

Abstract
H. pylori infection causes gastritis, peptic ulcers and gastric cancer. Eradicating H. pylori prevents ulcers, but to what extent this prevents cancer remains unknown, especially if given after intestinal metaplasia has developed. H. pylori infected wild-type (WT) mice do not develop cancer, but mice lacking the tumor suppressor p27 do so, thus providing an experimental model of H. pylori-induced cancer. We infected p27-deficient mice with H. pylori strain SS1 at 6–8 weeks of age. Persistently H. pylori-infected WT C57BL/6 mice served as controls. Mice in the eradication arms received antimicrobial therapy (omeprazole, metronidazole and clarithromycin) either “early” (at 15 weeks post infection, WPI) or “late” at 45 WPI. At 70 WPI, mice were euthanized for H. pylori determination, histopathology and cytokine/chemokine expression. Persistently infected mice developed premalignant lesions including high-grade dysplasia, whereas those given antibiotics did not. Histologic activity scores in the eradication groups were similar to each other, and were significantly decreased compared with controls for inflammation, epithelial defects, hyperplasia, metaplasia, atrophy and dysplasia. IP-10 and MIG levels in groups that received antibiotics were significantly lower than controls. There were no significant differences in expression of IFN-γ, TNF-α, IL-1β, RANTES, MCP-1, MIP-1α or MIP-1β among the three groups. Thus, H. pylori eradication given either early or late after infection significantly attenuated gastric inflammation, gastric atrophy, hyperplasia, and dysplasia in the p27-deficient mice model of H. pylori-induced gastric cancer, irrespective of the timing of antibiotic administration. This was associated with reduced expression of IP-10 and MIG.
Keywords: Helicobacter pylori, Gastric Cancer, Mouse Model, Inflammation, Chemokines
1. Introduction
Epidemiological and clinical studies as well as animal experiments demonstrate a causative link between chronic H. pylori infection and peptic ulcer disease as well as gastric adenocarcinoma and mucosa-associated lymphoid tissue lymphoma. Decades of chronic and severe inflammation in the gastric mucosa play an important role in this tumorigenic process [1, 2, 3]. H. pylori eradication by combining acid inhibition with a proton pump inhibitor (PPI) and at least two antibiotics has become a standard treatment in clinical practice for patients with gastritis and peptic ulcers [4], though increasing antibiotic resistance and H. pylori reinfection remain challenging obstacles to high eradication rates currently [5, 6].
Cohort studies and randomized controlled trials have demonstrated that H. pylori eradication not only prevents peptic ulcers but also slows the histological progression from chronic gastritis to gastric adenocarcinoma in patients with tumor-associated infection [7]. Although the incidence of stomach cancer is generally declining in the developed world, coincident with improved sanitation and a falling prevalence of H. pylori colonization, gastric cancer remains a major public health problem in regions with a high prevalence of H. pylori infection such as South East Asia, Eastern Europe, and Central and South America [8, 9].
Gastric cancer is recognized to be a multistep and multifactorial process that in most cases is preceded by a decades-long, stepwise progression of histological changes in the gastric mucosa from chronic gastritis through gastric atrophy, intestinal metaplasia, dysplasia and cancer [10, 11]. In retrospective sub-group analysis, it was noted that the beneficial effect of H. pylori eradication on lowering the incidence of gastric cancer depended upon eradicating H. pylori prior to the development of advanced pre-neoplastic changes, and that intestinal metaplasia may be the “point of no return” beyond which reversal of “Correa’s cascade” is no longer possible [7, 12]. With the designation of H. pylori as a definite carcinogen in 1994 [13] and the acceptance of this designation by public health authorities in high gastric cancer regions, ethical considerations now preclude recruitment of a comparator “placebo” arm of subjects who are not offered eradication therapy in clinical trials.
Because gastric cancer is a relatively rare consequence of H. pylori infection in humans and, with the limitations of performing appropriately powered long-term placebo-controlled H. pylori eradication studies in humans, mouse models of H. pylori infection may help us address the uncertain questions in this field. For example, is antibiotic therapy warranted in decreasing the incidence of gastric neoplasia even when given relatively late during the natural history of persistent H. pylori infection? In particular, is eradicating H. pylori at all beneficial if the precancerous lesion of intestinal metaplasia has already developed?
Several antimicrobial treatment studies have been previously conducted in rodent models of Helicobacter infection, usually using either a mouse-adapted H. pylori strain or the closely related Helicobacter species H. felis. The animal studies have included C57/BL6 mice, though they rarely develop neoplastic changes even when colonized by H. pylori for as long as 80 weeks [14], and hypergastrinemic INS-GAS mice in which spontaneous gastric carcinogenesis is accelerated by Helicobacter infection [15]. In Mongolian gerbils, results between different laboratories have been quite variable, reflecting in part the outbred nature of the animals [16, 17]. We have developed a mouse model of H. pylori-associated gastric carcinogenesis that mimics the slow progression towards gastric cancer observed in humans. In our previous study, we reported that wild type mice did not develop gastric cancer following experimental infection with the mouse-adapted H. pylori SS1 strain [18] while 7 out of 12 infected mice lacking the tumor suppressor p27 developed gastric dysplasia or carcinoma at the 60 week timepoint after infection [19]. Moreover, we observed marked gastric inflammation in this novel p27 deficient model of gastric cancer, and the development of pseudopyloric metaplasia of the corpus (the murine equivalent of intestinal metaplasia) [20] as early as 30 weeks post infection [19]. In the present study we have used this experimental model to recapitulate the H. pylori-induced gastric mucosal damage observed in humans and to investigate the effects of antibiotic eradication on preventing H. pylori-associated gastric cancer.
In particular, this study was designed to examine whether, and at what stage, H. pylori eradication might prevent gastric cancer in a long term H. pylori infection model, and to examine some potential mechanisms involved.
2. Materials and Methods
2.1 Mice, H. pylori Infection, Experimental Design
This study was approved by Rhode Island Hospital’s Animal Care and Use Committee. The experimental outline is shown in Figure 1. In brief, p27-deficient mice on a C57BL/6 background were gavaged at 6–8 weeks of age with H. pylori SS1 of approximately 109 bacterial colony forming units (CFU) in (200 μl) volume on three occasions over 5 days as described previously [19]. The H. pylori SS1-infected p27-deficient mice were then divided into three groups. Two groups of mice (18 each) were treated with an H. pylori eradication regimen - either at 15 weeks post infection (WPI) or at 45 WPI; the third group (15 mice) served as a non-treated control.
Figure 1. Experimental outline.
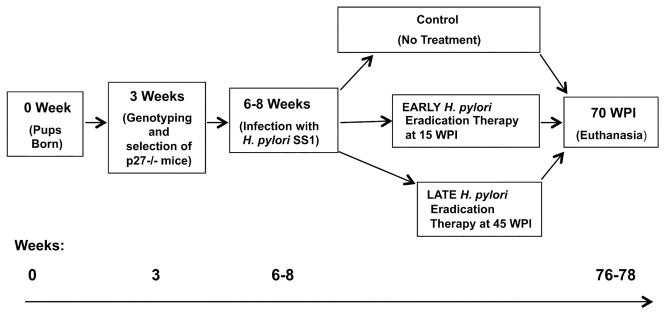
See Materials and Methods for details. WPI = Weeks post infection.
The eradication therapy was a triple regimen of omeprazole (400 μmol/kg/day), metronidazole oral suspension (14.2 mg/kg/day), and clarithromycin granules for oral suspension (7.15 mg/kg/day) administered by gavage twice daily for seven days with a 30 to 60 minute interval between omeprazole and antibiotics [21, 22]. All mice were euthanized and their stomachs were removed at 70 WPI, or sooner if their condition met Rhode Island Hospital’s Animal Care and Use Committee criteria for signs of distress. Stomachs were opened along the greater curvature and cut into several linear strips for DNA, RNA, protein assays, H. pylori quantitative culture and histologic analysis.
2.2 Histologic Evaluation
Longitudinal gastric strips from the lesser curvature were fixed in 10% neutral-buffered formalin, processed and stained with hematoxylin and eosin and scored by a veterinary pathologist who was blinded to the experimental details using the scoring criteria described previously [20]. As mucous metaplasia and hyalinosis may develop spontaneously in mice [23], these sub-scores were excluded from the calculation of the total histological activity index (HAI).
2.3 H. pylori Bacterial Load Assessment
H. pylori colonization in the mouse stomach was evaluated by quantitative culture and real time PCR.
2.3.1 H. pylori Quantitative Culture
One strip of gastric tissue from each mouse was weighed, homogenized in 250μl brucella broth with 20% glycerol (Catalog # 297466, BD Diagnostics, Sparks, MD, USA), and subsequently diluted 10, and 100-fold in brucella broth. A 25 μl homogenate from each dilution was cultured on agar plates containing 5% de-fibrinated horse blood, 3.3 μg/ml Polymyxin B sulfate, 100 μg/ml Vancomycin Hydrochloride, 200 μg/ml Bacitracin, 50 μg/ml Amphotericin B, and 10.7 μg/ml Naladixic Acid (Sigma-Aldrich, St. Louis, MO) as described previously and incubated for 7–10 days [15]. H. pylori colonies were identified by positive urease and oxidase reactions and then counted. After correcting for the initial dilution, the final results were expressed as CFU/gram gastric tissue.
2.3.2 DNA Extraction and H. pylori Quantitative Polymerase Chain Reaction (qPCR)
For PCR, DNA was extracted from one strip of gastric tissue with the QIAamp DNA Mini Kit (QIAGEN Sciences Inc., Germantown, MD). DNA concentration was measured and the quality for downstream experiments verified on a NanoDrop™ 2000 spectrophotometer (Thermo Scientific Inc., Waltham, MA). H. pylori DNA was quantified by amplification of the Helicobacter species-specific antigen (SSA) gene [24] with SYBER Green I QPCR mastermix reagents (QIAGEN, Valencia, CA) as previously described [19]. The threshold for positivity for H. pylori by quantitative PCR was considered to be at least 10 gene copies of H. pylori DNA, in accordance with Lee et al [20].
2.4 Cytokine and Chemokine Protein Measurement by Multiplex Bead Array
Cytokine and chemokine protein levels in the mouse stomachs were measured by a multiplex magnetic bead array kit customized by EMD Millipore (Billerica, MA) to include nine cytokines and chemokines: Interferon-gamma (IFN-γ), Tumor necrosis factor-alpha (TNF-α), Interleukin-1 beta (IL-1β), regulated upon activation, normal T cells-expressed and secreted chemokine (RANTES), monocyte chemoattractant protein-1 (MCP-1), monokine induced by gamma interferon (MIG), Interferon gamma-induced protein 10 (IP-10), macrophage inflammatory protein 1-alpha (MIP-1α) and macrophage inflammatory protein 1-beta (MIP-1β). One gastric strip from each mouse was homogenized and total protein concentration measured by BCA protein assay kit (Pierce Biotechnology, Rockford, IL). The multiplex bead arrays were performed according to the manufacturer’s instructions with a minimum detectable concentration varying from 0.8 to 11.9 pg/ml. Data were normalized to total protein concentration in each sample and expressed as pg/mg protein.
2.5 Gastric mRNA expression and serum protein levels of IP-10 and MIG
2.5.1 Gastric mucosal IP-10 and, MIG mRNA Expression
Total RNA was extracted from one strip of gastric tissue with the RNeasy mini Kit (QIAGEN, Germantown, MD). First strand cDNA generated from 2 μg total RNA was diluted by 1:10 with DNase RNAse free water and applied for real-time PCR. IP-10 and MIG quantitative PCR amplification and cycling program were performed as previously described [25] by using specific primer sets and SYBER Green I QPCR mastermix reagents (QIAGEN, Valencia, CA). IP-10 and MIG gene expression was analyzed via the delta-delta CT method, normalized to GAPDH and expressed as fold changes.
2.5.2 Determination of Serum IP-10 and MIG Concentrations
At euthanasia one milliliter whole blood was drawn from each mouse by cardiac puncture, and the serum collected and stored at −20 °C until analysis. Serum IP-10, MIG protein levels were determined using mouse IP-10 and mouse MIG quantikine ELISA kits (R & D Systems, Inc. Minneapolis, MN, USA) following the manufacturer’s instructions for each. All samples were run in duplicates of 50 μl diluted serum per mouse. The assay range of mouse IP-10 and MIG are 31.2 ~ 2000 pg/ml with a sensitivity of 4.2 pg/ml and 7.8 pg/ml, respectively.
2.6 Statistics
All statistical analysis were performed using Graphpad Prism 5.0 software (San Diego, CA), with differences between groups considered significant when P < 0.05. Histopathological scores were compared by a Kruskal-Willis one-way analysis of variance (ANOVA) followed by Dunn’s multiple comparison test. H. pylori colonization levels, and cytokine and chemokine expression were compared by ANOVA with Newman-Keuls test.
3. Results
3.1 Evaluation of H. pylori Eradication
Overall, the triple antibacterial therapy led to undetectable H. pylori colonization in the treated groups compared with infected control mice that did not receive H. pylori therapy (P<0.001 vs control, Figure 2 A). The mucosal bacterial load decreased markedly in the antibiotic-treated mice compared to the untreated mice, as measured by H. pylori quantitative PCR (P<0.05 vs control, Figure 2 B). There were no statistical differences in H. pylori infection eradication rates, and in bacterial loads between the two H. pylori eradication groups (15 WPI and 45 WPI).
Figure 2. H. pylori bacterial load assessment.
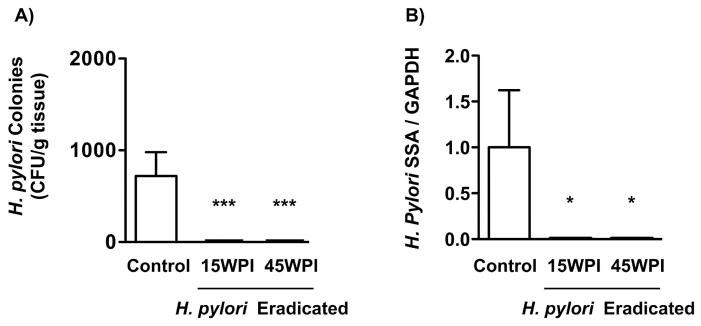
A) H. pylori colonization was evaluated by quantitative culture 70 weeks after H. pylori SS1 infection. The two groups given H. pylori eradication therapy had no detectable H. pylori colonies. Data were expressed as mean ± SEM (CFU/g tissue). SEM: standard error of mean, CFU: colony forming units. ***P<0.001 vs H. pylori non-eradicated control.
B) A longitudinal strip of stomach tissue from each mouse was harvested to measure bacterial load by quantitative PCR analysis. H. pylori non-eradicated group showed a significantly higher bacterial load in gastric mucosa in comparison with the two antimicrobial treated groups. Data were expressed as mean ± SEM. SEM: standard error of mean, ***P<0.001 vs H. pylori non-eradicated controls.
Final analyses were performed on a total of 39 mice that completed the protocol and were euthanized at 70 WPI (10 mice in H. pylori non-eradicated control group, 15 mice in 15 WPI and 14 mice in 45 WPI H. pylori-eradicated groups). These final numbers excluded mice that had died due to complications from gavage (N=2), spontaneous deaths after gavage but before the end of the study period (N=7) and mice that were not successfully eradicated of H. pylori (N=3)
3.2 Histological Evaluation
Hematoxylin and eosin (H&E) staining of mice stomach tissue demonstrated that the mice treated with antibiotics to eradicate H. pylori exhibited no or only mild lesions (Figure 3B, 3C) whereas the H. pylori infected control group (Figure 3A) developed premalignant changes including high-grade dysplasia, consisting of irregular glands with altered and complex architecture, variable cell sizes and hyperchromatic nuclei (Figure 3). In comparison with the H. pylori infected but untreated control group, antimicrobial treatment attenuated histological changes with decreased scores for inflammation (Figure 4A, P<0.01 for 15 WPI, p<0.001 for 45 WPI), and epithelial defects (Figure 4B, p<0.01 for 45 WPI). Similar trends (p<0.05 for 15 WPI, P<0.01 for 45WPI) were noted for oxyntic atrophy (Figure 4C), hyperplasia (Figure 4D), and pseudopyloric metaplasia (Figure 4E) which is the murine analog of human intestinal metaplasia. Importantly, mice in the H. pylori non-eradicated control group had significantly higher dysplasia scores compared to the two eradicated groups (Figure 4F, P<0.05). There were no statistical differences in any of the histological comparisons between the two H. pylori eradication groups (Figure 4). Both H. pylori eradicated groups had significantly lower overall disease scores as reflected by the histological activity index (HAI), a combination of all the previous six individual criteria scores (Figure 4G, p<0.05 for 15 WPI, p<0.01 for 45 WPI vs controls).
Figure 3. Representative gastric histopathology in H. pylori-infected p27 knockout mice.
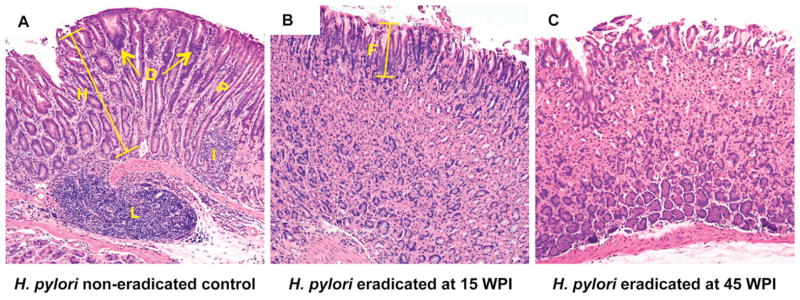
A) A p27-deficient mouse infected with H. pylori SS1 strain for 70 weeks without receiving eradication treatment. There is an intense inflammatory cell infiltrate with lymphoid follicle formation and a replacement of the normal gastric glands by irregular dysplastic glands lacking the specialized chief and parietal cells. Arrows and D: dysplasia, height bar and H: hyperplasia (compared to normal foveolar length, “F” in panel B), I: inflammation, P: pseudopyloric metaplasia, L: lymphoid follicle.
B), C) p27-deficient mice that received H. pylori eradication therapy at 15 weeks (B) or 45 weeks (C) post infection. These mice have much less inflammation and retention of a normal gastric epithelial architecture, with no dysplasia. F: normal superficial foveolar epithelium.
Hematoxylin and eosin staining, original magnification × 100.
Figure 4. Gastric histology scores in H. pylori-infected p27–deficient mice with or without eradication treatment.

The individual lesion scores of every mouse in each group were evaluated and compared for, A) Inflammation; B) Epithelial defects; C) Atrophy; D) Hyperplasia; E) Metaplasia; F) Dysplasia; and G) Histological Activity Index. Histological activity index is a combination of all the six individual criteria scores. In general, p27-deficient mice in the control group developed advanced pathological lesions, while the two eradicated groups showed a milder gastric pathology. Each solid triangle represents one mouse. The bar represents the median score in each group. * P<0.05, ** P<0.01, ***P<0.001 vs H. pylori non-eradicated controls.
3.3 Proinflammatory Cytokine and Chemokine Protein Expression
To evaluate the effects of H. pylori eradication on the levels of several proinflammatory cytokines and chemokines, the mucosal levels of nine cytokines and chemokines were measured by a customized multiplex magnetic bead array kit. There were no significant differences in protein levels of IFN-γ, TNF-α, IL-1β, RANTES, MCP-1, MIP-1 α and MIP-1 β among the three groups. However, IP-10 levels in the 15WPI (21.8 ± 2.5 pg/mg protein) and 45WPI (27.1 ± 6.3 pg/mg protein) eradication groups were significantly lower than the non-eradicated control group (79.1 ± 19.7 pg/mg protein) (Figure 5, P<0.001 for 15 WPI, P<0.05 for 45 WPI vs control). MIG levels in the non-eradicated control group (146.2 ± 42.9 mg/mg protein) were also markedly higher than the two groups that received antimicrobial treatment (19.6 ± 2.8 pg/mg protein for 15 WPI; 20.9 ± 3.1 pg/mg protein for 45 WPI) (Figure 5, P<0.001 for 15WPI, P<0.01 for 45WPI vs control).
Figure 5. Proinflammatory cytokine and chemokine expression in gastric mucosa.

Multiple proinflammatory cytokines and chemokines expression levels in the stomach tissue were analyzed. There are no significant differences in protein levels of IFN-γ, TNF-α, IL-1β, RANTES, MCP-1, MIP-1 α and MIP-1 β among the three groups, while IP-10 and MIG levels were significantly elevated in the non-eradicated group when compared to the two groups that received H. pylori eradication therapy. Data expressed as mean ± SEM (pg/mg protein), SEM: standard error of mean. * P<0.05, ** P<0.01, ***P<0.001 vs H. pylori non-eradicated controls. NS: not significantly different.
3.4 IP-10 and MIG Gene Expression in Gastric Mucosa and Serum
A) IP-10 and MIG mRNA expression in the stomachs of H. pylori untreated and treated groups of p27 deficient mice were assessed by quantitative real-time PCR and normalized to the levels of GAPDH expression. In comparison with the non-eradicated control mice, the two H. pylori-eradicated groups showed significantly decreased expression of both IP-10 and MIG mRNA (Figure 6A, * P<0.05, ***P<0.001 vs H. pylori non-eradicated control mice). There was no significant difference between the two H. pylori eradicated groups.
Figure 6. Gastric mRNA and serum protein levels of IP-10 and MIG.
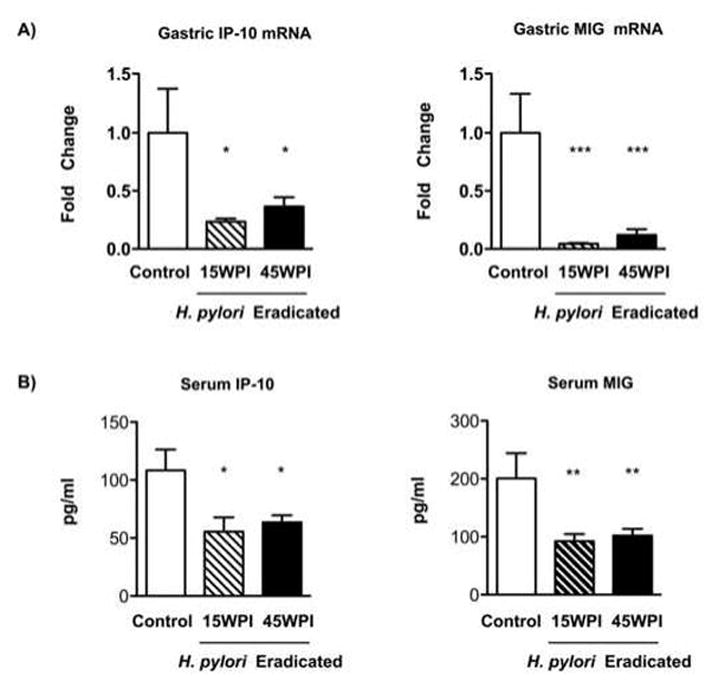
A) Gastric IP-10 and MIG mRNA expression were assessed by quantitative real-time PCR and normalized to the levels of GAPDH expression. Data expressed as average fold changes ± SEM in each group compared with the H. pylori non-eradicated control group. SEM: standard error of mean. * P<0.05, ***P<0.001 vs H. pylori non-eradicated control mice.
B) Serum protein concentrations of IP-10 and MIG were measured by ELISA. Results expressed as mean ± SEM (pg/ml). SEM: standard error of mean. * P<0.05, ** P<0.01 vs H. pylori non-eradicated control group.
B) Protein levels of IP-10 and MIG in serum were measured by IP-10 and MIG ELISA kits, respectively. Compared to the H. pylori non-eradicated control mice (IP-10: 108.4 ± 17.93 pg/ml, MIG: 200.8 ± 43.37 pg/ml), IP-10 (15WPI: 55.51 ± 12.24 pg/ml, 45WPI: 63.45 ± 6.28 pg/ml) and MIG levels (15WPI: 92.21 ± 12.5 pg/ml, 45WPI: 102 ± 11.74 pg/ml) were significantly lower in the two H. pylori eradicated groups (Figure 6B, * P<0.05, ** P<0.01 vs H. pylori non-eradicated control group). There was no significant difference between the two H. pylori eradicated groups.
4. Discussion
H. pylori infection causes gastritis, peptic ulcers and gastric cancer. Eradicating H. pylori cures peptic ulcers and prevents their recurrence, [1] but it is not known to what extent this will contribute to the prevention of gastric cancer development. Subgroup analysis of several underpowered clinical trials indicates that H. pylori eradication may be relatively ineffective when given relatively late in the natural history of infection, for example after the preneoplastic stage of intestinal metaplasia has developed [7].
Our results in the present study show that bacterial eradication given either at an early or a late stage of H. pylori infection can exert positive effects on gastric mucosal repair by significantly reducing the severity of inflammation, glandular atrophy, hyperplasia, and dysplasia in H. pylori-infected p27-deficient mice. As reported by Garhart et al, H. pylori colonization decreased in parallel with gastric inflammation over time [26]. At the very late stage at which the mice were euthanized (70 weeks post infection) both bacterial load and gastric inflammation in the non-treated control group remained significantly higher than in either of the two eradicated groups given H. pylori eradication therapy, though the magnitude of the decrease was smaller than has been reported by others, which may reflect the very late time-point that we examined [20]. Consistent with previous findings in other rodent models [20, 27], antimicrobial treatment decreased epithelial defects (45 WPI vs Control), reduced gastric atrophy and hyperplasia, restored glandular architecture and attenuated gastric premalignant lesions in both groups of H. pylori eradicated p27 knock-out mice. However, unlike the findings by Cai et al in C57BL/6 mice, the p27-deficient mice that received H. pylori eradication at a late time point were equally protected from developing dysplasia [27]. In contrast to most of the evidence from human studies [7], there were no significant differences regarding the histopathological scores between the group that received H. pylori eradication therapy relatively early (at 15 WPI) versus relatively late (at 45 WPI) - the latter time-point being chosen based upon our prior study [19] to include mice with pseudopyloric metaplasia. Extrapolating from our current study, H. pylori eradication may be beneficial for gastric cancer prevention in humans even after intestinal metaplasia has already developed.
The experimental model system that we employed here has also allowed us to investigate how H. pylori eradication may protect the gastric mucosa from neoplastic transformation. H. pylori-infection generally induces a T helper 1 (Th1)-skewed pro-inflammatory response in the gastric mucosa [28]. In previous human and animal studies, significantly increased mRNA levels of IFN-γ, TNF-α, IL-1β, RANTES, MIP-1α, MIP-1β and the CXC chemokines growth-regulated oncogene alpha (GRO-α), Interleukin-8 (IL-8), IP-10 and MIG have been demonstrated during H. pylori infection [29, 30]. After bacterial eradication, IFN-γ, TNF-α, IL-1β, IL-8/CXCL8 mRNA expression were reported to be downregulated [31, 32, 33], but less is known about dynamic changes of other chemotactic cytokines before and after H. pylori eradication, especially at the protein level.
Multiple cytokines and chemokine proteins were measured in murine gastric tissue using a cytometric bead array. Unlike previous mRNA expression studies, our results show no notable reductions at the protein level for three proinflammatory cytokines (IFN-γ, TNF-α, IL-1β) or four CC chemokines (RANTES, MCP-1, MIP-1α, MIP-1β) previously thought to be important in H. pylori pathogenesis in the two groups that received eradication therapy compared with the persistently infected control group. This could be due to the experimental model we used here since p27-deficient mice are known to have a dysregulated immune system, related to the importance of this critical cyclin-dependent kinase (CDK) inhibitor in lymphocyte development and responsiveness and in CD4 and CD8 memory T cell differentiation [34, 35, 36, 37]. In p27 knockout mice, numbers of IFN-γ, TNF-α and IL-2 producing antigen-specific CD4 T cells are significantly higher compared with wild type mice, though no significant differences in the percentage of antigen-specific CD4 T cells were reported [33]. Thus p27 deficiency may lead to generalized immune hyperresponsiveness, including in the gastric mucosa, even in the absence of antigen (in this instance, H. pylori), such that inflammatory cytokine and chemokine concentrations were not lowered by H. pylori eradication. As an alternative explanation for the equality of IFN-γ, TNF-α, IL-1β, RANTES, MCP-1, MIP-1α, MIP-1β among the control and H. pylori eradicated groups, it should be emphasized that the measurements were conducted at 70 weeks post infections, which is a very late time-point compared with other rodent studies. Although H. pylori-induced gastric mucosal inflammation is persistent, bacterial loads in mice subside with time, as does IFN-γ secretion in parallel [26, 38]. In Mongolian gerbils infected with H. pylori strain 7.13, gastric mucosal mRNA levels of IL-1β and TNF-α peaked at 1 month and 6 months post-infection respectively before dropping markedly at 12 months [17].
Unlike many of the other cytokines and chemokines evaluated, MIG and IP-10 protein levels in gastric mucosa were significantly lower in the mice receiving H. pylori eradication therapy, and this was concordant with parallel decreases in both gastric mRNA and serum concentrations in the antibiotic treated mice. MIG and IP-10 (alternatively termed CXCL9 and CXCL10) are chemokines known to be induced by IFN-γ. They function to recruit leukocytes to the sites of infection and inflammation in Th1 immune responses via binding to a common chemokine G protein-coupled receptor, CXCR3 [39, 40]. CXCL9 and CXCL10 require CXCR3’s carboxyl-terminal domain and an interaction of CXCR3 with β-arrestin 1 to internalize CXCR3, thereby inducing its biological functions [39]. The CXCR3/CXCL9, CXCL10 axis plays an important role in the recruitment of immune cells in infectious diseases [40]. In response to H. pylori infection, CXCR3 and its ligands CXCL9 and CXCL10 are up-regulated in human and murine gastric mucosa [30, 41], and the proinflammatory cytokines IFN-γ and TNF-α synergistically stimulated CXCL9 and CXCL10 in human gastric epithelial cell lines [42]. Recent studies have implicated both IP-10 and MIG in gastric cancer: their expression was elevated in gastric cancer tissue, and plasma levels of IP-10 and MIG fell sharply after gastric cancer resective surgery [43, 44]. Our findings support the concept that gastric mucosal IP-10 and MIG may promote or serve as biomarkers of gastric cancer because they both fell in the groups of mice treated by H. pylori eradication, in association with inhibition of gastric cancer progression, though it is conceivable that this may be due to H. pylori eradication alone.
In conclusion, the present study suggests that eradication therapy given either early or late after H. pylori infection significantly lowers gastric inflammation, gastric atrophy, hyperplasia, and dysplasia in the p27-deficient mice model of H. pylori-induced gastric cancer. Our results suggests that for humans, H. pylori eradication therapy may significantly ameliorate gastric inflammation and suppress the malignant potential in the gastric mucosa, even when given relatively late in the natural history of the disease. Finally, because reduced IP-10 and MIG expression in the gastric mucosa are associated with post eradication protection in p27-deficient mice, the role of these chemokines in H. pylori-associated gastric carcinogenesis is worthy of further study, in both rodent models and clinical studies.
Acknowledgments
The authors appreciate the assistance of Matthew Leon. The research was funded by the National Institutes of Health (CA 125126) to SFM.
Footnotes
Conflict of interest statement
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Songhua Zhang, Email: songhua_zhang@brown.edu.
Dong Soo Lee, Email: cmcdj9502@catholic.ac.kr.
Rhiannon Morrissey, Email: rhiannon.morrissey@salve.edu.
Jose R. Aponte-Pieras, Email: jose_aponte@alumni.brown.edu.
Arlin B. Rogers, Email: Arlin.Rogers@tufts.edu.
Steven F. Moss, Email: cmcdj9502@catholic.ac.kr.
References
- 1.McColl KE. Clinical practice Helicobacter pylori infection. N Engl J Med. 2010;362(17):1597–604. doi: 10.1056/NEJMcp1001110. [DOI] [PubMed] [Google Scholar]
- 2.Moss SF, et al. Mechanisms of disease: Inflammation and the origins of cancer. Nat Clin Pract Oncol. 2005;2:90–7. doi: 10.1038/ncponc0081. [DOI] [PubMed] [Google Scholar]
- 3.Wroblewski LE, et al. Helicobacter pylori in gastric carcinogenesis: mechanisms. Gastroenterol Clin North Am. 2013;42:285–98. doi: 10.1016/j.gtc.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chey WD, et al. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol. 2007;102:1808–25. doi: 10.1111/j.1572-0241.2007.01393.x. [DOI] [PubMed] [Google Scholar]
- 5.Morgan DR, et al. Risk of recurrent Helicobacter pylori infection 1 year after initial eradication therapy in 7 Latin American communities. JAMA. 2013;309:578–86. doi: 10.1001/jama.2013.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camargo MC, et al. The problem of Helicobacter pylori resistance to antibiotics: a systematic review in Latin America. Am J Gastroenterol. 2014;109:485–95. doi: 10.1038/ajg.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuccio L, et al. Meta-analysis: can Helicobacter pylori eradication treatment reduce the risk for gastric cancer? Ann Intern Med. 2009;151:121–8. doi: 10.7326/0003-4819-151-2-200907210-00009. [DOI] [PubMed] [Google Scholar]
- 8.Jemal A, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 9.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–44. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 10.Correa P, et al. A model for gastric cancer epidemiology. Lancet. 1975;306:58–60. doi: 10.1016/s0140-6736(75)90498-5. [DOI] [PubMed] [Google Scholar]
- 11.Correa P. Human gastric carcinogenesis: a multistep and multifactorial process-First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992;52:6735–40. [PubMed] [Google Scholar]
- 12.Mera R, et al. Long term follow up of patients treated for Helicobacter pylori infection. Gut. 2005;54:1536–40. doi: 10.1136/gut.2005.072009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schistosomes liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum; IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; Lyon. 7–14 June 1994; 1994. pp. 1–241. [PMC free article] [PubMed] [Google Scholar]
- 14.Kim DH, et al. Long-term evaluation of mice model infected with Helicobacter pylori: focus on gastric pathology including gastric cancer. Aliment Pharmacol Ther. 2003;18(Suppl 1):14–23. doi: 10.1046/j.1365-2036.18.s1.4.x. [DOI] [PubMed] [Google Scholar]
- 15.Fox JG, et al. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer Res. 2003;63:942–50. [PubMed] [Google Scholar]
- 16.Franco AT, et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci U S A. 2005;102:10646–51. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugimoto M, et al. Gastric mucosal interleukin-17 and -18 mRNA expression in Helicobacter pylori-induced Mongolian gerbils. Cancer Sci. 2009;100:2152–9. doi: 10.1111/j.1349-7006.2009.01291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee A, et al. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology. 1997;112:1386–97. doi: 10.1016/s0016-5085(97)70155-0. [DOI] [PubMed] [Google Scholar]
- 19.Kuzushita N, et al. p27kip1 deficiency confers susceptibility to gastric carcinogenesis in Helicobacter pylori-infected mice. Gastroenterology. 2005;129:1544–56. doi: 10.1053/j.gastro.2005.07.056. [DOI] [PubMed] [Google Scholar]
- 20.Rogers AB, et al. Helicobacter-based mouse models of digestive system carcinogenesis. Methods Mol Biol. 2009;511:267–95. doi: 10.1007/978-1-59745-447-6_11. [DOI] [PubMed] [Google Scholar]
- 21.Lee CW, et al. Helicobacter pylori eradication prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2008;68:3540–8. doi: 10.1158/0008-5472.CAN-07-6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Zanten SJ, et al. Gastric transitional zones, areas where Helicobacter treatment fails: results of a treatment trial using the Sydney strain mouse model. Antimicrob Agents Chemother. 2003;47:2249–55. doi: 10.1128/AAC.47.7.2249-2255.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee CW, et al. Wild-type and interleukin-10-deficient regulatory T cells reduce effector T-cell-mediated gastroduodenitis in Rag2−/− mice, but only wild-type regulatory T cells suppress Helicobacter pylori gastritis. Infect Immun. 2007;75:2699–707. doi: 10.1128/IAI.01788-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mikula M, et al. Quantitative detection for low levels of Helicobacter pylori infection in experimentally infected mice by real-time PCR. J Microbiol Methods. 2003;55:351–59. doi: 10.1016/s0167-7012(03)00166-0. [DOI] [PubMed] [Google Scholar]
- 25.Komatsu K, et al. Pathogenesis of herpetic stromal keratitis in CCR5- and/or CXCR3-deficient mice. Curr Eye Res. 2008;33:736–49. doi: 10.1080/02713680802344716. [DOI] [PubMed] [Google Scholar]
- 26.Garhart CA, et al. Clearance of Helicobacter pylori Infection and Resolution of Postimmunization Gastritis in a Kinetic Study of Prophylactically Immunized Mice. Infect Immun. 2002;70:3529–38. doi: 10.1128/IAI.70.7.3529-3538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai X, et al. Helicobacter felis eradication restores normal architecture and inhibits gastric cancer progression in C57BL/6 mice. Gastroenterology. 2005;128:1937–52. doi: 10.1053/j.gastro.2005.02.066. [DOI] [PubMed] [Google Scholar]
- 28.Wilson KT, et al. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology. 2007;133:288–308. doi: 10.1053/j.gastro.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 29.Yamaoka Y, et al. Chemokines in the gastric mucosa in Helicobacter pylori infection. Gut. 1998;42:609–17. doi: 10.1136/gut.42.5.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eck M, et al. CXC chemokines Gro (alpha)/IL-8 and IP-10/MIG in Helicobacter pylori gastritis. Clin Exp Immunol. 2000;122:192–9. doi: 10.1046/j.1365-2249.2000.01374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romero-Gallo J, et al. Effect of Helicobacter pylori eradication on gastric carcinogenesis. Lab Invest. 2008;88:328–36. doi: 10.1038/labinvest.3700719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Resnick MB, et al. Global analysis of the human gastric epithelial transcriptome altered by Helicobacter pylori eradication in vivo. Gut. 2006;55:1717–24. doi: 10.1136/gut.2006.095646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goll R, et al. Alterations in antral cytokine gene expression in peptic ulcer patients during ulcer healing and after Helicobacter pylori eradication. Scand J Immunol. 2008;67:57–62. doi: 10.1111/j.1365-3083.2007.02037.x. [DOI] [PubMed] [Google Scholar]
- 34.Zhang S, et al. Cytokine-stimulated T lymphocyte proliferation is regulated by p27Kip1. J Immunol. 2000;165:6270–77. doi: 10.4049/jimmunol.165.11.6270. [DOI] [PubMed] [Google Scholar]
- 35.Singh A, et al. Regulation of memory CD8 T-cell differentiation by cyclin-dependent kinase inhibitor p27Kip1. Mol Cell Biol. 2010;30:5145–59. doi: 10.1128/MCB.01045-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jatzek A, et al. p27 (Kip1) negatively regulates the magnitude and persistence of CD4 T cell memory. J Immunol. 2012;189:5119–28. doi: 10.4049/jimmunol.1201482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jatzek A, et al. T-cell intrinsic and extrinsic mechanisms of p27Kip1 in the regulation of CD8 T-cell memory. Immunol Cell Biol. 2013;91:120–9. doi: 10.1038/icb.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamaoka Y, et al. Natural history of gastric mucosal cytokine expression in Helicobacter pylori gastritis in Mongolian gerbils. Infect Immun. 2005;73:2205–12. doi: 10.1128/IAI.73.4.2205-2212.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colvin RA, et al. Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J Biol Chem. 2004;279:30219–27. doi: 10.1074/jbc.M403595200. [DOI] [PubMed] [Google Scholar]
- 40.Groom JR, et al. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89:207–15. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hahm KB, et al. Effect of long-term administration of rebamipide on Helicobacter pylori infection in mice. Aliment Pharmacol Ther. 2003;18(Suppl 1):24–38. doi: 10.1046/j.1365-2036.18.s1.3.x. [DOI] [PubMed] [Google Scholar]
- 42.Kraft M, et al. IFN-gamma synergizes with TNF-alpha but not with viable H. pylori in up-regulating CXC chemokine secretion in gastric epithelial cells. Clin Exp Immunol. 2001;126:474–81. doi: 10.1046/j.1365-2249.2001.01634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rajkumar T, et al. Identification and validation of genes involved in gastric tumorigenesis. Cancer Cell Int. 2010;10:45. doi: 10.1186/1475-2867-10-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee HJ, et al. CXC chemokines and chemokine receptors in gastric cancer: From basic findings towards therapeutic targeting. World J Gastroenterol. 2014;20:1681–1693. doi: 10.3748/wjg.v20.i7.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]