

Abstract
Background
Erythropoietin is a multifunctional cytokine which regulates the number of erythrocytes circulating in mammalian blood. This is crucial in order to maintain an appropriate oxygen supply throughout the body. Stimulation of primary human erythroid progenitors (PEPs) with erythropoietin (Epo) leads to the activation of the mitogenic kinases (MEKs and Erks). How this is accomplished mechanistically remained unclear.
Results
Biochemical studies with human cord blood-derived PEPs now show that Ras and the class Ib enzyme of the phosphatidylinositol-3 kinase (PI3K) family, PI3K gamma, are activated in response to minimal Epo concentrations. Surprisingly, three structurally different PI3K inhibitors block Ras, MEK and Erk activation in PEPs by Epo. Furthermore, Erk activation in PEPs is insensitive to the inhibition of Raf kinases but suppressed upon PKC inhibition. In contrast, Erk activation induced by stem cell factor, which activates c-Kit in the same cells, is sensitive to Raf inhibition and insensitive to PI3K and PKC inhibitors.
Conclusions
These unexpected findings contrast with previous results in human primary cells using Epo at supraphysiological concentrations and open new doors to eventually understanding how low Epo concentrations mediate the moderate proliferation of erythroid progenitors under homeostatic blood oxygen levels. They indicate that the basal activation of MEKs and Erks in PEPs by minimal concentrations of Epo does not occur through the classical cascade Shc/Grb2/Sos/Ras/Raf/MEK/Erk. Instead, MEKs and Erks are signal mediators of PI3K, probably the recently described PI3K gamma, through a Raf-independent signaling pathway which requires PKC activity. It is likely that higher concentrations of Epo that are induced by hypoxia, for example, following blood loss, lead to additional mitogenic signals which greatly accelerate erythroid progenitor proliferation.
Background
Erythropoietin (Epo) is a multifunctional cytokine [1-4]. It has been known for a long time as a crucial regulator during all stages of definitive erythropoiesis. More recently, Epo was shown to have an important role in the survival of neurons after stress and injury [5-7]. Epo drives not only the proliferation of already committed early erythroid progenitor cells (burst-forming unit-erythroid; BFU-E), but also, and prominently, the proliferation and differentiation of later stage cells (colony-forming unit-erythroid; CFU-E) towards mature erythrocytes [1,8].
Much of the circulating Epo is produced in the kidneys where blood oxygen levels are monitored, but other sites of Epo production – for example, liver and brain – are also known [9]. Several well-characterized signaling molecules such as the hypoxia-induced transcription factor HIF-1α and the 'stress kinase' p38α are key players in regulating Epo expression [9-11]. Epo concentrations of 25–50 mU/ml are found in umbilical cord blood at birth. In adults, Epo is typically present at 10–30 mU/ml, but levels can rise up to 3–10 U/ml as a consequence of severe blood loss.
Epo binds a transmembrane receptor protein (EpoR) that lacks intrinsic enzymatic activity and associates instead with tyrosine kinases like Jak2 [2,3,8,12-14]. Targeted disruptions of the genes for Epo or the EpoR in mice leads to a complete loss of the definitive embryonal erythropoiesis [15,16]. Other important clues regarding molecules relevant for Epo-induced signaling have come from disruptions of genes for Jak2, SHP2, PLC-γ, STAT5a/b, and GATA-1 and -2 [17-21]. While these knockout studies have provided considerable insight into key players in Epo-induced signaling, mice are not an ideal system for extensive biochemical analyses because the number of erythroid progenitors that can be readily obtained from them is not sufficient.
Therefore, most biochemical studies aiming to unravel the detailed molecular mechanisms of EpoR signaling have so far been carried out with cell lines expressing an endogenous or stably transfected EpoR (UT-7, SKT6, HEL, F-36P, HCD57, JE-2, AS-E2, K562, Friend cells, Ba/F3-EpoR, 32D-EpoR, FDCP-EpoR, etc.). Moreover, many of these experiments have been done with 'pathophysiological' concentrations of Epo above 1 U/ml.
In the many cell lines analyzed, a plethora of diverse signaling molecules appears to be crucial for Epo signaling. It is obvious that most discrepancies in the essential signaling proteins reported reflect the genomic instability of the various cancer cell lines, as well as distinct pre-set wiring diagrams of EpoR-transfected hematopoietic progenitor cells. Thus, these findings are important in defining candidate pathways potentially involved in vivo, but they do not necessarily represent actual signals induced upon Epo stimulation of primary human erythroid progenitors (PEPs). Consequently, although EpoR signaling has been intensely studied, many of its aspects are still unknown or remain puzzling. For example, it is certain that a large complex of signaling proteins is assembled on the EpoR upon Epo stimulation of various Epo-responsive cell lines as well as in vivo, and it is also known that many components of this complex directly associate with phosphorylated tyrosines of the EpoR. On the other hand, these tyrosines can be spatially separated from the Jak2 binding site on the EpoR or even abolished without apparent severe disruption of EpoR function [22-26].
Phosphatidylinositol-3 kinases (PI3Ks) [27-29] have been implicated in EpoR signaling by precipitation experiments which focussed on the formation of complexes between the p85/p110 PI3Ks (α and β isoforms) and the EpoR [13,30-32] and by the use of inhibitors like LY294002 and wortmannin [33-37], which are mechanistically and structurally distinct [38,39] but lack prominent selectivity for specific class I PI3K family members. The results from these experiments have not yet provided clear evidence of an essential function for p85/p110 PI3Ks in the activation of MEKs and Erks upon stimulation of cells with Epo. Only recently, caffeine has been described as an inhibitor of PI3K activity with a clear preference for certain PI3K family members in vitro [40]. In combination with isoform-specific PI3K activity assays, this now allows one to look at specific PI3K proteins more closely.
For many years it was difficult to perform in-depth biochemical studies with PEPs since methods for their efficient in vitro expansion were quite limited. Fortunately, it has now become possible to recapitulate the progression of hematopoietic progenitor cells into the erythroid lineage under serum-free conditions using defined cytokine cocktails [41]. This permits massive amplification and erythroid differentiation of CD34-positive cells isolated from human cord blood, bone marrow or peripheral blood, thereby facilitating biochemical analyses like GTPase and kinase assays. The PEPs generated by this procedure were shown to be 98% CD36 (thrombospondin receptor)-positive as well as CD71 (transferrin receptor)-high and greatly, but not exclusively, dependent on the presence of Epo for proliferation [41]. First biochemical experiments from this study showed that upon stimulation of starved PEPs with Epo, the EpoR, Jak2 and STAT5 were phosphorylated [41].
The aim of the current study was to analyze in more detail how MEKs and Erks are activated in PEPs upon stimulation with minimal concentrations of Epo. As anticipated, it was found that Ras is activated in these cells. However, Raf family kinases (c-Raf1, A-Raf, B-Raf), the classical effector proteins of Ras are not crucial for the subsequent activation of MEKs and Erks by Epo, while stimulation of c-Kit by stem cell factor (SCF) activates Erks in a Raf-dependent manner. Importantly, Ras, MEK and Erk activation by Epo clearly depends on PI3K activity. Inhibitor studies and lipid kinase assays implicate PI3K gamma (PI3Kγ) (p101/p110γ) as the prime candidate for the Ras, MEK and Erk-activating signal transducer.
Results
PI3 kinase-dependent activation of Ras, MEK and Erk by minimal concentrations of Epo
In initial experiments it was determined which concentrations of Epo are necessary to activate STAT5 and Erks in PEPs. For this, the cells were stimulated with increasing concentrations of Epo for different lengths of time and then analyzed with activation-specific phospho-epitope antibodies for STAT5, MEKs and Erks. Activation (that is, GTP-loading) of the Ras GTPase was also analyzed by using affinity precipitation with a GST-fusion protein that contains the Ras binding domain (RBD) of the kinase c-Raf1. Activation states were further investigated in normally growing and mock-stimulated PEPs. Since Epo-induced signals commonly peaked at around 10 min, PEPs were harvested at that time in all subsequent studies. While Ras, MEKs and Erks were substantially activated at 0.3 U/ml Epo (Figure 1), activation of STAT5 was already seen at 0.01 U/ml Epo. In contrast to Ras, activation of the Rac or Rap1 GTPases upon Epo-stimulation was not observed (our unpublished data).
Figure 1.
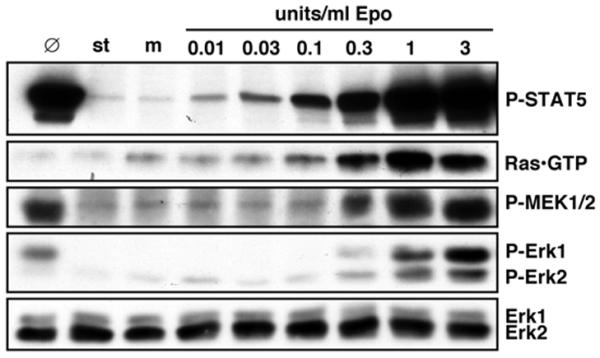
Dose-dependent activation of STAT5, Ras, MEKs and Erks by Epo in primary erythroid progenitors (PEPs). Starved (st) PEPs were mock-stimulated (m) or stimulated with various concentrations of Epo for 10 min as indicated. 100 μg of total cell protein was then separated by SDS-PAGE and immunoblotted with anti-P-STAT5, anti-P-MEK1/2, anti-P-Erk1/2 or anti-Erk1/2. GTP-loaded Ras was precipitated with GST-c-Raf1 RBD from 500 μg total cell protein and immunoblotted with anti-Ras. Ø indicates protein extracts from non-starved and non-treated PEPs (cultured in the presence of IL-3, IL-6, SCF and Epo).
Since MEKs are reputed to signal directly upstream of Erks, it was investigated and confirmed that the widely-used MEK inhibitor compound U0126 blocked Erk activation by Epo (data not shown). During the course of these experiments, another inhibitor LY294002 (LY) that inhibits PI3Ks, which was not expected to affect Erk activation, was also tested. Surprisingly, not only the phosphorylation of the kinase Akt, a target of PI3Ks, on Ser 473 was inhibited (Figure 2A), but also a block of Erk activation was seen with LY, albeit at higher concentrations. Dose-dependent Erk inhibition was further observed with the structurally and mechanistically distinct PI3K inhibitor wortmannin (WM), again at concentrations somewhat higher than those required to suppress Epo effects on Akt Ser473 phosphorylation (Figure 2A). Phospho-Erk inhibition by LY and WM was also not found when PEPs were stimulated with low concentrations of the c-Kit ligand SCF, suggesting that this is an Epo-specific signal (data not shown). WM also inhibited the phosphorylation of the Akt targets GSK3α and GSK3β and the activation of MEKs (Figure 2B). In contrast, effects on tyrosine phosphorylations of STAT5 (Figure 2A) or EpoR by WM were not detected (Figure 2B, top panel), and autophosphorylation of tyrosines 1007 and 1008 in the EpoR-associated kinase Jak2 was not inhibited (data not shown).
Figure 2.
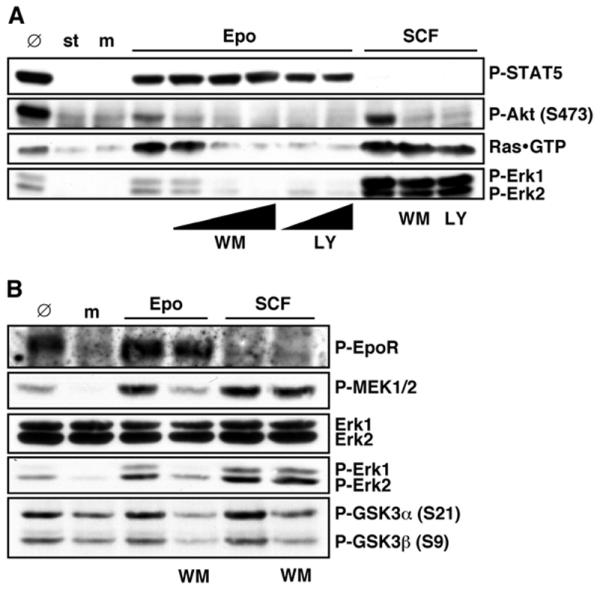
Structurally and mechanistically distinct PI3 kinase inhibitors prevent Epo-induced activation of Ras, MEKs and Erks. (A) Starved (st) PEPs were mock-stimulated (m) or pretreated with 0, 25, 50 or 100 nM wortmannin (WM) for 30 min or with 30 or 100 μM LY294002 (LY) for 1 h and then stimulated with 0.3 U/ml Epo where indicated. For comparison, PEPs starved and pretreated with 100 nM WM or 100 μM LY where indicated were stimulated with 25 ng/ml stem cell factor (SCF) for 10 min to activate c-Kit signaling. 100 μg total cell protein were immunoblotted with P-STAT5, P-Akt or P-Erk1/2 antibodies as indicated. GTP-loaded Ras was precipitated with GST-c-Raf1 RBD from 500 μg total cell protein and immunoblotted with anti-Ras. Ø indicates non-starved and non-treated PEPs. (B) PEPs pretreated with 100 nM WM for 30 min where indicated were mock-stimulated (m) or treated with 0.3 U/ml Epo or 25 ng/ml SCF for 10 min. 100 μg total cell protein were immunoblotted with P-MEK1/2, Erk1/2, P-Erk1/2 or P-GSK3α/β antibodies as indicated. Phosphorylated EpoR was immunoprecipitated with anti-phosphotyrosine mAb (4G10) from 500 μg cell protein and immunoblotted with anti-EpoR. Ø indicates non-starved and non-treated PEPs.
Ras is activated upon Epo treatment of PEPs (Figure 2A) and often upstream of MEKs. Consequently, the effects LY and WM have on GTP-loading of Ras (Figure 2A) were also investigated. Both inhibitors fully blocked Ras activation by Epo but not by SCF, indicating that Ras is downstream of a PI3K activity in Epo-stimulated PEPs. This mode of signal transmission from PI3K to Ras is distinct from signaling routes described for many other cell types or stimuli but not unprecedented, since the PI3Ks can, for example, induce the release of intracellular calcium, which is known to regulate Ras via Pyk2 and Ras-GRFs [42,43].
PI3 kinase gamma (PI3Kγ) is activated by Epo
PI3Ks are a diverse family of differentially regulated enzymes, which consists of three classes or subfamilies [29]. Of these, class I PI3Ks are frequently involved in the signal transmission of growth factors, hormones and cytokines; which type of class I PI3Ks might be responsible for transmitting Epo signals was therefore analyzed. The regulatory p85 subunits of the class Ia PI3Ks (α, β and δ) are known to translocate to the cell membrane in response to various stimuli. For this reason it was initially investigated if this occurred following Epo stimulation of PEPs, but no translocation was detectable. In activated p85/p110 PI3Ks, the p85 subunits are often phosphorylated on tyrosines and/or associated with various other tyrosine-phosphorylated proteins. Active p85/p110 PI3Ks can therefore be precipitated with antibodies against phosphotyrosine and activity detected by phosphatidylinositol (PI) kinase assay. Analyses of PEPs stimulated with Epo or SCF showed that SCF leads to a strong p85/p110 PI3K activation which can be inhibited by pretreatment with WM. In comparison with SCF, only low PI3K activation was detectable after Epo treatment (Figure 3A,3B). We were unable to detect significant activation of PI3Kα or β in PEPs treated with 0.3 U/ml Epo upon immunoprecipitation of the catalytical subunits. Furthermore, there are at present no isoform-specific inhibitors of PI3Ks available. Hence, it is not possible to analyze the class Ia p85/p110 PI3Ks in more detail at this stage.
Figure 3.
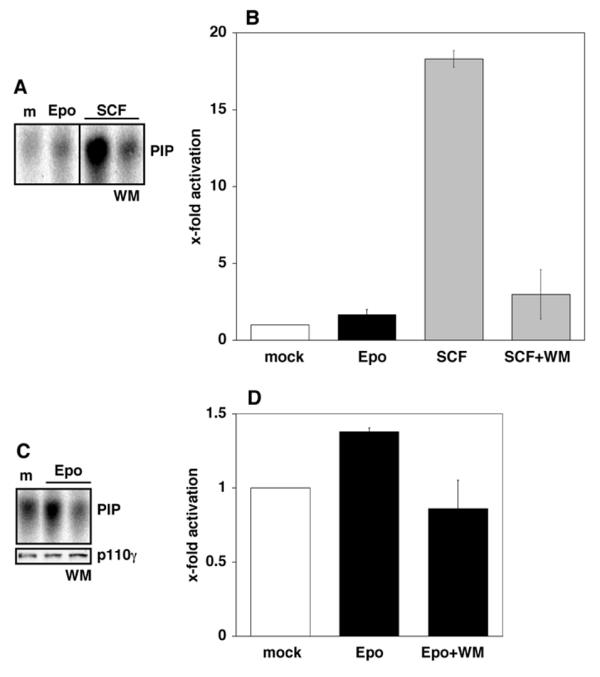
Strong activation of class Ia PI3 kinases by SCF, but PI3Kγ activation by Epo. Starved PEPs were mock-stimulated (m), stimulated with 0.3 U/ml Epo or with 25 ng/ml SCF with or without pretreatment with 100 nM WM as indicated. (A, B) The tyrosine-phosphorylated p85 regulatory subunits of activated class Ia PI3K enzymes (PI3Kα, β, δ) were immunoprecipitated from 500 μg total cell proteins with anti-phosphotyrosine mAb (4G10). To detect PI3K inositol kinase activity, immunoprecipitates were incubated with phosphatidylinositol (PI) and 32P-γ-ATP. Phosphatidylinositol phosphate (PIP) generated by active PI3Ks was separated from ATP by thin layer chromatography (TLC) and analyzed by phosphoimaging. A representative example of the results from the phosphoimaged experiments is shown in (A) and the quantitative analysis of the results obtained with PEPs from three different cord blood samples in (B). Epo and SCF significantly activate class I PI3Ks (pEpo < 0.01; pSCF < 0.001) and WM significantly inhibits the SCF-induced activation (p < 0.01). (C, D) Starved PEPs were mock stimulated (m), stimulated with 0.3 U/ml Epo with or without pretreatment with 100 nM WM as indicated. PI3 kinase activity was detected as in (A) and (B) but using anti-p110γ to immunoprecipitate the catalytic subunit of PI3Kγ. A representative example of the results from the phosphoimaged experiments is shown in (C) (upper panel) and the quantitative analysis of the results obtained with PEPs from three different cord blood samples in (D). Significant PI3Kγ activation (p < 0.001) and inhibition by WM (p < 0.001) was determined. Equal PI3Kγ immunoprecipitation was confirmed by western blot (C, lower panel).
The class Ib PI3K, PI3Kγ, is not regulated by p85 or similar subunits and instead interacts with a distinct regulatory protein, p101. There is at present no evidence that PI3Kγ is regulated by tyrosine phosphorylation. Immunoprecipitation of the catalytic subunit PI3Kγ p110 from protein extracts of mock-stimulated or Epo-stimulated PEPs showed a moderate but statistically significant and highly reproducible activation of PI3Kγ upon Epo treatment. This effect was inhibited in the presence of WM (Figure 3C,3D).
It has recently been shown that caffeine differentially inhibits class I PI3Ks in vitro and that PI3Kγ is the least sensitive isoform [40]. Pretreatment of PEPs with caffeine prior to Epo stimulation showed that Erk activation is only abolished at very high concentrations of caffeine (10 mM) (Figure 4A). As expected, SCF-induced Erk activation was not affected by caffeine, but phosphorylation of Akt and its target GSK3α/β was blocked.
Figure 4.
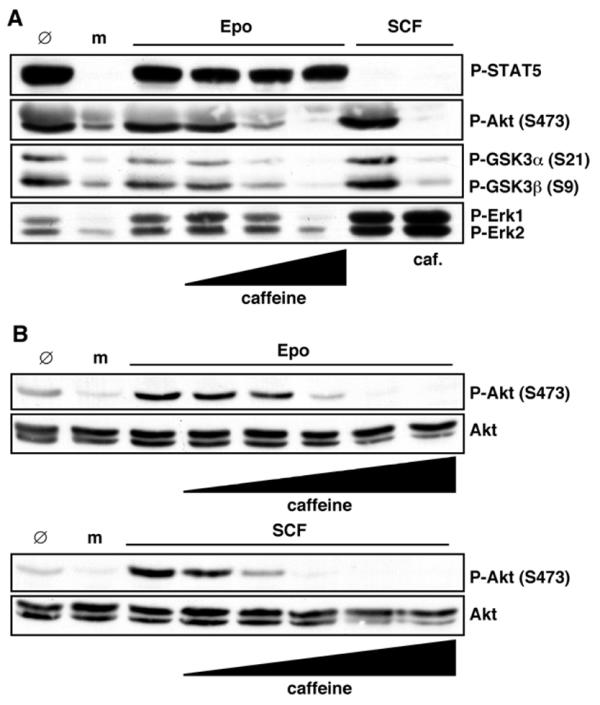
The PI3 kinase inhibitor caffeine inhibits Epo-induced Akt, GSK3α/β and Erk1/2 activation. Starved PEPs were mock-stimulated (m) or treated with 0.3 U/ml Epo or 25 ng/ml SCF as indicated. Some of the samples were pretreated with caffeine as detailed below. 100 μg cell protein were immunoblotted with P-STAT5, P-Akt, P-GSK3 α/β, P-Erk1/2 or Akt antibodies. Ø-lanes represent non-starved and non-treated PEPs. (A) Cells were pretreated with 0.1, 1 or 10 mM caffeine for 1 h before Epo stimulation and with 10 mM before SCF stimulation where indicated or (B) with 0.1, 0.3, 1, 3 or 10 mM caffeine for 1 h.
Since the p85 regulated isoforms of class I PI3Ks (α, β, and δ) are more sensitive to caffeine than PI3Kγ, we speculated that Akt phosphorylation induced by SCF may be blocked by lower caffeine concentrations compared to Akt phosphorylation induced by Epo. This was subsequently confirmed by a more detailed titration of the caffeine concentrations used to pretreat the PEPs (Figure 4B). From these results it is concluded that PI3K activation is essential for activation of MEKs and Erks at threshold concentrations of Epo, and PI3Kγ appears to be the prime candidate for transducing these signals. This emerging EpoR signaling pathway could explain the puzzling finding that EpoR mutants devoid of docking sites for class Ia PI3K p85 SH2 domains and classical Ras activator modules (like Shc/Grb2/SoS) are still able to transmit Epo signals and apparently do not compromise the viability of the mutant mice [22-25]. Importantly, this hypothesis does not preclude the notion that higher concentrations of Epo lead to additional, biologically significant signals being transmitted via classical mitogenic and anti-apoptotic pathways to accelerate PEP proliferation and increase PEP survival.
Activation of B-Raf by Epo is not essential for MEK and Erk activation
c-Raf1 has recently been shown to play a role in regulating the differentiation of erythroid progenitors in mice [44]. The PI3K-dependent activation of Ras by Epo now raised the question of whether Raf kinases are also essential to mediate signaling in human PEPs from Ras to MEKs and Erks. In initial experiments, different phospho-specific antibodies that recognize phosphorylated epitopes in c-Raf1 (Ser259, Ser338, Tyr340Tyr341) showed no apparent changes in c-Raf1 phosphorylation (data not shown). Subsequently, coupled Raf-MEK kinase assays with kinase inactive GST-Erk1K63M as substrate were used to analyze the different Raf family kinases. c-Raf1 and, in particular, B-Raf were moderately activated upon Epo stimulation of PEPs. The activation of B-Raf was reduced by WM pretreatment of the PEPs (Figure 5). No Epo-induced activity changes were observed with immunoprecipitated A-Raf (our unpublished data).
Figure 5.
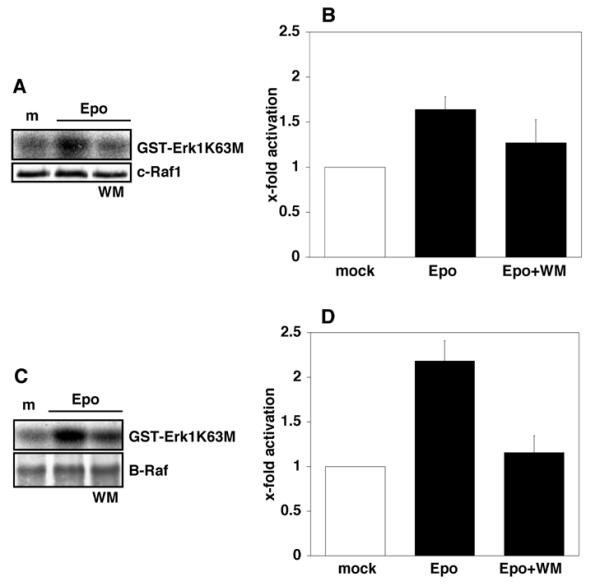
Activation of B-Raf by Epo is blocked by wortmannin. PEPs were mock-stimulated (m) or stimulated with 0.3 U/ml Epo or pretreated with 100 nM WM where indicated and then stimulated with Epo. (A, B) c-Raf1 was immunoprecipitated from 500 μg total cell protein with anti-c-Raf1. Precipitates were immunoblotted with anti-c-Raf1 for IP-control (A, lower panel) or incubated with GST-MEK and subsequently GST-ErkK63M and 32P-γ-ATP for coupled kinase assay. Proteins were separated by SDS-PAGE and phosphorylated GST-Erk1K63M analyzed by phosphoimaging. A representative example is shown in (A) (upper panel). Quantification of c-Raf1 activation from experiments with three different cord bloods is shown in B (pactivation < 0.001; pinhibition < 0.05). (C, D) B-Raf activation was analyzed as described in (A) and (B) but anti-B-Raf was used for immunoprecipitation and immunoblotting. Both activation and inhibition were statistically highly significant (pactivation < 0.001; pinhibition < 0.01).
To determine whether Raf kinases are crucial for MEK and Erk activation, PEPs were pretreated with the compound ZM336372 (ZM), a potent and specific Raf inhibitor [45]. B-Raf activation by Epo, as measured by the coupled in vitro kinase assay, was found to be fully blocked by pretreatment of PEPs with ZM (Figure 6A,6B). ZM pretreatment also suppressed SCF-induced phosphorylation of MEKs and Erks (Figure 6C). In contrast, Epo-mediated activation of MEKs and Erks was not affected by ZM pretreatment of PEPs. ZM treatment alone did not activate Erks in PEPs (data not shown). These data show that Raf kinases are not crucial for MEK and Erk activation in PEPs at low concentrations of Epo.
Figure 6.
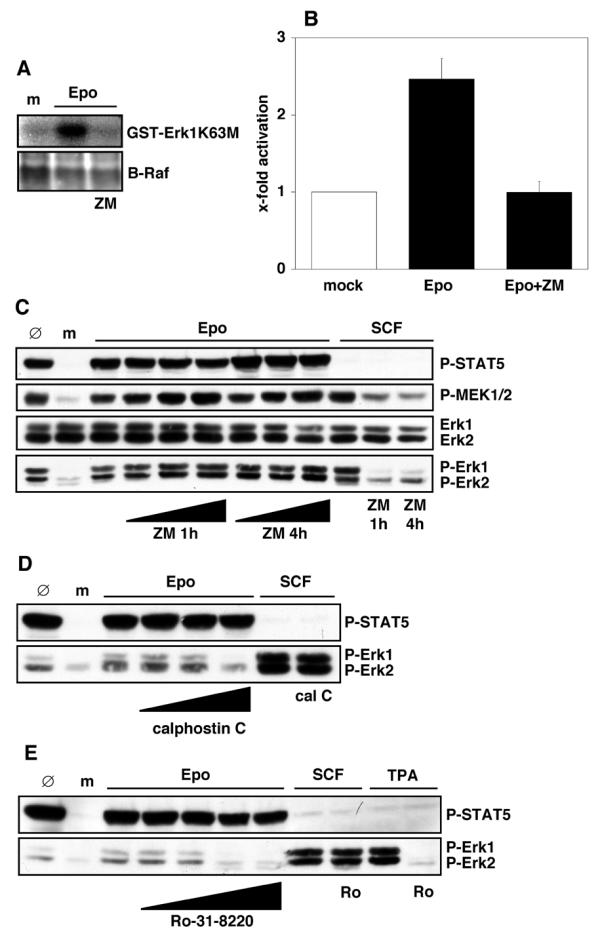
Inhibition of Erks by PKC inhibitors but not by Raf inhibitor. (A, B) Starved PEPs were mock-stimulated (m) or stimulated with 0.3 U/ml Epo with or without pretreatment with 10 μM ZM336372 (ZM) for 1 h. B-Raf was immunoprecipitated from 500 μg total cell protein with anti-B-Raf. Precipitates were immunoblotted with anti-B-Raf for IP-control (A, lower panel) or incubated with GST-MEK and subsequently GST-ErkK63M and 32P-γ-ATP for coupled kinase assay. Proteins were separated by SDS-PAGE and phosphorylated GST-Erk1K63M analyzed by phosphoimaging. A representative example is shown in (A) (upper panel). Quantification of B-Raf activation from experiments with three different cord bloods is shown in (B) (pactivation < 0.01; pinhibition < 0.05). (C) Starved PEPs were mock-stimulated (m) or pretreated with 0, 0.1, 1 or 10 μM ZM for 1 h or 4 h and stimulated with 0.3 U/ml Epo as indicated. Other cells were pretreated with 10 μM ZM and then stimulated with 25 ng/ml SCF. 100 μg total cell protein were immunoblotted with anti-P-STAT5, anti-P-MEK1/2, anti-Erk1/2 or anti-P-Erk1/2. Ø-lanes represent untreated PEPs. (D, E) Starved PEPs were mock-stimulated (m) or pretreated with PKC inhibitors and stimulated with 0.3 U/ml Epo or 25 ng/ml SCF. 100 μg of total cell protein were immunoblotted with anti-P-STAT5 and anti-P-Erk1/2. Inhibitor pretreatment was with 10, 100 or 1000 nM calphostin C for 1 h before Epo stimulation and with 1000 nM calphostin C before SCF stimulation. Other samples were pretreated with 0.1, 1, 10 or 100 μM Ro-31-8220 and stimulated with 0.3 U/ml Epo, or they were pretreated with 100 μM Ro-31-8220 where indicated and stimulated with 25 ng/ml SCF or 100 μM TPA for 10 min. 100 μg total cell protein were immunoblotted with anti-P-STAT5 or anti-P-Erk1/2. Ø-lanes represent untreated PEPs.
PKC inhibitors block Epo-induced activation of MEKs and Erks
An alternative mechanism to activate MEKs and Erks independent of Rafs is through protein kinase C (PKC) family enzymes. Since two members of this family, PKCα and PKCβ, are upregulated during the maturation of hematopoietic progenitor cells along the erythroid lineage [46], these kinases were analyzed by in vitro kinase assays after Epo stimulation. However, we were unable to observe an activation of PKCα or PKCβ when compared with control PEPs. Moreover, phospho-specific antibodies directed against phosphorylated epitopes of different PKC family members (PKCα, βII Thr638/Thr641; PKCδ Thr505; PKCδ Ser603; PKD/PKCμ Ser744/Ser748; PKD/PKCμSer916; PKCΘ Thr538; PKCζ/λ Thr403/Thr410) did not reveal Epo-induced changes (data not shown). Therefore, calphostin C and Ro-31-8220, two widely used and structurally distinct inhibitors which affect a broad spectrum of PKC family kinases, were tested for their ability to prevent Epo-stimulated activation of MEKs and Erks. Both inhibitors blocked Erk activation by Epo and by tetradecanoylphorbol 13-acetate (TPA), a strong activator for many PKCs, but not Erk activation induced by SCF (Figure 6D,6E, lower panels). Neither Epo-induced STAT5 phosphorylation (Figure 6D,6E, upper panels) nor tyrosine phosphorylation of Jak2 (data not shown) were affected.
Western blot analyses of total protein extracts showed that most isoforms of this large kinase family are expressed in PEPs (unpublished results). Therefore, it could not be rapidly assessed by in vitro kinase assays which PKC isoforms are activated by Epo. However, in many PKC signaling events an intracellular translocation of the activated PKCs to a distinct compartment occurs. Consequently, changes in the subcellular localization of various specific PKC isoforms expressed in PEPs (α, β, δ, ε, η, θ, λ, ι, μ, ζ) were analyzed by cell fractionation following mock treatment or stimulation with Epo or SCF, or after stimulation with TPA. Most PKC isoforms showed prominent translocation upon TPA treatment, but we were unable to detect changes after Epo stimulation (data not shown). Therefore, it remains to be determined which PKC isoform is essential for activation of MEKs and Erks.
Discussion
It is well known that Epo-induced signals synergize with those of other cytokines, growth factors and hormones in vivo. To dissect out the contribution made by Epo to the signaling in PEPs, Epo-induced signaling should be analyzed under serum-free culture conditions to yield results unbiased by the poorly understood actions of other factors contained at variable concentrations in serum. This source of variation was eliminated in the current study by the consistent use of a defined cytokine cocktail throughout all cell culturing. Another potential source of variability is the heterogeneous genetic background of the cord blood donors. However, this did not appear to greatly influence the signaling events that were observed in the current study.
The results of the experiments presented here implicate PI3Ks and, in particular, PI3Kγ as crucial mediators of signaling to MEKs and Erks at low Epo concentrations. This newly emerging EpoR signaling pathway is summarized and compared to c-Kit signaling in Figure 7. Since PKC activation influences MEK and Erk phosphorylation (Figure 6) it is possible that PKC kinases act as mediators between PI3Kγ and MAPKs. It is also conceivable that PKCs are activated in a pathway parallel to PI3Kγ and that these two pathways converge to activate MEKs.
Figure 7.
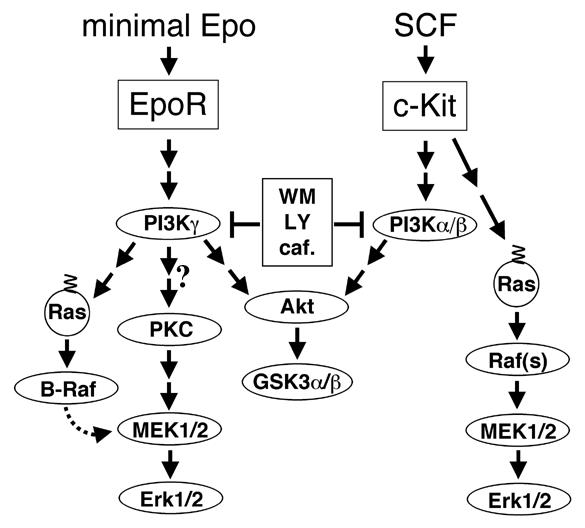
Schematic model of signaling events to MEKs and Erks induced by threshold concentrations of Epo. This basic Epo signal can be amplified or modulated by various other signaling pathways (not shown here) which become activated upon higher Epo concentrations and/or other factors and will often depend on SH2 domain interactions with the phosphorylated tyrosines in the cytoplasmic EpoR tail. PKCs could function as signal transducers for PI3Kγ, but it is also possible that PKCs are activated in a parallel pathway to PI3Kγ and that these two pathways converge to activate MEKs. B-Raf kinase does not significantly promote MAPK activation at low Epo concentrations, but since it is readily activated, it could play a role in signaling events induced by higher Epo concentrations.
Previous studies had shown that PI3Kγ functions in the signal transmission of the Gβγ subunits of heterotrimeric G protein-linked receptors to MAPKs [47-49]. Its regulatory subunit p101 associates tightly with Gβγs leading to a strong activation of PI3Kγ by Gβγs [50]. Roles for PI3Kγ in inflammation and allergies have been documented in other studies [51]. Furthermore, a direct in vitro interaction of Ras with PI3Kγ [52,53] has been reported. We have so far been unable to detect stable complex formations of the p110 or p101 subunits of PI3Kγ with Ras, Jak2 or the EpoR by co-immunoprecipitation experiments. Exactly how Epo stimulation of cells leads to an activation of PI3Kγ remains to be clarified. Studies by Mayeux and colleagues with Epo-responsive cell lines recently showed a link between EpoR signaling and heterotrimeric G proteins [54,55] but whether a similar link exists in PEPs is not yet certain.
In another set of experiments, relatively little elevation of tyrosine phosphorylation was detected upon stimulation of PEPs with 0.3 U/ml Epo. Nevertheless, it was possible to identify a 150 kDa phosphotyrosyl-protein as the inositolphosphatase p150 SHIP, which exists in a complex with Grb2 and Shc as determined by co-immunoprecipitations (our unpublished data). Whether or not this phosphorylation is functionally significant remains to be studied. However, the finding that SHIP is affected by Epo treatment raises an interesting possibility; it was recently shown for a variety of protein phosphatases that they become transiently inactivated by reversible oxidation upon recruitment to active receptor complexes. Moderate activation of PI3Kγ in combination with an inactivation of SHIP could lead to a massive increase in signal-transducing phosphoinositides.
Finally, PI3Kγ not only displays PI kinase activity but can also function as a protein kinase [48]. At present it is not clear whether the protein kinase activity of PI3Kγ plays a role in Epo signaling, for example in the activation of a PKC or Ras.
In summary, our results lead us to propose a new model for Epo signaling in which PI3Kγ provides a basal mechanism to transmit Epo signals to Ras, MEKs and Erks, possibly independent of the multiple tyrosines in the cytoplasmic tail of the EpoR. Whether PI3Kγ is activated through direct or indirect interactions with the EpoR remains to be determined.
The basal Epo signal might be modulated and amplified by other signaling pathways activated by higher concentrations of Epo which depend on phosphorylated EpoR tyrosines. If this model is correct, one would expect that the absence of the EpoR tyrosines would lead to an impaired response when the erythroid cell compartment is challenged by severe blood loss or hemolysis. While this manuscript was being prepared, a novel study by Wojchowski and co-workers reported exactly this finding in mice with a truncated and mutated EpoR devoid of the tyrosine residues which are known to serve as docking sites for various SH2 domain-containing signaling proteins [56].
Conclusions
Erythropoietin is a key regulator of erythropoiesis and drives progenitor cell proliferation as well as differentiation. The signaling mechanism through which Epo activates the mitogenic kinases in primary erythroid progenitors (PEPs) was until now largely unclear, in part because only very few in-depth biochemical studies with primary progenitors have been done so far. Our studies identify a novel signaling pathway from erythropoietin to the mitogenic MEK and Erk kinases that requires only minimal amounts of Epo and is thus believed to reflect the continuous signaling that occurs under conditions of blood homeostasis. Specifically, minimal levels of Epo which lead to a basal activation of the MEK and Erk kinases moderately activate the class Ib PI3 kinase isoform PI3Kγ. This is, to our knowledge, the first time that the activity of endogenously expressed PI3Kγ has been quantified. Three different PI3K inhibitors, which are structurally and mechanistically distinct, unexpectedly showed that PI3K activation is essential for MEK and Erk activation. Moreover, we report for the first time Epo-induced Ras activation in PEPs. Surprisingly, Ras activation is also dependent on PI3K activity. This differs greatly from the signaling mechanism for Ras in many other contexts, where class Ia PI3Ks are downstream of Ras. The new pathway described here could be independent of phosphotyrosyl-SH2 domain interactions that are seen in many receptor signaling pathways. This would explain why mice with a mutant Epo receptor that lacks all SH2 domain docking sites are still viable and have a normal hematocrit when unchallenged.
Methods
Purification and amplification of primary human erythroid progenitors
Primary human erythroid progenitors were purified as described in Freyssinier et al. [41]. Briefly, umbilical cord bloods (approximately 80 ml) were diluted 1:1 with phosphate buffered saline (PBS) containing 4% fetal bovine serum (FBS). After Histopaque-1077 (Sigma, Poole, UK) density gradient centrifugation, the low density cells were recovered and CD34+ cells purified by immunomagnetic cell separation (MACS, CD34+ isolation kit, 467-01; Miltenyi Biotec, Bergisch Gladbach, Germany). Selected CD34+ cells were cultured in 5% CO2 at 37°C for seven days in serum-free RM-B00 cell expansion medium (00201; RTM/MABIO-International, Tourcoing, France; does not contain dexamethasone,) with 10 ng/ml interleukin (IL)-3, 10 ng/ml IL-6 and 25 ng/ml SCF (all from PeproTech EC Ltd, London, UK). After seven days in culture, CD36+ cells were immunomagnetically isolated using a mouse anti-CD36+ mAb (0765; Immunotech, Marseille, France) and rat anti-mouse IgG1 microbeads (471-01; Miltenyi Biotec). CD36+ cells were then further cultured with IL-3, IL-6, SCF and 2 U/ml Epo (Janssen-CILAG, Saunderton, UK) for four days.
Cell stimulation, lysis and protein quantification
On day 7+4 CD36+ cells were starved for 6 h in RM-B00 medium and stimulated with 0.3 U/ml Epo, 25 ng/ml SCF or 100 μM TPA (Calbiochem, Nottingham, UK) for 10 min, unless indicated otherwise. Mock incubations were done with the appropriate solvents for cytokines or chemicals (H2O, RM-B00 medium and/or DMSO).
For lipid and protein kinase inhibition experiments, cells were pretreated with UO126 (Cell Signaling Technology/NEB, Hitchin, UK), wortmannin, caffeine (both Sigma), LY294002, ZM336372, calphostin C or Ro-31-8220 (all from Calbiochem) as indicated. Total cell protein extracts were prepared by solubilizing in Igepal CA-630 (syn. Nonidet P-40, NP-40; I 3021, Sigma) lysis buffer containing 1% (v/v) NP-40, 50 mM HEPES pH 7.5, 10% (v/v) glycerol, 150 mM NaCl, 2 mM EDTA, 50 mM sodium fluoride (NaF), 10 mM sodium pyruvate, 1 mM sodium vanadate (Na3VO4), 1 mM phenylmethylsulfonylfluoride (PMSF), 10 μg/ml aprotinin, 5 μg/ml antipain, 0.5 μg/ml leupeptin, 0.7 μg/ml pepstatin A, 1 × Complete™ inhibitors (1697498; Roche, Mannheim, Germany) and 3 mM dithiotreitol (DTT). After 30 min of nutation in the cold, lysates were cleared by centrifugation for 20 min at 2°C with 20,000 × g and supernatants further analyzed. Protein concentrations were determined with the Bradford dye binding assay for all extracts made.
Immunoblotting and immunoprecipitation
100 μg of total cell protein extract were separated by SDS polyacrylamide gel electrophoresis (SDS-PAGE). After membrane transfer (Hybond-P, Amersham Pharmacia Biotech, Little Chalfont, UK) proteins of interest were detected with antibodies against phospho-STAT5a/b (05-495; Upstate Ltd, Milton Keynes, UK), phosphotyrosine (4G10 hybridoma), Ras (R02120; BD Transduction Laboratories, Heidelberg, Germany), phospho-Erk1/2 (9101), phospho-MEK1/2 (9121), phospho-Akt (pS473; 9271), phospho-GSK3α/β (9331), Akt (9272) (all from Cell Signaling Technology/NEB) or Erk1/2 (sc-93; Santa Cruz, Heidelberg, Germany) and ECL (Amersham Pharmacia Biotech). To detect the tyrosine phosphorylated EpoR, phosphotyrosyl-proteins from 500 μg of total cell protein were immunoprecipitated with mAb 4G10 in IP-buffer (20 mM Tris HCl pH 7.5, 10 mM EDTA, 10 mM EGTA, 100 mM NaCl, 5% glycerol, 0.1% (v/v) Tween20, 10 μg/ml aprotinin, 1 mM PMSF, 5 μg/ml antipain, 0.5 μg/ml leupeptin, 0.7 μg/ml pepstatin A, 1 × Complete™ inhibitors, 3 mM DTT, 1 mM NaF, 1 mM Na3VO4) overnight in the cold. After SDS-PAGE and membrane transfer, EpoR was detected with polyclonal anti-EpoR (kind gift from Patrick Mayeux, Hospital Cochin, Paris, France) and ECL.
PI3 kinase assay
500 μg of total cell protein extract were immunoprecipitated in NP-40 lysis buffer with 4 μg anti-phosphotyrosine mAb (4G10) or with 4 μg PI3K p110γ polyclonal antibody (sc-7177 (H-199); Santa Cruz), respectively. Immunoprecipitates were washed three times with NP-40 lysis buffer, twice with 50 mM TrisHCl pH 7.5 plus 500 mM lithium chloride (LiCl), once with 20 mM TrisHCl pH 7.5 with 100 mM NaCl and 1 mM EDTA and once with 20 mM HEPES pH 7.5. 10 μg of lyophilised phosphatidylinositol (PI; P2517, Sigma) were then mixed with 10 μl of lipid kinase buffer (LKB; 20 mM TrisHCl pH 7.5, 100 mM NaCl, 1 mM EGTA) and sonicated for 6 × 5 sec at 30% power output with an MS72 sonotrode (Sonopuls GM70; Bandelin, Berlin, Germany), chilling PI on ice between sonications. Sonicated PI was then mixed with 30 μl LKB and added to the immunoprecipitates. After 5 min on ice, 10 μl LKB with 10 μCi 32P-γ-ATP (3000 Ci/mMol; Amersham, Little Chalfont, UK) and 2 mM magnesium chloride (MgCl2) were added and samples incubated at 37°C for 15 min. The reaction was stopped with 200 μl of 1 N hydrochloric acid (HCl). After extraction of the samples with 200 μl of methanol/chloroform (1:1), 50 μl of the organic phase were used for thin layer chromatography (TLC) on LK6D plates (4865-821; Whatman International Ltd, Maidstone, UK). Running buffer was methanol/chloroform/H2O/NH4OH (20:23:4:1). TLC plates were analyzed on a Storm860 phosphoimager (Amersham Pharmacia Biotech) with ImageQuant 5.2 (Molecular Dynamics, Sunnyvale, USA). To confirm equal PI3K precipitation independent of cell stimulation, parallel samples of immunoprecipitates from protein extracts analyzed in lipid kinase assay were separated by SDS-PAGE and blots probed with anti-p110γ mAb (804-230-L001; Alexis, Nottingham, UK) followed by ECL.
Raf-MEK coupled kinase assay
The coupled kinase assay was done essentially as described [57] with slight modifications. 500 μg of total cell protein extract were immunoprecipitated with 4 μg anti-c-Raf1 (R19120; BD Transduction Laboratories) or with 4 μg anti-B-Raf (07-453; Upstate) in buffer A (20 mM TrisHCl pH 8.0, 2 mM EDTA, 1% (v/v) TritonX-100, 10% glycerol). Immunoprecipitates were washed three times with buffer A and twice with buffer B (50 mM TrisHCl pH 7.5, 75 mM NaCl, 5 mM EGTA, 5 mM MgCl2). 0.2 μg activatable GST-MEK1 (gift from W. Kolch; or 14-420, Upstate) together with 5 μl buffer C (50 mM MgCl2, 2 mM ATP) and 10 μl buffer B were added to the washed immunoprecipitates, samples were mixed and incubated at 30°C for 20 min. The reaction was then disrupted by transferring samples into ice and adding 100 μl buffer D (buffer B plus 1 mM DTT and 1 mM Na3VO4). 25 μl of this reaction mix was subsequently incubated with 20 μg kinase-inactive GST-Erk1K63M (gift from Heidi Greulich, Boston), 2 μl buffer E (buffer B with 37.5 mM MgCl2 and 1.25 mM ATP) and 5 μCi 32P-γ-ATP at 30°C for 15 min. The reaction was stopped by adding SDS-PAGE sample buffer. After SDS-PAGE, Raf activity was determined by detecting GST-Erk1K63M phosphorylation with ImageQuant 5.2. Subsequently proteins were blotted and membranes probed with anti-c-Raf1 or anti-B-Raf.
Ras assay
Detection of GTP-loaded Ras has been previously described [58,59]. Cells were lysed in Mg2+-containing lysis buffer (MLB; 25 mM HEPES pH 7.5, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 10% glycerol, 1% NP-40, 0.25% (w/v) sodium deoxycholate, 1 mM Na3VO4, 10 μg/ml aprotinin, 0.5 μg/ml leupeptin) at 4°C for 15 min. 500 μg of MLB total cell protein were incubated with 25 μg GST-c-Raf1 RBD (amino acids 1–149) pre-coupled to GSH-beads (Amersham Pharmacia Biotech) at 4°C for 1 h. Precipitates were washed three times with MLB, separated by SDS-PAGE and immunoblotted with anti-Ras.
Statistical analysis of protein kinase assays
p-values were calculated using the Student's t-test (Microsoft Excel). Classification of values is p < 0.05, p < 0.01 or p < 0.001.
Authors' contributions
EKS co-designed and carried out the experiments and participated in the drafting of the manuscript. SF participated in the isolation and culture of haematopoietic stem cells from human cord blood samples. SMF co-designed and supervised the experiments, drafted and edited the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
We are grateful to Walter Kolch (Glasgow), Patrick Mayeux (Paris), Heidi Greulich (Boston), Ignacio Rubio, Tzvetanka Bondeva, Reinhard Wetzker (Jena) and Manfred Jücker (Hamburg) for reagents, valuable discussions and comments on the manuscript, to Adrian L. Harris (Oxford) and Georg Krohne (Würzburg) for support, and to Doug Higgs (Oxford), Ursula Klingmüller (Heidelberg) and Marc Lewitzky (Oxford) for critical reading of the manuscript.
Contributor Information
Enrico K Schmidt, Email: schmidt@ciml.univ-mrs.fr.
Serge Fichelson, Email: fichelso@cochin.inserm.fr.
Stephan M Feller, Email: stephan.feller@cancer.org.uk.
References
- Klingmuller U. The role of tyrosine phosphorylation in proliferation and maturation of erythroid progenitor cells--signals emanating from the erythropoietin receptor. Eur J Biochem. 1997;249:637–647. doi: 10.1111/j.1432-1033.1997.t01-1-00637.x. [DOI] [PubMed] [Google Scholar]
- Constantinescu SN, Ghaffari S, Lodish HF. The Erythropoietin Receptor: Structure, Activation and Intracellular Signal Transduction. Trends Endocrinol Metab. 1999;10:18–23. doi: 10.1016/S1043-2760(98)00101-5. [DOI] [PubMed] [Google Scholar]
- Wojchowski DM, Gregory RC, Miller CP, Pandit AK, Pircher TJ. Signal transduction in the erythropoietin receptor system. Exp Cell Res. 1999;253:143–156. doi: 10.1006/excr.1999.4673. [DOI] [PubMed] [Google Scholar]
- Lacombe C, Mayeux P. The molecular biology of erythropoietin. Nephrol Dial Transplant. 1999;14 Suppl 2:22–28. doi: 10.1093/ndt/14.suppl_2.22. [DOI] [PubMed] [Google Scholar]
- Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–647. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- Brines M. What evidence supports use of erythropoietin as a novel neurotherapeutic? Oncology (Huntingt) 2002;16:79–89. [PubMed] [Google Scholar]
- Goldman SA, Nedergaard M. Erythropoietin strikes a new cord. Nat Med. 2002;8:785–787. doi: 10.1038/nm0802-785. [DOI] [PubMed] [Google Scholar]
- Koury MJ, Sawyer ST, Brandt SJ. New insights into erythropoiesis. Curr Opin Hematol. 2002;9:93–100. doi: 10.1097/00062752-200203000-00002. [DOI] [PubMed] [Google Scholar]
- Maxwell P, Ratcliffe P. Regulation of expression of the erythropoietin gene. Curr Opin Hematol. 1998;5:166–170. doi: 10.1097/00062752-199805000-00003. [DOI] [PubMed] [Google Scholar]
- Tamura K, Sudo T, Senftleben U, Dadak AM, Johnson R, Karin M. Requirement for p38alpha in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell. 2000;102:221–231. doi: 10.1016/S0092-8674(00)00027-1. [DOI] [PubMed] [Google Scholar]
- Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O, Ihle JN. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74:227–236. doi: 10.1016/0092-8674(93)90414-L. [DOI] [PubMed] [Google Scholar]
- Klingmuller U, Wu H, Hsiao JG, Toker A, Duckworth BC, Cantley LC, Lodish HF. Identification of a novel pathway important for proliferation and differentiation of primary erythroid progenitors. Proc Natl Acad Sci U S A. 1997;94:3016–3021. doi: 10.1073/pnas.94.7.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu SN, Huang LJ, Nam H, Lodish HF. The erythropoietin receptor cytosolic juxtamembrane domain contains an essential, precisely oriented, hydrophobic motif. Mol Cell. 2001;7:377–385. doi: 10.1016/S1097-2765(01)00185-X. [DOI] [PubMed] [Google Scholar]
- Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995;83:59–67. doi: 10.1016/0092-8674(95)90234-1. [DOI] [PubMed] [Google Scholar]
- Lin CS, Lim SK, D'Agati V, Costantini F. Differential effects of an erythropoietin receptor gene disruption on primitive and definitive erythropoiesis. Genes Dev. 1996;10:154–164. doi: 10.1101/gad.10.2.154. [DOI] [PubMed] [Google Scholar]
- Qu CK, Shi ZQ, Shen R, Tsai FY, Orkin SH, Feng GS. A deletion mutation in the SH2-N domain of Shp-2 severely suppresses hematopoietic cell development. Mol Cell Biol. 1997;17:5499–5507. doi: 10.1128/mcb.17.9.5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socolovsky M, Fallon AE, Wang S, Brugnara C, Lodish HF. Fetal anemia and apoptosis of red cell progenitors in Stat5a-/-5b-/- mice: a direct role for Stat5 in Bcl-X(L) induction. Cell. 1999;98:181–191. doi: 10.1016/S0092-8674(00)81013-2. [DOI] [PubMed] [Google Scholar]
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood. 2001;98:3261–3273. doi: 10.1182/blood.V98.12.3261. [DOI] [PubMed] [Google Scholar]
- Liao HJ, Kume T, McKay C, Xu MJ, Ihle JN, Carpenter G. Absence of erythrogenesis and vasculogenesis in Plcg1-deficient mice. J Biol Chem. 2002;277:9335–9341. doi: 10.1074/jbc.M109955200. [DOI] [PubMed] [Google Scholar]
- Ohneda K, Yamamoto M. Roles of hematopoietic transcription factors GATA-1 and GATA-2 in the development of red blood cell lineage. Acta Haematol. 2002;108:237–245. doi: 10.1159/000065660. [DOI] [PubMed] [Google Scholar]
- Miller CP, Heilman DW, Wojchowski DM. Erythropoietin receptor-dependent erythroid colony-forming unit development: capacities of Y343 and phosphotyrosine-null receptor forms. Blood. 2002;99:898–904. doi: 10.1182/blood.V99.3.898. [DOI] [PubMed] [Google Scholar]
- Pircher TJ, Geiger JN, Zhang D, Miller CP, Gaines P, Wojchowski DM. Integrative signaling by minimal erythropoietin receptor forms and c-Kit. J Biol Chem. 2001;276:8995–9002. doi: 10.1074/jbc.M007473200. [DOI] [PubMed] [Google Scholar]
- Zang H, Sato K, Nakajima H, McKay C, Ney PA, Ihle JN. The distal region and receptor tyrosines of the Epo receptor are non-essential for in vivo erythropoiesis. Embo J. 2001;20:3156–3166. doi: 10.1093/emboj/20.12.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketteler R, Heinrich AC, Offe JK, Becker V, Cohen J, Neumann D, Klingmuller U. A functional green fluorescent protein-erythropoietin receptor despite physical separation of JAK2 binding site and tyrosine residues. J Biol Chem. 2002;277:26547–26552. doi: 10.1074/jbc.M202287200. [DOI] [PubMed] [Google Scholar]
- Fichelson S, Chretien S, Rokicka-Piotrowicz M, Bouhanik S, Gisselbrecht S, Mayeux P, Lacombe C. Tyrosine residues of the erythropoietin receptor are dispensable for erythroid differentiation of human CD34+ progenitors. Biochem Biophys Res Commun. 1999;256:685–691. doi: 10.1006/bbrc.1999.0340. [DOI] [PubMed] [Google Scholar]
- Djordjevic S, Driscoll PC. Structural insight into substrate specificity and regulatory mechanisms of phosphoinositide 3-kinases. Trends Biochem Sci. 2002;27:426–432. doi: 10.1016/S0968-0004(02)02136-9. [DOI] [PubMed] [Google Scholar]
- Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- Stein RC, Waterfield MD. PI3-kinase inhibition: a target for drug development? Mol Med Today. 2000;6:347–357. doi: 10.1016/S1357-4310(00)01770-6. [DOI] [PubMed] [Google Scholar]
- Mayeux P, Dusanter-Fourt I, Muller O, Mauduit P, Sabbah M, Druker B, Vainchenker W, Fischer S, Lacombe C, Gisselbrecht S. Erythropoietin induces the association of phosphatidylinositol 3'-kinase with a tyrosine-phosphorylated protein complex containing the erythropoietin receptor. Eur J Biochem. 1993;216:821–828. doi: 10.1111/j.1432-1033.1993.tb18203.x. [DOI] [PubMed] [Google Scholar]
- He TC, Zhuang H, Jiang N, Waterfield MD, Wojchowski DM. Association of the p85 regulatory subunit of phosphatidylinositol 3-kinase with an essential erythropoietin receptor subdomain. Blood. 1993;82:3530–3538. [PubMed] [Google Scholar]
- Kubota Y, Tanaka T, Kitanaka A, Ohnishi H, Okutani Y, Waki M, Ishida T, Kamano H. Src transduces erythropoietin-induced differentiation signals through phosphatidylinositol 3-kinase. Embo J. 2001;20:5666–5677. doi: 10.1093/emboj/20.20.5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang T, Prasad KS, Koury MJ, Brandt SJ. Mitogen-activated protein kinase mediates erythropoietin-induced phosphorylation of the TAL1/SCL transcription factor in murine proerythroblasts. Biochem J. 1999;343 Pt 3:615–620. doi: 10.1042/0264-6021:3430615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseyama Y, Sawada K, Oda A, Koizumi K, Takano H, Tarumi T, Nishio M, Handa M, Ikeda Y, Koike T. Phosphatidylinositol 3-kinase is involved in the protection of primary cultured human erythroid precursor cells from apoptosis. Blood. 1999;94:1568–1577. [PubMed] [Google Scholar]
- Cataldi A, Di Pietro R, Centurione L, Grilli A, Cutroneo G, Miscia S. Phosphatidylinositol-3-kinase activation and atypical protein kinase C zeta phosphorylation characterize the DMSO signalling in erythroleukemia cells. Cell Signal. 2000;12:667–672. doi: 10.1016/S0898-6568(00)00109-1. [DOI] [PubMed] [Google Scholar]
- Myklebust JH, Blomhoff HK, Rusten LS, Stokke T, Smeland EB. Activation of phosphatidylinositol 3-kinase is important for erythropoietin-induced erythropoiesis from CD34(+) hematopoietic progenitor cells. Exp Hematol. 2002;30:990–1000. doi: 10.1016/S0301-472X(02)00868-8. [DOI] [PubMed] [Google Scholar]
- Bouscary D, Pene F, Claessens YE, Muller O, Chretien S, Fontenay-Roupie M, Gisselbrecht S, Mayeux P, Lacombe C. Critical role for PI 3-kinase in the control of erythropoietin-induced erythroid progenitor proliferation. Blood. 2003;101:3436–3443. doi: 10.1182/blood-2002-07-2332. [DOI] [PubMed] [Google Scholar]
- Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, Williams RL. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell. 2000;6:909–919. doi: 10.1016/S1097-2765(00)00088-5. [DOI] [PubMed] [Google Scholar]
- Sui X, Krantz SB, Zhao ZJ. Stem cell factor and erythropoietin inhibit apoptosis of human erythroid progenitor cells through different signalling pathways. Br J Haematol. 2000;110:63–70. doi: 10.1046/j.1365-2141.2000.02145.x. [DOI] [PubMed] [Google Scholar]
- Foukas LC, Daniele N, Ktori C, Anderson KE, Jensen J, Shepherd PR. Direct effects of caffeine and theophylline on p110 delta and other phosphoinositide 3-kinases. Differential effects on lipid kinase and protein kinase activities. J Biol Chem. 2002;277:37124–37130. doi: 10.1074/jbc.M202101200. [DOI] [PubMed] [Google Scholar]
- Freyssinier JM, Lecoq-Lafon C, Amsellem S, Picard F, Ducrocq R, Mayeux P, Lacombe C, Fichelson S. Purification, amplification and characterization of a population of human erythroid progenitors. Br J Haematol. 1999;106:912–922. doi: 10.1046/j.1365-2141.1999.01639.x. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Cullen PJ, Lockyer PJ. Integration of calcium and Ras signalling. Nat Rev Mol Cell Biol. 2002;3:339–348. doi: 10.1038/nrm808. [DOI] [PubMed] [Google Scholar]
- Kolbus A, Pilat S, Husak Z, Deiner EM, Stengl G, Beug H, Baccarini M. Raf-1 antagonizes erythroid differentiation by restraining caspase activation. J Exp Med. 2002;196:1347–1353. doi: 10.1084/jem.20020562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Jackson CA, Eyers PA, Cohen P, Goedert M, Boyle FT, Hewitt N, Plant H, Hedge P. Paradoxical activation of Raf by a novel Raf inhibitor. Chem Biol. 1999;6:559–568. doi: 10.1016/S1074-5521(99)80088-X. [DOI] [PubMed] [Google Scholar]
- Myklebust JH, Smeland EB, Josefsen D, Sioud M. Protein kinase C-alpha isoform is involved in erythropoietin-induced erythroid differentiation of CD34(+) progenitor cells from human bone marrow. Blood. 2000;95:510–518. [PubMed] [Google Scholar]
- Lopez-Ilasaca M, Crespo P, Pellici PG, Gutkind JS, Wetzker R. Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase gamma. Science. 1997;275:394–397. doi: 10.1126/science.275.5298.394. [DOI] [PubMed] [Google Scholar]
- Bondeva T, Pirola L, Bulgarelli-Leva G, Rubio I, Wetzker R, Wymann MP. Bifurcation of lipid and protein kinase signals of PI3Kgamma to the protein kinases PKB and MAPK. Science. 1998;282:293–296. doi: 10.1126/science.282.5387.293. [DOI] [PubMed] [Google Scholar]
- Wetzker R, Bohmer FD. Opinion: Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat Rev Mol Cell Biol. 2003;4:651–657. doi: 10.1038/nrm1173. [DOI] [PubMed] [Google Scholar]
- Stephens LR, Eguinoa A, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, Smrcka AS, Thelen M, Cadwallader K, Tempst P, Hawkins PT. The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell. 1997;89:105–114. doi: 10.1016/S0092-8674(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Wymann MP, Bjorklof K, Calvez R, Finan P, Thomast M, Trifilieff A, Barbier M, Altruda F, Hirsch E, Laffargue M. Phosphoinositide 3-kinase gamma: a key modulator in inflammation and allergy. Biochem Soc Trans. 2003;31:275–280. doi: 10.1042/bst0310275. [DOI] [PubMed] [Google Scholar]
- Rubio I, Rodriguez-Viciana P, Downward J, Wetzker R. Interaction of Ras with phosphoinositide 3-kinase gamma. Biochem J. 1997;326 (Pt 3):891–895. doi: 10.1042/bj3260891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, Hawkins PT, Stephens L, Eccleston JF, Williams RL. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 2000;103:931–943. doi: 10.1016/S0092-8674(00)00196-3. [DOI] [PubMed] [Google Scholar]
- Guillard C, Chretien S, Jockers R, Fichelson S, Mayeux P, Duprez V. Coupling of heterotrimeric Gi proteins to the erythropoietin receptor. J Biol Chem. 2001;276:2007–2014. doi: 10.1074/jbc.M003527200. [DOI] [PubMed] [Google Scholar]
- Guillard C, Chretien S, Pelus AS, Porteu F, Muller O, Mayeux P, Duprez V. Activation of the mitogen-activated protein kinases Erk1/2 by erythropoietin receptor via a G(i )protein beta gamma-subunit-initiated pathway. J Biol Chem. 2003;278:11050–11056. doi: 10.1074/jbc.M208834200. [DOI] [PubMed] [Google Scholar]
- Li K, Menon MP, Karur VG, Hegde S, Wojchowski DM. Attenuated signaling by a phosphotyrosine-null Epo receptor form in primary erythroid progenitor cells. Blood. 2003;102:3147–53. doi: 10.1182/blood-2003-01-0078. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Meikle S, Yazici Z, Eulitz M, Kolch W. Regulation of Raf-1 activation and signalling by dephosphorylation. Embo J. 2002;21:64–71. doi: 10.1093/emboj/21.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SJ, Shalloway D. Cell cycle-dependent activation of Ras. Curr Biol. 1996;6:1621–1627. doi: 10.1016/S0960-9822(02)70785-9. [DOI] [PubMed] [Google Scholar]
- Posern G, Weber CK, Rapp UR, Feller SM. Activity of Rap1 is regulated by bombesin, cell adhesion, and cell density in NIH3T3 fibroblasts. J Biol Chem. 1998;273:24297–24300. doi: 10.1074/jbc.273.38.24297. [DOI] [PubMed] [Google Scholar]