

Abstract
Loss of the lysosomal ClC-7/Ostm1 2Cl−/H+ exchanger causes lysosomal storage disease and osteopetrosis in humans and additionally changes fur colour in mice. Its conversion into a Cl− conductance in Clcn7unc/unc mice entails similarly severe lysosomal storage, but less severe osteopetrosis and no change in fur colour. To elucidate the basis for these phenotypical differences, we generated Clcn7td/td mice expressing an ion transport-deficient mutant. Their osteopetrosis was as severe as in Clcn7−/− mice, suggesting that the electric shunt provided by ClC-7unc can partially rescue osteoclast function. The normal coat colour of Clcn7td/td mice and their less severe neurodegeneration suggested that the ClC-7 protein, even when lacking measurable ion transport activity, is sufficient for hair pigmentation and that the conductance of ClC-7unc is harmful for neurons. Our in vivo structure-function analysis of ClC-7 reveals that both protein-protein interactions and ion transport must be considered in the pathogenesis of ClC-7-related diseases.
Subject Categories: Membrane & Intracellular Transport; Molecular Biology of Disease
Keywords: acidification, anion transport, grey-lethal, lysosome, Wnt signalling
Introduction
Acidic luminal pH in endosomes and lysosomes influences their trafficking, enzymatic activities and transport of substances across their limiting membranes. Luminal acidification is accomplished by electrogenic vacuolar H+-ATPases that require an electric shunt, which in the classical model was thought to be mediated by chloride channels. Members of the CLC anion transporter gene family 1,2, five of which reside in endosomes or lysosomes, were thought to represent these channels. However, ClC-4 through ClC-7 are rather exchangers that couple Cl− influx to H+ efflux 3–7. Electrogenic Cl−/H+ exchange can support proton pumping 8,9 and might be even more efficient than Cl− channels in supporting vesicular acidification 6. However, lysosomal pH is normal in mice lacking ClC-7 6,10,11, and the strict coupling of Cl− flux to H+ countertransport suggested that vesicular CLCs accumulate Cl− into acidic compartments 12 as shown for lysosomes 6. To clarify the relative contributions of shunt conductance and proton coupling to their biological roles, we had generated Clcn5unc/unc and Clcn7unc/unc mice in which Cl− transport was uncoupled from H+ transport by single point mutations 6,8. Surprisingly, these unc mice 6,8 displayed grosso modo the same phenotypes as the respective null mice 10,13,14, that is, impaired renal endocytosis in Clcn5unc/unc mice and osteopetrosis associated with a lysosomal storage disorder and neurodegeneration in Clcn7unc/unc mice (Supplementary Table S1). Hence, a Cl− conductance cannot replace electrogenic Cl−/H+ exchange in many cellular functions.
ClC-7, together with its obligate β-subunit Ostm1 11, is expressed in virtually all tissues 14,15. It localizes to late endosomes and lysosomes and is inserted into the acid-secreting ruffled border of bone-resorbing osteoclasts 10,14. Loss of ClC-7 function causes osteopetrosis in mice 14, humans 14,16 and cattle 17 and entails lysosomal storage and neurodegeneration in mice 10. Decreased proteolytical capacity of lysosomes was demonstrated in Clcn7−/− proximal tubules 18. The unchanged steady-state pH of Clcn7−/− lysosomes 10,11 was explained by a lysosomal cation conductance that shunts H+-ATPase currents in parallel to ClC-7 6,19. By contrast, the osteopetrosis of Clcn7−/− mice was attributed to impaired acidification of the osteoclast resorption lacuna 14. Together with the H+-ATPase, ClC-7 is inserted by lysosomal exocytosis into the ruffled border of osteoclasts where it may shunt H+-ATPase currents 14. Finally, Clcn7−/− mice display grey fur in an agouti background. This phenotype might be linked to melanosomes, a lysosome-related compartment. Phenotypes virtually identical to those of Clcn7−/− mice are found in grey-lethal mice 11,20 which carry a mutation in the gene encoding Ostm1.
Whereas the phenotypes of Clcn5−/− and Clcn5unc/unc mice are nearly identical, some of the phenotypes of Clcn7−/− and Clcn7unc/unc mice differ in severity 6, that is, Clcn7unc/unc mice show less severe osteopetrosis and lack the coat colour phenotype 6,11 (Supplementary Table S1). Two hypotheses may be invoked to explain these differences. First, the shunt conductance provided by ClC-7unc may suffice to support some, but not all cellular functions. Second, the difference may be owed to lacking ClC-7 protein interactions in Clcn7−/−, but not in Clcn7unc/unc mice which express a correctly targeted ClC-7 mutant at normal levels 6.
Here, we generated a novel Clcn7td/td mouse model that expresses a transport-deficient point mutant of ClC-7. The ClC-7td mutant protein neither transports Cl− nor H+ to a measurable degree 7, but, like ClC-7unc, is expected to be fully interaction competent. Comparative analyses of these mice suggest that a pure Cl− conductance partially rescues the lack of Cl−/H+ exchange in osteoclasts, whereas normal pigmentation requires ClC-7 protein interactions, but not ClC-7 ion transport activity. Surprisingly, our study also shows that the Cl− conductance of ClC-7unc may have detrimental effects on CNS neurons.
Results
Transport-deficient ClC-7 mutant and Ostm1 are expressed normally
We generated mice in which the ‘proton glutamate’ E312 of ClC-7 was mutated to alanine (Supplementary Fig S1) abolishing both Cl− and H+ transport of ClC-7/Ostm1 7, hence our designation of this allele as ‘transport deficient’ (td). Homozygous Clcn7td/td mice were born at Mendelian ratio. Like Clcn7unc/unc and Clcn7−/− mice, they were growth retarded and most of them died within 6 weeks after birth. Surprisingly, a few Clcn7td/td mice survived more than 1 year (Supplementary Fig S2). The genetic background of Clcn7td/td mice cannot account for this difference as these mice were studied in comparable mixed genetic backgrounds. Clcn7+/td mice lacked an obvious phenotype. ClC-7td protein levels were undistinguishable from ClC-7 levels in wild-type (WT) mice (Fig 1A and B). Like in Clcn7unc/unc mice 6, neither the abundance of Ostm1 11 nor its processing by lysosomal proteases was changed in Clcn7td/td mice (Fig 1A). WT ClC-7 and ClC-7td similarly localized to Lamp-1-positive structures in primary fibroblasts (Fig 1C), suggesting unchanged interactions with the trafficking machinery. Ostm1 had left the endoplasmic reticulum (ER) and co-localized with ClC-7td in lysosomes (Fig 1C).
Figure 1. Normal expression of ClC-7td and Ostm1 in Clcn7td/td mice.
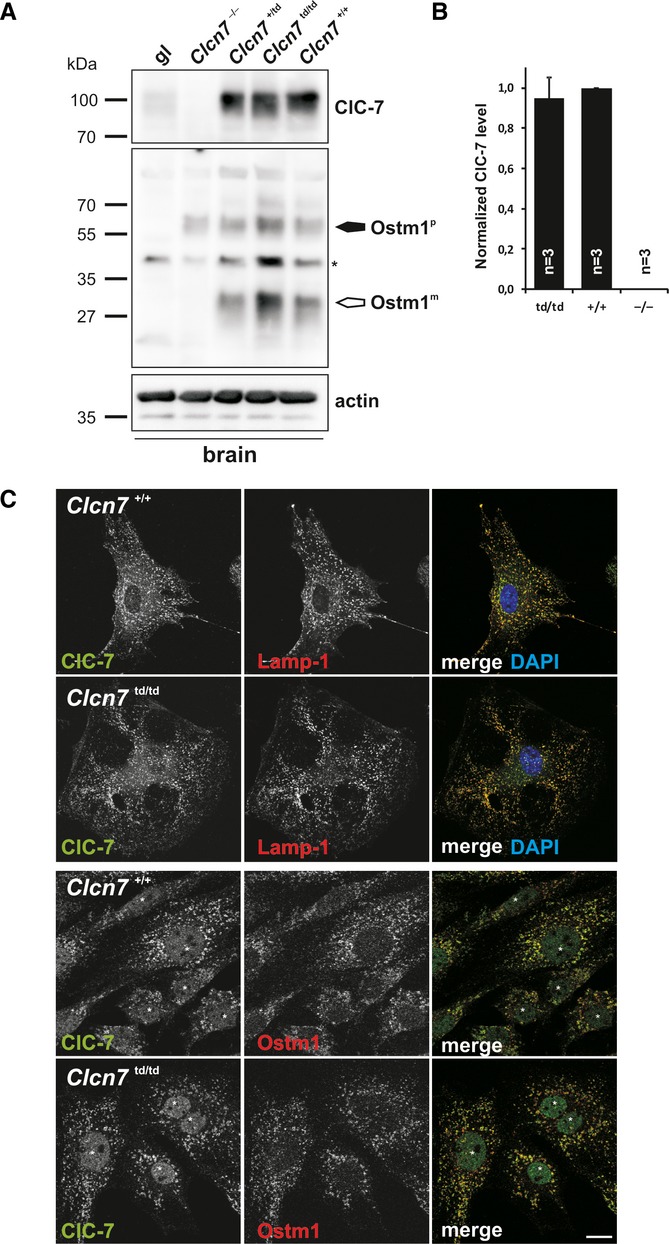
A ClC-7 and Ostm1 immunoblots of brain membranes. ClC-7 was drastically reduced in grey-lethal (gl) mice. Only the Ostm1 precursor Ostm1P (filled arrow) is present in Clcn7−/− tissue. Ostm1m, mature form (open arrow). Actin, loading control. Asterisk, non-specific band.
B Quantification of ClC-7 immunoblots (normalized to actin).
C Co-immunostaining of ClC-7 (green) with Lamp-1 (red, upper panels) or Ostm1 (red, lower panels) in primary fibroblasts. ClC-7td and Ostm1 co-localize in Lamp-1-positive structures. DNA stained with DAPI (upper panels). Asterisks, non-specific immunoreactivity in the nucleus (scale bar: 20 μm).
Lysosomal ion homeostasis in Clcn7td/td mice
Because ClC-7 may contribute to a countercurrent for the vacuolar H+-ATPase 7,14, we measured lysosomal pH of Clcn7td/td fibroblasts and found it to be unchanged (Supplementary Fig S3A). Measurements of lysosomal Cl− concentration with a dextran-coupled Cl−-sensitive ratiometric dye 6 revealed reduced lysosomal Cl− accumulation (Supplementary Fig S3B). Both results resemble those made with Clcn7−/− and Clcn7unc/unc mice 6,10.
Delayed neurodegeneration in Clcn7td/td mice
Like Clcn7−/− and Clcn7unc/unc mice, Clcn7td/td mice displayed progressive degeneration in the hippocampus (Fig 2). However, it appeared much later and was only detectable in the few surviving older mice. Neuronal cell loss was observed within the CA3 region and progressed to an almost complete loss of CA3 pyramidal cells at 10 months of age (Fig 2D and E). There was no detectable hippocampal cell loss in 4-week-old Clcn7td/td mice (Fig 2C). Nevertheless, pathological changes were observed, in particular in CA3 neurons and in some parts of the cortex. In those regions, lysosomal membrane proteins like Lamp-1 and ClC-7td itself were more intensely labelled and showed a broad distribution in neuronal somata rather than being stained in scattered puncta as in the WT (Fig 3A and B and Supplementary Fig S4). A similar observation was made for ClC-7unc (Fig 3A). Lysosomal storage was apparent 4 weeks after birth, including intracellular carbohydrate accumulation (Fig 2F) and increased levels of lysosomal acid phosphatase (Fig 2G). At P21, electron-dense osmiophilic material accumulated in lysosomal compartments of neuronal somata in the CA3, but not in the CA1 region (Fig 2H). The autophagy marker LC3-II was strongly increased in Clcn7−/− and Clcn7unc/unc, but not in Clcn7td/td mice (Fig 2I). In striking contrast to the early postnatal retinal degeneration of Clcn7−/− and Clcn7unc/unc mice, retinae of 3-week-old (Supplementary Fig S5A) and even 10-month-old Clcn7td/td mice (Supplementary Fig S5B) appeared normal. The increased survival of Clcn7td/td mice thus correlated with their delayed neurodegeneration.
Figure 2. Brain pathology of Clcn7td/td mice.

A–C Neuronal cell loss (arrowheads) in hippocampal CA3 region of Clcn7−/− but not of Clcn7td/td mice (Nissl staining).
D, E Incipient (D) and complete (E) CA3 neurodegeneration (arrowheads) of 5- and 10-month-old Clcn7td/td mice, respectively. Right panels: higher magnification not necessarily of section shown at left (scale bars: left, 400 μm; right, 100 μm).
F Strong PAS staining (arrowheads) in P28 CA3 neurons of Clcn7td/td but not of WT mice (scale bar: 100 μm).
G Increased lysosomal acid phosphatase activity in the cortex of P28 Clcn7td/td mice compared to WT (scale bar: 100 μm).
H Lysosomal storage material in CA3 pyramidal neuron somata shown by electron microscopy (N, nucleus; dotted line around soma). Right: higher magnification of a soma different from that at left (scale bars: left: 2 μm; right: 1 μm).
I Immunoblots showing an increase in the autophagic marker LC3-II in the brain of 3-week-old Clcn7−/− and Clcn7unc/unc versus Clcn7td/td and WT mice. Actin, loading control. Right panel: Quantification of immunoblots normalized to actin and LC3-I. Error bars denote s.d.
Figure 3. Abnormal lysosomal morphology in ClC-7 mouse models.
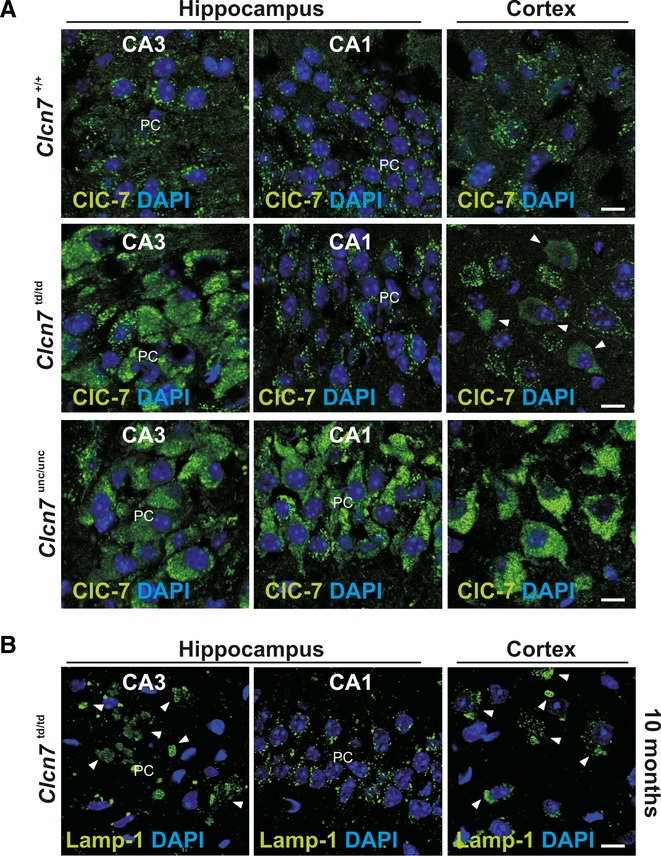
A ClC-7 immunolabelling in the somata of CA1 and CA3 pyramidal and of cortical neurons in Clcn7unc/unc, Clcn7td/td and WT mice. ClC-7td is abnormal in the CA3 region and partially in the cortex. Increased labelling intensity suggests larger ClC-7 amounts in Clcn7td/td CA3, and in Clcn7unc/unc CA3, CA1, and cortex (scale bar: 10 μm; PC, Purkinje cells).
B Abnormal Lamp-1 distribution in cortical and CA3 (arrowheads), but not CA1 neurons of 10-month-old Clcn7td/td mice. DNA stained with DAPI (scale bar: 10 μm).
Osteopetrosis of ClC-7td mice as severe as in ClC-7 KO
Immunolabelling of tibiae revealed that both ClC-7td and its β-subunit Ostm1 were normally expressed in Clcn7td/td osteoclasts (Supplementary Fig S6A). ClC-7 and ClC-7td similarly co-localized with the a3 subunit of the V-type H+-ATPase at the ruffled border (Supplementary Fig S6B). Unlike the milder osteopetrosis of Clcn7unc/unc mice 6, the osteopetrosis of Clcn7td/td (Fig 4A) was as severe as in Clcn7−/− 14 or grey-lethal (Ostm1−/−) mice 20. Bone density was similarly increased in Clcn7td/td and Clcn7−/− mice (Fig 4C). As observed for the other ClC-7 mouse models, teeth were formed in Clcn7td/td mice, but did not erupt (Fig 4B). Electron micrographs showed a partially deranged ruffled border membrane in Clcn7td/td mice (Fig 4D). Using the sealing zone, which laterally delimits the resorption lacuna between osteoclasts and bone matrix, as localization marker, we categorized ruffled borders in situ as absent, immature or mature (Fig 4D and E). 20% of osteoclasts from Clcn7−/− and Clcn7td/td mice totally lacked a ruffled border, and only about 40% showed a mature ruffled border (Fig 4E). All osteoclasts from Clcn7unc/unc mice formed ruffled border membranes, of which 70% appeared mature. Hence, the severity of osteopetrosis correlates with an impairment of ruffled border formation.
Figure 4. Osteopetrosis in Clcn7td/td mice.
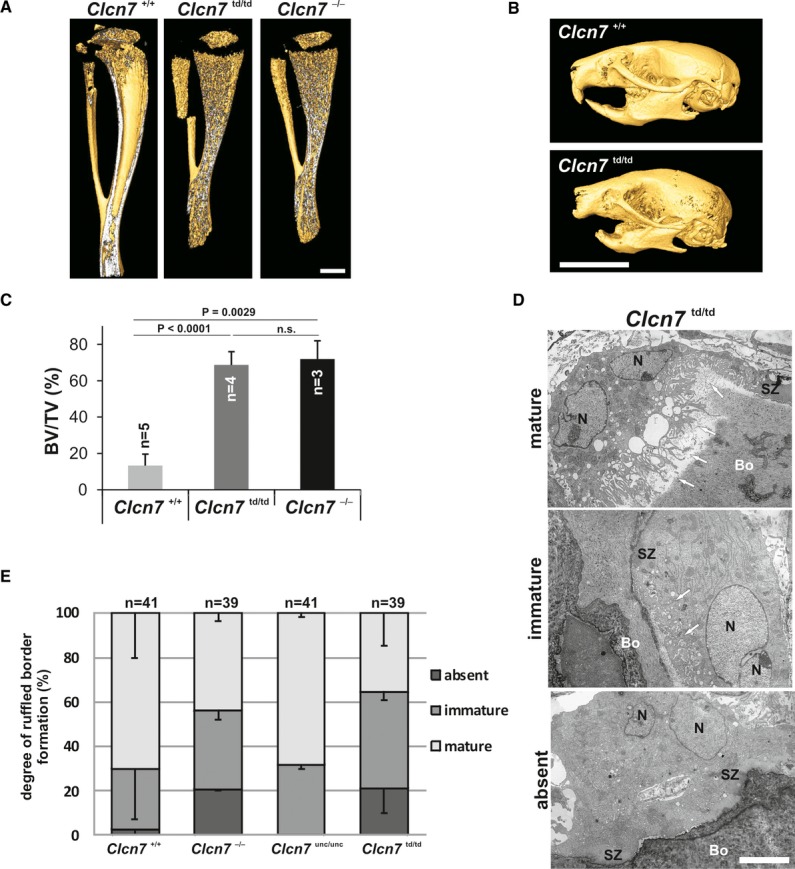
A Micro-CT revealed similar osteopetrosis of tibiae of 3-week-old Clcn7−/− and Clcn7td/td mice (scale bar: 1 mm).
B Micro-CT image of skull from P22 Clcn7td/td mouse showed impaired tooth eruption (scale bar: 10 mm).
C Similarly, increased bone volume fraction of proximal tibia metaphyseal trabecular bone in Clcn7td/td mice and Clcn7−/− mice. BV, bone volume; TV, tissue volume. Student’s t-test was applied; n.s., not significant. Error bars denote s.e.m.
D Electron microscopy showed mature, immature and lacking ruffled borders of Clcn7td/td osteoclasts despite the presence of a sealing zone (SZ). Arrows point at ruffled borders (scale bar: 5 μm) (Bo = bone; N = nucleus).
E Percentage of WT, Clcn7unc/unc, Clcn7td/td, Clcn7−/− osteoclasts exhibiting absent, immature or mature ruffled borders. Error bars denote s.e.m.
Coat colour phenotype is absent in ClC-7td mice
The pigments of hair and skin are synthesized in melanosomes, a lysosome-related compartment of melanocytes, and are then transferred to keratinocytes. The grey fur of Clcn7−/− or Ostm1−/− mice 14,20 thus agrees with the lysosomal localization of ClC-7/Ostm1. Surprisingly, the fur colour was changed neither in Clcn7unc/unc 6 nor in Clcn7td/td mice (Fig 5A) which express mutant full-length ClC-7 proteins that display or lack, respectively, a Cl− conductance. The agouti gene modulates the colour of the hair shaft, resulting in a band of yellow (owed to pheomelanin granules) in the otherwise dark (eumelanin) pigmented hair shaft. The pigment in the yellow band was clumped and reduced in Clcn7−/− and Ostm1−/− (gl) mice, whereas eumelanin granules were unchanged in their dark hair shafts (Fig 5B). Hair shaft pigmentation of Clcn7unc/unc and Clcn7td/td mice was unchanged compared to WT (Fig 5B).
Figure 5. ClC-7td mice lack a coat colour phenotype.
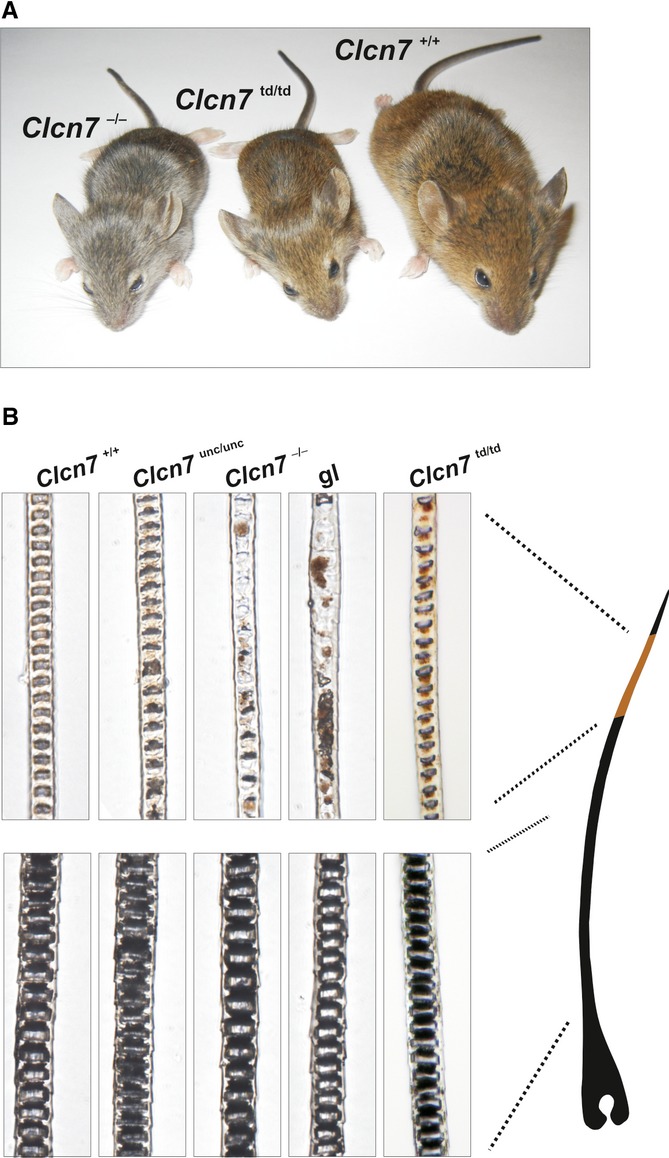
A Clcn7td/td mice are brown contrasting with the grey fur of Clcn7−/− mice.
B Upper panels: pheomelanin granules clump in hair shafts of Clcn7−/− and Ostm1−/− but not in Clcn7unc/unc, Clcn7td/td and WT mice. Normally distributed eumelanin granules in the dark hair shafts of all mouse models (lower panels).
Activation of Wnt signalling in primary fibroblasts and melanocytes
Melanocyte differentiation depends on Wnt signalling 21 and Ostm1 has been proposed to play a role in the canonical Wnt pathway 22 although the molecular mechanism remains obscure. Because the Ostm1 protein is absent or severely reduced in Ostm1−/− and Clcn7−/− mice, respectively 11, but unchanged in Clcn7unc/unc 6 and Clcn7td/td mice, we asked whether the difference in coat colour might be due to differences in Wnt signalling. Primary fibroblasts from WT, Clcn7−/− and Ostm1−/− mice were exposed to Wnt3a to activate canonical Wnt signalling and mRNA levels of the target gene axin2 23 were determined. Basal and Wnt3-stimulated axin2 expression was unchanged in Clcn7−/− and gl (Ostm1−/−) fibroblasts (Supplementary Fig S7A and B) and in Clcn7−/− melanocytes (Supplementary Fig S7C) compared to WT. Hence, differential activation of the Wnt signalling pathway is unlikely to contribute to the phenotypical differences of the present Clcn7 mouse models.
Discussion
Our analysis of Clcn7td/td, Clcn7−/− and Clcn7unc/unc mice 6,14 represents a novel in vivo structure-function analysis of ClC-7/Ostm1 that complements similar in vitro studies focusing on biophysical properties 7,24. Comparison of the pathologies of these mice (Supplementary Table S1) yielded a surprisingly complex picture of the roles of ClC-7 in lysosome, osteoclast and melanocyte biology. Phenotypes resulting from Clcn7 mutations cannot be assigned exclusively to a loss of ion transport activity.
As vesicular CLCs may shunt proton pump currents in the endolysosomal system and at the osteoclast resorption lacuna 2,8,9,14, we assumed that a similar shunt by the ClC-7unc Cl− conductance may rescue some of the pathologies of Clcn7−/− mice. Comparison of the present Clcn7td/td with Clcn7−/− and Clcn7unc/unc mice showed that this holds true for osteopetrosis, but not for the changed fur colour or neurodegeneration. The severity of osteopetrosis, which is less severe in Clcn7unc/unc mice than in the other two mouse models, correlated with the malformation of the ruffled border. Hence, the formation of this acid-secreting membrane does not only require the presence of the ClC-7/Ostm1 protein complex, but also its ion transport activity. A Cl− conductance can only partially substitute for 2Cl−/H+ exchange in osteoclast function.
Surprisingly, normal hair pigmentation did not require ClC-7/Ostm1 ion transport, but just the presence of the protein complex (Supplementary Table S1). Comparison of Clcn7td/td with Clcn7−/− mice showed that the ion transport-deficient ClC-7td/Ostm1 complex was also beneficial for neurons, but did not suffice to prevent lysosomal storage and CNS degeneration. Comparing Clcn7td/td with Clcn7unc/unc mice, both of which express correctly targeted ClC-7/Ostm1, revealed a detrimental effect of the ClC-7unc Cl− conductance on neurons, in stark contrast to its positive influence on osteoclasts. A toxic effect of the ClC-7unc conductance on neurons is also indicated by the fact that heterozygous Clcn7+/unc, but not Clcn7+/− mice display neurodegeneration 6. In all three genotypes (Clcn7−/−, unc/unc or td/td), lysosomal pH is normal and lysosomal Cl− concentrations are similarly decreased 6, eliminating these parameters as explanations for differences in lysosomal pathology. However, there may be differential effects on lysosomal voltage that may influence transmembrane transport processes and possibly membrane budding and fusion. Reductionist model calculations predict a lumen-positive potential (~20 mV) with a Cl− channel, but a lumen-negative potential with a 2Cl−/H+ exchanger 6. Moreover, the ClC-7 unc mutation (E245A) 7 not only uncouples Cl− transport from H+ transport, but also abolishes voltage- and time-dependent gating 3–5,7,24–26. WT ClC-7 almost lacks transport activity at cytoplasmic negative (i.e. lumen-positive) potentials. Currents increase steeply when cytoplasmic voltage exceeds approximately +20 mV 7. Hence, the unc mutation will robustly increase steady-state ClC-7 currents in lysosomes. Moreover, the slow gating of WT ClC-7/Ostm1 7 would prevent a full activation of ClC-7 during transient inside-negative voltage excursions that may occur, for example, upon NAADP-induced Ca2+ release 27,28. Intriguingly, many pathogenic CLCN7 mutations 7,17,29 accelerate ClC-7/Ostm1 gating, suggesting that early exchange currents may be pathogenic. ClC-7unc currents respond instantaneously to voltage and may thus be more harmful than those from accelerating mutants expressed in patients.
The beneficial effect of ClC-7td/Ostm1 on melanocytes and neurons raises the question whether it is totally transport deficient as assumed above. We cannot exclude that the mutant mediates currents below our detection limit of about 3% of WT. If small currents remain in ClC-7td mutants, they likely resemble ClC-7unc currents because similar mutations in EcClC-1 convert this bacterial 2Cl−/H+ exchanger into a pure Cl− conductance 30. As ClC-7unc currents are detrimental for neurons, we conclude that indeed the ClC-7td/Ostm1 complex itself, and not a putative ion transport activity, is beneficial for neurons and by extension for melanocytes. Identifying novel binding partners for ClC-7/Ostm1 that may explain these beneficial effects is a daunting task for future investigations.
Materials and Methods
Detailed methods can be found in Supplementary Materials and Methods. See Supplementary Table S3 for number of animals/cell lines used for experiments.
Mice
Clcn7−/− 14 and Clcn7unc/unc mice 6 have been described. Grey-lethal (Ostm1−/−) mice 20 were from Jackson Laboratories. Clcn7td/td mice were generated by homologous recombination using a construct in which the E312A mutation was inserted into exon 11 of Clcn7. Animals were housed under standard conditions in the MDC animal facility according to institutional guidelines and kept on a 12-h light/dark cycle. LAGeSo, Berlin, Germany, approved all experimental procedures.
Antibodies
Primary antibodies used can be found in Supplementary Table S2. Secondary antibodies were coupled to Alexa Fluor 488, 546 (Invitrogen) or HRP (Jackson ImmunoResearch).
Membrane preparation, tissue homogenates and immunoblot
Brain extracts were prepared from adult mice, blotted on PVDF membrane and probed according to standard procedures.
Histology and electron microscopy
Sections were stained with H&E, Nissl, periodic acid Schiff reagent (PAS), indicated antibodies and for lysosomal acid phosphatase activity. For EM, mice were perfused with 4% (w/v) PFA and 2.5% (v/v) glutaraldehyde in 0.1 M phosphate buffer (pH 7.4). 150-μm sagittal sections were prepared with a vibratome. Slices were postfixed in 2% (v/v) OsO4, dehydrated and embedded in epon. Semi-thin sections (0.5 μm) were labelled with toluidine blue. Ultrathin sections (60 nm) were stained with uranyl acetate and lead citrate.
Microcomputed tomography (CT)
PFA-fixed tibiae were analysed with a Skyscan 1172 μCT (Bruker-MicroCT) at 7 μm resolution. A ROI of 350 μm situated 200 μm below the growth plate comprising the secondary spongiosa was evaluated using the CTAn software with a lower grey threshold of 30 (Bruker-MicroCT). 3D reconstruction was done by the AMIRA software package (Visualization Sciences Group).
WNT stimulation of primary cells
Primary fibroblasts were starved > 6 h in DMEM containing 0.1% (w/v) BSA and stimulated with 80 ng/ml recombinant murine (rm) Wnt-3A (CF, R&D Systems) overnight in growth medium. Melanocytes were starved overnight in MEM Eagle containing 0.1% BSA and stimulated overnight with 80 ng/ml rmWnt-3A in MEM Eagle containing 10% (v/v) FBS (all Pan-Biotech) and 200 nM TPA.
Determination of lysosomal pH
Lysosomal pH was measured by ratiometric fluorescence imaging of the pH sensor Oregon Green dextran 488 (Invitrogen) as described 6.
Determination of relative lysosomal chloride concentrations
Lysosomal chloride was measured by ratiometric fluorescence live cell imaging of MEQ/TMR-dextran 6.
Acknowledgments
We thank M. Richter (FMP) for synthesizing the dextran-coupled Cl− indicator, K. Räbel and A. von Bock for technical assistance and H.J. Kaiser for writing the Fiji plug-in for the evaluation of lysosomal pH. This study was supported, in part, by grants from the Deutsche Forschungsgemeinschaft (JE164/7, JE164/9, SFB740 TP C5), an Advanced Investigator Grant from the European Research Council (ERC Grant Agreement no. 294435) and the Prix Louis-Jeantet de Médecine to TJJ.
Author contributions
SW generated knock-in mice, performed biochemical experiments, immunohistochemistry and measured lysosomal pH; SJ measured lysosomal [Cl−] and investigated Wnt signalling; SH performed electron microscopy, UK, and WLC performed μ-CT and bone characterization; TJJ planned the study; and TJJ, SW and SJ wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://embor.embopress.org
References
- Jentsch TJ. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit Rev Biochem Mol Biol. 2008;43:3–36. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]
- Stauber T, Weinert S, Jentsch TJ. Cell biology and physiology of CLC chloride channels and transporters. Compr Physiol. 2012;2:1701–1744. doi: 10.1002/cphy.c110038. [DOI] [PubMed] [Google Scholar]
- Scheel O, Zdebik A, Lourdel S, Jentsch TJ. Voltage-dependent electrogenic chloride proton exchange by endosomal CLC proteins. Nature. 2005;436:424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- Picollo A, Pusch M. Chloride / proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005;436:420–423. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]
- Neagoe I, Stauber T, Fidzinski P, Bergsdorf EY, Jentsch TJ. The late endosomal CLC-6 mediates proton/chloride countertransport in heterologous plasma membrane expression. J Biol Chem. 2010;285:21689–21697. doi: 10.1074/jbc.M110.125971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert S, Jabs S, Supanchart C, Schweizer M, Gimber N, Richter M, Rademann J, Stauber T, Komak U, Jentsch TJ. Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl− accumulation. Science. 2010;328:1401–1403. doi: 10.1126/science.1188072. [DOI] [PubMed] [Google Scholar]
- Leisle L, Ludwig CF, Wagner FA, Jentsch TJ, Stauber T. ClC-7 is a slowly voltage-gated 2Cl−/1H+-exchanger and requires Ostm1 for transport activity. EMBO J. 2011;30:2140–2152. doi: 10.1038/emboj.2011.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novarino G, Weinert S, Rickheit G, Jentsch TJ. Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis. Science. 2010;328:1398–1401. doi: 10.1126/science.1188070. [DOI] [PubMed] [Google Scholar]
- Günther W, Piwon N, Jentsch TJ. The ClC-5 chloride channel knock-out mouse - an animal model for Dent’s disease. Pflügers Arch. 2003;445:456–462. doi: 10.1007/s00424-002-0950-6. [DOI] [PubMed] [Google Scholar]
- Kasper D, Planells-Cases R, Fuhrmann JC, Scheel O, Zeitz O, Ruether K, Schmitt A, Poët M, Steinfeld R, Schweizer M, et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005;24:1079–1091. doi: 10.1038/sj.emboj.7600576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange PF, Wartosch L, Jentsch TJ, Fuhrmann JC. ClC-7 requires Ostm1 as a β-subunit to support bone resorption and lysosomal function. Nature. 2006;440:220–223. doi: 10.1038/nature04535. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ. Chloride and the endosomal-lysosomal pathway: emerging roles of CLC chloride transporters. J Physiol. 2007;578:633–640. doi: 10.1113/jphysiol.2006.124719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piwon N, Günther W, Schwake M, Bösl MR, Jentsch TJ. ClC-5 Cl−-channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature. 2000;408:369–373. doi: 10.1038/35042597. [DOI] [PubMed] [Google Scholar]
- Kornak U, Kasper D, Bösl MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling G, Jentsch TJ. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104:205–215. doi: 10.1016/s0092-8674(01)00206-9. [DOI] [PubMed] [Google Scholar]
- Brandt S, Jentsch TJ. ClC-6 and ClC-7 are two novel broadly expressed members of the CLC chloride channel family. FEBS Lett. 1995;377:15–20. doi: 10.1016/0014-5793(95)01298-2. [DOI] [PubMed] [Google Scholar]
- Cleiren E, Bénichou O, Van Hul E, Gram J, Bollerslev J, Singer FR, Beaverson K, Aledo A, Whyte MP, Yoneyama T, et al. Albers-Schönberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene. Hum Mol Genet. 2001;10:2861–2867. doi: 10.1093/hmg/10.25.2861. [DOI] [PubMed] [Google Scholar]
- Sartelet A, Stauber T, Coppieters W, Ludwig CF, Fasquelle C, Druet T, Zhang Z, Ahariz N, Cambisano N, Jentsch TJ, et al. A missense mutation accelerating the gating of the lysosomal Cl−/H+-exchanger ClC-7/Ostm1 causes osteopetrosis with gingival hamartomas in cattle. Dis Model Mech. 2014;7:119–128. doi: 10.1242/dmm.012500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartosch L, Fuhrmann JC, Schweizer M, Stauber T, Jentsch TJ. Lysosomal degradation of endocytosed proteins depends on the chloride transport protein ClC-7. FASEB J. 2009;23:4056–4068. doi: 10.1096/fj.09-130880. [DOI] [PubMed] [Google Scholar]
- Steinberg BE, Huynh KK, Brodovitch A, Jabs S, Stauber T, Jentsch TJ, Grinstein S. A cation counterflux supports lysosomal acidification. J Cell Biol. 2010;189:1171–1186. doi: 10.1083/jcb.200911083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub N, Benachenhou N, Rajapurohitam V, Pata M, Ferron M, Frattini A, Villa A, Vacher J. Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat Med. 2003;9:399–406. doi: 10.1038/nm842. [DOI] [PubMed] [Google Scholar]
- Goding CR. Mitf from neural crest to melanoma: signal transduction and transcription in the melanocyte lineage. Genes Dev. 2000;14:1712–1728. [PubMed] [Google Scholar]
- Feigin ME, Malbon CC. OSTM1 regulates β-catenin/Lef1 interaction and is required for Wnt/β-catenin signaling. Cell Signal. 2008;20:949–957. doi: 10.1016/j.cellsig.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig CF, Ullrich F, Leisle L, Stauber T, Jentsch TJ. Common gating of both CLC transporter subunits underlies voltage-dependent activation of the 2Cl−/1H+ exchanger ClC-7/Ostm1. J Biol Chem. 2013;288:28611–28619. doi: 10.1074/jbc.M113.509364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich T, Breiderhoff T, Jentsch TJ. Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J Biol Chem. 1999;274:896–902. doi: 10.1074/jbc.274.2.896. [DOI] [PubMed] [Google Scholar]
- Bergsdorf EY, Zdebik AA, Jentsch TJ. Residues important for nitrate/proton coupling in plant and mammalian CLC transporters. J Biol Chem. 2009;284:11184–11193. doi: 10.1074/jbc.M901170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459:596–600. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A, Ahuja M, Patel S, Brailoiu E, Muallem S. Convergent regulation of the lysosomal two-pore channel-2 by Mg2+, NAADP, PI(3,5)P2 and multiple protein kinases. EMBO J. 2014;33:501–511. doi: 10.1002/embj.201387035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barvencik F, Kurth I, Koehne T, Stauber T, Zustin J, Tsiakas K, Ludwig CF, Beil FT, Pestka JM, Hahn M, et al. CLCN7 and TCIRG1 mutations differentially affect bone matrix mineralization in osteopetrotic individuals. J Bone Miner Res. 2014;29:982–991. doi: 10.1002/jbmr.2100. [DOI] [PubMed] [Google Scholar]
- Accardi A, Walden M, Nguitragool W, Jayaram H, Williams C, Miller C. Separate ion pathways in a Cl−/H+ exchanger. J Gen Physiol. 2005;126:563–570. doi: 10.1085/jgp.200509417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.