

ABSTRACT
Defects in human lysosomal-trafficking regulator (Lyst) are associated with the lysosomal disorder Chediak–Higashi syndrome. The absence of Lyst results in the formation of enlarged lysosome-related compartments, but the mechanism for how these compartments arise is not well established. Two opposing models have been proposed to explain Lyst function. The fission model describes Lyst as a positive regulator of fission from lysosomal compartments, whereas the fusion model identifies Lyst as a negative regulator of fusion between lysosomal vesicles. Here, we used assays that can distinguish between defects in vesicle fusion versus fission. We compared the phenotype of Dictyostelium discoideum cells defective in LvsB, the ortholog of Lyst, with that of two known fission defect mutants (μ3- and WASH-null mutants). We found that the temporal localization characteristics of the post-lysosomal marker vacuolin, as well as vesicular acidity and the fusion dynamics of LvsB-null cells are distinct from those of both μ3- and WASH-null fission defect mutants. These distinctions are predicted by the fusion defect model and implicate LvsB as a negative regulator of vesicle fusion.
KEY WORDS: Chediak–Higashi syndrome, LvsB, Lyst, Endosome fusion
INTRODUCTION
The endo-lysosomal pathway is comprised of a complex network of traffic from the Golgi, plasma membrane and autophagosomes that converge through intricately regulated fusion and fission events. As early endosomes mature into lysosomes, they utilize fusion and fission events to acidify, sort cargo and acquire the lysosomal enzymes needed for the digestion of intra-luminal material (Ferris et al., 1987; Deng and Storrie, 1988; Mullock et al., 1998; Ward et al., 2000; Maniak, 2003; Mesaki et al., 2011). In professional phagocytes, like macrophages and Dictyostelium discoideum, indigestible material is exocytosed by a third, neutral post-lysosomal compartment (Padh et al., 1993; Sundler, 1997). The tight regulation of fusion and fission events along the endosomal pathway is integral for the correct processing of cargo that transits the endo-lysosomal pathway. Disruption of any of these steps can lead to devastating defects at the cellular and organismal level. This is shown by the severe phenotypes that are associated with lysosomal trafficking disorders, such as Hermansky–Pudlak, Griscelli and Chediak–Higashi syndrome (CHS), among others (Huizing et al., 2001). Although the underlying basis of these disorders is well understood, a consensus on the lysosomal defect contributing to the pathophysiology of CHS has yet to be reached.
CHS is a lethal autosomal recessive disorder that is characterized by the presence of abnormally large lysosome-related compartments in all cell types (Introne et al., 1999). Studies in human (CHS) and mouse (beige) cells have revealed that lesions in the mammalian lysosomal-trafficking regulator (Lyst) gene are responsible for the manifestations of CHS (Barbosa et al., 1996; Nagle et al., 1996). This discovery prompted the use of several model systems to investigate the molecular function of Lyst and its orthologs in order to understand the pathophysiology of CHS. These functional studies produced two distinct models for Lyst function. Initial characterization of beige mice suggested that Lyst functions to limit the rate of homotypic lysosome fusion (Oliver and Essner, 1975). This model for Lyst function was subsequently supported by many studies in humans (Stinchcombe et al., 2000), Dictyostelium (Harris et al., 2002; Kypri et al., 2007), mice (Willingham et al., 1981; Hammel et al., 1987; Hammel et al., 2010), cats (Collier et al., 1985) and Drosophila (Rahman et al., 2012). An alternative model suggested that Lyst functions to control lysosomal fission instead of fusion (Burkhardt et al., 1993). Studies in mice (Perou et al., 1997; Durchfort et al., 2012) and Dictyostelium (Charette and Cosson, 2007; Charette and Cosson, 2008) have attributed beige and LvsB mutant defects, respectively, to decreased lysosomal fission. Despite decades of research across a plethora of model systems, a unifying model for Lyst function has not been established.
Our research has focused on understanding the cellular mechanisms of the Lyst ortholog Large vacuolar sphere B (LvsB), in the simple soil amoebae Dictyostelium discoideum. Disruption of LvsB results in the accumulation of large acidic compartments, much like those observed in cells of CHS patients (Harris et al., 2002; Wang et al., 2002). Characterization of the LvsB-null phenotype has provided definitive evidence of an important role for LvsB during progression of the endo-lysosomal pathway (Kypri et al., 2007). However, similar to the conundrum of deciphering Lyst function in other systems, studies in Dictyostelium have proposed both the fusion and fission models for LvsB function. Studies published by Harris (Harris et al., 2002) and Kypri (Kypri et al., 2007) proposed that LvsB has a regulatory role in vesicle fusion. The fusion model for LvsB function was corroborated by recent evidence of a functional relationship between LvsB and the fusion regulatory GTPase Rab14 (Kypri et al., 2013). In contrast, Charette and Cosson (Charette and Cosson, 2007; Charette and Cosson, 2008) have described LvsB as a positive regulator of lysosomal fission. This discrepancy exists because many of the LvsB-null phenotypes described in these studies can be explained by either the fusion or fission regulatory model, and are therefore subject to interpretation.
The ambiguity of the LvsB-null phenotype is exemplified by its characteristic changes in endosomal membrane protein composition and luminal pH. In Dictyostelium, as a lysosome matures into a post-lysosome, the exocytic protein vacuolin accumulates on the vesicular membrane as the vacuolar (v)ATPase is removed and the lumen begins to de-acidify (Rauchenberger et al., 1997; Jenne et al., 1998; Carnell et al., 2011). Under normal conditions, lysosomes undergo homotypic fusion to exchange their contents, but are prevented from undergoing heterotypic fusion with post-lysosomes to preserve the identity of these distinct stages (Maniak, 2003). Owing to the coordination of lysosome to post-lysosome maturation, vacuolin is rarely observed on vesicular membranes until late stages of endosome maturation in wild-type cells, and the majority of vacuolin-labeled vesicles have a neutral luminal pH (Jenne et al., 1998; Kypri et al., 2007). In contrast, vacuolin frequently labels acidic vesicles that contain early endocytic markers in LvsB-null cells (Kypri et al., 2007). Both of these observations are used to support a model where LvsB inhibits heterotypic fusion between lysosomes and post-lysosomes, and the absence of LvsB allows fusion of neutral vacuolin-labeled post-lysosomes with earlier acidic lysosomes to form hybrid compartments. However, these observations can also be explained using the fission model for LvsB function. In this model, LvsB acts as a positive regulator of fission events that are necessary for the maturation of lysosomes into post-lysosomes. Thus, the absence of LvsB would cause delayed transition of acidic lysosomes to neutral post-lysosomes. This delay could, in theory, cause an accumulation of the post-lysosomal marker vacuolin on acidic late lysosomes that are still competent to undergo homotypic fusion with earlier endosomal compartments. Unfortunately, because fusion and fission events are so tightly interwoven during the progression of the endo-lysosomal pathway, most of the phenotypes described for LvsB-null cells and Lyst mutant cells are subject to inherent ambiguity. Consequently, it has been very difficult to ascribe a mechanism of function to Lyst or LvsB based on the currently published studies.
Without a consensus on the regulatory function of LvsB and Lyst, it is not possible to advance our understanding of how the loss of Lyst leads to the manifestation of CHS. Here, we have used a direct comparison of the LvsB-null phenotype with that of two known fission defect mutants to distinguish between the fusion and fission model for LvsB function. Our results reveal that the LvsB-null phenotype is distinct from that of both fission defect mutants, and supports a role for LvsB as a negative regulator of fusion. Additionally, we show that the phenotypic defects in cells expressing dominant-active Rab14 diverge from those of the fission defect mutants in a manner similar to what we found for LvsB-null cells. This is consistent with the proposed antagonistic relationship of LvsB with Rab14, and further substantiates the regulatory role of LvsB on vesicle fusion.
RESULTS
Fission defect mutants have a similar phenotype to LvsB-null cells
To distinguish between the two models for LvsB function, we wanted to compare specific aspects of the LvsB-null phenotype with two well-characterized fission defect mutants, the μ3-null and WASH-null cell lines (WASH is a WASP and SCAR homolog). The μ3 protein is a subunit of the adaptor protein 3 (AP3) complex in Dictyostelium. AP3 is a clathrin adaptor complex found on endosomal compartments (Bonifacino and Traub, 2003). Absence of Dictyostelium μ3 results in fission-mediated recycling defects during the early stages of endosome maturation (Charette et al., 2006; Charette and Cosson, 2008). The WASH protein is required for the removal of the vATPase from late lysosomes that are transitioning to the post-lysosomal stage. This WASH-dependent step occurs through actin-driven fission of small recycling vesicles (Carnell et al., 2011). Both of these mutant cell lines have a reported delay in the maturation of acidic lysosomes into neutral post-lysosomes as conjectured in the fission model for LvsB function. WASH and AP3 likely function beyond their roles in vesicle fission events. Therefore, we found it imperative to use both of these fission defect mutants in our comparative analyses in order to account for phenotypes associated with other, unique characteristics of each mutant.
To begin our comparative studies, we first determined the phenotype of these fission mutants with the same assays used to characterize the LvsB-null phenotype. As previously described, the characteristics and dynamics of vacuolin-labeled vesicles are perturbed in LvsB-null cells. These aspects of the LvsB-null phenotype can be visualized using GFP–vacuolin, which primarily labels neutral post-lysosomal compartments in wild-type cells, in conjunction with fluid-phase markers or the acidophilic dye Lysotracker Red, which preferentially accumulates in acidic lysosomal vesicles (Wubbolts et al., 1996). Consistent with previous studies, GFP-tagged vacuolin accumulated on dextran-labeled vesicles earlier in LvsB-null cells compared to wild-type cells (Fig. 1A–A″,B–B″,E). LvsB-null cells also contained a large proportion of acidic lysosomal vesicles labeled by GFP–vacuolin (47.8%±1.45 s.e.m.) (Fig. 2B–B″,E) compared to wild-type cells (11.1%±2.33 s.e.m.) (Fig. 2A–A″,E). The fission defect model predicts that vacuolin should accumulate on late acidic lysosomes that are delayed in their transition to the post-lysosomal stage. These vacuolin-labeled lysosomes should still be competent to fuse with earlier endosomes. In agreement with this model, we found that both μ3-null and WASH-null cells contained an increased proportion of GFP–vacuolin-labeled vesicles earlier than wild-type cells (Fig. 1C–C″,D–D″,E). In both cell lines, we also observed an increase in the percentage of acidic lysosomal vesicles labeled by GFP–vacuolin (40.6%±0.3 s.e.m. for the μ3 null; 30.1%±1.45 s.e.m. for the WASH null) (Fig. 2C–C″,D–D″,E) over wild-type cells. These observations show that the phenotype of LvsB-null cells demonstrated with these assays is similar to that of known fission mutants and that although the LvsB-null phenotype could be attributed to a defect in fusion (Kypri et al., 2007), it could also be interpreted as being caused by a defect in fission. Thus, our results emphasize the importance of studies that can distinguish between defects in fission and fusion.
Fig. 1.
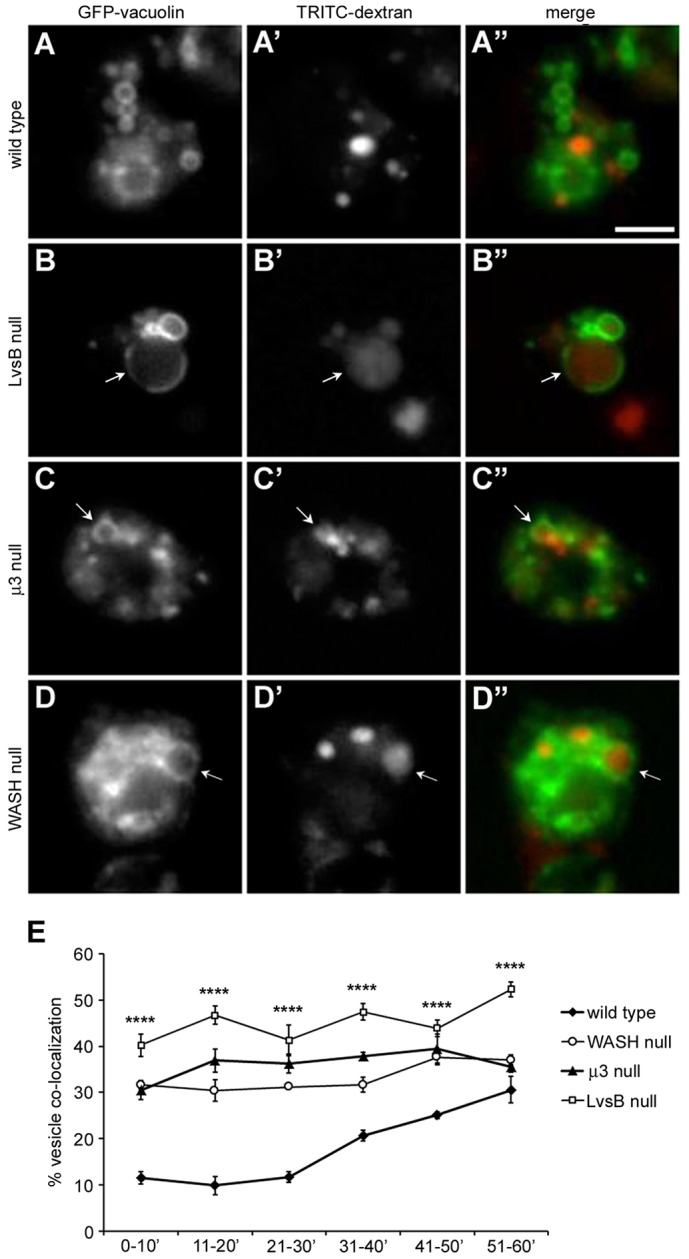
Vacuolin localizes to dextran-labeled vesicles at early time points in both LvsB-null and fission defect mutants. Cells were transfected with GFP–vacuolin and then given a 5-min pulse of TRITC–dextran to label a subpopulation of pinocytic vesicles. Cells were then washed and imaged continuously for 60 min. (A–D) Representative images selected from the 11–20-min time point are shown. (E) The percentage of GFP–vacuolin-labeled vesicles (>60 vesicles per experiment) containing TRITC–dextran was quantified for each 10-min interval and plotted as the mean±s.e.m. (n = 3). One-way ANOVA was used to calculate significant differences in the percentage of TRITC–dextran-labeled vesicles between cell lines for all time intervals shown. ****P<0.0001. During the first 30 min of imaging, the majority of wild-type cells maintained separation of their dextran and GFP–vacuolin-labeled vesicle populations (A–A″). In contrast, GFP–vacuolin was found to significantly overlap with TRITC–dextran at earlier time intervals in the LvsB-null (B–B″), μ3-null (C–C″) and WASH-null (D–D″) cell lines. Arrows indicate vesicles labeled by both TRITC–dextran and GFP–vacuolin. Scale bar: 5 µm.
Fig. 2.

LvsB-null and fission defect mutants have an increased occurrence of acidic vacuolin-labeled vesicles but the characteristics of acidic vesicles in LvsB-null cells are distinct from those in fission defect mutants. (A–D) GFP–vacuolin-expressing cells were incubated with Lysotracker Red then imaged live to visualize acidic compartments with and without GFP–vacuolin membrane localization. (E) Using similar live-cell images, we quantified the percentage of GFP–vacuolin vesicles with Lysotracker Red fluorescence and plotted the mean±range for two experiments. As previously described, there was an increased occurrence of acidic GFP–vacuolin vesicles in LvsB-null cells (B–B″) compared to wild-type cells (A–A″). The fission defects in μ3-null cells (C–C″) and WASH-null cells (D–D″) also resulted in a higher percentage of acidic GFP–vacuolin vesicles similar to that observed in the LvsB-null cell line. (F) The fluorescence intensity of Lysotracker Red was measured in GFP–vacuolin-positive (late lysosomal or hybrid lysosomal organelles) and in GFP–vacuolin-negative (normal lysosomal) populations and used as an indicator of relative vesicle acidity. The fluorescence intensity of Lysotracker Red for each population was normalized within each cell line to the average fluorescence of GFP–vacuolin-negative lysosomes and plotted as the mean±s.e.m. ns, not significant, *P<0.05, **P<0.01, ****P<0.0001 (two-tailed Student's t-test). Notice that the relative acidity of vacuolin-labeled compared with unlabeled vesicles in LvsB-null cells is very different from that seen in wild-type, μ3-null, and WASH-null cells. The higher acidity of vacuolin-labeled vesicles in μ3-null and WASH-null cells is consistent with their role in vesicle fission during lysosomal maturation. In contrast, the fluorescence of GFP–vacuolin-positive vesicles is significantly reduced when compared to GFP–vacuolin-negative vesicles in the LvsB-null cells. This is consistent with a role for LvsB in reducing fusion between acidic lysosomal and neutral post-lysosomal compartments. Arrows indicate vesicles labeled by both Lysotracker Red and GFP–vacuolin. Scale bar: 5 µm.
Acidity characteristics of mutant compartments suggest that LvsB has a fusion regulatory role
Although an increase in the number of acidic vesicles labeled by vacuolin can be explained by defects in either fusion or fission, the different mechanisms for how these vesicles arise should produce predictable differences in their characteristics. One such difference is the relative luminal acidity of vacuolin-positive acidic vesicles (hereafter referred to as hybrid lysosomal vesicles) when compared to the acidity of vacuolin-negative acidic vesicles (hereafter referred to as the normal lysosomal population). In the fission defect model, we propose that vacuolin accumulates on very late acidic lysosomes due to a delay in their maturation to post-lysosomes. This should result in hybrid lysosomal vesicles that are both labeled by vacuolin and have a luminal acidity that is higher than that of the normal lysosomal population because they still retain their proton pumps. In contrast, the hybrid lysosomal vesicles described in the fusion defect model arise from inappropriate fusion between acidic lysosomes and neutral, vacuolin-positive, post-lysosomes. This fusion should produce hybrid lysosomal vesicles that are labeled by vacuolin, but have lower luminal acidity compared to the normal lysosomal population. To obtain a comparative measure of hybrid vesicle acidity within each cell line, we incubated GFP–vacuolin-expressing cells with Lysotracker Red and then quantified the average red fluorescence signal of acidic vesicles with (hybrid lysosomal vesicles) or without (normal lysosomal population) GFP–vacuolin. The fluorescence of Lysotracker Red is not directly sensitive to pH changes, but its retention and concentration is known to correlate predictably with changes in vesicle acidity. Chen (Chen, 2002) has reported that the fluorescence intensity of Lysotracker-Red-labeled vesicles is significantly reduced in response to an ionophore-induced decrease in lysosomal pH. Given this correlation, we used Lysotracker Red fluorescence as an indicator of gross differences in acidity among populations of vesicles. In wild-type cells, the acidity of hybrid lysosomal vesicles was not significantly higher than the normal lysosomal population (Fig. 2A–A″,F). This result agrees with the described transition of lysosomes to post-lysosomes, where vacuolin arrives on the late lysosomal membrane just before the vATPase is removed (Carnell et al., 2011). Such a temporal overlap predicts that Lysotracker-Red-labeled vesicles that are labeled by GFP–vacuolin are late acidic lysosomes. Importantly, the results observed in μ3-null and WASH-null cells were consistent with their known defect in fission. The acidity of hybrid lysosomal vesicles in both μ3-null and WASH-null cells was significantly higher than their respective normal lysosomal populations (Fig. 2C–C″,D–D″,F). These results support our model of how these hybrid lysosomal vesicles form in fission defect mutants, but more importantly, they provide a baseline for comparison with the LvsB-null phenotype. In contrast to both wild-type and the fission defect mutants, the hybrid lysosomal vesicles in LvsB-null cells had a significantly lower acidity compared to their normal lysosomal population (Fig. 2B–B″,F). This decrease in fluorescence is not a function of increased vesicle size in these mutants. We compared Lysotracker Red fluorescence of hybrid lysosomal vesicles with similar diameter from μ3-null and LvsB-null cells and found that the LvsB-null hybrid lysosomes had a significantly lower average fluorescence (supplementary material Fig. S1). This result not only sets the LvsB-null phenotype apart from that of the fission defect mutants, but correlates with the predicted characteristics of hybrid lysosomal vesicles formed by inappropriate heterotypic fusion with post-lysosomes.
Formation of hybrid lysosomal compartments is different in LvsB-null cells than in fission defect mutants
To further compare the phenotypes of the LvsB-null and fission defect mutants, we modified a previously described pulse-chase assay with two differently labeled dextrans (Kypri et al., 2007). In this assay, cells were given a pulse of FITC–dextran followed by a 30-min chase and then a second pulse with TRITC–dextran. In wild-type cells, these maturation times correspond to FITC-labeled post-lysosomes and TRITC-labeled lysosomes (Padh et al., 1993) with very minimal colocalization of the two signals (5.6%±0.35 s.e.m.) (Fig. 3A–A″,F). Similar to previous results, LvsB-null cells had a significant increase in the number of vesicles containing both FITC– and TRITC–dextran (31.4%±0.99 s.e.m.) (Fig. 3B–B″,F). This phenotype can, again, be explained by a defect in either fusion or fission. In the fission model, delayed maturation of lysosomes into post-lysosomes would result in the delayed progression of the first pulse of FITC–dextran to post-lysosomes. Under these conditions, colocalization of the two pulses of dextran is indicative of the normal homotypic fusion events between lysosomes. To validate this interpretation, we showed that the maturation defects in both μ3-null and WASH-null cells resulted in an increased colocalization of the two dextran pulses (16.9%±0.12 s.e.m. for μ3 null and 21.3%±0.88 s.e.m. for WASH null) (Fig. 3C–C″,D–D″,F). Alternatively, in the fusion defect model, the absence of LvsB would result in the inappropriate heterotypic fusion of FITC–dextran-labeled post-lysosomes with earlier TRITC–dextran-labeled lysosomes.
Fig. 3.
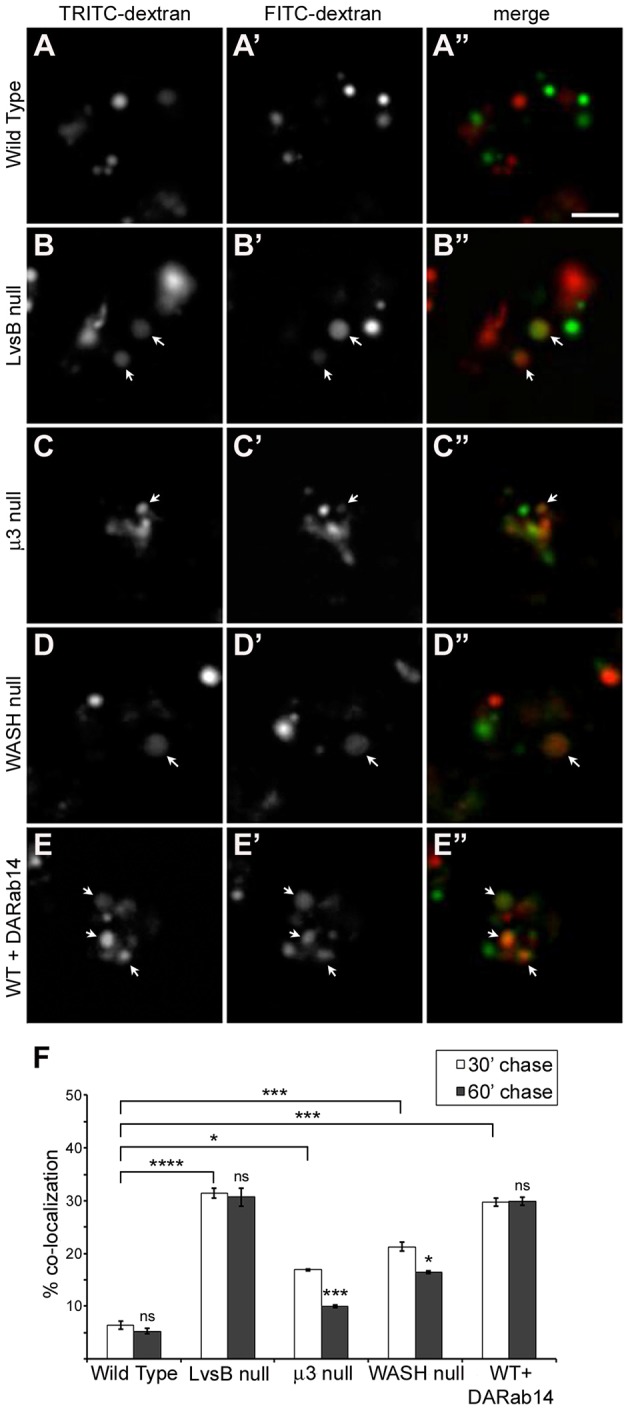
Longer maturation time causes reduced fusion of early and late endosome populations in fission defect mutants but not in the LvsB-null mutant. Cells were given a pulse of FITC–dextran followed by a 30-min (A–E) or 60-min chase then given a second pulse with TRITC–dextran. Colocalization of the two dextran signals was used as an indicator of fusion events between the two dextran-labeled populations. (F) The fraction of vesicles containing both fluid-phase markers was quantified for three independent experiments and shown as the mean±s.e.m. A one-way ANOVA indicated significant differences in heterotypic fusion between cell lines when a 30-min chase was applied (P<0.0001). ns, not significant, *P<0.05, ****P<0.0001 (two-tailed Student's t-test with Bonferroni correction). We observed significantly more fusion between dextran populations separated by a 30-min chase in the LvsB-null cells (B–B″) compared to wild-type cells (A–A″). Less dramatic increases in fusion were also evident in both the μ3-null (C–C″) and WASH-null (D–D″) fission defect mutants. This increased fusion was partially relieved in both the μ3 and WASH-null cell line with a 60-min chase. This reduction is indicative of a delay in the maturation of late lysosomes into post-lysosomes. In opposition to the fission defect model, LvsB-null cells experienced no reduction in fusion levels with a 60-min chase. This pattern of maturation-independent increased fusion is mimicked in DA-Rab14-expressing cells (E–E″). Arrows indicate vesicles labeled by both TRITC– and FITC–dextran. Scale bar: 5 µm.
To determine whether LvsB is a regulator of fusion or fission, we modified the dextran colocalization experiment to compare the amount of colocalization of the two dextran pulses using a 30-min or 60-min chase time. Each model predicts a different outcome when using longer chase times. If a defect in fission causes delayed delivery of the first pulse of FITC–dextran to post-lysosomes, then a longer chase time should allow more time for the FITC–dextran-labeled vesicles to mature. Prolonged maturation should result in a greater number of FITC–dextran vesicles reaching the post-lysosomal stage. These FITC-labeled post-lysosomes would no longer be competent to fuse with earlier TRITC–dextran vesicles leading to a decrease in colocalization between the two dextran-labeled vesicle populations. Consistent with this hypothesis, both the μ3-null and WASH-null mutants exhibited a significant decrease in colocalization of the two dextran pulses with a 60-min chase compared to a 30-min chase (decreased to 10%±0.29 s.e.m. for μ3 null and 16.5%±0.29 s.e.m. for WASH null) (Fig. 3F). Interestingly, the WASH mutant cells had a less significant decrease in dextran colocalization with a longer chase time than the μ3 mutant cells. This result is congruent with the severe delay in lysosomal maturation reported for WASH mutant cells compared to the relatively moderate delay reported for μ3 mutant cells (Charette and Cosson, 2008; Carnell et al., 2011). The fission defect model predicts that the LvsB-null cells should also have a decrease in dextran colocalization with increased chase times. Alternatively, the fusion model predicts that the 30-min chase time is sufficient to allow the first pulse of FITC–dextran to reach the post-lysosomes, and colocalization is the result of inappropriate fusion between early and late compartments. In this model, applying a longer chase time should not affect the stages of the endo-lysosomal pathway that are labeled by each dextran. Therefore, there should be no change in the amount of colocalization of the two markers. As predicted by the fusion defect model, in the LvsB-null cell line we observed no significant change in colocalization of the two dextran pulses with a 60-min chase (30.7%±1.68 s.e.m.) when compared to a 30-min chase (Fig. 3F). These results show another important difference in the LvsB-null phenotype from known fission defect mutants and imply that LvsB is a regulator of fusion rather than fission.
Formation of hybrid lysosomal compartments in LvsB-null cells is similar to that in cells expressing dominant-active Rab14
The small GTPase Rab14 acts as a promoter of homotypic fusion between lysosomes (Harris and Cardelli, 2002). We previously reported that LvsB has an antagonistic functional relationship with Rab14 based, in part, on phenotypic similarities shared by LvsB-null cells and cells expressing dominant-active (DA)-Rab14 (Kypri et al., 2013). Among these similarities, the expression of DA-Rab14 has been implicated in causing increased heterotypic fusion of lysosomes with post-lysosomes. However, we have shown here that many of the assays previously used to diagnose increased heterotypic fusion cannot reliably rule out defects in fission. Because of the known role of Rab14 in promoting homotypic fusion as well as strong evidence supporting a relationship between LvsB and Rab14, we were interested in comparing the vesicle fusion characteristics of the DA-Rab14 phenotype with the LvsB-null and fission defect mutants. To this end, we subjected DA-Rab14-expressing cells to the modified experiments with two dextran pulses described above. Consistent with previous studies, expression of DA-Rab14 caused a significant increase in the fusion of FITC-labeled vesicles with TRITC-labeled vesicles using a 30-min chase (29.8%±0.76 s.e.m.) (Fig. 3F) when compared to wild-type (5.6%±0.35 s.e.m.). This increased level of colocalization in DA-Rab14-expressing cells was not significantly reduced when a 60-min chase was applied (29.9%±0.76 s.e.m.) (Fig. 3E–E″,F). This observation is in agreement with the described role of Rab14 as a promoter of fusion and, by comparison with the trend observed in LvsB-null cells, reinforces the fusion regulatory model for LvsB function.
Endosome size is more severely perturbed in LvsB-null cells than in WASH-null cells
The fission model for Lyst and LvsB function predicts that the delayed maturation of lysosomes should cause an increase in vesicle size at the stage where maturation is delayed (Durchfort et al., 2012). Delaying the maturation of lysosomes is predicted to allow the continued flow of endosomes that are competent to undergo homotypic fusion. This should result in the ‘dumping’ of earlier endosomal contents into the delayed compartments and a consequential increase in their volume. Alternatively, the fusion model for LvsB function postulates that promiscuous fusion of post-lysosomes with earlier compartments in LvsB-null cells causes an increase in endosome size that is evident at the early endosomal stage and persists through the post-lysosomal stage. To test this theory, cells were given a 5-min pulse of TRITC–dextran and then chased for 15, 35 or 60 min. In wild-type cells, these chase times have been shown to correspond to lysosomes, the point of transition from lysosomes to post-lysosomes and post-lysosomes, respectively (Padh et al., 1993; Temesvari et al., 1996). The WASH-null mutant was selected for comparison because of its reported severe delay in maturation of lysosomes at the same stage that LvsB is suggested to function (Charette and Cosson, 2007; Carnell et al., 2011). To control for variance in vesicle size between different wild-type parental strains, we compared LvsB-null cells against NC4A2 wild-type cells, and WASH-null cells against AX2 wild-type cells. Surprisingly, the dextran endosomes in WASH-null cells showed no significant change in diameter when compared to wild-type cells after 15-, 35- or 60-min chase times (Fig. 4A–A″,C–C″,D). In contrast to the WASH-null fission mutant, the LvsB-null defect produced a significant increase in the average diameter of dextran-labeled endosomes at both the 15- and 35-min time points when compared to wild-type cells (Fig. 4A–A″,B–B″,D). At the 60-min time point, dextran labels the larger post-lysosomal vesicles in wild-type cells and reduces the difference in size that we observed between the labeled vesicles in wild-type and LvsB-null cells. The increase in endosome size at multiple time points observed in the LvsB-null cell line does not fit the fission model for how these morphological changes arise. However, promiscuous fusion of post-lysosomes with earlier vesicular compartments can explain an early increase in size that persists along the entire endo-lysosomal pathway. Additionally, the WASH-null defect did not induce a significant increase in vesicle size at any of the characterized time points despite the much longer delay in maturation that is reported for WASH-null cells as compared to LvsB-null cells (Charette and Cosson, 2008; Carnell et al., 2011). This, again, opposes the fission model for how enlarged endosomes arise in LvsB-null cells.
Fig. 4.

Increased vesicle size is evident earlier and is more severe in LvsB-null cells compared to WASH-null cells. (A–C) Endosomes were pulse-labeled with TRITC–dextran then imaged after 15 min (temporally consistent with the lysosomal stage), 35 min (temporally consistent with the lysosome to post-lysosome transition) and 60 min (temporally consistent with the post-lysosomal stage) of chase. (D) Dextran vesicle diameter for >80 vesicles/experiment was quantified from images similar to A–C and plotted as the mean±s.e.m. of three experiments. The LvsB-null cell line was compared with its parental NC4A2 wild-type cell line and the WASH-null line was compared with its parental AX2 wild-type cell line. One-way ANOVA indicated significant differences in vesicle diameter between cell lines at both 15 (P<0.001) and 35 min (P<0.0001) post labeling, but not at 60 min (P>0.01). ns, not significant, *P<0.05, **P<0.01 (two-tailed Student's t-test with Bonferroni correction). Surprisingly, despite a reported severe delay in lysosomal maturation, the WASH-null vesicles (C–C″) did not demonstrate a significant increase in diameter over those in wild-type cells (A–A″) at any of the three time points quantified. A significant increase in vesicle diameter is evident at the 15- and 35-min time points in LvsB-null cells (B–B″) compared to wild-type (A–A″). Both the magnitude and temporal pattern of increased endosome size in LvsB-null cells differs from the WASH-null fission defect mutant. These differences are consistent with the fusion model for LvsB function. Scale bar: 5 µm.
Defects in fission do not recapitulate the phagosome defect of LvsB-null cells
In addition to increased fusion between lysosomes and post-lysosomes, LvsB-null cells and wild-type cells expressing DA-Rab14 are characterized by the increased occurrence of multi-particulate phagosomes (Harris and Cardelli, 2002; Harris et al., 2002). It has been suggested that Rab14 promotes the homotypic fusion of phagosomes as well as the fusion of phagosomes with lysosomes for the digestion of phagocytosed material (Harris and Cardelli, 2002). As an antagonist of Rab14 function, it is not surprising that loss of LvsB leads to an increase in phagosome fusion (Harris et al., 2002). However, as previously discussed for lysosomes, the formation of multi-particulate phagosomes could be induced by a fission defect. There is a substantial amount of interplay between the phagocytic and pinocytic pathways in Dictyostelium and many proteins that are important for regulating the endo-lysosomal pathway are shared with maturing phagosomes. These proteins include Rab GTPases, such as Rab14 and Rab7, lysosomal hydrolytic enzymes, and the vATPase proton pump (Rupper and Cardelli, 2001; Gotthardt et al., 2002; Harris and Cardelli, 2002). Many of these shared proteins are delivered to phagosomes through fusion and intermingling of endo-lysosomal compartments with nascent and maturing phagosomes (Rupper and Cardelli, 2001; Clarke et al., 2010). Under these conditions, the fission defect model described above would predict that delayed progression of lysosomes to the post-lysosomal stage could also block progression of phagosomal compartments. A block of this nature has the potential to increase homotypic fusion of phagosomes to form multi-particulate phagosomes. To investigate this possibility, we used a phagosome fusion experiment in which cells were incubated with blue fluorescent beads for a short time, followed by a second pulse of red fluorescent beads. After a 30-min incubation time to allow the phagosomes to mature through fusion with endo-lysosomal compartments, the cells were fixed and immunostained for the p80 protein, which labels phagosomal, endo-lysosomal and plasma membranes (Ravanel et al., 2001). Immunostaining of p80-labeled phagosomal membranes allowed us to distinguish between phagosomes containing multiple beads, and clusters of closely adjacent phagosomes. Only phagosomes containing both blue and red beads were counted as multi-particulate to ensure that they arose through fusion events rather than by uptake of multiple beads in a single phagocytic cup. In agreement with previous studies (Harris and Cardelli, 2002; Harris et al., 2002), both the LvsB-null cell line and wild-type cells expressing DA-Rab14 displayed a significantly larger population of cells containing multi-particulate phagosomes (47.4%±1.65 s.e.m. for LvsB-null and 37.7%±1.84 s.e.m. for DARab14-expressing cells) (Fig. 5B–B‴,E–E‴,F) when compared to wild-type cells (19.2%±2.34 s.e.m.) (Fig. 5A–A‴,F). In contrast, we observed no significant difference in the occurrence of cells containing multi-particulate phagosomes in either the μ3- or WASH-null mutant lines compared to wild-type cells (22.5%±2.79 s.e.m. for μ3-null and 27.3%±0.78 s.e.m. for WASH-null cells) (Fig. 5C–C‴,D–D‴,F). This disparity between the LvsB-null cell line and the fission defect mutant lines coupled with the striking similarities that the LvsB mutant shares with cells expressing DA-Rab14 further validates the role of LvsB as an antagonist of Rab14-mediated fusion events. Furthermore, the absence of a phenotypic change in either of the fission mutants suggests that the LvsB-null phagosome phenotype cannot be directly attributed to a fission defect along the endo-lysosomal pathway.
Fig. 5.
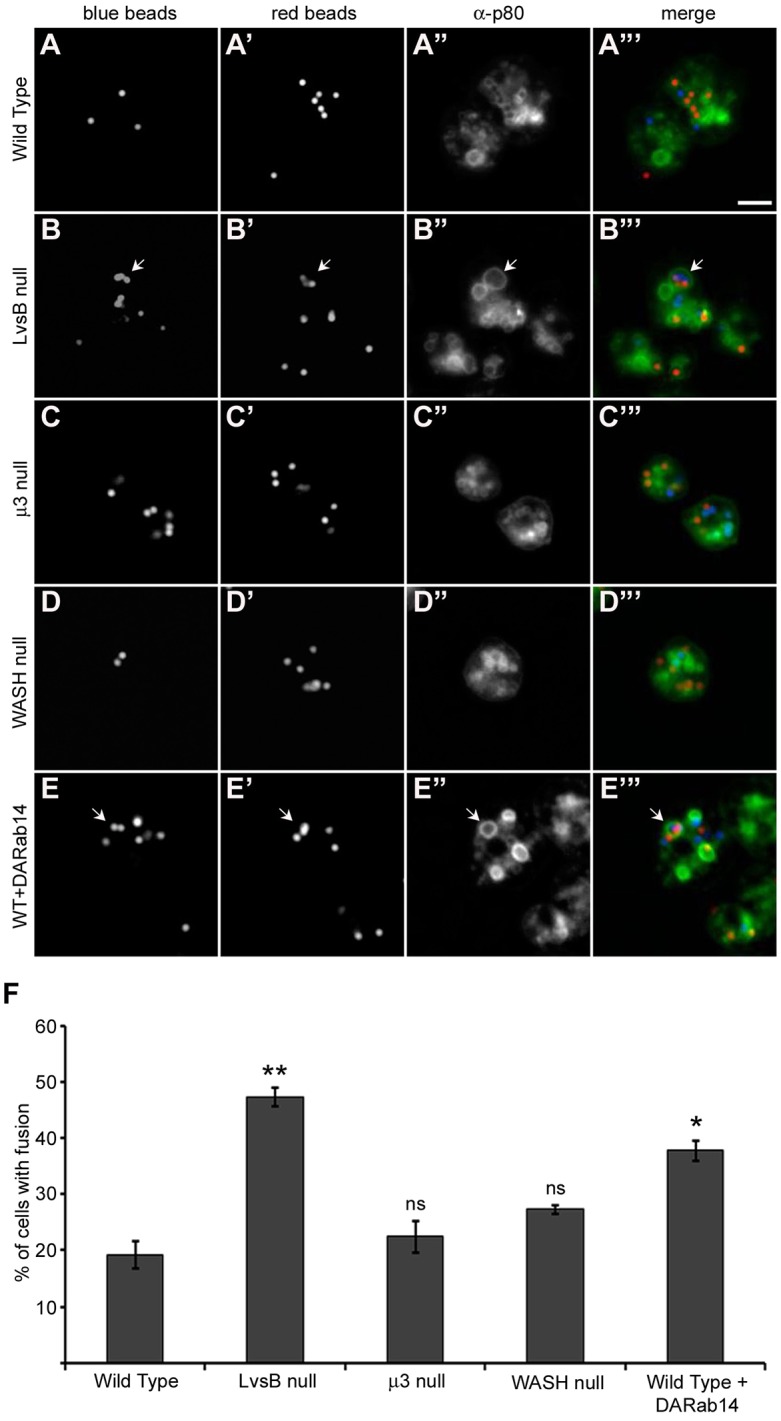
The phagosome defect of LvsB-null and DA-Rab14-expressing cells is not evident in the fission defect mutants. (A–E) Cells were given a pulse with blue fluorescent beads followed by a second pulse with red fluorescent beads to label adjacent populations of phagosomes. After a 30-min incubation to allow the labeled phagosomes to mature, cells were fixed, flattened and immunostained with anti-p80 antibody to label the limiting membranes of phagosomes. (F) The fraction of cells (>33 cells/experiment) with multi-particulate phagosomes containing both blue and red beads was plotted as the mean±s.e.m. of three experiments. A one-way ANOVA indicated significant differences in phagosome fusion between cell lines (P<0.0001). ns, not significant, *P<0.05, **P<0.01 (two-tailed Student's t-test with Bonferroni correction). The increased multi-particulate phagosome formation observed in both LvsB-null (B–B‴) and DA-Rab14-expressing (E–E‴) cells over wild-type cells (A–A‴) is consistent with previous studies and supports a fusion regulatory role for both proteins. The μ3-null (C–C‴) and WASH-null (D-D‴) fission defects did not induce significant increases in multi-particulate phagosome formation. This result suggests that a fission defect alone is not sufficient to cause the formation of multi-particulate phagosomes. Arrows indicate multi-particulate phagosomes. Scale bar: 5 µm.
Inappropriate heterotypic fusion is the major contributor to enlarged vesicle size in LvsB-null cells
The results of these comparative studies, as well as our previously published data, have centered on defects in heterotypic fusion between lysosomes and post-lysosomes. However, many studies have attributed the enlarged vesicular phenotype of Lyst or LvsB mutant cells to increased homotypic fusion between lysosome related organelles (Oliver and Essner, 1975; Willingham et al., 1981; Hammel et al., 1987; Stinchcombe et al., 2000; Harris et al., 2002; Hammel et al., 2010). This possibility is bolstered by the reported antagonistic relationship between LvsB and Rab14 in Dictyostelium (Kypri et al., 2013). Given that Rab14 functions to promote homotypic fusion amongst lysosomes, the absence of LvsB could result in unrestrained lysosome–lysosome fusion, producing enlarged lysosomal compartments. Previously published data showed that when the entire endo-lysosomal pathway is labeled with dextran, LvsB-null cells display a marked increase in average vesicle diameter compared to wild-type cells (Harris et al., 2002). Increased diameter of vesicles labeled by the post-lysosomal marker vacuolin in LvsB-null cells, along with the data reported in this study, implicate deleterious heterotypic fusion as responsible for the enlarged vesicle phenotype (Kypri et al., 2007). These studies cannot, however, exclude increased homotypic lysosome fusion as an additional factor promoting the generation of enlarged vesicles. To determine whether increased homotypic fusion of compartments preceding post-lysosomes also contributes to the LvsB-null phenotype, we compared the morphology of vesicles void of the post-lysosomal marker vacuolin with that of vacuolin-labeled vesicles in LvsB-null and wild-type cells. Vesicles that are labeled by vacuolin should encompass both the post-lysosomal and hybrid lysosomal vesicle population, whereas the population of vesicles not labeled by vacuolin will include the subset of lysosomes that have not undergone heterotypic fusion with post-lysosomes. For this, we used dextran to label the entire endo-lysosomal pathway in cells expressing GFP–vacuolin (Fig. 6A). The average diameter of dextran vesicles without GFP–vacuolin in the LvsB-null cell line was not significantly different than that of wild-type cells (1.04 µm±0.14 s.e.m. for LvsB-null and 0.62 µm±0.04 s.e.m. for wild-type; P>0.05) (Fig. 6B). This result translates to the conclusion that lysosomes that have not undergone mixing with post-lysosomes are not significantly enlarged in LvsB-null cells. In contrast, the average diameter of GFP–vacuolin-labeled dextran vesicles was significantly larger in LvsB-null cells compared to wild-type cells (1.93 µm±0.07 s.e.m. for LvsB-null and 0.87 µm±0.09 s.e.m. for wild-type cells; P<0.001) (Fig. 6B). This drastic increase in GFP–vacuolin-labeled vesicle size alongside a statistically insignificant difference in the size of vesicles that lack the post-lysosomal marker vacuolin in LvsB-null cells indicates that homotypic fusion of compartments upstream of post-lysosomes contributes minimally, if at all, to the enlarged vesicle phenotype in LvsB-null cells.
Fig. 6.

The morphology of GFP–vacuolin-labeled vesicles is most affected in LvsB-null cells. (A) Diagram of the endosomal pathway in a GFP–vacuolin-expressing wild-type and LvsB-null cell where the entire endosomal pathway is labeled. Live-cell images were used to measure the diameter of GFP–vacuolin negative vesicles (all vesicles including lysosomes that have not mixed with vacuolin-labeled post-lysosomes) and GFP–vacuolin-positive vesicles (hybrid lysosomal vesicles and post-lysosomes). (B) Results were plotted as the mean±s.e.m. for three experiments (>30 vesicles for each population/experiment). The average diameter of vesicles not labeled by GFP–vacuolin was not significantly elevated in the LvsB-null cell line compared to wild-type, whereas the hybrid lysosomal vesicle and post-lysosomal population demonstrated a twofold increase in average diameter. (C) Diagram of the endosomal pathway in a GFP–vacuolin-expressing wild-type and LvsB-null cell where acidic vesicles are labeled with Lysotracker Red. Live-cell images from the datasets described in Fig. 2 were used to measure the diameter of acidic GFP–vacuolin vesicles (hybrid lysosomal vesicles) and neutral GFP–vacuolin vesicles (post-lysosomes) in wild-type and LvsB-null cells. (D) Results were plotted as the mean±s.e.m. for 13 acidic vacuolin vesicles in wild-type cells and >35 vesicles for all other populations indicated. A one way ANOVA indicated significant differences in average diameter between populations (P<0.0001). The average diameter of both neutral and acidic vacuolin vesicles was significantly increased in LvsB-null cells. However, the hybrid lysosomes are most perturbed in LvsB-null cells. ns, not significant, *P<0.05, **P<0.01, *** P<0.001, ****P<0.0001 (two-tailed Student's t-test with Bonferroni correction).
One of the prominent phenotypic characteristics of the LvsB-null cell line reported here and in our previous work is the increased size of vesicles that are labeled by vacuolin (Kypri et al., 2007). In another study, Charette and Cosson (Charette and Cosson, 2007) reported that the size of neutral post-lysosomes (defined as vesicles containing p80 but lacking vATPase) was not significantly different in LvsB-null cells as compared to wild-type cells. Instead, an increase in the size of lysosomal vesicles (identified by the presence of both p80 and vATPase) was noted in LvsB-null cells. This population of vesicles labeled by both p80 and vATPase should encompass lysosomes as well as the ‘hybrid vacuolin-labeled lysosomes’ that we describe here. Taken together, these data predict that acidic vacuolin vesicles (hybrid lysosomes) primarily contribute to the increased size of the vacuolin vesicle population, whereas neutral vacuolin vesicles (presumably corresponding to p80-positive and vATPase-negative vesicles) should be similar in size to those in wild-type cells because they have not undergone heterotypic fusion with lysosomes. To test this hypothesis, we compared the average diameter of acidic GFP–vacuolin vesicles that were labeled by Lysotracker Red (hybrid lysosomes) and neutral GFP–vacuolin vesicles (post-lysosomes) in both LvsB-null cells and wild-type cells (Fig. 6C). Analysis of variance indicated a significant difference in the average diameter of the neutral and acidic vacuolin-labeled vesicle populations of wild-type and LvsB-null cells (P<0.0001). In wild-type cells, the small population of vacuolin-labeled acidic vesicles that was present did not have a significant difference in average diameter compared to neutral vacuolin-labeled vesicles (1.26 µm±0.045 s.e.m. for neutral vacuolin-labeled vesicles and 1.46 µm±0.186 s.e.m. for acidic vacuolin-labeled vesicles; P>0.05) (Fig. 6D). Surprisingly, the average diameter of both the neutral and acidic vacuolin-labeled vesicle populations in LvsB-null cells (1.93 µm±0.152 s.e.m. for neutral vacuolin-labeled vesicles and 3.12 µm±0.183 s.e.m. for acidic vacuolin-labeled vesicles) were significantly larger than respective populations in wild-type cells (P<0.001 for neutral vacuolin-labeled vesicles and P<0.00001 for acidic vacuolin-labeled vesicles) (Fig. 6D). In contrast to wild-type cells, the average diameter of the acidic vacuolin vesicle population was significantly larger than that of the neutral vacuolin vesicle population in LvsB-null cells (P<0.0001) (Fig. 6D). Importantly, the reduced size of neutral vacuolin vesicles (post-lysosomes) as compared to acidic vacuolin vesicles (hybrid lysosomes) in LvsB-null cells is consistent with their lack of participation in heterotypic fusion with earlier acidic compartments. These results suggest that heterotypic fusion between lysosomes and post-lysosomes is the major contributor to the enlarged vesicle phenotype of LvsB-null cells.
DISCUSSION
The complex intermingling of regulated fusion and fission events required for the maturation of vesicles along the endo-lysosomal pathway has hampered our understanding of the regulatory role of mammalian Lyst and its Dictyostelium ortholog LvsB. Studies in several model systems have led to two differing models for Lyst/LvsB function. Both the fusion and fission models are well supported in the literature. We attribute this discrepancy in interpretation of Lyst function to the inability of the methods used in most of the currently published studies to decipher between abnormalities caused by defects in fusion or fission. In this study, we used two well-characterized fission defect mutants to show that defects in fission can produce similar phenotypic characteristics to those that had been previously attributed to increased heterotypic fusion in LvsB-null cells. These results attest to the importance of designing studies that can reliably distinguish between defects in fusion and fission when characterizing regulators of endo-lysosomal trafficking. To this end, we also demonstrated four properties of LvsB-null cells that distinguish them from fission defect mutants and are consistent with a fusion regulatory model for LvsB function. First, the hybrid lysosomal compartments found in the LvsB-null cell line are significantly less acidic than the normal lysosomal population suggesting that they arise from fusion of acidic and neutral compartments and not from delayed maturation of lysosomes to the post-lysosomal stage. This conclusion is supported by the reported study of lysosome de-acidification dynamics in LvsB mutant cells (Kypri et al., 2007). Second, the amount of fusion between pulse-labeled vesicles decreases with increased maturation time in the fission defect mutants but not in LvsB-null cells. This observation is indicative of increased heterotypic fusion between lysosomes and post-lysosomes in the absence of LvsB and is mimicked by cells overexpressing the fusion promoting DA-Rab14 construct. Third, the WASH-null fission defect does not produce the same pattern or severity of increased endosome size that we observed in LvsB-null cells. LvsB-null cells contain aberrantly large vesicles at early and late times after endocytosis, which is consistent with uncontrolled fusion between early and late compartments. Finally, we showed that the fission defect mutants do not have the multi-particulate phagosome phenotype that characterizes both LvsB mutant and DA-Rab14-expressing cells. These phenotypic distinctions suggest that the primary cause of abnormal endolysosomal vesicles in LvsB mutant cells is inappropriate heterotypic fusion of post-lysosomes with earlier acidic vesicles. This study cannot, however, determine whether this aberrant fusion is a primary or secondary effect of the loss of LvsB function.
It is entirely possible that the function of LvsB/Lyst is far more complex than what is described by either the fusion or fission model. This possibility is suggested by the results of a two-hybrid screen that was used to identify Lyst-binding partners (Tchernev et al., 2002). Many of the proposed Lyst-interacting proteins identified in that study, such as HRS, CALM and CK2β, are implicated in the regulation of vesicle fusion (Allende and Allende, 1995; Peters and Mayer, 1998; Schekman, 1998; Tsujimoto and Bean, 2000). This ‘regulator of fusion regulators’ model for Lyst function is further supported by our recently published results showing an antagonistic relationship between LvsB and the fusion-promoting GTPase Rab14 in Dictyostelium (Kypri et al., 2013). We have shown here that the kinetics of increased fusion are also common to both the DA-Rab14 and LvsB mutant phenotypes. This characteristic is predictably distinct from the fission defect mutants, and correlates with opposing fusion regulatory roles for LvsB and Rab14. We have also shown that homotypic lysosome fusion contributes very minimally to the LvsB-null enlarged vesicle phenotype and that hybrid vacuolin-labeled lysosomal compartments are primarily affected in the absence of LvsB. This suggests that LvsB most likely antagonizes Rab14 function only at the late lysosomal or early post-lysosomal stage and not when Rab14 is needed to promote homotypic lysosome fusion. Based on these studies it is intriguing to consider LvsB and Lyst as a hub for regulating fusion events at the transition of lysosomes to the post-lysosomal stage. The localization of LvsB to late lysosomes and post-lysosomes in Dictyostelium puts it in the right place at the right time to mediate fusion regulation during this important transition (Kypri et al., 2007).
Adding to the possible complexity of Lyst function are the findings of Durchfort (Durchfort et al., 2012). Although our comparative analyses have shown strong evidence that the LvsB mutant phenotype arises from inappropriate fusion, they presented strong evidence that the mouse Lyst homolog promotes fission from drug-induced enlarged endosomal compartments. In reconciling these findings with the fusion model, we postulate that Lyst might also regulate fusion through fission-mediated recycling of fusion machinery as a lysosome matures into a fusion-restricted post-lysosome. The idea that both phagosome and lysosome fusion dynamics could be perturbed by a defect in recycling of fusion machinery is bolstered by the fact that phagosomes and lysosomes share multiple SNAREs (Gotthardt et al., 2002). This could explain why studies that focus specifically on fission show evidence that Lyst promotes fission, but inappropriate fusion defects are implicated in studies using Lyst mutant cells where exocytosis is the rate-limiting step. In agreement with this hypothesis, the strongest phenotypic effects of CHS arise due to decreased or aberrant function of specialized secretory cells that function through regulated exocytosis of post-lysosomal secretory vesicles (Introne et al., 2009). Additionally, a study comparing cells of beige and wild-type mice showed an 18-fold increase in mast cell secretory granule size, which can be retained for months before exocytosis, but only a moderate, 23%, increase in secretory granule size of pancreatic acinar cells, which are retained for just hours (Hammel et al., 1987; Hammel et al., 2010). With this model in mind, it would be interesting to investigate the recycling dynamics of SNAREs that function along both the endosomal and phagosomal pathways, such as Vamp7a and Vti1, in the LvsB-null cell line.
Because of homology with LvsB, we suggest that the enlarged lysosome-related compartments in CHS patients are a product of inappropriate fusion. This fusion regulatory model for Lyst function is not a new one, but has been widely disputed. Taking the vast number of functional Lyst homolog studies into account with our own comparative analysis of LvsB, we believe that Lyst is a negative regulator of fusion events, and that it helps to stop fusion of vesicles as they mature beyond the lysosomal stage. Although a few studies have provided insight into how Lyst might influence fusion regulation, the degree of separation between Lyst and the fusion machinery is yet undefined. As we collaborate to understand the mechanisms contributing to Lyst-mediated regulation, it will be interesting to see whether Lyst prevents fusion directly, indirectly or both.
MATERIALS AND METHODS
Cell lines and cultures
Dictyostelium discoideum cell lines were maintained in HL-5 medium supplemented with 0.6% penicillin-streptomycin at 18°C on Petri dishes. The media for maintenance of mutant and plasmid-transfected lines were supplemented with 5 µg/ml of blasticidin, and/or 10–15 µg/ml gentamycin accordingly. The wild-type strain NC4A2 was used for all controls and for the previous generation of the LvsB-null cell line (Kypri et al., 2007), except where explicitly noted. The WASH-null cell line (a gift from Thierry Soldati, Université de Genève, Switzerland) was generated in AX2. The AP3 μ3-null cell line (a gift from Pierre Cosson, Centre Medical Universitaire de Geneve, Switzerland) was derived from DH1-10.
Plasmid expression and antibodies
The GFP–vacuolin-B expression vector was constructed and provided by Marcus Maniak (Universitaet Kassel, Germany). The Flag–Rab14-Q67L (dominant active) expression vector was previously constructed (Kypri et al., 2013) from a template vector provided by the laboratory of James Cardelli (Louisiana State University Health Sciences Center, LA). The anti-p80 (H161) monoclonal antibody that was used to label the limiting membrane of phagosomes was generously provided by Pierre Cosson.
Endosome labeling
For morphological studies, live-cell images were taken of GFP–vacuolin-B-expressing cells after labeling of the entire endo-lysosomal pathway. Cells (1×106 cells/ml) were allowed to attach in two-well chambers (Nalge-Nunc Int., Rochester, NY) for 20 min and then switched to low-fluorescence medium (Liu et al., 2002) for 1 h at 18°C. Cells were then incubated in 2 mg/ml TRITC–dextran (64 kDa; Sigma-Aldrich, St Louis, MO) for 1 h at 18°C. The cells were washed four times with low fluorescence medium and imaged using GFP and Texas Red filters on a Nikon inverted epi-fluorescence microscope TE200 (Nikon Instruments, Dallas, TX). A Photometrics cooled CCD camera driven by Metamorph software was used to collect multi-dimensional acquisitions of different fields of cells continuously for 10 min. ImageJ software was used to generate merged images and to measure dextran vesicle diameter for GFP–vacuolin-B-positive and -negative populations.
For temporal endosome studies, visualization of maturing endosomes was performed using pulse labeling and subsequent live-cell imaging of Dictyostelium in two-well chambers. GFP–vacuolin-expressing cells (1×106 cells/ml) were allowed to attach in chambers as above and then switched to low-fluorescence media for 1 h at 18°C. Cells were then pulsed with 2 mg/ml TRITC-dextran for 5 min. The cells were then washed 4 times with low fluorescence media and imaged using a Nikon inverted epi-fluorescence microscope. Images were taken of different fields of cells over the course of 60 min using GFP and Texas Red filter sets. ImageJ software was subsequently used for quantification of colocalization.
Labeling of acidic compartments
To visualize the acidity characteristics of different populations of vesicles, live cell imaging was done in GFP–vacuolin-B-expressing cells exposed to a membrane permeable acidophilic fluorophore. Cells (1×106) were allowed to attach in chambers for 20 min and then switched to low-fluorescence media and incubated for 1 h at 18°C. Attached cells were then incubated with 1 µM Lysotracker red DND-99 (Invitrogen, Eugene, OR) for 10 min at room temperature. After being washed twice with low-fluorescence media, cells were imaged using an inverted Nikon epi-fluorescence microscope to collect multi-dimensional acquisitions as previously described. Different fields of cells were imaged continuously for only 10 min after being washed to prevent Lysotracker Red from artificially raising the alkalinity of acidic compartments. ImageJ software was used to generate merged images for quantification of colocalization as well as to quantify fluorescence intensity of acidic compartments. Background subtraction of fluorescence was done on a cell-by-cell basis.
In vivo endosome fusion assay
To assess fusion between different temporal populations of vesicles, cells were subjected to two pulses of differently labeled dextran separated by a specified chase time. Cells (1×106) were allowed to attach in chambers for 20 min and then switched to low-fluorescence medium and incubated for 1 h at 18°C. Attached cells were given a 5-min pulse of 4 mg/ml FITC–dextran (59 kDa; Sigma-Aldrich, St Louis, MO) and then washed three times before chasing for 30 or 60 min in low-fluorescence medium. Following the chase, cells were given a 5-min pulse of 2 mg/ml TRITC–dextran then washed four times in low-fluorescence medium before being imaged. Continuous live images of different fields were taken for 10 min using an inverted Nikon epi-fluorescence microscope to collect multi-dimensional acquisitions as previously described. Colocalization of the two dextran signals was quantified using ImageJ software.
Phagosome fusion assay
Phagosome fusion was assessed by pulse labeling of consecutive phagosomal populations with blue or red fluorescent beads. Cells were collected and re-suspended to 1.5×106 cells/ml then 5 ml of the re-suspension was allowed to recover in HL-5 medium by shaking at 160 rpm for 15 min at 18°C. Cells were given a 10-min pulse of 1-µm blue fluorescence (excitation 350 nm, emission 440 nm) Carboxylate FluoSpheres (Invitrogen, Eugene, OR) diluted 1∶2500 in the shaking culture. Cells were then collected by centrifugation and washed by re-suspending in 5 ml of ice-cold HL-5 medium and shaking for 2 min at 4°C. Following the wash, cells were collected, resuspended in 5 ml of room temperature HL-5 medium, and given a 10-min pulse of 1∶2500 diluted 1-µm red fluorescence (excitation 580nm, emission 605 nm) Carboxylate FluoSpheres (Invitrogen, Eugene, OR) while shaking at 160 rpm. Cells were collected and washed twice in ice-cold HL-5 medium before being re-suspended in 2.5 ml of room temperature HL-5 medium. 200 µl of the cell re-suspension was pipetted onto a coverslip, and cells were allowed to attach for 30 min at 18°C. Cells were briefly flattened using a thin 2% PCR agarose (Biorad) overlay before being fixed for 5 min at −20°C with 1% formaldehyde in methanol. Following fixation, cells were washed briefly in phosphate-buffered saline (PBS) and then immunostained.
For immuno-labeling of phagosomal membranes, cells were incubated with anti-p80 primary antibody for 1 h at 37°C in the dark. Cells were washed three times with PBS before being incubated with FITC-conjugated goat anti-mouse-IgG secondary antibody (30 µg/ml; Molecular Probes) for 1 h at 37°C in the dark. Cells were washed three times in PBS then rinsed briefly with distilled water and mounted on microscope slides with mounting medium (MOWIOL, Calbiochem, EMD Biosciences). The slides were dried in the dark at room temperature and imaged the following day. Phagosome fusion was quantified from merged images generated using ImageJ software.
Statistical analysis
For all results where more than two cell lines were compared, a one-way analysis of variance (ANOVA) was used to determine statistically significant differences present among multiple cell lines. Post-hoc statistical analysis was done using a two-tailed Student's t-test with Bonferroni correction such that all reported P-values were multiplied by the total number of pairwise comparisons made within the corresponding set of results.
Supplementary Material
Acknowledgments
We would like to thank the members of the De Lozanne and O'Halloran laboratories (at the University of Texas at Austin) for their comments and help throughout the development of this project.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
K.F. performed all experiments and analyzed data. Both authors designed the study and wrote, revised and approved the manuscript.
Funding
This research was supported by grants from the National Institutes of Health [grant numbers GM48745 and AI078136]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.138123/-/DC1
References
- Allende J. E., Allende C. C. (1995). Protein kinases. 4. Protein kinase CK2: an enzyme with multiple substrates and a puzzling regulation. FASEB J. 9, 313–323 [DOI] [PubMed] [Google Scholar]
- Barbosa M. D., Nguyen Q. A., Tchernev V. T., Ashley J. A., Detter J. C., Blaydes S. M., Brandt S. J., Chotai D., Hodgman C., Solari R. C. et al. (1996). Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature 382, 262–265 10.1038/382262a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino J. S., Traub L. M. (2003). Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 10.1146/annurev.biochem.72.121801.161800 [DOI] [PubMed] [Google Scholar]
- Burkhardt J. K., Wiebel F. A., Hester S., Argon Y. (1993). The giant organelles in beige and Chediak-Higashi fibroblasts are derived from late endosomes and mature lysosomes. J. Exp. Med. 178, 1845–1856 10.1084/jem.178.6.1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnell M., Zech T., Calaminus S. D., Ura S., Hagedorn M., Johnston S. A., May R. C., Soldati T., Machesky L. M., Insall R. H. (2011). Actin polymerization driven by WASH causes V-ATPase retrieval and vesicle neutralization before exocytosis. J. Cell Biol. 193, 831–839 10.1083/jcb.201009119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charette S. J., Cosson P. (2007). A LYST/beige homolog is involved in biogenesis of Dictyostelium secretory lysosomes. J. Cell Sci. 120, 2338–2343 10.1242/jcs.009001 [DOI] [PubMed] [Google Scholar]
- Charette S. J., Cosson P. (2008). Altered composition and secretion of lysosome-derived compartments in Dictyostelium AP-3 mutant cells. Traffic 9, 588–596 10.1111/j.1600-0854.2008.00706.x [DOI] [PubMed] [Google Scholar]
- Charette S. J., Mercanti V., Letourneur F., Bennett N., Cosson P. (2006). A role for adaptor protein-3 complex in the organization of the endocytic pathway in Dictyostelium. Traffic 7, 1528–1538 10.1111/j.1600-0854.2006.00478.x [DOI] [PubMed] [Google Scholar]
- Chen C. S. (2002). Phorbol ester induces elevated oxidative activity and alkalization in a subset of lysosomes. BMC Cell Biol. 3, 21 10.1186/1471-2121-3-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke M., Maddera L., Engel U., Gerisch G. (2010). Retrieval of the vacuolar H-ATPase from phagosomes revealed by live cell imaging. PLoS ONE 5, e8585 10.1371/journal.pone.0008585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier L. L., King E. J., Prieur D. J. (1985). Aberrant melanosome development in the retinal pigmented epithelium of cats with Chediak-Higashi syndrome. Exp. Eye Res. 41, 305–311 10.1016/S0014-4835(85)80021-X [DOI] [PubMed] [Google Scholar]
- Deng Y. P., Storrie B. (1988). Animal cell lysosomes rapidly exchange membrane proteins. Proc. Natl. Acad. Sci. USA 85, 3860–3864 10.1073/pnas.85.11.3860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durchfort N., Verhoef S., Vaughn M. B., Shrestha R., Adam D., Kaplan J., Ward D. M. (2012). The enlarged lysosomes in beige j cells result from decreased lysosome fission and not increased lysosome fusion. Traffic 13, 108–119 10.1111/j.1600-0854.2011.01300.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris A. L., Brown J. C., Park R. D., Storrie B. (1987). Chinese hamster ovary cell lysosomes rapidly exchange contents. J. Cell Biol. 105, 2703–2712 10.1083/jcb.105.6.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotthardt D., Warnatz H. J., Henschel O., Brückert F., Schleicher M., Soldati T. (2002). High-resolution dissection of phagosome maturation reveals distinct membrane trafficking phases. Mol. Biol. Cell 13, 3508–3520 10.1091/mbc.E02-04-0206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel I., Dvorak A. M., Galli S. J. (1987). Defective cytoplasmic granule formation. I. Abnormalities affecting tissue mast cells and pancreatic acinar cells of beige mice. Lab. Invest. 56, 321–328 [PubMed] [Google Scholar]
- Hammel I., Lagunoff D., Galli S. J. (2010). Regulation of secretory granule size by the precise generation and fusion of unit granules. J. Cell. Mol. Med. 14, 1904–1916 10.1111/j.1582-4934.2010.01071.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris E., Cardelli J. (2002). RabD, a Dictyostelium Rab14-related GTPase, regulates phagocytosis and homotypic phagosome and lysosome fusion. J. Cell Sci. 115, 3703–3713 10.1242/jcs.00050 [DOI] [PubMed] [Google Scholar]
- Harris E., Wang N., Wu Wl W. L., Weatherford A., De Lozanne A., Cardelli J. (2002). Dictyostelium LvsB mutants model the lysosomal defects associated with Chediak-Higashi syndrome. Mol. Biol. Cell 13, 656–669 10.1091/mbc.01-09-0454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizing M., Anikster Y., Gahl W. A. (2001). Hermansky-Pudlak syndrome and Chediak-Higashi syndrome: disorders of vesicle formation and trafficking. Thromb. Haemost. 86, 233–245 [PubMed] [Google Scholar]
- Introne W., Boissy R. E., Gahl W. A. (1999). Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol. Genet. Metab. 68, 283–303 10.1006/mgme.1999.2927 [DOI] [PubMed] [Google Scholar]
- Introne W., Westbroek W., Golas G. A., Adam D. (2009). Chediak-Higashi Syndrome. GeneReviews Pagon R, Bird T, Dolan C, edSeattle, WA: University of Washington [Google Scholar]
- Jenne N., Rauchenberger R., Hacker U., Kast T., Maniak M. (1998). Targeted gene disruption reveals a role for vacuolin B in the late endocytic pathway and exocytosis. J. Cell Sci. 111, 61–70 [DOI] [PubMed] [Google Scholar]
- Kypri E., Schmauch C., Maniak M., De Lozanne A. (2007). The BEACH protein LvsB is localized on lysosomes and postlysosomes and limits their fusion with early endosomes. Traffic 8, 774–783 10.1111/j.1600-0854.2007.00567.x [DOI] [PubMed] [Google Scholar]
- Kypri E., Falkenstein K., De Lozanne A. (2013). Antagonistic control of lysosomal fusion by Rab14 and the Lyst-related protein LvsB. Traffic 14, 599–609 10.1111/tra.12058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Mirschberger C., Chooback L., Arana Q., Dal Sacco Z., MacWilliams H., Clarke M. (2002). Altered expression of the 100 kDa subunit of the Dictyostelium vacuolar proton pump impairs enzyme assembly, endocytic function and cytosolic pH regulation. J. Cell Sci. 115, 1907–1918 [DOI] [PubMed] [Google Scholar]
- Maniak M. (2003). Fusion and fission events in the endocytic pathway of Dictyostelium. Traffic 4, 1–5 10.1034/j.1600-0854.2003.40101.x [DOI] [PubMed] [Google Scholar]
- Mesaki K., Tanabe K., Obayashi M., Oe N., Takei K. (2011). Fission of tubular endosomes triggers endosomal acidification and movement. PLoS ONE 6, e19764 10.1371/journal.pone.0019764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullock B. M., Bright N. A., Fearon C. W., Gray S. R., Luzio J. P. (1998). Fusion of lysosomes with late endosomes produces a hybrid organelle of intermediate density and is NSF dependent. J. Cell Biol. 140, 591–601 10.1083/jcb.140.3.591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle D. L., Karim M. A., Woolf E. A., Holmgren L., Bork P., Misumi D. J., McGrail S. H., Dussault B. J., Jr, Perou C. M., Boissy R. E. et al. (1996). Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat. Genet. 14, 307–311 10.1038/ng1196-307 [DOI] [PubMed] [Google Scholar]
- Oliver C., Essner E. (1975). Formation of anomalous lysosomes in monocytes, neutrophils, and eosinophils from bone marrow of mice with Chédiak-Higashi syndrome. Lab. Invest. 32, 17–27 [PubMed] [Google Scholar]
- Padh H., Ha J., Lavasa M., Steck T. L. (1993). A post-lysosomal compartment in Dictyostelium discoideum. J. Biol. Chem. 268, 6742–6747 [PubMed] [Google Scholar]
- Perou C. M., Leslie J. D., Green W., Li L., Ward D. M., Kaplan J. (1997). The Beige/Chediak-Higashi syndrome gene encodes a widely expressed cytosolic protein. J. Biol. Chem. 272, 29790–29794 10.1074/jbc.272.47.29790 [DOI] [PubMed] [Google Scholar]
- Peters C., Mayer A. (1998). Ca2+/calmodulin signals the completion of docking and triggers a late step of vacuole fusion. Nature 396, 575–580 10.1038/25133 [DOI] [PubMed] [Google Scholar]
- Rahman M., Haberman A., Tracy C., Ray S., Krämer H. (2012). Drosophila mauve mutants reveal a role of LYST homologs late in the maturation of phagosomes and autophagosomes. Traffic 13, 1680–1692 10.1111/tra.12005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauchenberger R., Hacker U., Murphy J., Niewöhner J., Maniak M. (1997). Coronin and vacuolin identify consecutive stages of a late, actin-coated endocytic compartment in Dictyostelium. Curr. Biol. 7, 215–218 10.1016/S0960-9822(97)70093-9 [DOI] [PubMed] [Google Scholar]
- Ravanel K., de Chassey B., Cornillon S., Benghezal M., Zulianello L., Gebbie L., Letourneur F., Cosson P. (2001). Membrane sorting in the endocytic and phagocytic pathway of Dictyostelium discoideum. Eur. J. Cell Biol. 80, 754–764 10.1078/0171-9335-00215 [DOI] [PubMed] [Google Scholar]
- Rupper A., Cardelli J. (2001). Regulation of phagocytosis and endo-phagosomal trafficking pathways in Dictyostelium discoideum. Biochim. Biophys. Acta 1525, 205–216 10.1016/S0304-4165(01)00106-4 [DOI] [PubMed] [Google Scholar]
- Schekman R. (1998). Membrane fusion. Ready…aim…fire! Nature 396, 514–515 10.1038/24986 [DOI] [PubMed] [Google Scholar]
- Stinchcombe J. C., Page L. J., Griffiths G. M. (2000). Secretory lysosome biogenesis in cytotoxic T lymphocytes from normal and Chediak Higashi syndrome patients. Traffic 1, 435–444 10.1034/j.1600-0854.2000.010508.x [DOI] [PubMed] [Google Scholar]
- Sundler R. (1997). Lysosomal and cytosolic pH as regulators of exocytosis in mouse macrophages. Acta Physiol. Scand. 161, 553–556 10.1046/j.1365-201X.1997.00262.x [DOI] [PubMed] [Google Scholar]
- Tchernev V. T., Mansfield T. A., Giot L., Kumar A. M., Nandabalan K., Li Y., Mishra V. S., Detter J. C., Rothberg J. M., Wallace M. R. et al. (2002). The Chediak-Higashi protein interacts with SNARE complex and signal transduction proteins. Mol. Med. 8, 56–64 [PMC free article] [PubMed] [Google Scholar]
- Temesvari L. A., Bush J. M., Peterson M. D., Novak K. D., Titus M. A., Cardelli J. A. (1996). Examination of the endosomal and lysosomal pathways in Dictyostelium discoideum myosin I mutants. J. Cell Sci. 109, 663–673 [DOI] [PubMed] [Google Scholar]
- Tsujimoto S., Bean A. J. (2000). Distinct protein domains are responsible for the interaction of Hrs-2 with SNAP-25. The role of Hrs-2 in 7 S complex formation. J. Biol. Chem. 275, 2938–2942 10.1074/jbc.275.4.2938 [DOI] [PubMed] [Google Scholar]
- Wang N., Wu W. I., De Lozanne A. (2002). BEACH family of proteins: phylogenetic and functional analysis of six Dictyostelium BEACH proteins. J. Cell. Biochem. 86, 561–570 10.1002/jcb.10254 [DOI] [PubMed] [Google Scholar]
- Ward D. M., Pevsner J., Scullion M. A., Vaughn M., Kaplan J. (2000). Syntaxin 7 and VAMP-7 are soluble N-ethylmaleimide-sensitive factor attachment protein receptors required for late endosome-lysosome and homotypic lysosome fusion in alveolar macrophages. Mol. Biol. Cell 11, 2327–2333 10.1091/mbc.11.7.2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham M. C., Spicer S. S., Vincent R. A., Jr (1981). The origin and fate of large dense bodies in beige mouse fibroblasts. Lysosomal fusion and exocytosis. Exp. Cell Res. 136, 157–168 10.1016/0014-4827(81)90047-1 [DOI] [PubMed] [Google Scholar]
- Wubbolts R., Fernandez-Borja M., Oomen L., Verwoerd D., Janssen H., Calafat J., Tulp A., Dusseljee S., Neefjes J. (1996). Direct vesicular transport of MHC class II molecules from lysosomal structures to the cell surface. J. Cell Biol. 135, 611–622 10.1083/jcb.135.3.611 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.