

Abstract
The KCC2 cotransporter establishes the low neuronal Cl− levels required for GABAA and glycine (Gly) receptor-mediated inhibition, and KCC2 deficiency in model organisms results in network hyperexcitability. However, no mutations in KCC2 have been documented in human disease. Here, we report two non-synonymous functional variants in human KCC2, R952H and R1049C, exhibiting clear statistical association with idiopathic generalized epilepsy (IGE). These variants reside in conserved residues in the KCC2 cytoplasmic C-terminus, exhibit significantly impaired Cl−-extrusion capacities resulting in less hyperpolarized Gly equilibrium potentials (EGly), and impair KCC2 stimulatory phosphorylation at serine 940, a key regulatory site. These data describe a novel KCC2 variant significantly associated with a human disease and suggest genetically encoded impairment of KCC2 functional regulation may be a risk factor for the development of human IGE.
Keywords: cation-chloride cotransporters, epilepsy, GABA, KCC2, kinase
Introduction
The K+-Cl− cotransporter KCC2 (SLC12A5) is the main Cl− extrusion mechanism in mature neurons and is essential for type A GABA (GABAA) and glycine (Gly) receptor-mediated Cl− currents underlying fast synaptic inhibition 1. KCC2 deficiency in model organisms results in network hyperexcitability 2–6. KCC2 functional down-regulation has been implicated in neurological disorders featuring GABAergic disinhibition 7. However, no mutations in KCC2 have been described in human disease.
The idiopathic generalized epilepsies (IGEs) are primarily genetic in origin and include rare Mendelian diseases and more common familial forms which manifest as complex traits 8. However, the genetic architecture of IGE is not well understood. A recent exome-sequencing study attempted to identify IGE variants of large effect by taking a variant-based approach and performing large-scale genotyping on selected rare exome-sequencing variants 9. Individual variants contributing significantly to disease were not identified because, as the authors noted, the impact of any individual single-nucleotide variant in IGE is small. A gene-based approach seeking multiple rare alleles within the same gene might be an alternative approach for identifying genetic risk factors for IGE 9.
The cytoplasmic C-terminus of KCC2 is an important regulatory region of transporter function [reviewed in 1, 10–12; e.g., 13] (Fig 1). We employed a targeted DNA-sequencing approach to screen this region in KCC2 (amino acids 894 1086; NP_065759), along with the homologous region in KCC3 (SLC12A6) (amino acids 979 1570; NP_598408), and the N-terminal regulatory region in NKCC1 (SLC12A2) 14,15 (amino acids 150 252; NP_001037) for genetic variations in a 380-patient French-Canadian cohort of IGE (see Supplementary Methods). We hypothesized functionally relevant genetic variation in these regions might alter transporter activity and contribute to IGE by altering neuronal Cl− homeostasis, and consequently, GABA/glycine activity.
Figure 1. Identified KCC2 (SLC12A5) variants in human IGE.

A DNA chromatograms illustrating the detection of c.2855 G > A (p.R952H) and c.3145 C > T (p.R1049C) via Sanger sequencing.
B, C Evolutionary conservation of amino acids p.R952 and p.R1049.
D Schematic representation of KCC2 (human). Orange dots indicate the positions of the known critical phospho-regulatory residues p.T906, p.S940, p.T1007, and p.Y1087 (reviewed in 10). Pink region denotes the KCC2 ‘ISO’ domain, required for hyperpolarizing GABAergic transmission 13. Red dots depict the two identified IGE mutations, p.R952 and p.R1049.
E The modeled structure of the human KCC2 C-terminal domain, based on homology modeling by I-TASSER using a prokaryotic member of the cation-chloride cotransporter family (PDB code 3g40) (see Materials and Methods for details). Color scheme same as in (D). Note the proximity of the KCC2 IGE variants to important regulatory residues and domains.
Results
After Sanger sequencing the C-terminus of KCC2, an enrichment of non-synonymous (NS) alleles was discovered in IGE cases compared to controls (Table 1; see Supplementary Methods for IGE clinical details). A trend was first noticed when comparing the total number of NS alleles identified in IGE cases (n = 8/760) to Quebec controls (n = 2/950; P = 0.028; Table 1). In order to validate this association, an additional 739 Quebec controls were screened, bringing the total number of control alleles to 2428. A total of 6 NS alleles were detected in the control cohort (n = 6/2428), improving the P-value to 7.50 × 10−3 (Table 1). When considering the total number of C-terminal NS alleles that were identified in the European American (EA) cohort from the Exome Variant Server (EVS), a P-value of 2.2 × 10−4 was generated. The P-value became more significant when all EVS alleles were considered (P = 7.7 × 10−5; Supplementary Table S1). Overall, NS alleles in this important regulatory region are associated with IGE. Specifically, two different NS KCC2 variants were identified, R1049C and R952H (Fig 1 and Table 1). All variants were heterozygous.
Table 1.
KCC2 (SLC12A5) variants detected in the IGE cohort
| Non-synonymous variants detected in the ‘hotspot’ | Detection of variants in IGE cohort | Control data | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Quebec population controls (Phase 1: 475 controls) | Quebec population controls (Total: 1,214 controls) | |||||||||||
| mRNA (NM_020708.4) | Protein (NP_065759) | Number of probands | Number of alleles | Allele frequency (%) | Number of alleles | Allele frequency (%) | P-value | Odds ratio | Number of alleles | Allele frequency (%) | P-value | Odds ratio |
| c.2855 G>A | p.R952H | 5/380 | 5/760 | 0.66 | 2/950 | 0.21 | 0.253 | 3.14 CI95 [0.5–33.0] | 5/2,428 | 0.21 | 0.065 | 3.21 CI95 [0.7–14.0] |
| c.3145 C>T | p.R1049C | 3/380 | 3/760 | 0.39 | 0/950 | 0.00 | 0.088 | Inf CI95 [0.5-Inf] | 1/2,428 | 4.12 × 10−4 | 0.044 | 9.61 CI95 [0.8–503.6] |
| Total number of non-synonymous alleles detected | 8/380 | 8/760 | – | 2/950 | – | 0.028 | 5.04 CI95 [1.0–48.8] | 6/2,428 | – | 7.50 × 10−3 | 4.29 CI95 [1.3–15.1] | |
R1049C is a novel KCC2 variant detected in 3 of the 380 IGE cases (Table 1; Supplementary Table S2). This variant is previously unreported and was not detected in the first 950 Quebec alleles that were screened but was detected in one of the 2428 Quebec control alleles (Table 1). This extremely rare variant is enriched in our IGE cohort (P = 0.044; Table 1). Furthermore, R1049 is a highly conserved residue (Fig 1), and a substitution for a cysteine at that position is predicted to be highly pathogenic using multiple in silico bioinformatics programs (Table 2). Moreover, the parental DNA was available for two of the three probands with R1049C. In both cases, the parents were unaffected, and the variant was maternally inherited.
Table 2.
Predicted pathogenicity of the IGE SLC12A5 (KCC2) variants. See also Supplementary Table S2
| Variant Name | Predicted Pathogenicity | ||||
|---|---|---|---|---|---|
| mRNA (NM_020708.4) | mRNA (NP_065759) | Mutation Taster | Panther | SIFT | |
| c.2855 G>A | p.952 R>H | Disease causing* | P (probability): 0.7174 | No prediction data | Not tolerated |
| c.3145 C>T | p.1049 R>C | Disease causing* | P (probability): 0.9999 | Pdeleterious = 0.94634 | Not tolerated |
Predicted specifically to disrupt the function of the last cytoplasmic domain.
The R952H variant was detected in five IGE cases, corresponding to an allele frequency of 0.66% (Table 1; Supplementary Table S2). After screening the first 950 control alleles, an allele frequency of 0.21% was determined (P = 0.253). The same allele frequency was calculated upon increasing the number of control alleles to 2428, which improved the P-value to 0.065 (Table 1). This rare variant appears to be more frequent in Quebec than in other populations; for example, it is reported in only 0.07% of EA alleles and 0.05% of all alleles on the EVS (Supplementary Table S1). Nevertheless, R952H is enriched in these Quebec IGE cases compared to Quebec controls. R952 shows strong evolutionary conservation (Fig 1), and histidine substitution at this site is predicted to have deleterious effects on protein function (Table 2). Thus, similar to R1049C, R952H appears to be an IGE risk variant. Furthermore, DNA was available from the parents of three probands with R952H, all of which were unaffected. The variant was inherited in all cases (maternally in two and paternally in one case).
KCC3 and NKCC1, CCCs like KCC2, have been implicated in seizures in humans and model organisms 16–20. Given the conservation of regulatory mechanisms regulating the CCCs 21–23, we screened these regions of KCC3 and NKCC1 in our IGE patient cohort (see Supplementary Methods for details). No KCC3 or NKCC1 coding variants were detected, thereby highlighting the specificity of the association of KCC2 variation with human IGE.
We next determined whether the identified IGE variants affect KCC2 function by recording the reversal potential of glycine receptors (GlyR, EGly), which is principally determined by intracellular Cl− concentration ([Cl−]i). GlyRs were co-expressed with IGE KCC2 variant or WT KCC2 in N2a cells. To visualize cells expressing different KCC2 species, all KCC2 constructs were conjugated at their N-termini with mCherry fluorescent protein (mCherry-KCC2) (Fig 2A) 24. Using the gramicidin-perforated patch-clamp technique, we found that cells expressing mCherry-KCC2 exhibited a strong shift in EGly toward negative (more hyperpolarized) values compared to controls (Fig 2B,C; −38.1 ± 3.7 mV and −89.4 ± 3.2, respectively, see Supplementary Table S3). mCherry-KCC2R1049C and mCherry-KCC2R952H also produced a hyperpolarizing shift of EGly; however, the absolute values were more positive (i.e., less hyperpolarized) compared to WT mCherry-KCC2 (Fig 2C). The difference between EGly in cells expressing wild-type mCherry-KCC2 and either mutant was reproducible and highly significant (P < 0.01).
Figure 2. Effect of IGE variants on KCC2-dependent Cl− reversal potential.
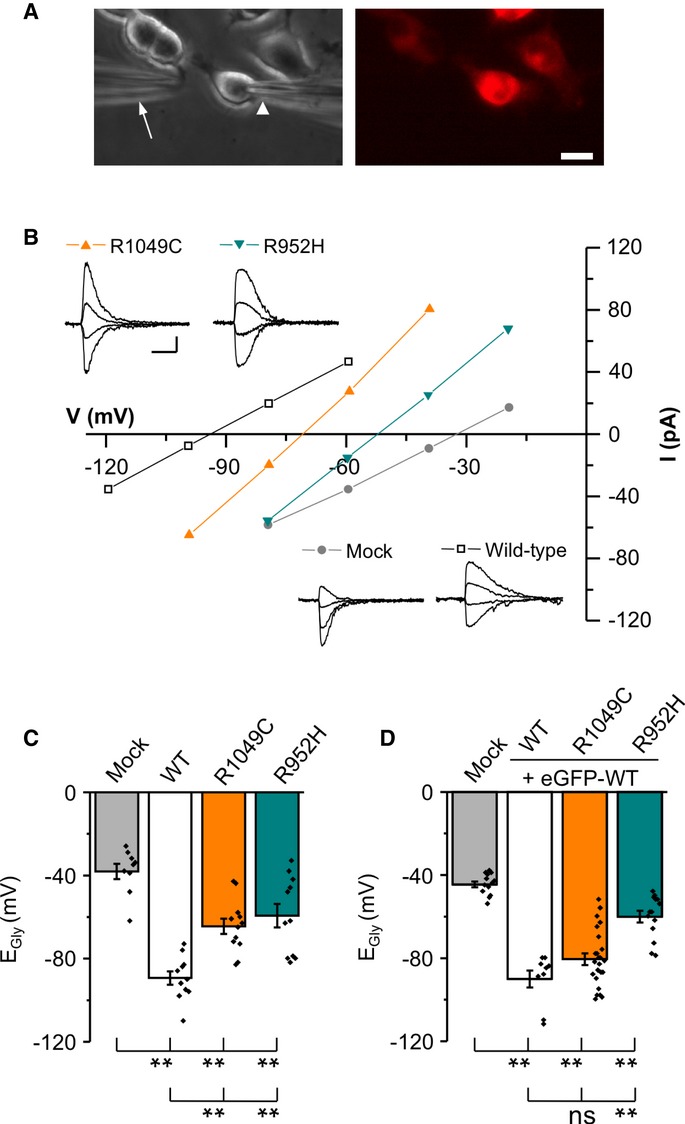
A Images of cells illustrating the position of the patch (arrowhead) and application pipette (arrow) used to deliver jet pulses of glycine (left). The mCherry-KCC2 positive cells were identified by their red fluorescence (right). Scale bar, 10 μm.
B Traces show examples of gramicidin patch-clamp recording obtained from N2a cells co-transfected with GlyR and an mCherry-KCC2 construct. Scale bars, 20 pA (vertical) and 500 ms (horizontal). Current amplitudes of GlyR currents (inset) were plotted against holding membrane potential. The current intercepts the voltage axis at EGly.
C Summary of all experiments similar to (B) (mean ± s.e.m., pooled data from 4 cultures, 2–3 cells per culture and condition). **P < 0.01, one-way ANOVA test. Mock denotes cells transfected with mCherry and GlyR; see Supplementary Table S2 for detailed statistics.
D Summary of all experiments similar to B, but obtained from cells co-transfected with a 50:50 mixture of WT eGFP-KCC2 and one of the indicated mCherry-KCC2 IGE mutants (mean ± s.e.m., pooled data from 4 cultures, 5–8 cells per culture and condition). **P < 0.01, one-way ANOVA test.
We non-invasively corroborated these results by measuring KCC2-mediated Cl− extrusion using Cl−-Sensor, a genetically encoded, ratiometric indicator of [Cl−]i 25. Cl−-Sensor was expressed in N2a cells with GlyR and WT or IGE variant mCherry-KCC2 (Fig 3A). Ratiometric recording of fluorescence was performed using excitation at 430 and 500 nm (R430/500) 26. Given the favorable weak endogenous Cl−-extruding mechanisms in N2a cells, once pre-loaded with Cl− using a 3-minute co-application of KCl and glycine, N2a cells maintain a relatively elevated Cl− level for more than 30 min 26 (Fig 3B). The mean ± s.e.m. half decay time of the fluorescence ratio was 26.7 ± 5.2 min, n = 4 (Fig 3D). In contrast to mock-transfected cells, cells expressing mCherry-KCC2 showed much faster ratiometric fluorescence recovery (see black trace in Fig 3B). The mean ± s.e.m. half decay time was 4.5 ± 0.4 min, (n = 4, P < 0.01, Fig 3D). Consistent with results obtained using gramicidin patch-clamp recording, mCherry-KCC2R952H or mCherry-KCC2R1049C resulted in a significantly slower recovery of the ratiometric fluorescence as compared to WT mCherry KCC2 (Fig 3B and D; Supplementary Table S4).
Figure 3. Effect of IGE variants on KCC2-mediated Cl− extrusion capacity.
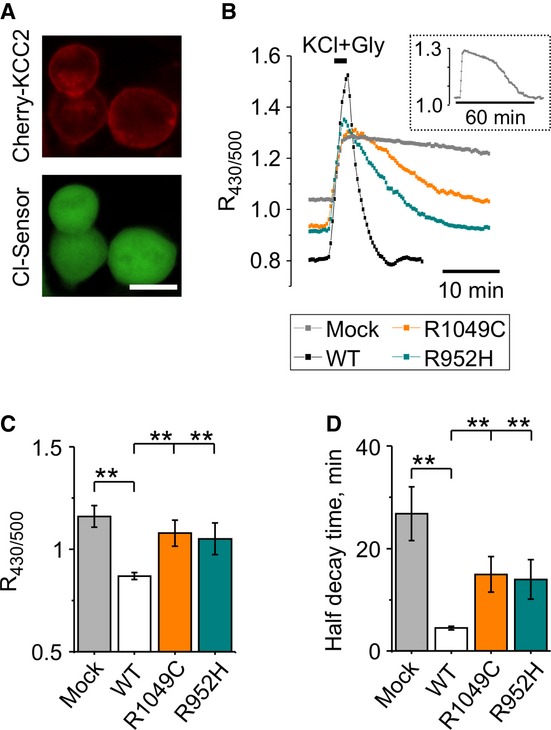
A Fluorescence signals recorded from N2a cells co-transfected with Cl-Sensor (green), GlyR (non-fluorescent), and mCherry-KCC2 (red, Obj 20×, NA 0.45, 500 nm excitation, scale bar = 20 μm).
B Representative traces of Cl-Sensor fluorescence ratio recording from N2a cells expressing different KCC2 constructs as indicated. Horizontal bar indicates the time of application of 100 mM KCl and 50 μM glycine. The ordinate axis indicates the ratio of Cl-Sensor fluorescence measured at 430 and 500 nm excitation wavelengths (R430/500). The inset illustrates the full record of R430/500 fluorescence from mock-transfected cells shown in the main plot.
C, D Columns show mean ± s.e.m. of the basal R430/500 level (C) and half decay time of Cl− extrusion after glycine + KCl application (D). n = 4 – 7 experiments. **P < 0.01, one-way ANOVA test (See Supplementary Tables S3 and S4 for details).
Measurements of the basal level of fluorescence ratio in cells expressing WT or IGE variant KCC2 constructs also supported our gramicidin-perforated patch results. Expression of mCherry-KCC2 produced a significant decrease in the basal level of R430/500 from 1.16 ± 0.05 to 0.87 ± 0.01 arbitrary units (AU) (Fig 3B and C). Expression of mCherry-KCC2R952H or mCherry-KCC2R1049C produced intermediate values that were significantly lower than those measured with mCherry-KCC2 (P < 0.05 for both mutants) and did not differ from the control mock-transfected cells (P > 0.05; Fig 3C, Supplementary Table S5). Thus, both identified IGE KCC2 mutants extrude Cl− with much lower efficacy relative to WT KCC2, resulting in cells with a higher basal level of intracellular Cl−, and consequently, less hyperpolarized responses to glycine.
We modeled the effects of the two IGE KCC2 variants in conditions mimicking their heterozygous nature by performing gramicidin-perforated patch-clamp recordings in cells expressing equal proportions of WT eGFP-KCC2 and either of the IGE variant mCherry-KCC2 constructs (see Supplementary Methods for details) (Fig 2D). Recordings were performed exclusively from cells showing both a green and red fluorescence signal of equivalent intensity. Similar to experiments in cells expressing KCC2 R952H alone (see Fig 2C), cells co-expressing WT KCC2 and KCC2 R952H exhibited significantly less hyperpolarizing values of EGly relative to WT KCC2 (Fig 2D; Supplementary Table S3). Cells co-expressing WT KCC2 and KCC2 R1049C also showed a trend toward less hyperpolarized EGly values; however, this was not statistically different from WT KCC2, and the variability coefficient was much higher (Fig 2D; Supplementary Table S3).
We next studied whether the identified IGE KCC2 variants exhibit alterations in KCC2 surface expression by expressing WT or IGE variant KCC2 constructs harboring a pHluorine tag in the second putative transmembrane domain of KCC2 (KCC2-pHext; see Supplementary Methods for details) in cultured hippocampal neurons (Fig 4). WT KCC2-pHext exhibited the expected strong membrane-localized fluorescence (Fig 4B; Supplementary Table S6); in contrast, no signal was detected in neurons expressing a control KCC2 construct harboring an eGFP tag on the intracellular N-terminal domain (eGFP-KCC2). Neurons expressing KCC2-pHext R1049C showed transporter membrane localization similar to neurons expressing WT KCC2-pHext (Fig 4B and C; Supplementary Table S5). In contrast, neurons expressing KCC2-pHext R952H exhibited a consistent >twofold decrease in surface KCC2-pHext signal (Fig 4B and C; Supplementary Table S5).
Figure 4. Effect of IGE KCC2 variants on KCC2 surface expression in rat primary hippocampal cultures.
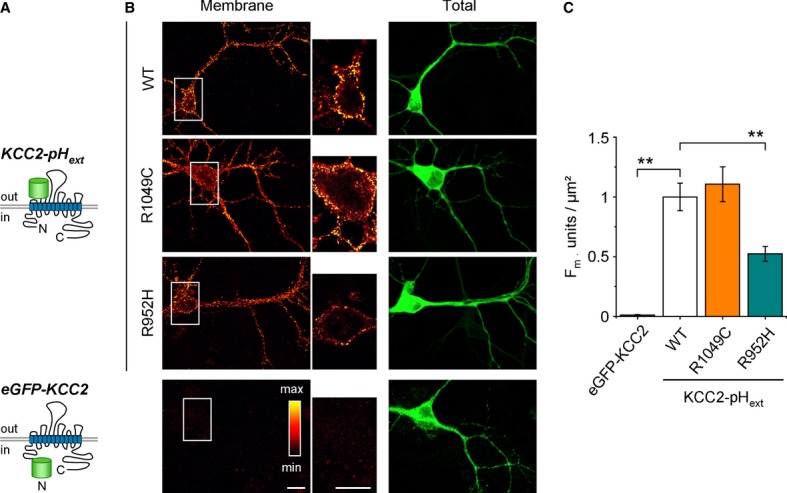
A Schematic presentation of transfected constructs.
B Representative images illustrating surface labeling (left column) and total expression (right column) of WT KCC2-pHext (top, raw image) and the R952H mutant of KCC2-pHext (middle, raw image). As a control for the efficacy of surface labeling, we used a KCC2 construct with eGFP linked to the intracellular N-terminal domain (bottom, raw image). Scale bars, 20 μm.
C Normalized mean ± s.e.m. of the fluorescence intensity (Fm, left plot) of the surface-labeled KCC2-pHext clusters (pooled data from 5 cultures, 5–8 cells per culture and condition). **P < 0.01, one-way ANOVA test. (See Supplementary Table S5 for details).
The regulatory phosphorylation of KCC2 is altered in several neurological disease models, resulting in impaired KCC2 function [reviewed in 10,11]. The stimulatory phosphorylation of KCC2 serine 940 (S940) promotes KCC2 activity via effects on both intrinsic transporter activity and trafficking to the plasma membrane, depending on the cellular context 27. Using phospho-specific antibodies 21,27 in Western blot assays with lysates derived from HEK293 cells expressing WT or IGE-mutant KCC2 constructs (see Supplementary Methods for details), we found that both KCC2 R952H and R1049C exhibited a reproducible and significant >50% decrease in S940 phosphorylation compared to WT KCC2, despite having unaltered levels of total protein expression (Fig 5A and B). These results suggest IGE KCC2 variants exhibit impaired function in part from a decrease in stimulatory S940 phosphorylation.
Figure 5. Effect of IGE KCC2 variants on serine 940 (S940) regulatory phosphorylation.
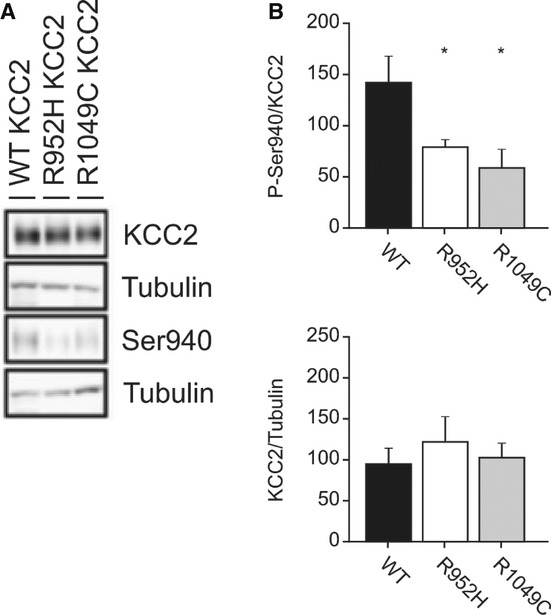
A HEK293 cells were nucleofected with the indicated WT, R952H, and R1049C KCC2 constructs. After 3 days, cells were lysed, and Western blotting was performed using anti-phospho-S940 KCC2 and total anti-KCC2 antibodies, as described in Materials and Methods. A representative Western blot is shown.
B Quantification of total phosphorylation of S940 relative to total KCC2 expression for each construct. 4 separate nucleofections were performed. *P < 0.05, one-way ANOVA test; All pairwise multiple comparison procedures (Tukey’s test). Error bars represent the mean ± s.e.m.
Discussion
The significant enrichment of KCC2 C-terminal NS genetic variants in IGE cases relative to controls (Table 1); the evolutionary conservation of the involved residues (Fig 1B and C); the predicted pathogenicity of their alteration in IGE (Table 2); and the impact of the detected IGE KCC2 variants on transporter function, trafficking, and/or S940 regulatory phosphorylation (Figs 2, 3, 4 and 5) suggest that genetically encoded impairment of KCC2 functional regulation might be a risk factor or contribute to the pathogenesis of human IGE. Notably, while our paper was in revision, Puskarjov et al 28 reported the KCC2-R952H variant co-segregating with febrile seizures in a single Australian family. In their study, KCC2-R952H was also shown to have reduced Cl− extrusion capacity and decreased surface expression relative to WT KCC2. In addition, Puskarjov et al 28 showed KCC2-R952H has a compromised ability to induce dendritic spines. These data suggest that KCC2-R952H is also a susceptibility variant for febrile seizures in addition to IGE and further strengthen the genetic link between KCC2 and human epilepsy.
IGE is genetically heterogeneous, and the difficulty of identifying single variants as risk factors has been demonstrated by 9. Heinzen et al did, however, report a list of variants that were found exclusively in IGE cases as potential risk factors (which did not include KCC2 variants), but suggested that the impact of an individual variant in IGE is small and that gene-based approaches might be more successful 9. We have demonstrated that targeting a particular gene and taking a ‘functional domain screening’ approach, followed by detailed physiological validation, might be a complimentary approach in gaining insight into the genetic predisposition of IGE.
The empirical risk of IGE is compatible with an oligogenic cause of disease, suggesting that variants in multiple genes collectively contribute to the disorder 29–31. Klassen et al 32 recently highlighted the complexity of IGE genetics. The authors noted that ion channel mutations are an important cause of disorders that affect the brain and other tissues so they decided to sequence 237 ion channel genes in 152 individuals with idiopathic epilepsy and 139 healthy controls. After developing variant profiles, three major conclusions were drawn: (i) complex combinations of common and rare ion channel variants are seen in epilepsy cases and controls; (ii) healthy individuals carry variants in known epilepsy genes; and (iii) epilepsy individuals carry more than one variant in known epilepsy genes. Thus, the authors concluded that, in most cases, causality cannot be assigned to any one variant, but rather results from an individual’s variant pattern, indicating an oligogenic mechanism 32. They ultimately suggest that computational modeling of biologic networks is needed to improve risk predictions. Additionally, the authors state that these variant patterns can even explain the silencing of pathogenic alleles and why such variants (e.g., KCC2 R952H and R1049C) can be present in control individuals as well as inherited from unaffected parents 32. Ultimately, population-based case/control studies are an approach that can implicate novel disease loci and provide a statistical measure of genetic risk. Studying a gene or gene region in this manner enables one to group variants together for statistical analysis, which minimizes multiple testing and makes it easier to reach levels of significance. Therefore, we conclude that NS variants in the C-terminus of KCC2 increases risk for IGE.
IGE KCC2 variants impair transporter Cl− extrusion capacity and render EGly less hyperpolarized compared to WT KCC2 (Figs 2 and 3), consistent with in silico prediction models. Decreased KCC2-mediated Cl− efflux in individuals carrying the IGE KCC2 variants would be anticipated to increase intracellular [Cl−], raising the Cl− reversal potential (ECl) to less hyperpolarized potentials and compromising GABAAR- and/or GlyR-mediated hyperpolarizing inhibition. These effects of IGE KCC2 variants would be similar in nature, though much less potent in magnitude given their heterozygous state, to model organisms with complete knockout 2–5 or perhaps mild dysfunction (~30%) of KCC2 functional expression 33,34, and humans with loss-of-function mutations in multiple GABAAR subunits in Mendelian IGE syndromes 35.
Our two IGE KCC2 mutants compromise KCC2 function likely by distinct mechanisms, including decreasing transporter plasmalemmal expression (R952H) or lowering the intrinsic activity of transporters at the cell surface (R1049C), consistent with the known trafficking-dependent and trafficking-independent mechanisms of KCC2 regulation encoded within the KCC2 C-terminus 10–12. IGE KCC2 variants, by changing C-terminal protein structure, might alter the function of key regulatory domains (e.g., the so-called ISO domain, encoded in amino acids 1022-1037, which is required for isotonic KCC2-mediated hyperpolarizing GABAergic transmission 13) by disrupting the binding of associated molecules. The significant inhibitory effect of R952H when co-expressed with WT KCC2 in a 1:1 ratio in neurons suggests a dominant-negative effect on transporter function, consistent with the known dimerization and oligomerization of KCC2 molecules 12,36. Compared to R952H, a different mechanism is likely involved with R1049C, given results in 1:1 co-expression experiments.
Interestingly, both IGE mutants, despite having different effects on KCC2 trafficking, nonetheless exhibit decreased S940 phosphorylation. This result is interesting, suggesting specific relevance for residue S940 in IGE pathogenesis. In this regard, it is notable that brief exposure to glutamate, which is associated with seizure activity in humans 37, causes a rapid inhibition of KCC2 activity via S940 dephosphorylation that results in a prolonged loss of hyperpolarizing GABAA-mediated currents in cultured hippocampal neurons 27,38. Further investigation into these mechanisms of KCC2 functional regulation and phosphorylation will be important topics of future detailed biochemical study and may reveal novel insights not only into IGE pathogenesis, but also into potential ways to modulate KCC2 activity for therapeutic benefit in treatment-resistant seizures 39,40 and possibly other pathologies 7,11.
Materials and Methods
Recruitment and diagnosis of affected individuals
A total of 380 individuals with idiopathic generalized epilepsy (IGE) from the Quebec population of Canada were studied. Ethics approval for the recruitment and genetic analysis of these individuals were granted by the ethics committee at the CRCHUM (Centre de recherche du Centre hospitalier de l’Université de Montréal) and Montreal Neurological Institute. Upon recruitment, informed consent was obtained from all participants and a blood sample was collected for the genetic analysis. Recruitment was based on a referral from a neurologist or pediatrician in Quebec, Canada. All patients were from the greater Montreal and Quebec City regions. The diagnosis of IGE was based on a detailed clinical interview, full neurological examination, and an electroencephalography (EEG) recording. Patients were diagnosed according to International League Against Epilepsy (ILAE) criteria 41.
Population Controls
A total of 475 unrelated individuals who were recruited to the Rouleau laboratory as Quebec population controls were Sanger sequenced during Phase 1 of the project following the same protocol as IGE cases. In an effort to validate the association that was identified in Phase 1, an additional 516 population control samples from the Rouleau laboratory were Sanger sequenced. Furthermore, 223 unrelated individuals with French-Canadian ancestry who have never been diagnosed with a neurodevelopmental or neurodegenerative disorder and were previously exome sequenced were used as additional controls [96 of the exome-sequenced samples were provided by the CARTaGENE project 42; an additional 74 and 53 exome-sequenced samples were provided by the laboratories of Dr. Majewski and Dr. Guy Rouleau, respectively].
Data within the chromosome 20 interval between 44,683,555 and 44,685,942 base pairs (hg19) that correspond to the nucleotides in exons 21 25 [NM_020708.4] and encode the C-terminus of KCC2 (SLC12A5) (encompassing amino acids 894 1086 [NP_065759]) were analyzed for variants. Samples were captured using either the Agilent capture kits v3 and v4 or the Illumina TruSeq Exome Enrichment and TruSeq DNA LT Sample Prep v2 kits. Sequencing was performed using the Illumina Highseq2000 sequencing platform at the Genome Quebec Innovation Center achieving a sequencing coverage averaging 70X. Exome read files were aligned to the human reference genome (hg19) using Burrow’s Wheeler Aligner (BWA). PCR duplicates were removed, and only properly paired and uniquely mapped reads were kept. Realignments around indels and recalibration were performed with GATK tools. The variants were called using SAMtools/BCFtools and annotated using Annovar.
Please see Supplementary Methods for further materials and methods.
Acknowledgments
KK is supported by a Harvard-MIT Neuroscience grant and the Manton Center for Orphan Disease Research at Children’s Hospital Boston. GR is supported by the Canadian Institutes of Health Research.
Author contributions
KTK, NDM, BL, PAD, PA, JM, TZD, SJM, IM, and GAR conceived and designed the experiments. PC provided samples. KTK, NDM, PF, LS, BL, YS, PLT, CB, AL, DS, ADL, AH, HN, and IM performed the experiments. KTK, NDM, PF, LS BL, AK, YS, PLT, BW, DS, ADL, AH, PA, HN, JM, TZD, SJM, IM, and GAR analyzed the data. KTK, NDM, AK, PAD, BW, TZD, SJM, IM, and GAR wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://embor.embopress.org
References
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Tanis JE, Bellemer A, Moresco JJ, Forbush B, Koelle MR. The potassium chloride cotransporter KCC-2 coordinates development of inhibitory neurotransmission and synapse structure in Caenorhabditis elegans. J Neurosci. 2009;29:9943–9954. doi: 10.1523/JNEUROSCI.1989-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellemer A, Hirata T, Romero MF, Koelle MR. Two types of chloride transporters are required for GABA(A) receptor-mediated inhibition in C. elegans. EMBO J. 2011;30:1852–1863. doi: 10.1038/emboj.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Lundy MY, Ranga R, Tanouye MA. Mutations in the K+/Cl− cotransporter gene kazachoc (kcc) increase seizure susceptibility in Drosophila. J Neurosci. 2006;26:8943–8954. doi: 10.1523/JNEUROSCI.4998-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Mercado A, Fajilan AA, Lee AW, Hsu R, Mount DB, Tanouye MA. Seizure sensitivity is ameliorated by targeted expression of K+-Cl− cotransporter function in the mushroom body of the Drosophila brain. Genetics. 2010;184:171–183. doi: 10.1534/genetics.109.109074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, Mount DB. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol. 2008;4:490–503. doi: 10.1038/ncpneuro0883. [DOI] [PubMed] [Google Scholar]
- Gardiner M. Genetics of idiopathic generalized epilepsies. Epilepsia. 2005;46(Suppl 9):15–20. doi: 10.1111/j.1528-1167.2005.00310.x. [DOI] [PubMed] [Google Scholar]
- Heinzen EL, Depondt C, Cavalleri GL, Ruzzo EK, Walley NM, Need AC, Ge D, He M, Cirulli ET, Zhao Q, et al. Exome sequencing followed by large-scale genotyping fails to identify single rare variants of large effect in idiopathic generalized epilepsy. Am J Hum Genet. 2012;91:293–302. doi: 10.1016/j.ajhg.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamma I, Chevy Q, Poncer JC, Levi S. Role of the neuronal K-Cl co-transporter KCC2 in inhibitory and excitatory neurotransmission. Front Cell Neurosci. 2012;6:5. doi: 10.3389/fncel.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Deeb TZ, Puskarjov M, Silayeva L, Liang B, Kaila K, Moss SJ. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 2013;36:726–737. doi: 10.1016/j.tins.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina I, Friedel P, Rivera C, Kahle KT, Kourdougli N, Uvarov P, Pellegrino C. Current view on the functional regulation of the neuronal K+-Cl− cotransporter KCC2. Front Cell Neurosci. 2014;8:27. doi: 10.3389/fncel.2014.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acton BA, Mahadevan V, Mercado A, Uvarov P, Ding Y, Pressey J, Airaksinen MS, Mount DB, Woodin MA. Hyperpolarizing GABAergic transmission requires the KCC2 C-terminal ISO domain. J Neurosci. 2012;32:8746–8751. doi: 10.1523/JNEUROSCI.6089-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darman RB, Forbush B. A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J Biol Chem. 2002;277:37542–37550. doi: 10.1074/jbc.M206293200. [DOI] [PubMed] [Google Scholar]
- Gimenez I. Molecular mechanisms and regulation of furosemide-sensitive Na-K-Cl cotransporters. Curr Opin Nephrol Hypertens. 2006;15:517–523. doi: 10.1097/01.mnh.0000242178.44576.b0. [DOI] [PubMed] [Google Scholar]
- Loscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology. 2013;69:62–74. doi: 10.1016/j.neuropharm.2012.05.045. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Staley KJ. Neonatal seizures and neuronal transmembrane ion transport. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4th edn. Bethesda, MD: National Center for Biotechnology Information (US); 2012. pp. 1–12. [Internet] [PubMed] [Google Scholar]
- Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev. 2005;85:423–493. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- Howard HC, Mount DB, Rochefort D, Byun N, Dupre N, Lu J, Fan X, Song L, Riviere JB, Prevost C, et al. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet. 2002;32:384–392. doi: 10.1038/ng1002. [DOI] [PubMed] [Google Scholar]
- Boettger T, Rust MB, Maier H, Seidenbecher T, Schweizer M, Keating DJ, Faulhaber J, Ehmke H, Pfeffer C, Scheel O, et al. Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J. 2003;22:5422–5434. doi: 10.1093/emboj/cdg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Los Heros P, Alessi DR, Gourlay R, Campbell DG, Deak M, Macartney TJ, Kahle KT, Zhang J. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl− co-transporters. Biochem J. 2014;458:559–573. doi: 10.1042/BJ20131478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Rinehart J, Lifton RP. Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim Biophys Acta. 2010;1802:1150–1158. doi: 10.1016/j.bbadis.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, Risinger M, Pan W, Wu D, Colangelo CM, et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell. 2009;138:525–536. doi: 10.1016/j.cell.2009.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino C, Gubkina O, Schaefer M, Becq H, Ludwig A, Mukhtarov M, Chudotvorova I, Corby S, Salyha Y, Salozhin S, et al. Knocking down of the KCC2 in rat hippocampal neurons increases intracellular chloride concentration and compromises neuronal survival. J Physiol. 2011;589:2475–2496. doi: 10.1113/jphysiol.2010.203703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markova O, Mukhtarov M, Real E, Jacob Y, Bregestovski P. Genetically encoded chloride indicator with improved sensitivity. J Neurosci Methods. 2008;170:67–76. doi: 10.1016/j.jneumeth.2007.12.016. [DOI] [PubMed] [Google Scholar]
- Friedel P, Bregestovski P, Medina I. Improved method for efficient imaging of intracellular Cl(−) with Cl− sensor using conventional fluorescence setup. Front Mol Neurosci. 2013;6:7. doi: 10.3389/fnmol.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Deeb TZ, Walker JA, Davies PA, Moss SJ. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat Neurosci. 2011;14:736–743. doi: 10.1038/nn.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puskarjov M, Seja P, Heron SE, Williams TC, Ahmad F, Iona X, Oliver KL, Grinton BE, Vutskits L, Scheffer IE, et al. A variant of KCC2 from patients with febrile seizures impairs neuronal Cl− extrusion and dendritic spine formation. EMBO Rep. 2014;15:723–729. doi: 10.1002/embr.201438749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinlein OK. Genetic mechanisms that underlie epilepsy. Nat Rev Neurosci. 2004;5:400–408. doi: 10.1038/nrn1388. [DOI] [PubMed] [Google Scholar]
- Lu Y, Wang X. Genes associated with idiopathic epilepsies: a current overview. Neurol Res. 2009;31:135–143. doi: 10.1179/174313209X393942. [DOI] [PubMed] [Google Scholar]
- Saint-Martin C, Gauvain G, Teodorescu G, Gourfinkel-An I, Fedirko E, Weber YG, Maljevic S, Ernst JP, Garcia-Olivares J, Fahlke C, et al. Two novel CLCN2 mutations accelerating chloride channel deactivation are associated with idiopathic generalized epilepsy. Hum Mutat. 2009;30:397–405. doi: 10.1002/humu.20876. [DOI] [PubMed] [Google Scholar]
- Klassen T, Davis C, Goldman A, Burgess D, Chen T, Wheeler D, McPherson J, Bourquin T, Lewis L, Villasana D, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145:1036–1048. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo NS, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- Zhu L, Polley N, Mathews GC, Delpire E. NKCC1 and KCC2 prevent hyperexcitability in the mouse hippocampus. Epilepsy Res. 2008;79:201–212. doi: 10.1016/j.eplepsyres.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maljevic S, Krampfl K, Cobilanschi J, Tilgen N, Beyer S, Weber YG, Schlesinger F, Ursu D, Melzer W, Cossette P, et al. A mutation in the GABA(A) receptor alpha(1)-subunit is associated with absence epilepsy. Ann Neurol. 2006;59:983–987. doi: 10.1002/ana.20874. [DOI] [PubMed] [Google Scholar]
- Uvarov P, Ludwig A, Markkanen M, Soni S, Hubner CA, Rivera C, Airaksinen MS. Coexpression and heteromerization of two neuronal K-Cl cotransporter isoforms in neonatal brain. J Biol Chem. 2009;284:13696–13704. doi: 10.1074/jbc.M807366200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993;341:1607–1610. doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- Lee HH, Walker JA, Williams JR, Goodier RJ, Payne JA, Moss SJ. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J Biol Chem. 2007;282:29777–29784. doi: 10.1074/jbc.M705053200. [DOI] [PubMed] [Google Scholar]
- Deeb TZ, Maguire J, Moss SJ. Possible alterations in GABAA receptor signaling that underlie benzodiazepine-resistant seizures. Epilepsia. 2012;53:79–88. doi: 10.1111/epi.12037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeb TZ, Nakamura Y, Frost GD, Davies PA, Moss SJ. Disrupted Cl− homeostasis contributes to reductions in the inhibitory efficacy of diazepam during hyperexcited states. Eur J Neurosci. 2013;38:2453–2467. doi: 10.1111/ejn.12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anon. Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia. 1989;30:389–399. doi: 10.1111/j.1528-1157.1989.tb05316.x. [DOI] [PubMed] [Google Scholar]
- Hodgkinson A, Idaghdour Y, Gbeha E, Grenier JC, Hip-Ki E, Bruat V, Goulet JP, de Malliard T, Awadalla P. High-resolution genomic analysis of human mitochondrial RNA sequence variation. Science. 2014;344:413–415. doi: 10.1126/science.1251110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.