

Abstract
The adaptive immune response starts when CD4+ T cells recognize peptide antigens presented by class II molecules of the Major Histocompatibility Complex (MHCII). Two outstanding features of MHCII molecules are their polymorphism and the ability of each allele to bind a large panoply of peptides. The ability of each MHCII molecule to interact with a limited, though broad, range of amino acid sequences, or “permissive specificity” of binding, is the result of structural flexibility. This flexibility has been identified through biochemical and biophysical studies, and molecular dynamic simulations have modeled the conformational rearrangements that the peptide and the MHCII undergo during interaction. Moreover, there is evidence that the structural flexibility of the peptide/MHCII complex correlates with the activity of the “peptide-editing” molecule DM. In light of the impact that these recent findingshave on our ability to predict MHCII epitopes, a review of the structural and thermodynamic determinants of peptide binding to MHCII is proposed.
Introduction
Molecules of the class IIMajor Histocompatibility Complex (MHCII) are trans-membrane heterodimeric proteins expressed on the surface of Antigen Presenting Cells (APCs) and are fundamental in initiating or propagating an immune response by presenting antigenic peptides to CD4+ T lymphocytes. When complexed with peptides, MHCII molecules are important in establishing the naïve T cell repertoire as part of thymic selection, whereas in the periphery they are involved in homeostatic maintenance and antigen-driven selection of T cells. MHCII are loaded with antigenic fragments derived from proteins that gain access to endosomal compartments, providing a way for CD4+ T cells to respond to exogenous antigens internalized by APCs through phagocytosis, macropinocytosis, receptor-mediated endocytosis and other mechanisms(1).
MHCII molecules are highly polymorphic proteins formed through the non-covalent association of a 32 kilodaltonα -chain and a 29 kilodaltonβ-chain. Thus far, between human and murine, about 80 pMHCII crystal structures have been determined. In all of them, α and β chains features similar conformation: a membrane-distal domain, an Immunoglobulin (Ig)-like membrane-proximal domain and a trans-membrane sequence for anchoring. The two membrane-distal domains, α1 and β1, form a single, groove-shaped, peptide-binding site composed of two α-helices loops supported by a platform of eight anti-parallel β-sheets. The great degree of polymorphism characterizing MHCII molecules is concentrated in the binding groove, imposing some structural constraints to the number (and chemistry) of sequences that can bind each allele (hence the specificity of peptide binding)(2). However, the analysis of naturally processed peptides eluted from cell-bound MHCII revealed that each allele is able to bind a broad range of amino acid sequences featuring different structures and affinities(3-6). The latter observation would support the hypothesis that the MHC diversity is the result of the selection of those alleles that can present various peptides from many different pathogens, limiting the possibility of immunological escape. This permissiveness can extend to peptides with structural-chemical characteristics that would appear to rule out binding whatsoever, but which nevertheless can play an important role in T cell response(7-10). Outlining the details of the interaction between MHCII and peptide that can account for permissive specificity would lead us to a better model of epitope selection and provide important insights into the receptor-ligand interaction at large. We have a general understanding of the intracellular process that leads a certain pMHCII complex to be presented (11)(Figure 1). Newly formed MHCII molecules complex in the endoplasmic reticulum with the chaperone protein invariant chain (Ii) for stable assembly. Ii contains an unstructured segment, known as CLIP, that fits in the binding groove as peptide-surrogate to stabilize the heterodimer. A trimerization domain located near the C-terminus of Ii chain allows three Ii/MHCII complexes to form a nonameric assembly. The cytoplasmatic domain of Ii contains a targeting/sorting signal sequence under whose influence the nonamers traverse the Golgi and are transported to multivescicular/multilamellar late endosomal compartments known as MHC class II compartments (MIIC). Upon arrival in the MIIC, the Ii molecule is cleaved by proteases, leaving CLIP in the binding groove. For many alleles, CLIP self-release would be too slow, preventing a significant number of MHCII from being loaded with exogenous peptides before reaching the cell surface. Indeed, CLIP removal is facilitated by the action of an accessory molecule, HLA-DM (DM), allowing for antigenic peptides to bind MHCII. DM is a non-polymorphic MHC class II molecule that contains its own endosomal targeting signal and whose narrow groove is unable of binding peptides. Rather, DM would act as a peptide editor in the context of the epitope selection process. Indeed, DM's exchange role is not limited to CLIP, but it can promote the exchange of antigenic peptides to select for a kinetically stable pMHCII repertoire(12, 13).
Figure 1. Cellular trafficking of MHCII proteins within an APC.

MHC/Ii complexes are synthesized in the ER, and directed through the trans-Golgi, where the majority is sorted to the endosomal compartment. In the endosome, Ii is cleaved, leaving CLIP bound to the peptide-binding groove. Endocytosed antigens enter the APC and are degraded through the endosomal/lysosomal pathway via the action of proteases. MHCII may bind antigenic peptides in the endosome, but the majority traffics to the lysosome/MIIC where accessory molecules for peptide loading such as DM and DO (not represented) reside. Once bound to the peptide, MHCII can be translocated to the surface of an APC for T cell recognition. In addition to the canonical MIIC pathway, there is also evidence that MHCII/Ii complexes can sort directly from the trans-Golgi to the cell surface. From there they may directly bind antigen at the cell surface, or access endosomal compartments through receptor-mediated internalization.
In B cells, specific dendritic cell subsets and thymic epithelial cells, the peptide loading of class II molecule is modified by the expression of the non-classical class II molecule HLA-DO. In the currently accepted model, DO expression inhibits the presentation of peptides acquired by cell via fluid phase endocytosis. Specifically in B cells, DO enhances the presentation of antigens internalized by B cell receptor(14).
The peptide/MHCII structures crystallized so far provide some indications to the rules governing peptide binding. These structures suggested a rigid docking model of interaction, where structural and charge complementarities would determine binding properties. In the last decade a significant number of studies have begun to speculate that the typical “key and lock” model probably is not adequate to explain the complexity of this interaction. The most straightforward evidence for the inadequacy of such models is the poor accuracy of all the MHCII epitope prediction algorithms built around them(15, 16). Experimental observations coupled with molecular dynamic studies clearly indicate a characteristic flexibility of the MHCII binding groove as it swings between bound, partially filled and empty state. Analysis of peptide binding to and release from murine and human MHCII also indicate that the interaction of peptides and MHCII undergoes cooperativity and entropy enthalpy compensation, clearly indicating that the system is structurally dynamic. This reviewfocuses on the structural and thermodynamic determinants of peptide binding to MHCII with particular attention to the modifications both the peptide and the binding groove undergo during interaction. Finally, the impact of system flexibility on the outcome of epitope selection and how immunoinformatics can incorporate this new evidence in peptide binding prediction will be highlighted.
Structural properties of peptides bound to class II
Before detailed crystallographic structures of both class I MHC (MHCI) and MHCII molecules became available, many laboratories attempted to gain insight into the nature of the peptide/MHC interaction analyzing the peptides that could be eluted from purified MHC preparation. Through HPLC analysis and Edman degradation, patterns of amino acid or “binding motifs” were deduced. In general, peptides eluted from MHCI were 8-11 amino acids in length(17), while MHCII-bound peptides were much more variable in length (12-24 amino acids)(18). MHCI-bound peptides had a preference for hydrophobic amino acids at relative positions 2 and 9, while MHCII-bound where more much variable in sequence, with a preference for hydrophobic amino acids at position 1 and 9, and allele- specific preferences at positions 4 and 6. These observations led to the hypothesis that the side-chains at these positions in the peptide were important for the interaction with the MHC molecule. Thus, MHC/peptide binding was initially interpreted in terms of fitting between these “anchor” residues and“pockets” within the MHCII structure. Conversely, this model predicted that side chains that did not serve as anchors (i.e. did not interact directly with MHC) were available to contact the TCR(4, 19, 20).
When the crystal structure of an MHCI/peptide complex became available, many of the predictions made by the functional studies were confirmed(21). The structure showed endogenous peptides bound to a binding groove formed by the α1 and α2 domains of the molecule. Subsequent higher resolution structures identified pockets in the MHCI molecule with which peptide side chains at position 2 and 9 could interact, providing a molecular explanation for the binding motifs identified through elution studies. These pockets are named for the residue in the peptide sequence that interacts with them; i.e. the P4 pocket interacts with the 4th peptide side chain relative to the side chain interacting with the P1. Furthermore, MHC polymorphic residues were concentrated in the areas that formed the pockets in the structure, lending further support to the concept that the MHC pockets provided for peptide specificity, and thus were a key determinant of immunogenicity.
Based on the MHCI HLA-A2 structure, a hypothetical model of an MHCII structure was generated(22), which was later confirmed by the structure of HLA-DR1 bound to an endogenous peptide(23). In contrast to the MHCI structures, the peptide-binding site was intermolecular, being formed by the α1 and β1 domains (Figure 2). However, the overall structure was similar, with a binding site characterized by a “floor” consisting of eight strands of anti-parallel β sheet, and two α helices: one each from the α and β chain. With the advent of baculoviral systems to produce peptide-free “empty” DR1, crystals of DR1 complexed to a high affinity peptide ligand from influenza virus hemagglutinin (HA) could be produced(24). These structures revealed key details about the nature of peptide/MHCII interaction process and defined the nature of the binding strategies adopted by both the peptide and MHCII to form a complex: peptide hydrophobic side chain encapsulation into non-polar pockets, H-bond network and ionic interactions.
Figure 2. Overall structure of an MHCII/peptide complex.

Presented is the structure of the human MHCII molecule HLA-DR3 in complex with CLIP. The orientation is such that the membrane distal α1 and β1 domains are located at the top, and the membrane proximal α2 and β2 domains are at the bottom. CLIP is presented in full atomic detail with carbon in yellow, nitrogen in blue and oxygen in red. The peptide interacts with the DR3 binding groove mainly through four residues (magenta asterisks). All known class II-associated peptides adopt a similar extended conformation, although peptides bound to I-A alleles tend to dip lower in the center of the binding groove relative to peptides bound to either I-E or HLA-DR alleles. Coordinates are from ref. 26. The model was visualized with PyMol (60).
Hydrophobic peptide side chain and non-polar pockets
the peptides adopt a type II polyproline helix as they interact with the binding groove; this conformation causes the peptide to twine in a specific fashion, with the encapsulation of peptide side chains in polymorphic pockets located at both ends of the MHCII protein(25). Generally, these pockets accommodate the side chains of peptide residues at the extremities of the peptide-binding core and have been identified as “major anchors”. In addition to these largely solvent-inaccessible interactions, smaller pockets or shelves in the center of the binding groove are recognized as minor or auxiliary anchoring sites. Depending on the allele, they may contain charged side chains possibly involved in ionic interactions with the peptide. Side chains projecting at regular intervals from the peptide are encapsulated within these pockets. The pocket-side chain interaction increases the specificity of binding and correlates to the burial of otherwise solvent-exposed areas(24).
The individual pockets are numbered according to the position of the peptide side chain they accept, with the first pocket located at the N-terminal of the binding site named P1. Other 3-4 pockets can be individuated, in general at P4, P6, P7 and P9. Usually P1 and P9 are the deepest pockets, whereas the intermediate positions are shallower or sometimes are referred to as “shelves”, thus featuring some degree of solvent exposure(24, 26).
H-bond network
In structural studies with the murine MHCII allele I-Ad bound to peptides derived from the ovalbumin protein (OVA), it was found that a high affinity peptide/MHCII interaction could be achieved without peptide side chains filling three of the major pockets in the MHCII molecule. These results were extended by the structure of the I-Ab/Ea structure, in which the four major pockets in the MHCII were largely unfilled due to the presence of Ala side chains in the peptide at that position(27). An even more extreme example was provided by the structure of I-Au/MBP1-11, in which the peptide only interacts with the COOH-terminal half of the peptide-binding groove, leaving the P1 pocket completely exposed to solvent. Despite this rather low affinity interaction, the MBP 1-11 peptide is the dominant epitope in experimental autoimmune encephalomyelitis, a mouse model of the human autoimmune disease multiple sclerosis(28). Taken together, these results suggested that complimentary anchor/pocket interactions are not an absolute requirement for MHCII/peptide affinity and immunogenicity. One possible explanation accounting for such a high affinity interaction in the absence of viable “anchoring” residues was indicated in the presence of a highly conserved hydrogen bond network between main chain atoms of the peptide and the MHCII. Because the hydrogen bond network forms to the main chain atoms of the bound peptide, it is thought to provide a sequence-independent contribution to binding energy(24). Actually, although some of the MHCII residues that contribute to this network are highly conserved (β81 His, (β82 Asn, α53 main chain atoms (Ser, Leu, Arg)), the pattern of hydrogen bonding can also be specific to a given MHCII/peptide combination due to polymorphic differences. For example, in the structure of CLIP bound to DR3(26), residue β30 Tyr forms a hydrogen bond to the amide nitrogen at P7 that cannot form in DR1 due to the presence of a Cys residue at position β30. In addition to main chain atoms, peptide side chains can form hydrogen bonds to MHCII residues as shown by the structure of DR1 bound to a peptide derived from HLA-A2.
Some insight into the function of the hydrogen bond network in MHCII/peptide interactions was initially provided by the discovery of β chain mutations, which affected MHCII transport within the cell and surface expression(29). Subsequent work by Sant and co-workers indicated the β80 and β82 region as critical for these trafficking defects, and further experiments showed that specific mutation of the highly conserved hydrogen bond donors β81 and β82 could prevent MHCII molecules from binding to peptides in the endosome, thus leading to their degradation(30). More detailed in vitro analyses indicated that the loss of the β81 hydrogen bond could result in higher rates of peptide dissociation from MHCII, an effect that could be seen with the loss of hydrogen bonds across the peptide-binding groove, including those mediated indirectly through water molecules(31). Similar increases in peptide dissociation rates can be observed through the chemical disruption of hydrogen bonds by incubating peptide/MHCII complexes in –OH containing compounds such as 4-Chlorophenol. Thus, H-bond interactions seem to beas important as the pocket/“anchor” interactions in terms of peptide/MHCII binding(32). However, due to the conserved nature of the network across alleles, H-bonds have been indicated as preferential target of DM action(33-35); a similar model would account for the evidence that DM promotes release of peptides that are very different in sequence. Nevertheless, the role of the H-bond network in the context of DM-mediated peptide exchange is still matter of debate.
Flexibility of the peptide-binding groove
The contacts observed in the available experimental MHCII structures have suggested an interpretation for the MHCII peptide binding specificity and sequence dependence. However the mechanistic details of the peptide loading process and the structure of the peptide-free MHCII molecules (empty form) are less well understood. So far, it has not been possible to crystallize MHCII in empty form probably due to the enhanced conformational heterogeneity of the protein in the absence of a peptide. Various biophysical and biochemical techniques, however, have been used to characterize the peptide-free form. Several studies indicate that MHCII molecules undergo a conformational transition during peptide binding or exchange from an enhanced conformational heterogenic state to a more rigid and stable state(36-38). Peptide-free DR1 has been shown to have a larger hydrodynamic radius than the peptide loaded form (29 vs. 35 Å), as well as a decrease in helicity as measured by circular dichroism. These differences are reversed upon binding peptide(36). Analysis of peptide binding to and release from MHCII has been flanked by studies aimed to provide the structural foundation for the complexity of the peptide binding interaction. Early kinetic studies of peptide/MHC interactions revealed that many different peptides bind with moderate or high stability to any given MHC protein, with half-times of minutes to months for peptide dissociation. In addition, they found that peptide binding to MHC appeared to be very slow, with association rate constants of only 1–100 M-1 s-1, at least 7 orders of magnitude below the diffusion limit(39). In attempting to understand the rate-limiting step in the process of peptide binding, two significant discoveries were made.
First, it was found thatduring SDS-PAGE, certain peptide/MHCII complexes, though identical in covalent composition, could adopt two conformations: a “floppy” or slower migrating form, and a “compact” faster migrating form. For the murine I-Ek MHCII allele, the “floppy” form could be induced by low pH, and the stable form could be generated over time by high affinity ligand binding. Although the initial complex of peptide with MHCII (“floppy” form) had fast dissociation rates, these low affinity interactions could preserve MHCII from aggregating and accelerate the formation of the stable conformer(40, 41). In addition, conformational variants of peptide/MHCII complexes have been identified using photoaffinity-labeled peptides, N- and C- terminal amino acid extensions, conformational-specific antibodies and in NMR experiments with 19F-labeled peptide. On the basis of the discovery of these isomers, it has been hypothesized that the short-lived complex might be a kinetic intermediate in the formation of the long-lived complex and that slow interconversion of the complexes is responsible for the slow apparent association rate of peptide to MHC. Whereas interconversion between the two forms is still debated, further work has confirmed the presence of peptide/MHC complex isomers using NMR spectroscopy and demonstrated the ability of the different isomers to trigger distinct T cell responses(39, 42-46).
More recently, however, it was demonstrated that it is the presence of distinct isomers of empty (i.e., devoid of peptide) MHC, not peptide/MHC complexes, that largely explains the slow apparent association rate of peptide to MHC. One isomer of empty MHC (inactive) does not bind peptide detectably. The other (active) binds peptide rapidly, approximately 1000-fold faster than previously estimated. The peptide-receptive, or active, form is formed by dissociation of previously bound peptide and rapidly inactivates in the absence of added peptide. At equilibrium, the inactive form of empty MHC predominates, only slowly converting to the active form. This slow conversion of the inactive to the active form accounts for the slow binding of peptide to MHC observed in previous studies.(47)
Simulation analyses of peptide binding to DR1 and DR3 have been performed in the attempt to visualize the conformational changes the MHCII undergoes as it shifts between the different states(48, 49). The amino-terminal region of the peptide-binding site of HLA-DR1 seems to be the most flexible and the one mainly involved in the structural modifications. Indeed, upon simulated removal of the peptide, the α50-59 region of DR would fill the amino-terminal end of the peptide-binding site occupying, in part, the area where the antigenic peptide is usually found. A sharp kink would form at Gly α58, allowing the region α50-59 to fold into the binding site, taking the place of the bound peptide in the P1 to P4 region. Smaller but still significant changes are observed in the helical regions that flank α50-59, in the adjacent α46-49 loop, and in β-subunit helical regions. After moving into the peptide-binding site, the main chain of the α50-59 region would be involved in the formation of all the hydrogen bonds in this region lost upon removal of the peptide. In the conserved arrangement observed in MHCII/peptide crystal structures, the backbone of a bound antigenic peptide forms six hydrogen bonds with the side chains of non-polymorphic DR1 residues Gln α9, Arg β71, His β81 and Asn β82. In the molecular dynamics model of the peptide-free form of DR1, each of these hydrogen-bonding interactions is observed, by direct hydrogen bonding between the main chain atoms of DR1 α53-57 and side chains of Gln α9, Arg β71, His β81 and Asn β82(48) (Figure 3).
Figure 3. Structural rearrangement of the MHC binding groove between peptide bound and peptide free states.

Diagram of the DR1 binding site bound to HA (top panel) and in empty state (bottom panel). The figure is adapted from the model proposed in ref. 48. The N-terminal region of the binding site is expected to undergo the most significant modifications. In particular, the α50-59 region of DR is expected to fold into the amino-terminal end of the groove, taking the place of the bound peptide in the P1 to P4 region.
Movement of the α50-59 region into the peptide-binding site also would result in the occupancy of the P1-P4 side-chain binding pockets. In the molecular dynamics model of the peptide-free form of DR1, the side chain of Phe α54 is expected to bind into the P1 pocket. As recalled above, the P4 pocket is shallower than the P1 pocket and open at the end, and in DR1 exhibits a weaker preference for residues with some aliphatic character. In the model of the peptide-free form of DR1, the side chain of Gln α57 would bind into the P4 pocket, essentially identical to Gln at the corresponding position in a bound peptide(48).
The predicted conformational changes in the peptide-free model might account for the peptideaverse form of DR1. It is plausible that upon peptide release, the binding groove is not obstructed and therefore could bind subsequent peptide directly, whereas once the peptide binding groove is engaged by the α50-59 region, the protein would be in the peptide-averse form, and would have to undergo a conformational change to allow space for the peptide to enter the binding groove.
The evidence that MHCII molecules must bind a peptide for efficient cellular trafficking and presentation, and to prevent aggregation of averse empty forms, led to the hypothesis that peptides are a critical determinant of MHCII structure. Indeed, peptide binding to DR1 results in a condensation of the DR1 molecule around the peptide, whose initiation requires the peptide to interact with the P1 pocket, though full conversion to the compact form is dependent on H-bondingnetwork formation across the groove(37).
In keeping with the evident flexibility of the peptide-binding groove, the idea of peptide/MHCII binding as typical docking reaction between a ligand and a receptor has changed in favor of a “folding” model(50, 51). As a direct consequence, the outcome of the peptide binding process and of the overall epitope selection would rely on the contribution of each source of binding energy to the folding of the peptide and the MHCII groove to reach a stable, low-energy conformational state.
Thus, the question as to whether a given peptide can bind to a given allele can be addressed by quantifying the extent of these structural modifications, and to this aimone can take advantage of the observation that protein folding is a cooperative process(52). This is the phenomenon for which multipoint binding between reactants usually display considerably different binding affinities than one would expect from simply summing the association energies of all the respective parts. Measuring cooperativity during a folding reaction is a relatively easy process and by performing ad hoc substitution within the peptide and/or the MHCII, cooperative effects have been quantified during complex formation or peptide release and, with those, magnitude of folding.
The interaction of peptides with MHCII is a cooperative event
Andersonand Gorskistarted to investigate the possibility that peptide binding to MHCII is a distributive process by studying the contribution to the interaction of peptide side chains that reside in positions with an intermediate solvent accessibility and can play a role as a TCR contacts rather than anchors/pockets(53). The DR1/HA complex was chosen as model, and side chain substitutions in the sequence of the HA peptide at P2, P3, P7 and P10 positions were introduced. These residues show accessibility to solvent ranging from 42% to 15%. Crystallographic analysis indicates that P2 and P3 constitute TCR contacts, and also P7 may interact with the TCR. Mutations postulated to prevent H-bond formation were performed, as this is the main source of interaction between those peptide positions and the MHCII binding groove. The most striking result was that a peptide carrying disruptive substitutions at all four positions was unable to bind, though containing all the major anchoring side chains (P1/P4/P6/P9). The loss of binding for some peptides carrying three out of the four possible mutations was greater than the one observed when the major anchor at P1 is changed to Ala, which is considered a dramatic substitution itself. An interesting observation made in that initial series of experiments is that multiple substitutions show a greater negative impact on complex stability than expected from single substitutions. This phenomenon led the authors consider the possibility that not only the binding energy is determined by the overall sequence of the peptide, but also that this is specifically a cooperative interaction, which would indicate that MHCII/peptide complex formation is a folding event.
This possibility was explored by applying the mutant cycle engineered by Fersht and colleagues(54). This approach consists in introducing multiple substitutions in the sequence of the reactants and assessing their binding parameters. If the effect on the binding free energy of the double (or triple) mutation is not equal to the sum of the effects of the single mutations then the two (or three) residues are coupled (cooperative). Initially, the substitutions performed within the peptide concerned the residues with intermediate solvent exposure. One substitution was also introduced in the MHCII protein. The His at position β81 was indeed mutated into an Asp, whose side chain is postulated to assume a rotamer incompatible with the formation of the H-bond usually present between the wild type residue and the peptide backbone at P-1. Cooperative effects were observed on both peptide binding to and release from DR1. Similar results, though to a lesser extent, were observed when a mutation was performed at P7, which has intermediate solvent exposure but has a hydrophobic property(50).
Subsequently the focus was shifted on anchor/pocket interactions, to analyze whether they also undergo cooperativity. Indeed, one alternative model could consider these interactions reflecting initial nucleation events, and they may be upstream of the folding process and not show cooperativity. The results of that investigation showed that cooperative effects also impact the interaction between peptide hydrophobic side chain and MHCII pockets. Cooperativity was also observed between pocket positions and positions with intermediate solvent accessibility, indicating that the hydrophobic interactions participate in the overall folding process(51).
On the basis of these results, a structuralmodel was outlined to explain stable peptide binding recalling the nucleation-condensation mechanism. The initial event of the process would involve scanning for hydrophobic side chain to fill the P1 pocket, since it is known that filling this pocket is a primary requirement, at least for human MHCII. This first rearrangement would trigger the involvement of the adjacent, partially disordered, α1/β1 helical regions and it would facilitate the formation of the other pocket/side chain interactions. At first, the nucleus would be stabilized by the interactions in the P(–1) to P2 region, via H-bonds between His β81 and Asnβ82, as well as the residues from α51 to α53, and the bound peptide. Then, the onrush of cooperative interactions would: 1) induce the folding of the helices; 2) induce the other hydrophobic pocket interactions; 3) stabilize both types of binding energy; 4) induce the folding of the peptide into a polyproline type II conformation. Stabilization of the nucleus would require the correct conformation of contacts by a significant fraction of the structures on the C-terminal region, both from the peptide and the MHCII. This model would explainwhy P1 occupancy, needed for promoting conversion to a condensed form of the MHCII, is not sufficient for high affinity binding, which requires main chain interactions in the P5-P9 region. At this point the whole molecule would reach the transition state, with mutual cooperative interactions strengthening the nucleus, to which the close conformer and final structure would rapidly follow(51).
Thermodynamic correlates of peptide/MHCII complex flexibility
The analysis of cooperativity in peptide/MHCII interaction has led to the important finding that cooperative effects increase exponentially with modification of peptide Kd or complex stability(50, 51). Due to the nature of the mutations adopted in those experiments, cooperative effects were contributing exponentially to the loss of complex stability or peptide affinity as disruptive substitutions were added to the system. This specific trend can be visualized in a cooperativity
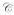 versus affinity Kd plot, in which a Kd range can be identified, for which little or no
can be measured, followed by a steep increase of
with peptide Kd increasing (Figure 4 - for convenience, this plot is sometimes converted to the equivalent linear ln-ln graph shown in the inset). This behavior was interpreted as a further indication of system flexibility, in that it was apparent that an energy range exists, in which the folding of the complex barely changes in spite of significant modifications of the chemistry of the peptide.
versus affinity Kd plot, in which a Kd range can be identified, for which little or no
can be measured, followed by a steep increase of
with peptide Kd increasing (Figure 4 - for convenience, this plot is sometimes converted to the equivalent linear ln-ln graph shown in the inset). This behavior was interpreted as a further indication of system flexibility, in that it was apparent that an energy range exists, in which the folding of the complex barely changes in spite of significant modifications of the chemistry of the peptide.
Figure 4. Thermodynamic model of peptide/MHCII complex formation and DM activity.

Peptide binding can be envisaged as a folding process, quantifiable in terms of Cooperativity
. The impact of cooperative effects on peptide affinity has been investigated applying the mutant cycle approach to the DR/HA system. Due tothe disruptive nature of the modifications, the observed Cooperativity can be interpreted as lack of folding. Cooperativity affects complex formation in an exponential fashion, indicating that disrupting interaction after interaction has an amplified effect on the ability of the complex to fold into a stable conformer. At the left side of the curve, an affinity range can be identified, for which null or little cooperativity can be measured (Compensatory Range, CR framed in the plot). The broader is this range, the greater is the ability of the system to compensate any lack of interactions (H-bonds, Hydrophobic or salt bridges) with the residual flexibility (phenomenon of entropy-enthalpy compensation). To the extent that DM interacts and destabilizes complexes featuring greater residual entropy, thus reducing the compensatory range for stable complexion, this approach may also be used to identify the susceptibility to DM of a complex on the basis of its thermodynamic profile.
To gain a better understanding of the biophysics behind this trend, the thermodynamics of the cycle-mutated HA/DR1 system was analyzed(55). Importantly, in the case of MHCII, a complete characterization of the binding energetics and correlation of thermodynamic data with the structures involved was lacking. These studies have provided the fundamental know-how for the development of structure-based binding prediction strategies. Changes of all thermodynamic parameters, including free energy of binding (ΔG), enthalpy (ΔH) and entropy (ΔS) of binding and heat capacity (ΔCp) were measured.
Themain finding of that analysis indicated that the peptide/MHCII system is characterized by isothermal entropy-enthalpy compensation (iEEC). This is a phenomenon by which the entropy and the enthalpy of intermolecular complexation compensate each other, and the trade-off between intermolecular motion and enthalpic interactions accounts for this observation(56). The observation of iEEC in the context of the peptide/MHCII system suggested that a peptide whose enthalpic contribution to binding (which is what most epitope prediction models consider) is poor, might nevertheless bind if the entropic contribution can compensate, hence permissive specificity. A similar analysis conducted on MHCI has shown that the major factor in determining the stability of the complex is enthalpy, which is partially compensated by entropy(57). Therefore, also in the case of class I molecules, EEC appears to be the physical-chemical basis for the broad specificity in interaction with peptide antigens, adding one more element of similarity to those already known between the MHCI- and MHCII-restricted epitope selection.
However, the detected compensatory mechanism is not complete, therefore as the peptide sequence worsens in terms of enthalpic contributions, the residual flexibility is not able to maximize the few interactions available. This evidence provided an explanation for the exponential trend by which cooperativity affects binding Kd. The range of peptide affinity with null or little cooperativity identifies those complexes in which, irrespective of the number of interactions, the residual motility is sufficient to optimize the few available sources of binding. The compensatory mechanism identified in this range would be responsible for the specificity and permissiveness of peptide binding. For values of peptide affinity beyond this range, the paucity of interactions is not sufficiently balanced by the increase of conformational mobility. As a result, the effect of cooperativity increases dramatically as evidenced by the lack of folding upon introduction of disruptive mutations in the complex. This phenomenon would reduce the probability for stable interaction of these sequences and would provide the thermodynamic foundation for negative selectivity of peptide binding.
The partitioned approach applied to the thermodynamic analysis of pMHCII complex enabled for a more refined model explaining the interaction of peptides with MHCII. The sequestration of peptide hydrophobic side chains into regions of the groove with a greater-than-average exposure of hydrophobic residueswould constitutethe binding trigger. This initial step would be driven by the entropically dominated solvation free energy. The reorganization of the solvent would be partially compensated by the increased enthalpy of H-bonding network between water molecules. The ability of the peptide to promote desolvation of the binding groove would depend on steric and electrostatic complementarity as well as total hydrophobic surface. Once enough water molecules are displaced to allow an initial association, the intermolecular H-bond network would be responsible for the tightening of the complex. Thus, multiple weak interactions between peptide and MHCIIare responsible for the kinetic stability of the final structure. Every interaction point would undergo a balancing between the favorable enthalpy associated with the formation of that interaction and the unfavorable entropy of restraining the conformational mobility of the system. In a specular way, the inability of forming an interaction would be compensated by a gain in conformational mobility(58).
Consequently, the global bound state of a given peptide/MHCII dyad is a collection of various differently complexed states in equilibrium. In other words, the macrostate of the peptide/MCHII system is characterized by a probability distribution of possible states across a certain statistical ensemble of all microstates. This distribution describes the probability of finding the system in a certain microstate. For highly enthalpic complexes (left side of the flat portion of the curve in Figure 4), the probability of finding all the viable binding points engaged in interactions is the greatest. Complexes represented by the right side of the flat portion of the same plot feature greater residual entropy. For these complexes, the microstates accessible to the system are significantly more numerous. Because of the association between the thermodynamic signature of a complex and its representation on the curve in Figure 4, the power law of
vs. Kd can be read as the probability distribution for a peptide to fold the MHCII groove and itself into a low energy complex. Consequently, the function associated to the curve can be adopted as mathematical tool to implement peptide-binding prediction.
One intriguing question following the above thermodynamic model of peptide binding to MHCII concerns the structure of the resulting complex. It seems evident that the extent of “tightness” generated by the relative enthalpic and entropic contributions determines the probability with which a complex assumes a stable conformer. For complexes relying on asignificantentropic component, the number of possible microstates is greater than for an isoenergetic complex in which the enthalpic component determines the net kinetic stability. For these highly entropic dyads, the incompletely folded structures are more numerous. Thus, it appears plausible that the thermodynamic signature of the complex may have a structural counterpart. Considering that the greater conformational flexibility of the complex is located on the N-terminal side, one could speculate that the multiple microstates will differ in particular in this region. This consideration might have an important consequence in the context of DM“peptide-editing” activity. The hypothesis by which DM interacts with complexes featuring a flexible P1 pocket(59) could be interpreted under the thermodynamic model as DM targeting high-entropy complexes. Validating such a hypothesis would provide the thermodynamic foundation for a unifying theory of peptide binding and DM-mediated peptide exchange, facilitating the inclusion of DM action in an epitope prediction algorithm (Figure 4).
Conclusions and Perspectives
Prediction of MHCII-restricted antigens is an important goal for measuring immune correlates of protection after vaccination and for vaccine development. However, currently available algorithms based on docking models of peptide binding suffer from low accuracy, excluding actual epitopes or including experimental poor binders. These limitations might be resolved with a model of pMHCII complex formation that considers folding and iEEC, allowing inclusion of permissive specificity and DM activity in epitope prediction.
The work performed on the peptide/MHCII system has advanced our understanding of the mechanisms underlying the recognition of ligands with different chemistry by the same receptor, as well as the relationships between molecular flexibility/rigidity, binding cooperativity and the role of entropy therein. These are outstanding questions from the biophysical/biochemical standpoint, and only in recent times have we started to appreciate the impact of flexibility and entropy on molecular recognition. Additional immunological relevance lies in the areas of Ag-Ab interaction, recognition by the TCR of its ligand and binding of type I interferons to IFNAR. As such, delving into the peptide/MHCII interaction will further our understanding of binding interaction mechanisms that can be extended to other systems.
Acknowledgments
I wish to specially thank Dr. Jack Gorski for his remarkable mentorship and for his inspiring creative thinking. Funding for the research described here was from NIH grant R01AI63016 to Dr. Gorski. This work was supported by National Institute of General Medical Sciences of the National Institutes of Health under Award Number P20GM103395 by the Pfizer-sponsored Aspire Award Number WS1907326. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health or Pfizer.
Reference List
- 1.Benacerraf B. Role of MHC gene products in immune regulation. Science. 1981;212(4500):1229–38. doi: 10.1126/science.6165083. Epub 1981/06/12. [DOI] [PubMed] [Google Scholar]
- 2.Nelson CA, Fremont DH. Structural principles of MHC class II antigen presentation. Rev Immunogenet. 1999;1(1):47–59. [PubMed] [Google Scholar]
- 3.Chaturvedi P, Yu Q, Southwood S, Sette A, Singh B. Peptide analogs with different affinites for MHC alter the cytokine profile of T helper cells. Int Immunol. 1996;8(5):745–55. doi: 10.1093/intimm/8.5.745. [DOI] [PubMed] [Google Scholar]
- 4.Engelhard VH. Structure of peptides associated with class I and class II MHC molecules. Annu Rev Immunol. 1994;12:181–207. doi: 10.1146/annurev.iy.12.040194.001145. [DOI] [PubMed] [Google Scholar]
- 5.Lippolis JD, White FM, Marto JA, Luckey CJ, Bullock TN, Shabanowitz J, et al. Analysis of MHC class II antigen processing by quantitation of peptides that constitute nested sets. J Immunol. 2002;169(9):5089–97. doi: 10.4049/jimmunol.169.9.5089. [DOI] [PubMed] [Google Scholar]
- 6.Suri A, Lovitch SB, Unanue ER. The wide diversity and complexity of peptides bound to class II MHC molecules. Curr Opin Immunol. 2006;18(1):70–7. doi: 10.1016/j.coi.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Ferlin WG, Mougneau E, Hugues S, Appel H, Jang MH, Cazareth J, et al. Self-peptides that bind with low affinity to the diabetes-associated I-A(g7) molecule readily induce T cell tolerance in non-obese diabetic mice. Eur J Immunol. 2004;34(10):2656–63. doi: 10.1002/eji.200425413. [DOI] [PubMed] [Google Scholar]
- 8.Kawamura K, McLaughlin KA, Weissert R, Forsthuber TG. Myelin-reactive type B T cells and T cells specific for low-affinity MHC-binding myelin peptides escape tolerance in HLA-DR transgenic mice. J Immunol. 2008;181(5):3202–11. doi: 10.4049/jimmunol.181.5.3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muraro PA, Vergelli M, Kalbus M, Banks DE, Nagle JW, Tranquill LR, et al. Immunodominance of a low-affinity major histocompatibility complex-binding myelin basic protein epitope (residues 111-129) in HLA-DR4 (B1*0401) subjects is associated with a restricted T cell receptor repertoire. J Clin Invest. 1997;100(2):339–49. doi: 10.1172/JCI119539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patil NS, Pashine A, Belmares MP, Liu W, Kaneshiro B, Rabinowitz J, et al. Rheumatoid arthritis (RA)-associated HLA-DR alleles form less stable complexes with class II-associated invariant chain peptide than non-RA-associated HLA-DR alleles. J Immunol. 2001;167(12):7157–68. doi: 10.4049/jimmunol.167.12.7157. [DOI] [PubMed] [Google Scholar]
- 11.Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nature reviews Immunology. 2011;11(12):823–36. doi: 10.1038/nri3084. Epub 2011/11/15. [DOI] [PubMed] [Google Scholar]
- 12.Sadegh-Nasseri S, Chen M, Narayan K, Bouvier M. The convergent roles of tapasin and HLA DM in antigen presentation. Trends Immunol. 2008;29(3):141–7. doi: 10.1016/j.it.2008.01.001. Epub 2008/02/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schulze MS, Wucherpfennig KW. The mechanism of HLA-DM induced peptide exchange in the MHC class II antigen presentation pathway. Curr Opin Immunol. 2012;24(1):105–11. doi: 10.1016/j.coi.2011.11.004. Epub 2011/12/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Busch R, Rinderknecht CH, Roh S, Lee AW, Harding JJ, Burster T, et al. Achieving stability through editing and chaperoning: regulation of MHC class II peptide binding and expression. Immunol Rev. 2005;207:242–60. doi: 10.1111/j.0105-2896.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- 15.Lin HH, Zhang GL, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC-II peptide binding prediction servers: applications for vaccine research. BMC Bioinformatics. 2008;9(Suppl 12):S22. doi: 10.1186/1471-2105-9-S12-S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaves FA, Lee AH, Nayak JL, Richards KA, Sant AJ. The utility and limitations of current Web-available algorithms to predict peptides recognized by CD4 T cells in response to pathogen infection. J Immunol. 2012;188(9):4235–48. doi: 10.4049/jimmunol.1103640. Epub 2012/04/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jardetzky TS, Lane WS, Robinson RA, Madden DR, Wiley DC. Identification of self peptides bound to purified HLA-B27. Nature. 1991;353(6342):326–9. doi: 10.1038/353326a0. [DOI] [PubMed] [Google Scholar]
- 18.Chicz RM, Urban RG, Lane WS, Gorga JC, Stern LJ, Vignali DA, et al. Predominant naturally processed peptides bound to HLA-DR1 are derived from MHC-related molecules and are heterogeneous in size. Nature. 1992;358(6389):764–8. doi: 10.1038/358764a0. [DOI] [PubMed] [Google Scholar]
- 19.Kropshofer H, Max H, Muller CA, Hesse F, Stevanovic S, Jung G, et al. Self-peptide released from class II HLA-DR1 exhibits a hydrophobic two-residue contact motif. J Exp Med. 1992;175(6):1799–803. doi: 10.1084/jem.175.6.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Max H, Halder T, Kropshofer H, Kalbus M, Muller CA, Kalbacher H. Characterization of peptides bound to extracellular and intracellular HLA-DR1 molecules. Hum Immunol. 1993;38(3):193–200. doi: 10.1016/0198-8859(93)90540-h. [DOI] [PubMed] [Google Scholar]
- 21.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. Structure of the human class I histocompatibility antigen, HLA-A2. Nature. 1987;329(6139):506–12. doi: 10.1038/329506a0. Epub 1987/10/08. [DOI] [PubMed] [Google Scholar]
- 22.Brown JH, Jardetzky T, Saper MA, Samraoui B, Bjorkman PJ, Wiley DC. A hypothetical model of the foreign antigen binding site of class II histocompatibility molecules. Nature. 1988;332(6167):845–50. doi: 10.1038/332845a0. [DOI] [PubMed] [Google Scholar]
- 23.Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, et al. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature. 1993;364(6432):33–9. doi: 10.1038/364033a0. Epub 1993/07/01. [DOI] [PubMed] [Google Scholar]
- 24.Stern LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG, Strominger JL, et al. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 1994;368(6468):215–21. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 25.Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Strominger JL, et al. Crystallographic analysis of endogenous peptides associated with HLA-DR1 suggests a common, polyproline II-like conformation for bound peptides. Proc Natl Acad Sci U S A. 1996;93(2):734–8. doi: 10.1073/pnas.93.2.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh P, Amaya M, Mellins E, Wiley DC. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature. 1995;378(6556):457–62. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]
- 27.Scott CA, Peterson PA, Teyton L, Wilson IA. Crystal structures of two I-Ad-peptide complexes reveal that high affinity can be achieved without large anchor residues. Immunity. 1998;8(3):319–29. doi: 10.1016/s1074-7613(00)80537-3. Epub 1998/04/07. [DOI] [PubMed] [Google Scholar]
- 28.Lee C, Liang MN, Tate KM, Rabinowitz JD, Beeson C, Jones PP, et al. Evidence that the autoimmune antigen myelin basic protein (MBP) Ac1-9 binds towards one end of the major histocompatibility complex (MHC) cleft. J Exp Med. 1998;187(9):1505–16. doi: 10.1084/jem.187.9.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chervonsky AV, Gordon L, Sant AJ. A segment of the MHC class II beta chain plays a critical role in targeting class II molecules to the endocytic pathway. Int Immunol. 1994;6(7):973–82. doi: 10.1093/intimm/6.7.973. Epub 1994/07/01. [DOI] [PubMed] [Google Scholar]
- 30.Tan LJ, Ceman S, Chervonsky A, Rodriguez-Paris J, Steck TL, Sant AJ. Late events in the intracellular sorting of major histocompatibility complex class II molecules are regulated by the 80-82 segment of the class II beta chain. Eur J Immunol. 1997;27(6):1479–88. doi: 10.1002/eji.1830270626. Epub 1997/06/01. [DOI] [PubMed] [Google Scholar]
- 31.McFarland BJ, Beeson C, Sant AJ. Cutting edge: a single, essential hydrogen bond controls the stability of peptide-MHC class II complexes. J Immunol. 1999;163(7):3567–71. Epub 1999/09/22. [PubMed] [Google Scholar]
- 32.Bandyopadhyay A, Arneson L, Beeson C, Sant AJ. The relative energetic contributions of dominant P1 pocket versus hydrogen bonding interactions to peptide:class II stability: implications for the mechanism of DM function. Mol Immunol. 2008;45(5):1248–57. doi: 10.1016/j.molimm.2007.09.011. Epub 2007/11/06. [DOI] [PubMed] [Google Scholar]
- 33.Narayan K, Chou CL, Kim A, Hartman IZ, Dalai S, Khoruzhenko S, et al. HLA-DM targets the hydrogen bond between the histidine at position beta81 and peptide to dissociate HLA-DR-peptide complexes. Nat Immunol. 2007;8(1):92–100. doi: 10.1038/ni1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stratikos E, Wiley DC, Stern LJ. Enhanced catalytic action of HLA-DM on the exchange of peptides lacking backbone hydrogen bonds between their N-terminal region and the MHC class II alphachain. J Immunol. 2004;172(2):1109–17. doi: 10.4049/jimmunol.172.2.1109. [DOI] [PubMed] [Google Scholar]
- 35.Sant AJ, Beeson C, McFarland B, Cao J, Ceman S, Bryant PW, et al. Individual hydrogen bonds play a critical role in MHC class II: peptide interactions: implications for the dynamic aspects of class II trafficking and DM-mediated peptide exchange. Immunol Rev. 1999;172:239–53. doi: 10.1111/j.1600-065x.1999.tb01369.x. Epub 2000/01/13. [DOI] [PubMed] [Google Scholar]
- 36.Zarutskie JA, Sato AK, Rushe MM, Chan IC, Lomakin A, Benedek GB, et al. A conformational change in the human major histocompatibility complex protein HLA-DR1 induced by peptide binding. Biochemistry. 1999;38(18):5878–87. doi: 10.1021/bi983048m. [DOI] [PubMed] [Google Scholar]
- 37.Sato AK, Zarutskie JA, Rushe MM, Lomakin A, Natarajan SK, Sadegh-Nasseri S, et al. Determinants of the peptide-induced conformational change in the human class II major histocompatibility complex protein HLA-DR1. J Biol Chem. 2000;275(3):2165–73. doi: 10.1074/jbc.275.3.2165. [DOI] [PubMed] [Google Scholar]
- 38.Carven GJ, Stern LJ. Probing the ligand-induced conformational change in HLA-DR1 by selective chemical modification and mass spectrometric mapping. Biochemistry. 2005;44(42):13625–37. doi: 10.1021/bi050972p. Epub 2005/10/19. [DOI] [PubMed] [Google Scholar]
- 39.Sadegh-Nasseri S, McConnell HM. A kinetic intermediate in the reaction of an antigenic peptide and I-Ek. Nature. 1989;337(6204):274–6. doi: 10.1038/337274a0. [DOI] [PubMed] [Google Scholar]
- 40.Sadegh-Nasseri S, Germain RN. A role for peptide in determining MHC class II structure. Nature. 1991;353(6340):167–70. doi: 10.1038/353167a0. [DOI] [PubMed] [Google Scholar]
- 41.Dornmair K, Rothenhausler B, McConnell HM. Structural intermediates in the reactions of antigenic peptides with MHC molecules. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 1):409–16. doi: 10.1101/sqb.1989.054.01.050. [DOI] [PubMed] [Google Scholar]
- 42.Kasson PM, Rabinowitz JD, Schmitt L, Davis MM, McConnell HM. Kinetics of peptide binding to the class II MHC protein I-Ek. Biochemistry. 2000;39(5):1048–58. doi: 10.1021/bi9921337. [DOI] [PubMed] [Google Scholar]
- 43.Schmitt L, Boniface JJ, Davis MM, McConnell HM. Conformational isomers of a class II MHC-peptide complex in solution. J Mol Biol. 1999;286(1):207–18. doi: 10.1006/jmbi.1998.2463. [DOI] [PubMed] [Google Scholar]
- 44.Schmitt L, Boniface JJ, Davis MM, McConnell HM. Kinetic isomers of a class II MHC-peptide complex. Biochemistry. 1998;37(50):17371–80. doi: 10.1021/bi9815593. [DOI] [PubMed] [Google Scholar]
- 45.Rabinowitz JD, Vrljic M, Kasson PM, Liang MN, Busch R, Boniface JJ, et al. Formation of a highly peptide-receptive state of class II MHC. Immunity. 1998;9(5):699–709. doi: 10.1016/s1074-7613(00)80667-6. [DOI] [PubMed] [Google Scholar]
- 46.Witt SN, McConnell HM. Formation and dissociation of short-lived class II MHC-peptide complexes. Biochemistry. 1994;33(7):1861–8. doi: 10.1021/bi00173a032. [DOI] [PubMed] [Google Scholar]
- 47.Joshi RV, Zarutskie JA, Stern LJ. A three-step kinetic mechanism for peptide binding to MHC class II proteins. Biochemistry. 2000;39(13):3751–62. doi: 10.1021/bi9923656. [DOI] [PubMed] [Google Scholar]
- 48.Painter CA, Cruz A, Lopez GE, Stern LJ, Zavala-Ruiz Z. Model for the peptide-free conformation of class II MHC proteins. PLoS One. 2008;3(6):e2403. doi: 10.1371/journal.pone.0002403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yaneva R, Springer S, Zacharias M. Flexibility of the MHC class II peptide binding cleft in the bound, partially filled, and empty states: a molecular dynamics simulation study. Biopolymers. 2009;91(1):14–27. doi: 10.1002/bip.21078. [DOI] [PubMed] [Google Scholar]
- 50.Anderson MW, Gorski J. Cooperativity during the formation of peptide/MHC class II complexes. Biochemistry. 2005;44(15):5617–24. doi: 10.1021/bi048675s. [DOI] [PubMed] [Google Scholar]
- 51.Ferrante A, Gorski J. Cooperativity of hydrophobic anchor interactions: evidence for epitope selection by MHC class II as a folding process. J Immunol. 2007;178(11):7181–9. doi: 10.4049/jimmunol.178.11.7181. [DOI] [PubMed] [Google Scholar]
- 52.Dornmair K, McConnell HM. Refolding and reassembly of separate alpha and beta chains of class II molecules of the major histocompatibility complex leads to increased peptide-binding capacity. Proc Natl Acad Sci U S A. 1990;87(11):4134–8. doi: 10.1073/pnas.87.11.4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderson MW, Gorski J. Cutting edge: TCR contacts as anchors: effects on affinity and HLA-DM stability. J Immunol. 2003;171(11):5683–7. doi: 10.4049/jimmunol.171.11.5683. [DOI] [PubMed] [Google Scholar]
- 54.Horovitz A, Fersht AR. Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J Mol Biol. 1990;214(3):613–7. doi: 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- 55.Ferrante A, Gorski J. Enthalpy-entropy compensation and cooperativity as thermodynamic epiphenomena of structural flexibility in ligand-receptor interactions. J Mol Biol. 2012;417(5):454–67. doi: 10.1016/j.jmb.2012.01.057. Epub 2012/02/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lumry R. Uses of enthalpy-entropy compensation in protein research. Biophys Chem. 2003;105(2-3):545–57. doi: 10.1016/s0301-4622(03)00065-6. [DOI] [PubMed] [Google Scholar]
- 57.Kang J, Auerbach JD. Thermodynamic characterization of dissociation rate variations of human leukocyte antigen and peptide complexes. Mol Immunol. 2009;46(15):2873–5. doi: 10.1016/j.molimm.2009.05.184. Epub 2009/07/01. [DOI] [PubMed] [Google Scholar]
- 58.Hunter CA, Tomas S. Cooperativity, partially bound states, and enthalpy-entropy compensation. Chem Biol. 2003;10(11):1023–32. doi: 10.1016/j.chembiol.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 59.Sadegh-Nasseri S, Natarajan S, Chou CL, Hartman IZ, Narayan K, Kim A. Conformational heterogeneity of MHC class II induced upon binding to different peptides is a key regulator in antigen presentation and epitope selection. Immunol Res. 2010;47(1-3):56–64. doi: 10.1007/s12026-009-8138-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: 2002. [Google Scholar]