

Abstract
Mutations of Wnt/β-catenin signaling pathway play essential roles in development and cancer. Although β-catenin and adenomatous polyposis coli (APC) gene mutations are well established and are known to drive tumorigenesis, discoveries of mutations in other components of the pathway lagged which hinders the understanding of cancer mechanisms. Here we report that δ-catenin (gene designation: CTNND2), a primarily neural member of the β-catenin superfamily which promotes canonical Wnt/β-catenin/LEF-1-mediated transcription, displays exonic mutations in human prostate cancer and promotes cancer cell survival adaptation and metabolic reprogramming. When overexpressed in cells derived from prostate tumor xenografts, δ-catenin gene invariably gave rise to mutations leading to sequence disruptions predicting functional alterations. Ectopic δ-catenin gene integrating into host chromosomes is locus non-selective. δ-Catenin mutations promote tumor development in mouse prostate with probasin promoter (ARR2PB)-driven, prostate-specific expression of myc oncogene, while mutant cells empower survival advantage upon overgrowth and glucose deprivation. Reprogramming energy utilization accompanies the down-regulation of glucose transporter-1 (Glut-1) and Poly (ADP-ribose) polymerase (PARP) cleavage while preserving tumor type 2 pyruvate kinase (PKM2) expression. δ-Catenin mutations increased β-catenin translocation to the nucleus and HIF-1α expression. Therefore, introducing δ-catenin mutations is an important milestone in prostate cancer metabolic adaptation by modulating β-catenin and HIF-1α signaling under glucose shortage to amplify its tumor promoting potential.
Keywords: Prostate cancer, somatic mutations, host chromosomal targeting, glucose deprivation, metabolic reprogramming, survival adaptation
Introduction
Mutations, such as that of β-catenin and adenomatous polyposis coli (APC) genes of the Wnt/wingless signaling pathway, play pivotal roles in human diseases including cancer (1). Mutations contribute to genetic instability, a major underlying hallmark of cancer, which propels human tumor progression (2). Such mutations in oncogene β-catenin and tumor suppressor APC endow cancer cells with the ability to outgrow or outlive their neighboring cells not affected by mutations (1). However, the understanding of other genes in the Wnt/wingless pathway is less clear. Many genes and their proteins, upon modifications, can elicit opposing growth-promoting and suppressive functions at different cancer stages. Yet, little is known as to how these processes are controlled at the genetic level.
δ-Catenin/NPRAP/Neurojungin (gene designation: CTNND2) is primarily a neuronal protein expressed in brain (3-5). However, this initially described neural specific member of the β-catenin/armadillo superfamily is commonly overexpressed in cancers of peripheral tissues, including prostate, esophageal, breast, lung and ovarian cancers (6-9). While the mechanisms of δ-catenin regulation in neurologic disorders and cancer are beginning to emerge, studies show that δ-catenin promotes canonical Wnt/β-catenin/LEF-1-mediated transcription (10). δ-Catenin genelocus (5p15.2) is highly susceptible for generating single nucleotide polymorphism (SNP). Compelling recent evidences have linked CTNND2 SNP or mutations to cancer, myopia, cortical cataract and Alzheimer's disease (11, 12). Chromothripsis and focal copy number alterations in 5p12-5p15 also determine poor outcomes in malignant melanoma (13). In cancer cells, δ-catenin can exert both pro- and anti-growth effects and is correlated with poor patient survival (9, 14-15). Although the mechanisms by which these paradoxical functions are controlled genetically and how they promote cancer pathogenesis have not been well established, δ-catenin is a potential cancer biomarker and could be an important target for therapeutic interventions.
Here we take the advantage of a serendipitous discovery of induced CTNND2 mutations in cells derived from prostate cancer xenografts, which lead to sequence disruptions predicting functional alterations. We further revealed a broad spectrum of exonic mutations in δ-catenin associated with human prostatic adenocarcinoma. We found that the integration of ectopic δ-catenin gene into the host chromosomes is not locus selective. δ-Catenin mutations promote ARR2PB-driven, myc-induced mouse prostate tumor development, while mutant cells empower survival advantage upon overgrowth and glucose deprivation. We found that reprogramming energy utilization accompanies the down-regulation of glucose transporter-1 (Glut-1) and PARP cleavage, preserving PKM2, β-catenin translocation to the nucleus and increases in HIF-1α expression. We conclude that the introduction of CTNND2 gene variation is an important milestone in prostate cancer metabolic adaptation with the potential of being a target element for prostate cancer treatment.
Results
Ectopically expressed δ-catenin is invariably mutated in prostate cancer cells
We overexpressed δ-catenin into CWR22-Rv1 cells derived from human primary prostatic tumor xenografts and PC-3 cells of prostate cancer bone metastasis. Stable cell lines were successfully established. But to our surprise, the full-length δ-catenin gradually and invariably gave way to faster migrating variants, which eventually stabilized at ∼ 100 kDa on SDS-PAGE when cells were cultured with repeated interruption of medium replenishments (Fig 1A). We initially regarded this variant as being derived either from a truncated cDNA contamination or proteolysis due to unfavorable culture conditions. However, mapping with antibodies against epitopes covering the entire protein length ruled out cDNA contamination as the cause because it would have represented a nonexistent cDNA. We then applied a panel of protease inhibitors to determine whether the presumptive proteolysis can be reduced or prevented. E64D, E64D plus leupeptin (which inhibits most peptidases), or A-acetyl-cysteine (NAC) as anti-autophage/oxidant agent (18) did not inhibit this variant (Fig 1B). E-cadherin and p120ctn, two adherens junction proteins that are co-localized with δ-catenin and are known to undergo proteolysis (19, 20), were quite stable under the same culture condition (Fig 1C). The phenomenon that fast migrating protein bands became dominant over time was also observed in PC-3 cells stably transfected with the full-length δ-catenin ectopically (Fig 1D). Furthermore, once cell cultures with the variant were established, the truncated protein could be observed from cells lysed soon after re-plating. Therefore, protein degradation due to culture aging cannot be the major mechanism of generating this variant.
Figure 1. Mutations and formation of δ-catenin variants.

(A). Ectopically expressed full-length δ-catenin gradually changes to truncated variants in human prostate cancer xenograft CWR22-Rv1 in culture. FBS: fetal bovine serum with high glucose; dFBS1 and dFBS2: the first and second rounds of treatment with FBS and glucose deprivation. Rv/C: control cells; Rv/δ: cells expressing full-length δ-catenin. Numbers underneath the gels: ratio of truncated variant to full-length δ-catenin in Rv/δ cells. (B). Protease inhibition did not prevent the formation of δ-catenin variant. Rv/δ cells from dFBS2 culture were either untreated (UnT), treated with E64D+leipeptin for 2 or 4 hours, or treated with NAC for 4 hours. (C). Rv/δ cells from dFBS2 culture displaying fast migrating δ-catenin variant but not E-cadherin or p120ctn proteolysis. (D). Ectopically expressed full-length δ-catenin in transient (Trans) transfected human prostate cancer bone metastasis PC-3 cells (PC-3/δ) gave rise to truncated variants that migrated faster on SDS-gel after the δ-catenin overexpressing cells became stable cell lines (Stable) in culture. (E). Example of δ-catenin mutations in Rv/δ cells from dFBS2 culture. The lower panel indicates mutation and the upper panel indicates the normal control sequence. Molecular weights are on the left.
A closer examination of δ-catenin cDNA isolated from the stable cell lines that express the mixed full-length and truncated δ-catenin bands showed sequence variations. This surprising result prompted the hypothesis that δ-catenin gene may be genetically targeted by cancer cells. We then sequenced the entire δ-catenin cDNA stably expressed in CWR22-Rv1 cells. We discovered that the cells expressing mainly the truncated δ-catenin variant displayed frame-shifting mutations that lead to premature termination (Fig 1E). Remarkably, the predicted mutant protein variants derived from some of these premature terminations (e.g. exon 16 in Fig 1E) fell in the close range of the size of the fast migrating protein band on SDS-gels. To demonstrate that the mutations in δ-catenin can indeed generate the fast migrating protein band, we performed a mutagenesis experiment. We generated the identical mutation as the naturally occurring frame-shifting mutation described in Figure 1E, and then transiently transfected it into CWR22-Rv1 and PC-3 cells. Western blot analysis confirmed that the fast migrating protein band was at the same size of the predicted molecular weight for the truncated protein as the mutation would cause (Fig S1). These findings highlighted the potential of malignant tumor environment in inducing genetic mutations in an overexpressed gene.
There are hot-spot mutation cluster regions in CTNND2 exons in human primary prostatic adenocarcinomas
The above data prompted the question of whether the mutations are the consequences of δ-catenin overexpression in cultures in vitro or reflect an event that also occurs in human prostate cancer in vivo (6, 7) where δ-catenin expression is increased. Therefore, we sequenced and analyzed the entire coding region of δ-catenin gene (exon 1-22) isolated from human prostatic adenocarcinomas. Most functional mutations occurred after exon 9 (Fig 2A). Some mutations that are either mis-sense or frame-shifting occurred after exon 13 (Fig 2A, and B-G, bottom panels). So far, gene variations in δ-catenin coding region have been identified in 24 of 40 pathologically confirmed prostatic adenocarcinomas (Fig 2; Table S1). Little or no mis-sense or frame-shifting mutations were found in four exonic DNAs from clinically disease-free normal specimens (Fig 2B-G, top panels). We also did not observe significant mutations at the amino-terminus corresponding to exons 1-9 (Fig 2A). These data strongly suggested that human δ-catenin gene contains hot-spot mutation cluster regions (MCRs) in prostate cancer.
Figure 2. Mutation cluster regions (MCRs) ofδ-catenin coding sequences in human prostate cancer.

(A). Schematic illustration of MCRs in δ-catenin coding region. The entire coding region of δ-catenin gene was sequenced. Recurrent and non-recurrent mutations were identified. Shown here are mutations in exons 10, 13, 14, 19, 21, and 22 (recurrent) as well as 20 (non recurrent). No mutations were observed from exons 1 to 9. (B-G). Sequence chromatograms showing examples of somatic δ-catenin exon mutations. Some mutations lead to the disruption of protein sequences. Note: The lower panels indicate the tumor and the upper panels indicate the matched normal control.
Examination of Sanger cancer gene mutation database found similar trends of sequence variation in δ-catenin coding region in other cancer types (http://cancer.sanger.ac.uk/cosmic/). For example, hot-spot MCRs are located at exons 10-14 and 17-22 in cancers including lung, prostate, pancreas, skin, intestine and upper gastrointestinal tract cancers, as well as leukemia (Fig 3A, Fig S2). The F1172F (c.3516C>T) mutation in exon 22 in human skin cancer, while it is a same-sense mutation, is identical to that in prostate cancer. A number of mutations predicted significant impact, as they either lead to premature termination or potential alteration of protein functions (Fig 3B). Searching for commonly mutated genes in cancer exerts significant impact on effective cancer diagnosis and therapeutic design (16, 17). Our findings clearly support δ-catenin as one such somatically mutated gene in prostate cancer.
Figure 3. MCR of δ-catenin showing functional impacts.
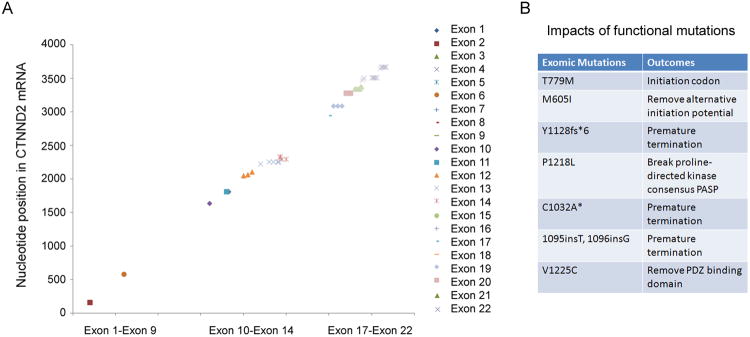
(A). Summary of MCR of δ-catenin coding region in different cancer types. Note: δ-catenin mutations were clustered around exons 10-14 and exons 17-22. There are few mutations in the exon 1-9. The data reflect MCR from eight different cancer types. (B).δ-Catenin exonic mutations in cancer predictive of functional impacts. Seven exomic mutations are listed here as examples to show the potential to alter protein functions, including premature termination.
CTNND2 mutations are not locus-selective and promote prostate tumor development in ARR2PB driven myc-expressing transgenic mice
We then cloned CWR22-Rv1 cells that expressed only the truncated δ-catenin variant (Fig 4A). As expected, full-length δ-catenin was distributed at the cell-cell junction (Fig 4B, GFP: arrows, Rv/δ) (5, 10). However, the truncated δ-catenin was exclusively cytoplasmic (Fig 4B, GFP: arrowhead, Rv/M1) and did not co-localize with the cell-cell junction protein p120ctn (Fig 4B, p120ctn: arrows) (5). We also examined CWR22-Rv1 cells stably transfected with cDNAs with amino-terminal 280 amino acid truncated (ΔN) or carboxyl-terminal 207 amino acid truncated (ΔC) δ-catenin (Fig 4C). Although the truncation was further toward the carboxyl-terminus, ΔC also showed cytoplasmic localization as Rv/M1 (Fig 4B and C, Compare Rv/M1 and ΔC).
Figure 4. Cancer cells are capable of altering ectopic δ-catenin gene sequence and function.

(A). The establishment of δ-catenin truncation variant clones. CWR22-Rv1 cells displaying both full-length and truncation variants (H) were subcloned. Shown here are six examples of clones expressing only the truncation variant (M1 to M6). (B). Distribution of GFP-tagged full-length δ-catenin and its induced truncation variant in comparison to a closely related protein p120ctn. Arrows: cell-cell junction; Arrowheads: truncated δ-catenin distribution. Bar: 10 μm. (C). Distribution of full-length δ-catenin and its amino- or carboxyl-terminal truncation mutants. Upper panel: δ-catenin cDNA construct illustration. Arrows: cell-cell junction; Arrowheads: cytoplasm. Bar: 10 μm. (D). FISH probe specific to mouse δ-catenin cDNA detected multiple hybridization signal (Arrows) in Rv/δ but not in Rv/C cell chromosomal spreads. Asterisks: intact Rv/δ cells. Bar: 0.5 μm.
It is widely known that ectopically transferred genes do not insert into a specific chromosome in host cells (21). FISH analysis showed that a biotin-tagged DNA probe specific to δ-catenin DNA sequence labeled multiple host chromosomes in δ-catenin transfected cells (Fig 4D, Rv/δ) but not in vector transfected control cells (Fig 4D, Rv/C). These observations suggest that the generation of δ-catenin mutations was not relying upon its bona fide chromosomal locus of 5p15.2. Cancer cells must have developed the capacity to target an overexpressed δ-catenin gene by altering its DNA sequences genetically (22).
We found that while Rv/δ cells grew faster as previously reported (14), Rv/M1 cells grew slower than either Rv/CorRv/δ cells. Likewise, Rv/ΔC cells showed a similar cell death rate to Rv/C, while apoptosis of Rv/δ cells was reduced (Fig S3). These results suggest that carboxyl-terminal deletion of δ-catenin did not confer growth advantage under the normal culture condition in which the supplies of glucose and serum factors were abundant. However, when cultures grew until they reached beyond full confluency, Rv/M1 cells displayed increased cell numbers (Fig 5A). The tumor promoting effects of δ-catenin truncation can also be demonstrated in vivo. Mice with δ-catenin gene disrupted in exon 9 (23) and myc oncogene conditionally expressed in the prostate (24) showed increased tumor size and progression from prostatic intraepithelial neoplasia (PIN, Myc/δ+/+ and Myc/δ/+/-) to adenocarcinomas (Myc/δ-/-) (Fig 5B; Fig S4; Table S2). The effects of δ-catenin gene disruption on prostatic tumorigenesis were dependent on the Myc oncogene because δ-catenin mutant mice without Myc expression did not develop PIN or adenocarcinomas (Fig 5C). In the Myc/δ-/- mice, both Ki67 indexed cell proliferation and TUNNEL positive apoptosis progressively increased when compared to WT (No Myc/δ+/+), Myc/δ-cat+/+ and Myc/δ-cat+/- mice (Fig 5D).
Figure 5. δ-Catenin truncation variant promotes overgrowth in vitro and in vivo.

(A). δ-Catenin truncation variant promotes overgrowth. The numbers of Rv/C, Rv/δ, and Rv/M1 cells were counted without fresh FBS replenishment after full confluence of cultures. *: p<0.05. (B). Myc-induced tumor development was increased in mutant mice expressing a δ-catenin exon 9 disruption cassette. Inserts: mouse prostates. 400 ×. (C). Histological analysis of No Myc/δ-/- mouse prostate exhibits 1 to 2 layers of epithelial cells, characteristic of normal-appearing prostate glands. Magnification: 100×. Insets: 400×. DLP: Dorsal lateral prostate. VP: Ventral prostate. (D). Quantifications of proliferative and apoptotic cells following immunostaining with anti-Ki67, a marker of cell proliferation, and TUNEL assay, which detects cells undergoing apoptosis in 6-month old mice bearing ARR2PB-driven Myc expression with various δ-catenin expression background. Note that the number of Ki67 positive proliferative cells is dramatically increased and approximately four-fold greater than that for TUNEL staining in Myc/δ-/-. Values represent mean ± S.E.M for a total of 500 cells counted from 2 sets of independent experiments. p< 0.01 * relative to wild type (No Myc/δ+/+) Ki67 and TUNEL.
δ-Catenin mutations lead to survival adaptation under glucose deprivation and reprogram energy utilization
Recent studies highlighted the ability of cancer cells to adapt to glucose-deprived conditions by introducing mutations, e.g., in Ras expressing tumor cells (22). When grown in culture with high glucose, Rv/M1 and Rv/C cells had similar death indexes whereas that of Rv/δ cells was reduced (Fig 6A, High glucose). However, under glucose deprivation, the death index of Rv/M1 cells reduced precipitously whereas that of Rv/C cells increased dramatically (Fig 6A, Low glucose). Rv/M1 cells were visibly more resilient to glucose starvation than Rv/C cells (Fig 6B). According to Warburg hypothesis, cancer cells require much less energy to survive than they do for growth under densely packed conditions (25, 26). δ-Catenin mutations clearly empowered prostate cancer cells with an increase in such capabilities of survival under glucose deprivation.
Figure 6. δ-Catenin mutations promote metabolic reprogramming to increase cancer cell survival and adaptation under glucose deprivation.

(A). Rv/M1 cells show survival advantage under low glucose but not normal growth condition with high glucose. The dying, floating cells versus the healthy, adherent cells were counted separately and their ratio was obtained as death index. *: p< 0.05 (High glucose); **: p< 0.05 (Low glucose). (B). Phase images of cells that grew for seven days under either high glucose or low glucose conditions. Rv/M1 cells showed increased survival under glucose deprivation. Bar: 50 μm. (C). Rv/δ and Rv/M1 cells showed increased OCR/ECAR when compared to Rv/C under the normal growth condition with high glucose. Rv/M1 cells maintained higher OCR/ECAR, whereas Rv/C and Rv/δ cells showed much lower OCR/ECAR during glucose deprivation.*: p< 0.05 (High glucose); **: p< 0.05 (Low glucose). (D). Rv/δ and Rv/M1cells preserved mitochondrial respiration functions and unleashed its utilization under glucose starvation. Seahorse XF24 analyzer acquisition showed an example of real-time OCR responses to the 2, 4-DNP, 2-DG, and rotenone treatments.
Our previous study showed that Rv/δ cells increased expression of hexokinase II, a key enzyme for phosphorylation of glucose in entering glycolysis, which is increased in prostate cancer (14, 27). To determine how δ-catenin mutations may reprogram energy metabolism to promote survival under nutrient deprived conditions, we examined the ability of transfected cells to utilize energy. Under the normal growth condition, Rv/δ and Rv/M1 cells showed increased ratio of oxygen consumption rate (OCR) over glycolysis-mediated extracellular acidification rate (ECAR) when compared to Rv/C cells (Fig 6C, High glucose). However, under glucose deprivation, Rv/M1 cells displayed higher OCR/ECAR than that of either Rv/C or Rv/δ cells (Fig 6C, Low glucose).
After the cultures were maintained with low glucose for a week, Rv/C cells suffered massive cell death (Fig 6B, Low glucose-Rv/C). Correspondingly, the OCR of Rv/C cells did not respond to 2, 4 DNP (Fig 6D), indicating their reliance on glucose for mitochondrial respiration (28). Rv/δ cell death was moderate, while Rv/M1 cell death was minimal (Fig 6B, Low glucose-Rv/δ and Rv/M1). Under this condition, Rv/δ and Rv/M1 responded to 2, 4 DNP and displayed an OCR uptrend (Fig 6D), which resembled that of Rv/C, Rv/δ and Rv/M1 cells under normal culture condition (Fig S5). Therefore, Rv/δ and Rv/M1 cells preserved mitochondrial respiration capability and can unleash its utilization under glucose starvation. Continuing with 2-DG treatment, which inhibits glycolysis (28), Rv/C and Rv/δ cells increased their relative OCR levels, but Rv/M1 cells showed a sharp reduction in OCR. Corresponding to the very low cell death observed under the low glucose condition, the deficient OCR response of Rv/M1 following 2-DG treatment suggests the potential adaptation mechanisms by which δ-catenin mutations promote cell survival (Fig 6B). All cells responded to rotenone, which inhibits mitochondrial NADH dehydrogenase/complex I and, consequentially, blocks mitochondrial respiration.
While the tumor-selective pyruvate kinase, tumor type 2 (PKM2) was reduced in Rv/C cells upon glucose deprivation for one week, PKM2 expression remained strong in Rv/δ and Rv/M1 cells (Fig 7A; Fig S6A). The expression of mitochondrial complexes was not significantly changed in Rv/M1 compared to Rv/C and Rv/δ cells (Fig 7A). However, compared to Rv/C and Rv/δ, Rv/M1 cells showed reduced expression of cleaved PARP, a critical enzyme in the DNA repair defect-initiated apoptosis (Fig 7B; Fig S6B). Bcl-2 expression was increased in Rv/δ and Rv/M1 cells when compared to Rv/C cells with or without glucose deprivation (Fig 7B; Fig S6A). Glucose transporter-1 (Glut-1) showed a distinct shift of protein migration pattern of Glut-1H and Glut-1L variants from high glucose to low glucose culturing condition (Fig 7B). Both variants showed a reduced expression under glucose deprivation in Rv/M1 cells (Fig 7B).
Figure 7.

δ-Catenin mutations alter protein expression and distribution in the Wnt/β-catenin and hypoxia pathways. (A). Rv/δ and Rv/M1 cells preserved PKM2 expression but did not alter mitochondrial respiratory complex proteins under glucose deprivation. GAPDH was used as loading control. (B). Rv/M1 cells suppressed PARP cleavage and altered Glut-1 expression under glucose deprivation. Both Rv/δ and Rv/M1 cells showed increased Bcl-2 expression than that of Rv/C cells with or without glucose deprivation. (C). There were only moderate changes in HIF-1α expression but significant changes in β-catenin expression and nuclear translation among Rv/C, Rv/δ and Rv/M1 cells when grown in normal medium with high glucose. (D). There were significant increases in β-catenin and HIF-1α expression and nuclear translocation in Rv/M1 cells when compared to Rv/C cells under glucose deprivation.
To determine whether the changes in cell growth and survival properties involved Wnt/β-catenin signaling, we found that with high glucose in the medium, β-catenin expression and translocation to the nucleus was increased in Rv/δ and Rv/M1 in comparison to that of Rv/C (Fig 7C; Fig S6C). But there were no meaningful differences in HIF-1α expression among Rv/C, Rv/δ, and Rv/M1 cells (Fig 7C; Fig S6D, Glucose). However, there was a remarkable increase in both β-catenin and HIF-1α expression and translocation to the nucleus in Rv/M1 compared to Rv/C cells during glucose deprivation (Fig 7D; Fig S6, No Glucose). These results are consistent with our previous studies (14), but further support that the carboxyl-terminus of δ-catenin is important for prostate cancer expansion under the normal growth condition. Eliminating these sequences reassigned energy utilization for survival over proliferation-required biomass production under nutrition deprivation (26).
Discussion
Our studies identified that δ-catenin (CTNND2) gene displays somatic mutations that are highly inducible when overexpressed, likely representing and predicting an important milestone in prostate cancer cell survival and metabolic adaptation during densely-packed tumorigenesis.
The molecular mechanism of inducible mutagenesis of δ-catenin in prostate cancer is not clear at present. Our studies showed that the generation of truncated δ-catenin protein species is not due to protein degradation. We also showed that the ectopic δ-catenin gene integrating into host chromosomes is not locally adherent to its native gene locus at Chr5p15.2. It is possible that an increased expression of δ-catenin ignited a feedback reaction onto replicating chromosomes where ectopic δ-catenin cDNA was inserted, and a low fidelity replication due to destabilized genome in δ-catenin transfected cells allowed un-conserved base pairing to proceed. These random hits towards the 3′-region of ectopic δ-catenin sequences were then followed by a functional selection to favor carboxyl-terminal truncation mutant cells while eliminating full-length δ-catenin expressing cells from the prostate cancer cell populations.
δ-Catenin is at the cross points of several signaling pathways that play critical roles in development and cancer. δ-Catenin interacts with classical cadherins at their juxtermembrane domain, an association motif shared with that for p120ctn which is another protein of the delta subfamily (p120ctn: CTNND1) of β-catenin/armadillo superfamily (5, 10, 31). This interaction is critical for regulating E-cadherin stability and in brain it involves the regulation by presenilin (PS) of Alzheimer's disease (32, 33). While the roles of β-catenin and its mutations in development and cancer by effecting Wnt/wingless signaling are well established, recent evidence placed δ-catenin in the same pathway. Indeed, δ-catenin promotes E-cadherin processing and activates β-catenin-mediated signaling in human prostate cancer (10). δ-Catenin was shown as a new member of the glycogen synthase kinase-3 (GSK-3) signaling complex that promotes β-catenin turnover (34). GSK-3 phosphorylates δ-catenin and negatively regulates its own stability via ubiquitination/proteosome-mediated proteolysis (35). Besides the interaction of δ-catenin and p120ctn with the canonical Wnt-GSK3-β-catenin-LEF1 signaling pathway, non-canonical Wnt signals are modulated by the Kaiso transcriptional repressor, which is also a downstream target of δ-catenin and p120ctn (36-38). Therefore, δ-catenin is intimately involved in Wnt signaling and promotes cancer cell growth and shows poor cancer survival when it is overexpressed.
When released from cadherins, δ-catenin clearly interacts functionally with actin regulatory network proteins, notably with Rho family small GTPases. Overexpression of δ-catenin induces branched cellular processes, which is reminiscent of RhoA inhibition (39). δ-Catenin overexpression decreased the binding between p190RhoGEF and RhoA, and significantly lowered the levels of GTP-RhoA (40, 41).
δ-Catenin mutant cells (Rv/M1) survived better than Rv/C cells under glucose deprivation but the expression of well-established mitochondrial complexes was not significantly changed. These data indicated that the changes in mitochondrial respiration due to an altered expression pattern of these complexes are less likely the mechanisms of enhanced survival of Rv/M1 cells. Seahorse XF24 metabolic analysis showed that Rv/M1 cells are sensitive to the treatment of 2-DG, an inhibitor of glycolysis. Although we do not yet understand the molecular mechanisms how the truncated δ-catenin leads to metabolic reprogramming, it is possible that C-terminal truncation reduced the ability of δ-catenin to inhibit small GTPase RhoA (40) and allowed cytokinesis to proceed. The removal of cytokinesis inhibition could result in reprogramming energy utilization in cell cycle progression and prevent cytokinesis checkpoint-initiated apoptosis.
In normal cells, we propose that the full-length δ-catenin displays overall tumor suppressive function. This means that overexpressing δ-catenin ectopically in a normal cell type would either inhibit or slow cell proliferation. This notion is supported by our studies as well as others (5; 15). In malignant cancers, δ-catenin expression increased cell growth while knockdown of δ-catenin reduced cell growth. This conclusion is supported d by our studies as well as others (9; 14; 42). This is also true in the current study when prostate cancer cells grow under abundant serum and high glucose condition.
However, as the cancer cells deregulate many gene's expression during transformation, some genes when their expressions being overly high, can be harmful for overall tumor growth if the nutrition supplies cannot match the demand of rapid growth in certain tumor areas. We hypothesize that the N-terminus of δ-catenin may promote cancer cell growth and survival (14), whereas the C-terminus of δ-catenin when overly active interacts with the small GTPase RhoA to inhibit cytokinesis as seen in p0071 (43). When the promoter of δ-catenin is activated in prostate cancer (6; 44) the cancer cells could balance the rate of cell cycle progression by sensing the nutrition supplies. When nutrition is abundant, δ-catenin N-terminal function dominates and its carboxyl-terminal interaction with RhoA is limited. When nutrition is limited, cancer cells adapt to inactivate the overexpressed δ-catenin C-terminus. This modulation may create a favorable situation for cancer cells to continue to grow and survive under the stressful condition of rapid overgrowth but avoid its elimination due to conflict between rapid cell cycle entrance and delayed cytokinesis.
In the case of Myc-driven prostate cancer mouse model, Myc overexpression promoted the proliferation and transformation. However, δ-catenin expression on promoting cell cycle entrance without proceeding through cytokinesis may not be favorable for tumorigenesis. In this case, the δ-catenin C-terminal truncation mutant may function to release this inhibition.
Recent studies of Rho GTPase links to mitochondrial enzyme glutaminase (GLS1) suggested that signaling via the Rho GTPases, together with NF-κB, can elevate mitochondrial glutaminase activity in cancer cells, thereby helping cancer cells meet their altered metabolic demands (45). There is also evidence that TSC2 inhibits cell growth by acting as a GTPase-activating protein toward Rheb, thereby inhibiting mTOR. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth (46). Despite the understanding of δ-catenin functions in Rho GTPase (40; 41; 47; 48) and Wnt/β-catenin signaling (10; 34; 35), the current study is the first to provide significant evidence of exonic mutations in δ-catenin coding region and demonstrate their consequences in cancer metabolic adaptations in tumor progression.
These studies thus raise questions for cancer mechanisms and drug targeting strategies. Cancer cells can apparently alter genetic composition and subsequent functions of foreign DNAs, potentially evading ectopic expression-based gene therapy. Additionally, targeting cancer metabolism may benefit by incorporating enforcers that inhibit genes promoting adaptation under metabolic stress. Finally, chromosomal machinery responsible for mutagenesis is likely not locally adhering to a specific gene locus and is deregulated in cancer homing for introduction of cancer driving and adapting mutations. We have now succeeded in generating innovative cell and mouse models which can potentially elucidate such mechanisms that can hold a key to reduce genomic rearrangements and make targeting cancer more effective.
Materials and Methods
Antibodies and reagents
Mouse anti-E-cadherin, anti-p120ctn, and anti-δ-catenin clone 30 (to amino acids 85-194) were from BD Biosciences (Palo Alto, CA). Monoclonal anti-GAPDH was from EMD Science. Rabbit, guinea pig, and mouse antibodies against different epitopes of mouse or human δ-catenin (epitopes on mouse 85-194, 292-309, 691-708, 1017-1035; human 434-530, 828-1022) were described previously (4, 5). Rabbit anti-PARP and anti-PKM2 were from Cell Signaling. Rabbit anti-HIF-1α and anti-Bcl-2 were from Santa Cruz Biotechnology whereas rabbit anti-β-catenin was from Sigma. Rabbit anti-Glut-1 and mouse anti-mitochondrial NADPH dehydrogenase/complex cocktail were from Abcam. All other chemicals were from sigma unless indicated otherwise.
Cell culture, mutagenesis, and transfection
Human prostate tumor xenograft CWR22-Rv1 or PC-3 cells were stably transfected with full-length δ-catenin (named as Rv/δ) or GFP vector alone (named as Rv/C) using FuGENE 6 (Roche Scientific, Gaithersburg, MD). For selection of pEGFP-δ-catenin transfected cells, cells were first selected in G418 containing medium. Then, pEGFP-δ-catenin transfected cells were further selected by GFP-based cell sorting using a FACS Vantage (BD Biosciences). The amino-terminal 280 amino acid deleted (Rv/ΔN) or carboxyl-terminal 207 amino acid deleted (Rv/ΔC) δ-catenin cDNA with pEGFP fusion were transfected similarly. The stable cell lines were maintained in RPMI1640 medium containing 25 mM glucose and supplemented with 10% FBS and 0.25% Gentamicin (G418) (Gibco). To spontaneously generate truncation variant, cell cultures were plated in high glucose medium with 10% FBS followed by repeated interruption of medium replenishment. Cells subjected to glucose deprivation were cultured in RPMI1640 medium with 0.25 mM or no glucose and supplemented with 10% dialyzed FBS. All cells were incubated at 37°C in 5% CO2 environment.
To generate the truncated δ-catenin with targeted mutation, we performed in vitro mutagenesis with the following strategy. We took part of δ-catenin sequence from the original pEGFP-δ-catenin vector and made the mutation to “TGGGCTGAGGAATGTGGCAGGC” from “TGGGCTGAGGATGTGGCAGGC” using primer sets as forward primer “AAGGGCTGGGCTGAGGAATGTGGCAGGCATGG” and reverse primer “CCATGCCTGCCACATTCCTCAGCCCAGCCCTT” by PCR. This mutation predicts a stop codon after 138 nucleotide sequence in the mutant δ-catenin. Mutated δ-catenin sequence was verified by sequencing.
Genomic DNA extraction from tissue and PCR amplification of δ-catenin gene
The archival paraffin blocks of radical prostatectomy (RP) cases were performed at the Brody School of Medicine. The clinical pathology analysis regarding Gleason score, tumor stage, and status of surgical margins were assessed as described (7). All 16 prostate cancer and clinically disease-free tissue samples were transferred to the laboratory according to the Institutional Research Board approved protocol # 06-0300. Additionally, two cases of commercial DNA from prostate cancer patients were obtained from Promega.
Genomic DNAs were extracted from fresh tissue using DNeasy Tissue Kit (Qiagen Science, Maryland) or from paraffin embedded primary prostatic adenocarcinoma samples and non-cancer prostate tissues.
PCR analysis was performed to amplify δ-catenin exon sequences using genomic DNA as templates from cells of prostate cancer tissue specimens. The PCR primers and reaction conditions used for detection of δ-catenin DNA sequences were listed as seen in Supplemental materials and methods.
PCR products sequence analysis
The purified PCR products using QIAquick PCR Purification Kit (Qiagen Science, Maryland) were sent for sequencing. The DNA sequences of amplified PCR fragments obtained from prostate cancer tissues were compared to human δ-catenin DNA sequence as reference published in the NCBI database using BLAST Program.
To conduct mutation cluster region (MCR) analysis, mutations deposited in the Sanger cancer sequencing database were combined with the ones in this study. Nucleotide position of the mutations was plotted against the exons in δ-catenin.
Genomic DNA extraction from cultured cell line and PCR amplification of mouse δ-catenin CDS
PCR analysis was performed to amplify δ-catenin exon sequence using genomic DNA as templates isolated from Rv/δ and Rv/C cell lines. The PCR primers and reaction conditions used for detection of mouse δ-catenin cDNA sequences were listed as seen in the Supplemental materials and methods.
Purified PCR products were sequenced and compared to mouse δ-catenin cDNA sequence as reference published in the NCBI database using BLAST Program.
δ-Catenin mutant and Myc transgenic mice
The generation of δ-catenin mutant mice (δ-Cat-/-, we designated it as No Myc/δ-/- in this study) was described earlier (23). This mutant showed expression of δ-catenin with the intact amino-terminal 435 amino acids after a targeted disruption of exon 9. The Myc transgenic mice (ARR2PB-Myc) showed tissue-specific expression of Myc in the mouse prostate, which was described previously (24). The animals were kept under pathogen-free conditions according to the guidelines of East Carolina University Animal Use Protocol. These two mouse strains were cross-bred, and the offspring were aged to determine if prostate lesion developed in the transgene-positive mice.
The genotypes were confirmed by collecting DNA from tail snips after three weeks post-birth. Mouse-tail DNA was subjected to a PCR-based screening assay for genotyping. For Myc transgenic mouse genotype (ARR2PB-Myc), primers were selected as follows: MO45 (5′AAACATGATGACTACCAAGCTTGGC 3′) is specific for the ARR2Pb promoter element, and MO47 (5′ATGATAGCATCTTGTTCTTAGTCTTTTTCTT AATAGGG 3′) is specific for the non-coding region upstream to the probasin promoter, which resulted in a PCR product of 100-200 base pairs. For δ-catenin mutants (No Myc/δ-cat-/-), two different sets of primers were used as follows: Primers for wild type (No Myc/δ-cat+/+) include F1 (GACGTTTGGTTTTCCGAATG) and R3 (AGAGTCAACGGAGGCACAAT), and primers for δ-catenin mutants (No Myc/δ-cat-/-) include Ex9forward (AACCGTTTCTGTGAGCAGGTCC) and GFP1300 reverse (GCTCGTCCATGCCGAGAGTG), resulting in PCR product 2000 and 800 base pairs, respectively.
For histological examination, mice were sacrificed at six months. The prostate tissues were dissected, fixed in 4% paraformaldehyde, and stored at -4°C. Serial tissue sections (5μm thick) were cut from paraffin-embedded blocks and placed on charged glass slides. H&E was performed using standard procedures. All slides were analyzed by light microscopy using a Carl Zeiss microscope equipped with Axiovision Rel 4.8 imaging software.
Immunofluorescent light microscopy
Cultured cells were plated on the coverslips and fixed in warm 4% paraformaldehyde. Following permeabilization in 0.2% Triton X-100, the cells were stained using mouse monoclonal anti-p120ctn. The cells were then incubated with the goat anti-mouse Cy3 conjugated secondary antibodies. The nuclei of the cells were stained with Hoechst 33258. The cover slips were mounted on slides using Anti-Fade medium (Invitrogen) and photographed under the Zeiss Axiovert inverted fluorescent microscope.
To detect cell proliferation and apoptosis, frozen mouse prostate sections were immunostained using anti-Ki67, a marker of cell proliferation and TUNEL assay, which detects cells undergoing apoptosis. Hoechst, a marker for nuclei, was used to stain all cells. Immunofluorescent density analyses were performed using a MetaMorph 4.6 imaging software system (Universal Imaging Corp., West Chester, PA). All data was presented as Mean±SEM and statistically evaluated with a t-test. The confidence level was set at 0.05.
Western blot with ECL detection
Cells for protein analysis were lysed in RIPA buffer (1% Triton x-100, 0.5% Deoxycholic Acid, 0.2% SDS, 150mM sodium chloride, 2mM EDTA) with complete protease inhibitor cocktail tablet (Roche, Germany) and pepstatin A. After removing cell debris by centrifugation, protein concentration was determined using BCA method. The proteins separated by SDS-PAGE were transferred to the nitrocellulose membrane (Optitran, Germany) for Western blot analyses. After incubation in appropriate primary and secondary antibodies, the membranes were developed with ECL detection reagents. Densitometry using Quantity One (Bio-Rad) was carried out to semi-quantitatively measure protein intensity on the Western blots. Statistical analysis was performed to obtain means and standard errors with p-values.
Chromosome spread preparation
Metaphase chromosomes were prepared following the method of Deng et al (49) with minor modification. Briefly, the cells were treated with nocodazole (0.01%) at 37°C for 3 h. After treated with hypotonic solution (0.8 % Sodium Citrate), the cells were prefixed in freshly prepared Carnoy's fixative (methanol: glacial acetic acid = 3:1 v/v) for 10 min. The cells were collected by centrifugation and washed with Carnoy's fixative three times. After the final centrifugation, the supernatant was removed completely and the cell pellet was resuspended in 0.3 ml of Carnoy's fixative, and the optimal cell spreading was accomplished.
Fluoroscence in situ hybridization (FISH)
To determine whether mouse δ-catenin cDNA integrates into multiple chromosomes after transfection and stabilization into cultured CWR22Rv-1 cells, biotin-labeled probe were designed for FISH analysis. The probe for oligonucleotide sequences was as follows and labeled with biotin at 5-terminal: TCGGCATGGACGAGCTGTACAAGTCCGGACTCAGATCTATGTTCGCCAGGAAGCAGTCGG.
FISH was carried out according to Huang et al (50) with some modifications. Briefly, the chromosomes spreads were digested at 37°C with 0.01% pre-warmed pepsin in 10mM HCl for 10 min. After prefixed with 37% formaldehyde for 5 min at room temperature, the chromosome spreads were denatured in a mixture containing 75% formamide and 2 × SSC at 75°C for 15 min, dehydrated with a chilled ethanol series, 70%, 90%, 100%, for 3 min each, and air-dried. The hybridization mixture (10 ng/μl probes, 10% dextran sulfate, and 50% deionized formamide in 2 × SSC) was denatured at 100°C for 15 min and chilled immediately by being put on ice for at least 5 min. Denatured probe was applied onto the slide and DNA-DNA in situ hybridization was carried out in a humidity chamber at 37°C overnight. Following hybridization, the slides were washed in 2 × SSC at 37 °Cfor 10 min, 0.2 × SSC at 53° C for 5 min and, finally, briefly equilibrated with 4× SSC/Tween 20 at room temperature. The slides were then blocked with 3% BSA in 4× SSC/Tween 20 solution at 37°Cfor 30 min. After washed with 4× SSC/Tween 20 at 45° C for 5 min, the slides were incubated with anti-avidin-Cy3 (1:500 dilution in 4 × SSC, 0.1% Tween 20, 1% BSA) at 37 °C for 30 min. Finally, chromosomes were counterstained with Hoechst and mounted with anti-fade medium before imaging acquisition and analysis under Zeiss Axiovert fluorescence light microscope equipped with the MatMorph software.
Cell growth with high glucose or with glucose deprivation
Cells under the normal growth condition with high glucose and FBS for seven days were harvested after trypsinization and mixed with trypan blue at a 1:1 dilution. They were counted using an automated cell counter (Invitrogen, Countness™). All experiments were performed in triplicate. Data are expressed as mean ± SEM. To determine growth after cells piled up, cells were plated in the FBS containing medium for two days and were replenished with fresh FBS, allowing them to grow to near conflency. Then, the cell numbers were counted following an additional five days without fresh FBS replenishment.
Some cell cultures grown under high glucose (25 mM) or low glucose (0.25 mM) with dialyzed FBS were divided into two groups: healthy adherent cells and detached dying cells. The ratio of detached to adherent cells was designated as the death index.
To determine cell death by flow cytometry, Rv/C, Rv/δ, Rv/ΔN, and Rv/ΔC cells were grown to 60% confluency, trypsinized, and centrifuged at 1000 rpm to pellet the cells. The supernatant was carefully removed and the cells were re-suspended in 0.7mL cold 100% ethanol and 0.3mL cold PBS. The cells were stained with propidium iodide before they were taken for flow cytometry analysis. The propidium iodide staining solution was made with 470uL PBS with 5uL RNase A and 25uL stain propidium iodide (1mg/mL in de-ionized water). The cells were incubated in propidium iodide stain for 1 hour. The cell counts were obtained using the software Cell Quest, with the parameters set to include apoptosis as sub-G1 populations. The results were analyzed by taking the mean and standard error results.
Metabolic measurement
Two days before the experimental analysis, Rv/C, Rv/δ, and Rv/M1 cells were plated (20,000 cells/well) onto custom XF24 microplate (Searhorse Bioscience, North Billerica, MA). Some cells were cultured in the presence of normal glucose (25 mM) and 10% FBS, while other cells were cultured in low glucose (0.25 mM) with dialyzed FBS. Cells were rinsed three times in unbuffered Dulbecco's modified essential medium (DMEM), and incubated for 1 h at 37°C without CO2 in 600 μL of unbuffered DMEM (pH 7.4) supplemented with 10mM glucose, 1mM sodium pyruvate. The cell glycolysis (ECR) and mitochondrial oxidative phosphorylation (OXPHOS) (OCR) were measured in real-time using a Seahorse Bioscience XF24 Extracellular Flux Analyzer (Billerica, MA). Following the basal rates of OCR and ECAR measurements, a series of injections of three compounds that affect bioenergetics was introduced which included: 100 μM 2, 4 dinitrophenol (2,4 DNP), 100 mM 2-deoxyglucise (2-DOG), and 1uM rotenone. Each cell type was plated and analyzed by quintuplicate while outliers were removed so the most consistent triplicate data were integrated.
Preparation of nuclear and cytoplasmic protein fractions
In order to prepare the cytoplasmic and nuclear fractions, cultured Rv/C, Rv/δ, and Rv/M cells were resuspended in buffer A containing 15 mM HEPES, pH 7.4, 80 mM KCl, 15 mM NaCl, 5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, and 25 mM sucrose with protease and phosphatase inhibitor cocktails. The cells were lysed in the buffer, homogenized in a syringe with 22 gauge needles, and centrifuged at 1,000 rpm for 10 min at 4 °C. The supernatants were again centrifuged at 21,000 rpm (Beckman coulter Optima™ Max-E ultracentrifuge) for 90 min at 4 °C. The supernatants were used as the cytoplasmic fractions, and the pellets were used as the nuclear fractions.
Supplementary Material
Acknowledgments
We thank Joani Zary Oswald for technical assistance. This work was supported, in part, by DOD grant PC040569 (to Q.L), NIH grants CA111891 and CA165202 (to Q.L), ES016888 (to Y.H.C), and DK073488 and DK074825 (to P.D.N).
Drs. Qun Lu, Yan-Hua Chen, and P. Darrell Neufer are funded by NIH.
Footnotes
Conflicts of Interests: Jongdee Nopparat, Dr. Jiao Zhang, Dr. Jian-Ping Lu, Dr. Donghai Zheng, Dr. Junmin Fan, Dr. Heng Hong, and Christi Boykin declare no conflicts of interests.
References
- 1.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Paffenholz R, Franke WW. Identification and localization of a neurally expressed member of the plakoglobin/armadillo multigene family. Differentiation. 1997;61:293–304. doi: 10.1046/j.1432-0436.1997.6150293.x. [DOI] [PubMed] [Google Scholar]
- 4.Zhou J, Liyanage U, Medina M, Ho C, Simmons AD, Lovett M, et al. Presenilin 1 interaction in the brain with a novel member of the Armadillo family. Neuroreport. 1997;8:2085–2090. doi: 10.1097/00001756-199705260-00054. [DOI] [PubMed] [Google Scholar]
- 5.Lu Q, Paredes M, Medina M, Zhou J, Cavallo R, Peifer M, et al. δ-Catenin, an adhesive junction-associated protein which promotes cell scattering. J Cell Biol. 1999;144:519–532. doi: 10.1083/jcb.144.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burger MJ, Tebay MA, Keith PA, Samaratunga HM, Clements J, Lavin MF, et al. Expression analysis of δ-catenin and prostate-specific membrane antigen: Their potential as diagnostic markers for prostate cancer. Int J Cancer. 2002;100:228–237. doi: 10.1002/ijc.10468. [DOI] [PubMed] [Google Scholar]
- 7.Lu Q, Dobbs LJ, Gregory CW, Lanford GW, Revelo MP, Shappell S, et al. Increased expression of delta-catenin/neural plakophilin-related armadillo protein is associated with the down-regulation and redistribution of E-cadherin and p120ctn in human prostate cancer. Hum Pathol. 2005;36:1037–1048. doi: 10.1016/j.humpath.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 8.Zheng M, Simon R, Mirlacher M, Maurer R, Gasser T, Forster T, et al. TRIO amplification and abundant mRNA expression is associated with invasive tumor growth and rapid tumor cell proliferation in urinary bladder cancer. Am J Pathol. 2004;165:63–69. doi: 10.1016/S0002-9440(10)63275-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang JY, Wang Y, Zhang D, Yang ZQ, Dong XJ, Jiang GY, et al. δ-Catenin promotes malignant phenotype of non-small cell lung cancer by non-competitive binding to E-cadherin with p120ctn in cytoplasm. J Pathol. 2010;222:76–88. doi: 10.1002/path.2742. [DOI] [PubMed] [Google Scholar]
- 10.Kim H, He Y, Yang I, Zeng Y, Kim Y, Seo YW, et al. δ-Catenin promotes E-cadherin processing and activates β-catenin-mediated signaling: Implications on human prostate cancer progression. Biochim Biophys Acta. 2012;1822:509–521. doi: 10.1016/j.bbadis.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu B, Jiang D, Wang P, Gao Y, Sun W, Xiao X, et al. Replication study supports CTNND2 as a susceptibility gene for high myopia. Invest Ophthalmol Vis ScI. 2011;252:8258–8261. doi: 10.1167/iovs.11-7914. [DOI] [PubMed] [Google Scholar]
- 12.Jun G, Moncaster JA, Koutras C, Seshadri S, Buros J, McKee AC, et al. δ-Catenin is genetically and biologically associated with cortical cataract and future Alzheimer-related structural and functional brain changes. PLoS ONE. 2012;7:e43728. doi: 10.1371/journal.pone.0043728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirsch D, Kemmerling R, Davis S, Camps J, Meltzer PS, Ried T, et al. Chromothripsis and focal copy number alterations determine poor outcome in malignant melanoma. Cancer Res. 2012;73:1454–1460. doi: 10.1158/0008-5472.CAN-12-0928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeng Y, Abdallah A, Lu JP, Wang T, Chen YH, Terrian DM, et al. δ-Catenin promotes prostate cancer cell growth and progression by altering cell cycle and survival gene profiles. Mol Cancer. 2009;8:8–19. doi: 10.1186/1476-4598-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westbrook TF, Martin ES, Schlabach MR, Leng Y, Liang AC, Feng B, et al. A genetic screen for candidate tumor suppressors identifies REST. Cell. 2005;121:837–848. doi: 10.1016/j.cell.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 16.Papadopoulos N, Kinzler KW, Vogelstein B. The role of companion diagnostics in the development and use of mutation-targeted cancer therapies. Nat Biotechnol. 2006;24:985–995. doi: 10.1038/nbt1234. [DOI] [PubMed] [Google Scholar]
- 17.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan H, Perry CN, Huang C, Iwai-Kanai E, Carreira RS, Glembotski CC, et al. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol. 2009;296:H470–479. doi: 10.1152/ajpheart.01051.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rios-Doria J, Day KC, Kuefer R, Rashid MG, Chinnaiyan AM, Rubin MA, et al. The role of calpain in the proteolytic cleavage of E-cadherin in prostate and mammary epithelial cells. J Biol Chem. 2003;278:1372–1379. doi: 10.1074/jbc.M208772200. [DOI] [PubMed] [Google Scholar]
- 20.Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. J Cell Biol. 2003;163:525–534. doi: 10.1083/jcb.200307111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baum C, von Kalle C, Staal FJ, Li Z, Fehse B, Schmidt M, et al. Chance or necessity? Insertional mutagenesis in gene therapy and its consequences. Mol Ther. 2004;9:5–13. doi: 10.1016/j.ymthe.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 22.Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Israely I, Costa RM, Xie CW, Silva AJ, Kosik KS, Liu X, et al. Deletion of the neuron-specific protein delta-catenin leads to severe cognitive and synaptic dysfunction. Curr Biol. 2004;14:1657–1663. doi: 10.1016/j.cub.2004.08.065. [DOI] [PubMed] [Google Scholar]
- 24.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–238. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 25.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 26.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: Cancer's double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–4786. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Chen JH, Lu Q. Pro-oncogenic and anti-oncogenic pathways: Opportunities and challenges of cancer therapy. Future Oncol. 2010;6:587–603. doi: 10.2217/fon.10.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeBusk LM, Boelte K, Min Y, Lin PC. Heterozygous deficiency of delta-catenin impairs pathological angiogenesis. J Exp Med. 2010;207:77–84. doi: 10.1084/jem.20091097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu Q. δ-Catenin dysregulation in cancer: Interactions with E-cadherin and beyond. J Pathol. 2010;222:119–123. doi: 10.1002/path.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kouchi Z, Barthet G, Serban G, Georgakopoulos A, Shioi J, Robakis NK. p120 catenin recruits cadherins to gamma-secretase and inhibits production of Abeta peptide. J Biol Chem. 2009;284:1954–1961. doi: 10.1074/jbc.M806250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JS, Bareiss S, Kim KK, Tatum R, Han JR, Jin YH, et al. Presenilin-1 inhibits delta-catenin-induced cellular branching and promotes delta-catenin processing and turnover. Biochem Biophys Res Commun. 2006;351:903–908. doi: 10.1016/j.bbrc.2006.10.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bareiss S, Kim K, Lu Q. Delta-catenin/NPRAP: A new member of the glycogen synthase kinase-3beta signaling complex that promotes beta-catenin turnover in neurons. J Neurosci Res. 2010;88:2350–2363. doi: 10.1002/jnr.22414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh M, Kim H, Yang I, Park JH, Cong WT, Baek MC, et al. GSK-3 phosphorylates delta-catenin and negatively regulates its stability via ubiquitination/proteosome-mediated proteolysis. J Biol Chem. 2009;284:28579–28589. doi: 10.1074/jbc.M109.002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dai SD, Wang Y, Zhang JY, Zhang D, Zhang PX, Jiang GY, et al. Upregulation of δ-catenin is associated with poor prognosis and enhances transcriptional activity through Kaiso in non-small-cell lung cancer. Cancer Sci. 2011;102:95–103. doi: 10.1111/j.1349-7006.2010.01766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim SW, Park JI, Spring CM, Sater AK, Ji H, Otchere AA, et al. Non-canonical Wnt signals are modulated by the Kaiso transcriptional repressor and p120-catenin. Nat Cell Biol. 2004;6:1212–1220. doi: 10.1038/ncb1191. [DOI] [PubMed] [Google Scholar]
- 38.Rodova M, Kelly KF, VanSaun M, Daniel JM, Werle MJ. Regulation of the rapsyn promoter by kaiso and delta-catenin. Mol Cell Biol. 2004;24:7188–7196. doi: 10.1128/MCB.24.16.7188-7196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim K, Sirota A, Chen Yh YH, Jones SB, Dudek R, Lanford GW, et al. Dendrite-like process formation and cytoskeletal remodeling regulated by delta-catenin expression. Exp Cell Res. 2002;275:171–184. doi: 10.1006/excr.2002.5503. [DOI] [PubMed] [Google Scholar]
- 40.Kim H, Han JR, Park J, Oh M, James SE, Chang S, et al. Delta-catenin-induced dendritic morphogenesis. An essential role of p190RhoGEF interaction through Akt1-mediated phosphorylation. J Biol Chem. 2008;283:977–987. doi: 10.1074/jbc.M707158200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim H, Oh M, Lu Q, Kim K. E-Cadherin negatively modulates delta-catenin-induced morphological changes and RhoA activity reduction by competing with p190RhoGEF for delta-catenin. Biochem Biophys Res Commun. 2008;377:636–641. doi: 10.1016/j.bbrc.2008.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang H, Dai SD, Zhang D, Liu D, Zhang FY, Zheng TY, Cui MM, Dai CL. Delta-catenin promotes the proliferation and invasion of colorectal cancer cells by binding to E-cadherin in a competitive manner with p120 catenin. Target Oncol. 2013 Feb 20; doi: 10.1007/s11523-013-0269-6. [DOI] [PubMed] [Google Scholar]
- 43.Wolf A, Keil R, Götzl O, Mun A, Schwarze K, Lederer M, Hüttelmaier S, Hatzfeld M. The armadillo protein p0071 regulates Rho signalling during cytokinesis. Nat Cell Biol. 2006;8:1432–1440. doi: 10.1038/ncb1504. [DOI] [PubMed] [Google Scholar]
- 44.Wang T, Chen YH, Hong H, Zeng Y, Zhang J, Lu JP, Jeansonne B, Lu Q. Increased nucleotide polymorphic changes in the 5′-untranslated region of delta-catenin (CTNND2) gene in prostate cancer. Oncogene. 2009;28(4):555–564. doi: 10.1038/onc.2008.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson KF, Erickson JW, Antonyak MA, Cerione RA. Rho GTPases and their roles in cancer metabolism. Trends Mol Med. 2013;19:74–82. doi: 10.1016/j.molmed.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 47.Gu D, Sater AK, Ji H, Cho K, Clark M, Stratton SA, Barton MC, Lu Q, McCrea PD. Xenopus delta-catenin is essential in early embryogenesis and is functionally linked to cadherins and small GTPases. J Cell Sci. 2009;122(Pt 22):4049–4061. doi: 10.1242/jcs.031948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martinez MC, Ochiishi T, Majewski M, Kosik KS. Dual regulation of neuronal morphogenesis by a delta-catenin-cortactin complex and Rho. J Cell Biol. 2003;162(1):99–111. doi: 10.1083/jcb.200211025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deng W, Tsao SW, Lucas JN, Leung CS, Cheung AL. A new method for improving metaphase chromosome spreading. Cytometry A. 2003;51:46–51. doi: 10.1002/cyto.a.10004. [DOI] [PubMed] [Google Scholar]
- 50.Huang X, Hu J, Hu X, Zhang C, Zhang L, Wang S, et al. Cytogenetic characterization of the bay scallop, Argopecten irradians irradians, by multiple staining techniques and fluorescence in situ hybridization. Genes Genet Syst. 2007;82:257–263. doi: 10.1266/ggs.82.257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.