

INTRODUCTION
Alzheimer’s Disease (AD) is a progressive and fatal brain disorder afflicting over five million Americans (2); yet there are currently no effective treatments that target the underlying molecular causes of the disease (3). Pathologically, the AD brain at end stage is characterized by atrophy of the hippocampus and cerebral cortex, and an accumulation of extracellular proteinaceous plaques composed of amyloid fibrils that result from the uncontrolled aggregation of the amyloid beta peptide (Aβ). Because the appearance of insoluble plaque is tightly linked to neurotoxicity and disease (4–11), amyloid fibrils were initially thought to be the molecular culprit responsible for AD. Recent studies, however, indicate a more decisive correlation between the levels of soluble Aβ oligomers and the extent of synaptic loss and cognitive impairment (12–16). Irrespective of the exact structure and oligomeric state of the toxic species, the overall link between AD and Aβ aggregation suggests that inhibitors of the aggregation process may lead to effective therapeutics (16–27).
Structural studies of Aβ have advanced significantly in recent years (28–32). These studies provide a foundation for structure-based design of compounds that bind Aβ oligomers and/or inhibit their formation (33). However, since the high-resolution structure of the toxic Aβ oligomer is not known, high throughput screening (HTS) followed by analysis of structure activity relationships (SAR) remain productive approaches for identifying inhibitors of Aβ aggregation.
In previous studies, we described a novel high throughput method to enable rapid screening of compounds for their abilities to inhibit the aggregation of Aβ42 (26). More recently, we implemented this screen on a library of 65,000 compounds, and discovered several small molecules that reduced aggregation substantially. Among these ‘hits’, a compound designated D737 (C25H20N2O, Fig. 1) was studied in depth and shown to (i) inhibit the formation of Aβ oligomers, (ii) reduce Aβ induced cytotoxicity, and (iii) enhance the longevity and climbing ability of transgenic fruit flies expressing human Aβ42 in their central nervous system (1).
Figure 1.
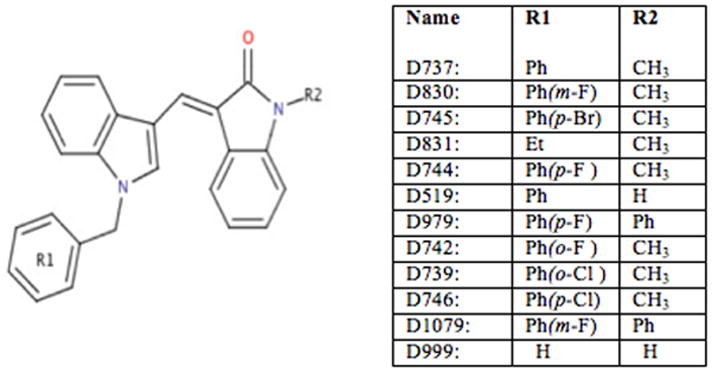
Compound D737 and its analogs.
Compound D737 has several properties considered important for Aβ binding. These include hydrophobicity and the presence of aromatic groups (17–24). For the current study, we probed structure/activity relationships for a small collection of eleven commercially available analogues of the original HTS hit (Fig. 1). This study has two goals: (i) To identify some of the chemical features of D737 that are important for activity; and (ii) To search for analogues that might be more active than the original D737 molecule. Our results show that alterations of a phenyl group at one end of D737 and a methyl group at the other end both affect activity. Two compounds (D744 and D830) with fluorine substitutions on an aromatic ring inhibited Aβ42 aggregation and increased the longevity of transgenic flies beyond that observed for compound D737.
METHODS AND MATERIALS
Compounds
D737 and analogs were purchased from ChemDiv (San Diego, CA). All compounds were first tested for intrinsic fluorescence at 512 nm (GFP wavelength) and 570 nm (MTT wavelength) and fluorescence was negligible. DMSO concentration did not exceed 1% in any experiments.
Aβ42-GFP Fusion Screen
As described in previous work (26), E. coli strain BL21 (DE3) harboring a vector encoding the Aβ42-GFP fusion protein was grown in LB supplemented with 35 mg/mL kanamycin. When cultures reached an OD600=0.8, 100 μL of culture was transferred to the wells of 96 well plates. D737 analogs were added to a final concentration of 50 μM, and protein expression was induced by adding isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. Samples were incubated with gentle agitation at 37°C. Following incubation for 5 hours, the fluorescence of each well was measured at 512 nm (excitation 490 nm) using a Varioskan plate reader. Results represent the average of 3 wells.
Synthetic Peptide
Aβ42 peptide was purchased from the Keck Institute at Yale University and purified on a C4 reverse phase column (Vydac). After purification, the peptide was snap frozen and lyophilized. Monomeric samples were prepared by adding trifluoroacetic acid (TFA) and sonicating for 15 minutes. Residual TFA was removed by hexafluoroiso-propanol and argon blow.
Cell Toxicity Assays
Rat pheochromocytoma (PC12) cells were cultured on collagen coated tissue treated petri dishes in 5% CO2 at 37°C in complete growth media (82.5% RPMI, 15% horse serum and 2.5% fetal bovine serum - ATCC). The cells were plated in 96 well plates to a concentration of 10,000 cells per well and allowed to attach to the plate overnight before adding peptide. Synthetic Aβ42 peptide at 200 μM was pre-incubated in PBS for 24 hours in the presence or absence of inhibitors. Aβ42 concentration was 20 μM and small molecule concentrations were 50 μM. Following this incubation, 10 μL Aβ42 (with or without compound) was added to cells. After 24 hours at 37°C, cell viability was evaluated using the MTT assay according to the supplier’s instructions (Roche). The lane marked “cells” indicates the viability of the PC12 cells without added peptide. This positive control is normalized to 100%. The lane marked “DMSO” is the negative control showing the reduced viability of cells that received Aβ42, but no added compound.
Fly Longevity Assay
Male flies carrying elav-Gal4 (on the X chromosome) were crossed with female flies carrying Aβ42 under UAS-Gal control to produce female progeny expressing Aβ42 in the central nervous system. Positive control flies were female carriers of elav-Gal4, which do not express peptide. Flies were reared at 29°C on medium with 20 μM D737 analogs or an equivalent amount of DMSO. For each class, vials containing 20 female flies each were collected and fed fresh food twice a week. The number of viable flies was recorded daily post eclosion. Survival rates were analyzed using Kaplan Meier statistics. Medial survival represents the day when 50% flies remain alive, and the student TTEST was used to generate P values.
Fly Climbing Assay
Locomotive ability was assayed as described in reference 45. Ten cm vials containing 20 flies each were tapped gently on the table. The number of flies that climbed to the top of the vial was recorded after 18 seconds. The fraction of flies that climbed to the top of the vial after 18 seconds was recorded 2–3 times per week.
RESULTS AND DISCUSSION
Analogs of D737 Inhibit Aggregation
In previous work, we described a high throughput screen to search for compounds that inhibit the aggregation of Aβ (26). This screen uses green fluorescent protein (GFP) as a reporter for the solubility (non-aggregation) of Aβ. Briefly, the 42-residue alloform of Aβ is linked upstream of GFP, and the Aβ42-GFP fusion protein is expressed in E coli. In the absence of inhibition, the Aβ42 portion of the fusion aggregates rapidly and causes the entire fusion protein to misfold and aggregate into an insoluble precipitate that does not fluoresce. However, inhibition of Aβ42 aggregation allows GFP to fold into its native green fluorescent structure (34). Compounds such as D737, which inhibit Aβ aggregation, yield green fluorescence, while compounds that are inactive (or toxic) do not produce fluorescence (26, 34, 35).
We used this assay to test analogues of D737 for their efficacy as inhibitors of Aβ aggregation. Assays were performed using E. coli cells transformed with a plasmid directing expression of the Aβ42-GFP fusion protein, as described previously (26, 34, 35). IPTG was added to induce expression, and cells were grown in 96 well plates containing 50 μM compound or DMSO control. After 5 hours of growth at 37°C, GFP fluorescence was measured (Fig. 2). Higher fluorescence indicates a compound inhibits Aβ aggregation, thereby enabling the folding and fluorescence of the Aβ42-GFP fusion (26, 34, 35). As shown in Figure 2, most of the analogues inhibit aggregation, albeit at lower levels than D737. One compound, D830, has similar activity as the D737 parent compound.
Figure 2.
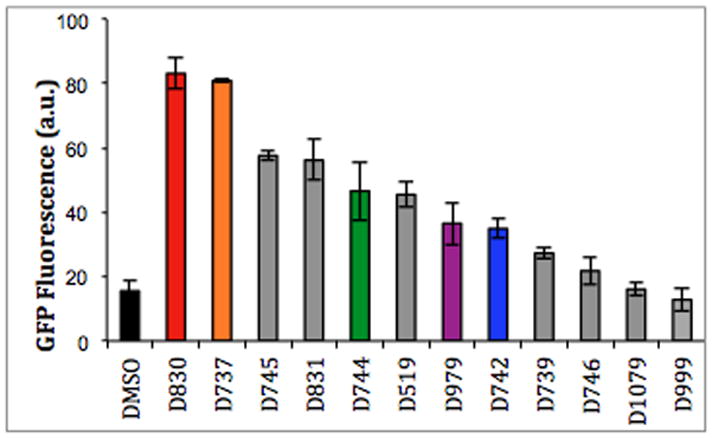
Fluorescence was measured for E. coli cells expressing the Aβ-GFP fusion protein.
Note: Compounds are color-coded consistently for comparison of Figures 2–6.
Our SAR studies focused on two parts of the D737 scaffold: The phenyl group at R1 and the methyl group at R2 (Fig. 1).
Modification or replacement of the phenyl group at R1
Halogen substitutions on aromatic rings are known to affect the binding properties of small molecules (36, 37). To probe the effect of halogen substitutions on the inhibitory activity of D737, fluorine, chlorine, and bromine were incorporated at the ortho, meta, and para positions of the R1 aromatic ring. As shown in Figure 2, while a fluorine substitution to the R1 phenyl group in the meta position (D830) retains essentially the same inhibition activity as D737, a para substitution (D744) slightly decreases inhibition of aggregation and inhibition is greatly reduced by a fluorine substitution in the ortho position (D742) of R1. The position of fluorine substitutions greatly affects the inhibition activity of D737 (m-F > p-F > o-F), which suggests that there are specific interactions that allow the aromatic R1 to fit into the chemical landscape of Aβ assemblies.
Next, we assayed compounds with bromine or chlorine on the phenyl group. Chlorine substitutions to the R1 phenyl group in the ortho position (D739) and para position (D746) do not significantly inhibit aggregation, which suggests that the larger chlorine atom may be unfavorable for peptide binding and inhibition. Given that chlorine substitutions are less active, one would expect that a larger bromine substitution would also be unfavorable for inhibition. Surprisingly, D745 (p-Br) is a very effective inhibitor. This suggests that in addition to size, factors such as solubility, electronegativity, and sterics may influence inhibitory activity
Lastly, in compound D831, an ethyl group replaces the aromatic phenyl group of D737. Though D831 still shows an ability to inhibit Aβ aggregation it permits less GFP fluorescence than D737, which suggests that the R1 phenyl ring is indeed involved for interrupting critical protein-protein interactions that allow Aβ assembly. These results demonstrate that both the identity and the position of halogen atoms on the R1 aromatic ring influence the activity of analogues of D737.
Deletion or replacement of a methyl group
To determine if the methyl group in the R2 position is optimal for activity, we replaced it with yet another aromatic phenyl group or hydrogen. In compound D519 the R2 methyl group is replaced with hydrogen and inhibition activity is slightly decreased. The diminished activity of compound D519 suggests that the methyl group on D737 may be more favorable for disrupting aggregation.
The methyl group of R2 is replaced with a phenyl group in compounds D979 and D1079; additionally, the R1 components of D979 and D1079 have p-F and m-F substitutions, respectively. This comparison serves a dual purpose; to determine if increased aromaticity favors or disfavors binding, and to confirm whether halogen additions to the aromatic ring in the R1 component do indeed require precise orientations. Interestingly, D1079 (m-F) allows for slightly less GFP fluorescence than Aβ42-GFP fusion without drug. On the other hand, compound D979, which has a phenyl group in the R2 position and a p-F substitution in the R1 component, inhibits Aβ aggregation. Somehow, the fluorine in the para position allows for more favorable protein-drug interactions and inhibition of aggregation. In comparison, D745, which has only one addition to D737, p-Br in the R1 component, is an active aggregation inhibitor. This result suggests that those compounds with substitutions in the para position of the R1 could be creating a more favorable environment, critical enough to balance the unfavorable environment created by adding a second aromatic group of R2.
Finally, D999 replaces both the R1 and R2 components of D737 with hydrogen and these substitutions seem to abrogate activity. Overall, we conclude that the aromatic group at the R1 position is critical for inhibition of Aβ aggregation, and a methyl group in the R2 component may be the optimal size for fitting into the Aβ structure (Figure 2).
Analogues of D737 Reduce Aβ42 Toxicity in Neuronal Cells
Inhibiting aggregation of the Aβ peptide has been suggested as an approach for reducing cytotoxicity (38–40). Therefore, we probed the ability of D737 and its analogues to prevent Aβ induced toxicity in cultured neuronal cells. Because Aβ oligomers are thought to be more toxic than fibrils, we allowed the peptide to aggregate at 37°C without shaking, conditions known to favor the formation of soluble oligomers (17, 18, 41).
Synthetic Aβ42 peptide (20 μM) was incubated with 50 μM of D737 or its analogs for 24 hours and then added to cultured PC12 cells. (Compounds were also tested alone for toxicity to PC12 cells and showed no apparent toxicity.) Following incubation for an additional 24 hours, cell viability was determined by the MTT assay (42). As shown in Figure 3, compounds that inhibited Aβ aggregation in the Aβ42-GFP assay (Fig. 2) also reduced Aβ induced cytotoxicity, thereby increasing cell viability. For example, the parent compound, D737, increased cell viability by about 30% at 100 μM and about 20% at 50 μM, 20 μM, or 5 μM concentrations (Fig 3 and Supp. Fig. 1). Additionally, compounds with fluorine substitutions on the R1 ring, including D744 (p-F) and D830 (m-F), also increase cell viability by 20–30%. In contrast, D999 and D1079, which were less effective at inhibiting aggregation in the Aβ–GFP screen (Fig. 2), are also less effective at reducing Aβ induced toxicity (Fig. 3).
Figure 3.
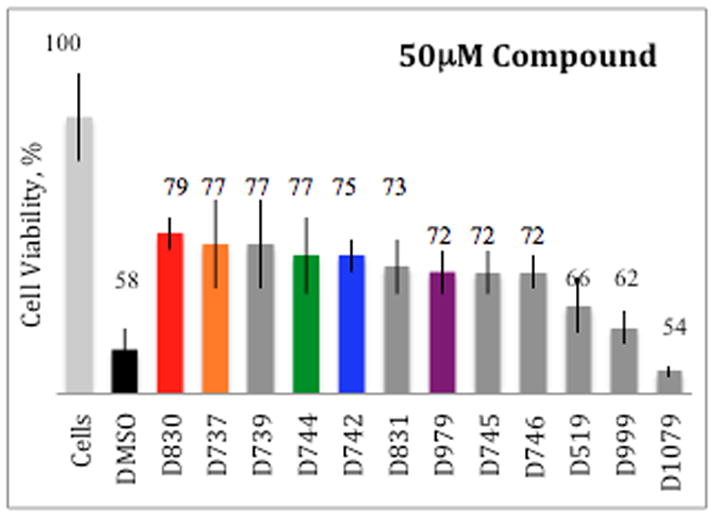
Analogues of compound D737 rescue PC12 cells from Aβ42 induced toxicity.
Although, overall, we observed a good correlation between the ability of a compound to inhibit aggregation in the Aβ42-GFP assay, and its efficacy in reducing Aβ induced cytotoxicity in PC12 cells (Fig. 4); two analogues -- D739 and D746 – were dramatically less active in one assay than another 20–100μM concentrations (Fig. 3). For instance, D746 (p-Cl) was not shown to significantly inhibit aggregation in the screen (Fig. 2) but increases cell viability by 8–14% at 100 μM, 50 μM, 20 μM, and 5 μM concentrations (Supp. Fig. 1). Likewise, D739 (o-Cl) is effective at high concentrations 21%, 19%, and 11% at 100 μM, 50 μM, and 20 μM respectively, but actually reduces cell viability at 5 μM unlike D737, which greatly reduce Aβ induced toxicity at 5μM (Suppl. Fig. 1). This result implies that D739 and D742 are only effective exceeding stoichiometric levels not employed during the HTS.
Figure 4.
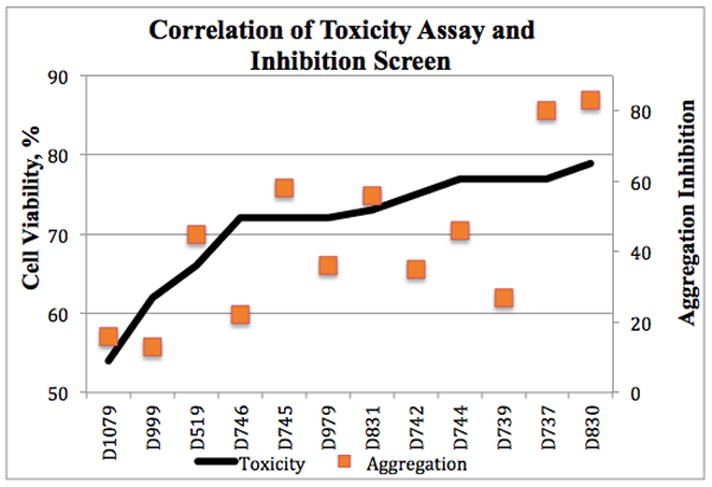
Correlation between the ability of a compound to inhibit Aβ42 aggregation and its efficacy in preventing Aβ42 induced toxicity.
Analogues of D737 Increase the Longevity of Transgenic Flies Expressing Aβ42
Perhaps the most important activities for SAR studies are those monitored in vivo. To test whether analogues of D737 are active in vivo, we used transgenic Drosophila melanogaster that express human Aβ42 in their central nervous system. In these flies, Aβ42 is expressed under control of a Gal4-UAS system. Male stocks carry the pan-neuronal elav-Gal4 on their X chromosome, while female stocks are homozygous for the autosomal UAS-regulated Aβ42 transgene. Both the male and female stocks are asymptomatic and can be propagated normally. Crossing these flies, however, produces female progeny that express Aβ42 in their CNS and display disease phenotypes, including reduced longevity and a gradual decline of locomotive ability (43, 44).
We used these transgenic flies to assess the activity in vivo of D737 and four analogues. Three of these analogues carry substitutions on the R1 phenyl group: Compounds D744, D830, and D742 contain fluorine at the para, meta and ortho positions of this aromatic ring. The fourth compound, D979, also has methyl to phenyl substitution at R2, was chosen as a negative control.
Flies were fed 20 μM compounds (or DMSO as a control) continuously from the embryonic stage through adult life. In our earlier studies we had determined that this concentration was optimal for D737 in extending life span, while higher doses slightly impaired longevity of the flies. As summarized in Table 1, all five compounds enhanced longevity beyond the DMSO control. Compound D737 increased the median lifespan by 1 week, while D744, D830, D979, and D742 increased lifespan by 9, 8, 5 and 3 days, respectively (Table 1). As shown in Figure 5, the most effective compound (D744) produced a lifespan curve for the transgenic flies that is fairly close to that of the positive control (female carriers that do not express Aβ42).
Figure 5.
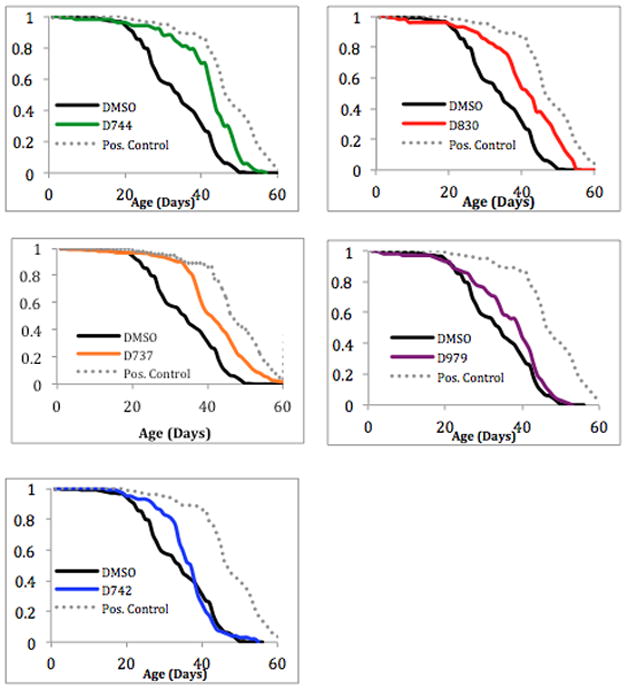
The effect of compound D737 and analogues on the lifespan of transgenic Drosophila expressing Aβ42 in the CNS.
Compounds D742, D744, and D830 differ from one another only by the position of the fluorine on the R1 aromatic ring, yet they have different impacts on lifespan (Table 1 and Fig. 5). This is consistent with results presented above using Aβ42-GFP to probe inhibition of aggregation (Fig. 2), and PC12 cells to measure inhibition of Aβ induced cytotoxicity (Fig. 3). In those assays, compound D742 was less active than D744, D830, or the parent compound D737. Likewise, in the Drosophila longevity assay, compounds D744, D830, and D737 increase median lifespan by 9, 8, 7 days, respectively, while D742 increase lifespan by only 3 days (Table 1).
Compound D979, with its methyl to phenyl substitution at R2, does not improve fly longevity significantly (P value = 0.25604). This is consistent with results shown above (Figs. 2–4), which suggested that replacing this methyl with an aromatic ring does not increase activity relative to D737.
In this study we learned that those compounds shown previously to inhibit Aβ42 aggregation and reduce Aβ42-induced cytotoxicity also improve the lifespan of flies expressing Aβ42. More importantly, we showed that our compounds are active in vivo, as both the parent compound, D737, and its analogues affect the lifespan and behavior of Drosophila. Remarkably, although compound D744 (p-F) was not the most effective in either the HTS or the cell viability assays, it was the most effective in increasing the lifespan of transgenic flies. This effect probably occurs because many other factors affect bioavailability when protein is expressed in vivo and in higher organisms such as challenges in crossing the blood brain barrier. Possibly the increased molecular hydrophobicity of D744 is responsible for improved efficacy.
Analogues of D737 Enhance the Climbing Ability of Transgenic Flies Expressing Aβ42
The locomotive ability of transgenic flies expressing Aβ42 was monitored in the presence or absence of D737 and the four analogues described above. Flies were fed the indicated compound (or DMSO) continuously from the embryonic stage through adult life. The flies were then tested for locomotive ability every few days. Flies (20 per vial) were gently tapped to the bottom of the vial, and after 18 seconds, the fraction of flies climbing to the top of the vial was recorded. As shown in Figure 6, flies receiving the DMSO control began to show a sharp decline in locomotive ability around 31 days post eclosion. For example after one month, only 23% of the control flies had the ability to climb (Fig. 6). Compound D737 and all four analogues improved climbing ability: After 31 days the following percentages and statistical significances of flies retained the ability to climb: D737 (45%, p value=0.0001), D744 (40%, p value=0.001), D830 (40%, p value=0.018), D742 (38%, p value=0.244) or D979 (33%, p value=0.034). In agreement with the results on fly longevity (Table 1), D737, D744, and D830 were more active, while compounds D742 and D979 showed the least activity in this assay for mobility in a simple animal model of AD.
Figure 6.
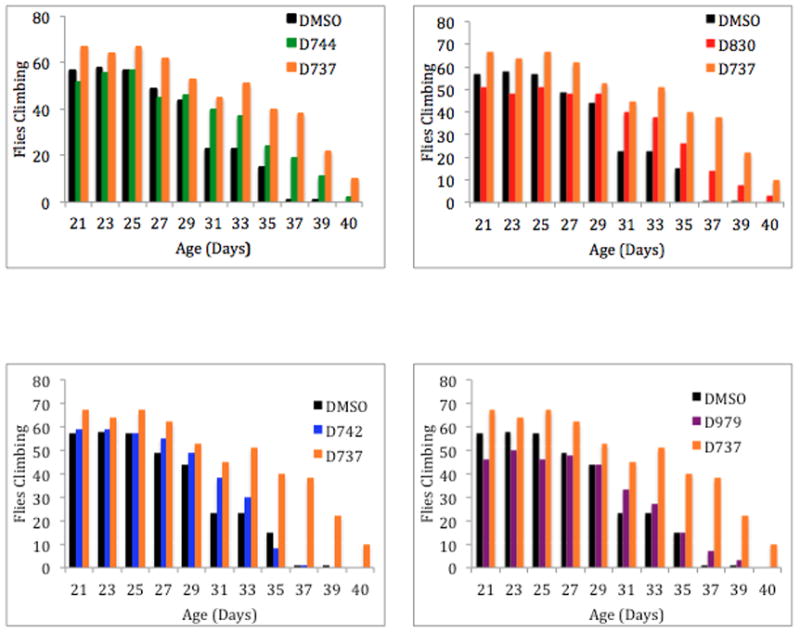
D737 analogs improve the locomotive ability of transgenic flies expressing Aβ42. Bar graphs represent weeks 3–5.
CONCLUSIONS
We chose 11 commercially available analogues to probe the importance of functional groups at the R1 and R2 positions of D737, a molecule that had been discovered in a HTS for inhibitors of Aβ aggregation. Five of these compounds (including D737) were also tested in vivo in a drosophila model of AD. Overall, we observed good correlations between inhibition of Aβ42 aggregation, reduction of Aβ42-induced cytotoxicity, and improved lifespan and locomotive ability of flies expressing Aβ42. In particular, assays in vivo suggest that a simple substitution in which fluorine is included on an aromatic ring can improve the efficacy of the original compound in a drosophila model of Alzheimer’s disease.
Supplementary Material
Analogues of compound D737 rescue PC12 cells from Aβ42 induced toxicity at concentrations 5–100 μM.
Acknowledgments
Supported by grant 5R21AG028462 from the NIH and Award IIRG-08-89944 from the Alzheimer’s Association to MHH, and by funding from the Howard Hughes Medical Institute to TS.
References
- 1.McKoy AF, Chen J, Schupbach T, Hecht MH. A novel inhibitor of amyloid β (Aβ) peptide aggregation: from high throughput screening to efficacy in an animal model of Alzheimer’s Disease. J Biol Chem. 2012;287:38992–39000. doi: 10.1074/jbc.M112.348037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.http://www.alz.org/downloads/facts_figures_2013.pdf
- 3.Sperling RA, Jack CR, Jr, Aisen PS. Testing the right target and right drug at the right stage. Sci Transl Med. 2011;3:111cm33. doi: 10.1126/scitranslmed.3002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clippingdale A, Wade J, Barrow C. The amyloid-beta peptide and its role in Alzheimer’s Disease. J Peptide Sci. 2001;7:227–249. doi: 10.1002/psc.324. [DOI] [PubMed] [Google Scholar]
- 5.Hardy J, Higgens G. Alzheimer’s Disease: the amyloid cascade hypothesis. Science. 1992;956:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 6.Alexandrescu A. Amyloid accomplices and enforcers. Protein Science. 2005;14:1–12. doi: 10.1110/ps.04887005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selkoe DJ, Shenk D. Alzheimer’s Disease: molecular understanding predicts amyloid based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- 8.Hardy J, Allsop D. Amyloid deposition as the central event in the etiology of Alzheimer’s Disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 9.Masters C, Simms G, Weiman N, Multhaup G, McDonald B, Beyreuther K. Amyloid plaque core protein in Alzheimer’s Disease and Down Syndrome. PNAS. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selkoe D. Alzheimer’s Disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 11.Walsh DM, Hartley DM. Amyloid beta protein fibrillogenesis structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 12.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid-β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 13.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 14.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β peptide. Nature Reviews Molec Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 15.Kayed R, Head E, Thompson J, McIntire T, Milton S, Cotman C, Glabe C. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–48. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 16.Hefti F, Goure WF, Jerecic J, Iverson KS, Walike PA, Krafft GA. Trends in Pharmacological Sciences. The case for soluble Aβ oligomers as a drug target in Alzheimer’s disease. 2013;34:261–266. doi: 10.1016/j.tips.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 17.Necula M, Kayed R, Milton S, Glabe C. Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J Biol Chem. 2007;282:10311–10324. doi: 10.1074/jbc.M608207200. [DOI] [PubMed] [Google Scholar]
- 18.Necula M, Breydo L, Milton S, Kayed R, van der Veer W, Glabe C. Methylene blue inhibits amyloid A-beta oligomerization by promoting fibrillization. Biochem. 2007;46:8850–5560. doi: 10.1021/bi700411k. [DOI] [PubMed] [Google Scholar]
- 19.Reinke A, Gestwicki J. Structure-activity relationships of amyloid beta aggregation inhibitors based on curcumin: influence of linker length and flexibility. Chem Biol Drug Des. 2007;70:206–215. doi: 10.1111/j.1747-0285.2007.00557.x. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, Shorter S. Direct and selective elimination of specific prions and amyloid by 4,5-dianilinophthalimide analogs. PNAS. 2008;105:7159–7164. doi: 10.1073/pnas.0801934105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klafki HW, Staufenbiel M, Kornhuber J, Wiltfang J. Therapeutic approaches to Alzheimer’s Disease. Brain. 2006;129:2840–2855. doi: 10.1093/brain/awl280. [DOI] [PubMed] [Google Scholar]
- 22.Citron M. Alzheimer’s Disease: strategies for disease modification. Nat Rev Drug Discovery. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 23.Porat Y, Abramowitz A, Gazit E. Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Design. 2006;67:27–37. doi: 10.1111/j.1747-0285.2005.00318.x. [DOI] [PubMed] [Google Scholar]
- 24.Hawkes C, Ng V, McLaurin J. Small molecule inhibitors of A-beta aggregation and neurotoxicity. Drug Dev Res. 2009;70:111–124. [Google Scholar]
- 25.Cheng PN, Liu C, Zhao M, Eisenberg D, Nowick JS. Amyloid β-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nat Chem. 2012;4:927–33. doi: 10.1038/nchem.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim W, Kim Y, Min J, Kim D, Chang Y, Hecht M. A high-throughput screen for compounds that inhibit aggregation of the Alzheimer’s peptide. ACS Chem Biol. 2006;1:461–469. doi: 10.1021/cb600135w. [DOI] [PubMed] [Google Scholar]
- 27.Chen J, Armstrong AH, Koehler A, Hecht MH. Small Molecule Microarrays Enable the Discovery of Compounds that Bind the Alzheimer’s Aβ Peptide and Reduce Cytotoxicity. J Am Chem Soc. 2010;132:17015–17022. doi: 10.1021/ja107552s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petkova AT, Ishii Y, Leapman R, Delaglio F, Tycko R. A structural model for Alzheimer’s beta-amyloid fibrils based on experimental constraints from solid state NMR. PNAS. 2002;26:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petkova AT, Yau W, Tycko R. Experimental constrains on quaternary structure in Alzheimer’s beta-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paravastua AK, Leapmanb RD, Yaua W-M, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. PNAS. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fitzpatrick, et al. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. PNAS. 2013;110:5468–5473. doi: 10.1073/pnas.1219476110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, Pensalfini A, Soriaga AB, Landau M, Teng PK, Cascio D, Glabe C, Eisenberg D. Atomic view of a toxic amyloid small oligomer. Science. 2012;335:1228–31. doi: 10.1126/science.1213151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang L, Liu C, Leibly C, Landau M, Zhao M, Hughes MP, Eisenberg DS. Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid beta. eLife 2013. 2013;2:e00857. doi: 10.7554/eLife.00857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waldo GS, Standish BM. Rapid protein folding assay using green fluorescent Protein. Nat Biotechnol. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- 35.Wurth C, Guimard NK, Hecht MH. Mutations that Reduce Aggregation of the Alzheimer’s Aβ42 Peptide: An Unbiased Search for the Sequence Determinants of Aβ Amyloidogenesis. J Molec Biology. 2002;319:1279–1290. doi: 10.1016/S0022-2836(02)00399-6. [DOI] [PubMed] [Google Scholar]
- 36.Araman H, Resnati G, Metrangolo P. Halogen bonding: fundamentals and applications. Springer Publishing; Berlin, Germany: 2008. [Google Scholar]
- 37.Zhu W, Wang Y, Yunxian L. Nonbonding interactions of organic halogens in biological systems: implications for drug discovery and biomolecular design. Phys Chem Chem Phys. 2010;12:4543–3441. doi: 10.1039/b926326h. [DOI] [PubMed] [Google Scholar]
- 38.Walsh DM, Selkoe DJ. A beta oligomers-a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 39.McLean CA, Cherny RA. Soluble pool of a beta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s Disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 40.Walsh DM, Selkoe DJ. Oligomers in the brain: the emerging role of protein aggregates in neurodegeneration. Pept Lett. 2004;11:213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- 41.Petkova AT, Leapman RD, Guo ZH, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 42.Mosmann T. Rapid colorometric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 43.Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie F, Gubb DC, Lomas DA. Intraneuronal amyloid beta non-amyloid aggregates and neurodegeneration a drosophila model of Alzheimer’s Disease. Neurosci. 2005;132:123–135. doi: 10.1016/j.neuroscience.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 44.Pandey U, Nichols C. Human disease models in Drosophila Melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev. 2011;63:411–436. doi: 10.1124/pr.110.003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scherzer-Attali R, Pellarin R, Convertino M, Frydman-Marom A, Egoz-Matia N, Peled S, Levy-Sakin M, Shalev D, Caflisch A, Gazit E, Segal D. Complete Phenotypic Recovery of an Alzheimer’s Disease Model by a Quinone-Tryptophan Hybrid Aggregation Inhibitor. PloS ONE. 2010;5(6):e11101. doi: 10.1371/journal.pone.0011101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analogues of compound D737 rescue PC12 cells from Aβ42 induced toxicity at concentrations 5–100 μM.