a non-functional ZF4 showed that this domain is largely dispensable for A20’s anti-inflammatory function 8. A20 ZF7 binds K63-linked and, even more efficiently, M1-linked polyubiquitin chains, allowing A20 to interfere with NF-κB signaling in a catalytic-independent manner 7,9. In addition, ZF7 is known to target specific signaling proteins for lysosomal degradation 10. These findings urge the generation of A20 ZF7 knockin mice.
The study of De and colleagues clearly demonstrates that A20 DUB activity does not participate in TNF- nor LPS-induced NF-κB signaling or cell death, nor in normal tissue homeostasis. Nevertheless, a similar recent study revealed slightly enhanced gene expression, NF-κB activation, and RIP1 ubiquitination in TNF-stimulated mouse embryonic fibroblasts (MEF) isolated from A20-C103A mice when compared to wild-type cells, although this was much less pronounced than in full knockout cells 8. The reason for this discrepancy between both studies is unclear, but may reflect differences in experimental conditions and cell types (MEFs versus bone marrow-derived macrophages). Also in that study, A20-C103A mice were found to be grossly normal and to develop little spontaneous disease, clearly indicating that the DUB activity is not singly responsible for all in vivo functions of A20. However, Lu et al reported that A20-C103A mice are more sensitive to experimentally induced colitis 8, which was unfortunately not investigated by De and colleagues 3. Such studies, together with the generation and characterization of mice expressing double-mutant A20 proteins (inactivating OTU and ZF4 or ZF7 simultaneously), will be crucial for deciphering how A20 exerts its pathophysiological roles.
References
- Lee EG, Boone DL, Chai S, et al. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catrysse L, Vereecke L, Beyaert R, et al. Trends Immunol. 2014;35:22–31. doi: 10.1016/j.it.2013.10.005. [DOI] [PubMed] [Google Scholar]
- De A, Dainichi T, Vendan Rathinam C, et al. EMBO Rep. 2014;15:775–783. doi: 10.15252/embr.201338305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz IE, O’Rourke KM, Zhou H, et al. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- Shembade N, Ma A, Harhaj EW. Science. 2010;327:1135–1139. doi: 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PC, Ovaa H, Hamon M, et al. Biochem J. 2004;378:727–734. doi: 10.1042/BJ20031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaug B, Chen J, Du F, et al. Mol Cell. 2011;44:559–571. doi: 10.1016/j.molcel.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TT, Onizawa M, Hammer GE, et al. Immunity. 2013;38:896–905. doi: 10.1016/j.immuni.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhelst K, Carpentier I, Kreike M, et al. EMBO J. 2012;31:3845–3855. doi: 10.1038/emboj.2012.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Soetandyo N, Wang Q, et al. Biochim Biophys Acta. 2009;1793:346–353. doi: 10.1016/j.bbamcr.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]