

Abstract
Background
Endometrial cancer is the most common gynecologic malignancy. Type II endometrial carcinoma is often poorly differentiated and patients diagnosed with Type II disease (~11%) are disproportionately represented in annual endometrial cancer deaths (48%). Recent genomic studies highlight mutations in chromatin regulators as drivers in Type II endometrial carcinoma tumorigenesis, suggesting the use of epigenetic targeted therapies could provide clinical benefit to these patients. We investigated the anti-tumor efficacy of the LSD1 inhibitor HCI2509 in two poorly differentiated Type II endometrial cancer cell lines AN3CA and KLE.
Methods
The effects of HCI2509 on viability, proliferation, anchorage-independent growth, global histone methylation, LSD1 target gene induction, cell cycle, caspase activation and TUNEL were assayed. KLE cells were used in an orthotopic xenograft model to assess the anti-tumor activity of HCI2509.
Results
Both AN3CA and KLE cells were sensitive to HCI2509 treatment with IC50s near 500 nM for cell viability. Inhibition of LSD1 with HCI2509 caused decreased proliferation and anchorage independent growth in soft agar, elevated global histone methylation, and perturbed the cell cycle in both cell lines. These effects were largely dose-dependent. HCI2509 treatment also caused apoptotic cell death. Orthotopic implantation of KLE cells resulted in slow-growing and diffuse tumors throughout the abdomen. Tumor burden was distributed log-normally. Treatment with HCI2509 resulted 5/9 tumor regressions such that treatment and regressions were significantly associated (p = 0.034).
Conclusions
Our findings demonstrate the anti-cancer properties of the LSD1 inhibitor HCI2509 on poorly differentiated endometrial carcinoma cell lines, AN3CA and KLE. HCI2509 showed single-agent efficacy in orthotopic xenograft studies. Continued studies are needed to preclinically validate LSD1 inhibition as a therapeutic strategy for endometrial carcinoma.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2407-14-752) contains supplementary material, which is available to authorized users.
Background
Endometrial carcinoma (EC) arises from the lining of the uterus and is the most commonly diagnosed invasive gynecologic malignancy, exceeding the incidence of cervical, ovarian, vaginal, and vulvar cancers combined [1, 2]. With 50,230 new cases and 8,590 deaths estimated in the U.S. for 2014 it is the fourth most prevalent cancer among women in developed countries, and the sixth worldwide [1, 3, 4]. Most patients present with low-grade early-stage disease, but patients diagnosed with more aggressive, high-grade, advanced disease that has spread beyond the uterus will progress within 1 year [5]. EC has been broadly classified into two subtypes based on differing clinico-pathologic characteristics. Over 80% of ECs are categorized as Type I endometroid adenocarcinomas [6, 7], while the remaining are Type II serous, clear-cell, poorly differentiated, and grade 3 endometrioid carcinomas [6, 7]. Type I malignancies are associated with extended periods of elevated estrogen exposure, obesity, and estrogen and progesterone receptor positivity. These cancers present and are diagnosed in earlier stages and are typically more differentiated, responsive to progesterone treatment, and consequently have a more favorable prognosis [6, 7]. Type I tumors are more common than Type II tumors in pre- and perimenopausal women [6]. On the other hand, Type II EC more frequently occurs in postmenopausal women and tumors are typically poorly differentiated [7]. Unlike Type I, Type II disease is unrelated to hyperestrogenic risk factors, diagnosed in later stages of the disease, and is clinically more aggressive. While representing only ~15% of all clinical cases Type II disease is responsible for around ~48% of endometrial cancer-related deaths, despite adjuvant chemotherapy and radiation, mainly due to metastasis and recurrent disease [7]. Better therapeutic strategies are needed for these patients.
No single hereditary risk factor plays a dominant role in endometrial cancer, which is driven by an interplay of genetic, environmental, and epigenetic factors. Several instances of epigenetic misregulation have been described in endometrial cancer. Specifically, alterations in DNA methylation have been broadly observed, with promoter hypermethylation leading to silencing of the progesterone receptor and other tumor suppressors like MLH1, APC, MGMT, and PTEN [8, 9]. Hypomethylation at the CD133 promoter has been observed in tumor initiating cells, suggesting epigenetic regulation does affect the mechanisms driving tumorigenicity and disease recurrence [10]. Additionally, the expression of various histone modifying enzymes are altered in endometrial cancer, including histone deacetylases as well as the histone methyltranferase EZH2. Their inhibition decreases proliferation and invasiveness in endometrial cancer cell lines [11–14]. Importantly, the advent of next generation sequencing has allowed further characterization of the molecular etiology of Type II EC, shedding more light on possible epigenetic targets and allowing for novel treatment options to be developed. Analysis of the genomic landscape of Type II EC identified somatic mutations in members of the nucleosome remodeling and deacetylase complex (NuRD), CHD4 and MBD3, as well as mutations in the chromatin and transcriptional regulators EP300, ARID1A, and TAF1 as candidate driver events [15–17]. While the functional significance of these mutations in Type II EC remains to be elucidated, these data underscore the significance of the interplay between genetic and epigenetic factors in the development, progression and prognosis of Type II EC.
Unlike genetic mutations, epigenetic changes, including DNA methylation and posttranslational modifications of histones, are dynamic and reversible through pharmacological intervention, such that the readers, writers, and erasers of epigenetic marks are emerging therapeutic targets [18, 19]. Patterns of histone lysine methylation are maintained in a more cell-type specific manner than DNA methylation or histone acetylation, and it is thought that pharmacologically modulating offending histone lysine methyltransferases or demethylases can confer increased therapeutic specificity and decreased dose-limiting off-target toxicities [20–23]. Lysine-specific demethylase 1 (LSD1) is a histone lysine demethylase with specificity for mono- and dimethylated histone H3 lysine 4 (H3K4) and lysine 9 (H3K9) [24, 25]. Methylation at H3K4 is generally considered to be permissive, while H3K9 methylation is repressive [26]. LSD1 is upregulated in several malignancies and associated with decreased differentiation, aggressive tumor biology, and poor prognosis [27–34]. HCI2509 is a small molecule inhibitor of LSD1 that has shown in vitro anti-tumor efficacy in triple negative breast cancer, and single-agent in vivo efficacy in both Ewing sarcoma and castration-resistant prostate cancer [35–38]. A cell line panel showed one Type II EC cell line, AN3CA, to be sensitive to treatment with HCI2509 [35]. In this investigation, we validate this result in another Type II cell line, KLE, and further evaluate the mechanism of action by testing whether HCI2509 causes global changes in histone methylation, modulates the LSD1 target gene HMOX1 and CDH1, and disrupts oncogenic transformation. More importantly, we also assess whether HCI2509 displays any anti-tumor efficacy in vivo. In order to most accurately represent disease spread mimicking human EC as well as more predictable therapeutic efficacy, we utilize an orthotopic xenograft mouse model to demonstrate the in vivo activity of HCI2509 against poorly differentiated Type II EC.
Methods
Antibodies and reagents
Immunodetection was performed with the following antibodies: anti-α-Tubulin (Calbiochem CP06), anti-LSD1 (Cell Signaling C69G12), anti-H3 (Cell Signaling Technology D2B12), anti-H3K4me3 (Cell Signaling Technology C42D8), anti-H3K9me2 (Cell Signaling Technology 9753), anti-H3K27me3 (Cell Signaling Technology C36B11). Propidium iodide (Sigma P4864), medroxyprogesterone 17-acetate (MPA; Sigma M1629). HCI2509 is previously described [35].
Cell culture, proliferation, colony formation assays, cell viability, and caspase 3/7 activation
Endometrial carcinoma cell lines AN3CA and KLE were obtained from ATCC and maintained in the DMEM/F12 supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. All experiments were performed prior to passage 10. Proliferation assays (3T5) and colony formation assays were performed as previously described [39, 40]. Cell viability and caspase activation were performed using Cell Titer-Glo and Caspase 3/7-Glo (Promega). The same vehicle (0.3% DMSO) was used for both HCI2509 and MPA in all in vitro treatments.
Western blots and quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR)
AN3CA and KLE cells were seeded in triplicate in 6-well dishes at a density of 3.5 × 105 cells/well or 2 × 105 cells/well, respectively. Cells were treated with varying concentrations of HCI2509 for 48 hours, harvested, and flash frozen for protein or RNA extraction. Total RNA was extracted from treated cells using an RNeasy Plus kit (Qiagen). cDNA was generated using qScript cDNA SuperMix (Quanta Bioscience). Template was then amplified, detected, and quantified using SYBR green fluorescence. Each replicate was normalized to the internal housekeeping gene (RPL19) and induction was calculated relative to the vehicle control. The following primers were used: RPL19_fwd 5′-ATGTATCACAGCCTGTACCTG-3′, RPL19_rev 5′-TTCTTGGTCTCTTCCTCCTTG-3′; HMOX1_fwd 5′-AACTTTCAGAAGGGCCAGGT-3′, HMOX1_rev 5′-GTAGACAGGGGCGAAGACTG-3′; CDH1_fwd 5′-TGCCCAGAAAATGAAAAAGG-3′, CDH1_rev 5′-GTGTATGTGGCAATGCGTTC-3′.
Cell cycle analysis
1 × 106 cells (KLE, AN3CA) were seeded in 10 cm dishes and treated with either vehicle alone or HCI2509 for the appropriate duration, trypsinized, centrifuged at 1000 rcf for 5 min, and fixed in ice cold 70% ethanol. Staining was performed by centrifuging 1.5 × 106 fixed cells at 770 rcf for 5 minutes, aspirating ethanol, and resuspending in 350 μL of staining buffer (4 mM citrate, 3% PEG8000, 50 μg/mL propidium iodide (PI), 180 units/mL RNase, 0.1% Triton X-100) incubating at 37°C for 20 minutes, and adding 350 μL of salting buffer (400 mM NaCl, 3% PEG8000, 50 μg/mL PI, 0.1% Triton X-100). Cells were analyzed on a BD FACSCanto with Software Diva vs6.1.3 (BD Biosciences San Jose CA).
TUNEL and fluorescence microscopy
9 × 104 AN3CA cells or 3 × 104 KLE cells were seeded onto glass coverslips in a 12-well dish. Cells were treated with either vehicle or 3 X EC50 HCI2509 for 72 hours to correlate with the caspase activation assay. Cells were fixed in formalin and stained with the DeadEnd Fluorescent TUNEL system (Promega). DNase treatment and no labeling reaction were used as positive and negative internal controls, respectively. Cells were then stained with AlexaFluor Phalloidin (1:100) (Molecular Probes) and DAPI (0.3 μM) (Molecular Probes). Fluorescent cell images were collected on a Zeiss Axioskop2 mot plus microscope with a 40X dry objective (NA 0.75 NeoFluor), Axiocam MR camera, and Axiovision v4.8.1 software (Carl Zeiss MicroImaging, Inc.).
In vivoxenograft studies
All xenograft experiments were performed in accordance with protocol 11–12001 approved by the University of Utah IACUC. Female nude mice (strain J:Nu) were purchased from Jackson Laboratory (Bar Harbor, ME) and housed under appropriate conditions. Mice were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine and surgical procedures were carried out in a clean room on a circulating water warming pad set to 38°C. A frontal midline incision was made to enter the peritoneal cavity and 2 × 106 KLE cells expressing luciferase were implanted into the bifurcation of the uterus in 50 μL of 1:1 DMEM/F12:Matrigel (Corning). Following tumor cell implantation, the peritoneum and skin were each sutured separately and recovery was assessed daily for 7 days by weight measurements and visual inspection. VivoGlo Luciferin (Promega) was resuspended in PBS at a concentration of 30 mg/ml and passed through a 0.22 μM filter. Mice were imaged on day 7 using an IVIS Spectrum (PerkinElmer). Images were acquired 10 minutes after intraperitoneal (IP) administration of 100 μL luciferin. Mice with detectable tumor on day 7 were randomized into three groups: Vehicle only (n = 7; 100 μL 1:1 PBS:PEG400 IP daily), HCI2509 30 mg/kg (n = 9; 100 μL suspension IP daily), or untreated (n = 3). Body weight was tracked three times per week and luminescence was tracked weekly for the entire treatment period of 35 days. At day 42 of the study, mice were sacrificed, organs including uteri harvested and weighed, and fixed in formalin prior to paraffin embedding.
Results
HCI2509 impairs viability, proliferation, and transformation in Type II endometrial cancer cell lines
We first validated previous data suggesting that Type II endometrial carcinoma cells were sensitive to LSD1 inhibition with HCI2509 [35]. Both AN3CA and KLE cell lines exhibited a dose-dependent decrease in cell viability after 96 hours of treatment with HCI2509 (Figure 1A, B) with EC50 values determined at 499 nM and 435 nM, respectively (Figure 1A, B). In separate experiments, treatment with medroxyprogesterone 17-acetate (MPA) showed no effect on cell viability, confirming that both cell lines exhibit resistance to hormone treatment (Figure 1A, B). Having determined the EC50 we next tested the effect of HCI2509 on population doubling times using a 3T5 proliferation assay in treatment conditions below and above the EC50 (Figure 1C, D). HCI2509 decreased proliferation rates in a dose dependent manner in both AN3CA and KLE cell lines. Interestingly, even the lowest tested treatment concentration (0.3 X IC50) resulted in cytostasis in KLE cells. At and above the IC50, both cell lines exhibited negative growth, suggesting cell death.
Figure 1.
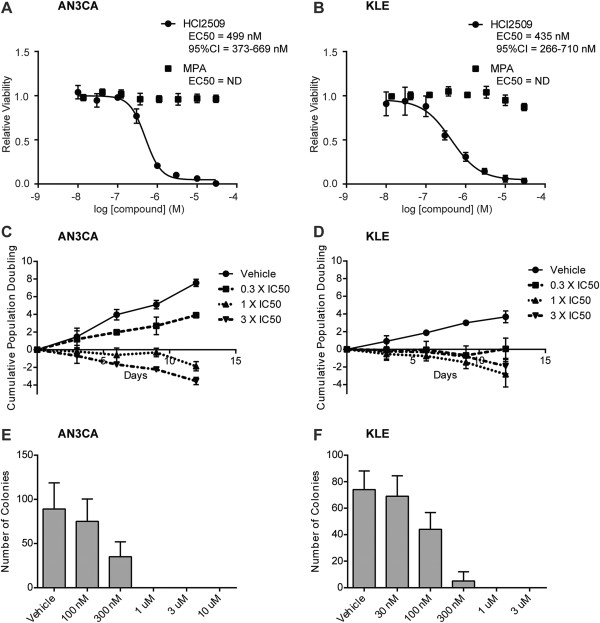
HCI2509 impairs cell viability, proliferation and transformation in Type II EC cell lines. (A, B) Dose–response curves showing the effects of 96-hour HCI2509 or medroxyprogesterone 17-acetate (MPA) treatment on cell viability of (A) AN3CA and (B) KLE cells normalized to vehicle controls. EC50s and 95% CI’s were calculated using GraphPad Prism 6.0 and are reported where the R2 > 0.9. Data points are reported as mean and standard deviation (n = 3). (C, D) Proliferation (3 T5) assays showing cell doubling times for (C) AN3CA and (D) KLE cells with vehicle and increasing doses of HCI2509. Data points are reported as mean and standard deviation (n = 3). (E, F) Quantification of colonies formed by (E) AN3CA or (F) KLE cells in soft agar with either vehicle or HCI2509 treatment at varying concentrations. Error bars indicate SD of duplicate assays.
In addition to the anti-proliferative effects seen in the 2-dimensional viability and proliferation assays, we also tested the ability of HCI2509 to impair anchorage-independent growth in soft agar. Cells were tested for colony formation at a range of concentrations spanning 30 nM to 10 μM. Based on the increased sensitivity of the KLE cells in the proliferation assay, the dose range tested in agars was shifted one half-log lower than that for AN3CA cells. HCI2509 impaired colony formation in both cells lines in a dose-dependent manner (Figure 1E, F). Above the viability EC50 for both cell lines, anchorage-independent growth was ablated, and at concentrations below the EC50 for KLE cells, colony formation was reduced, suggesting that HCI2509 impaired transformation at concentrations lower than those for which it induces cell death in KLE cells. AN3CA cells showed reduced colony formation near the viability EC50.
LSD1 inhibition results in global histone methylation changes and induction of LSD1 target genes
LSD1 is the primary histone demethylase for the cell and having demonstrated dose-dependent effects on viability, proliferation, and transformation, we next investigated whether HCI2509 treatment also caused dose-dependent increases in histone methylation marks. We evaluated both LSD1 histone substrates, H3K4 and H3K9. Analysis of H3K4me1 and H3K4me2 showed no effect of HCI2509 treatment on the monomethyl mark and accumulation of H3K4me2 in only AN3CA cells (Additional file 1: Figure S2A, B). We next asked whether at 48 hours impaired demethylation of H3K4 may result in accumulation of the H3K4 trimethyl mark. While trimethyllysine is not chemically accessible to LSD1, the effect of demethylation at promoter H3K4 is gene repression, and impaired demethylation at that mark may result in increased levels of the transcriptionally activating H3K4me3 chromatin. Additionally, H3K4me3 is depleted in an LSD1-dependent fashion during the epithelial-to-mesenchymal transition (EMT) [41]. HCI2509 treatment resulted in a dose-dependent increase in H3K4me3 in both cell lines (Figure 2A, B).
Figure 2.
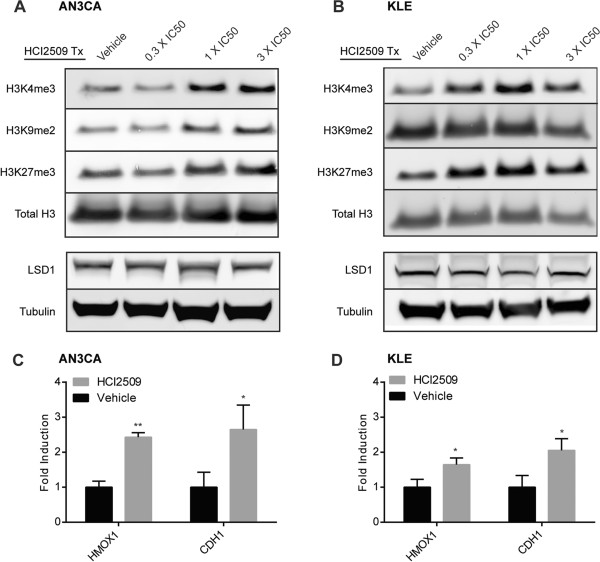
Treatment with HCI2509 causes changes in global histone methylation and induces LSD1 target genes. (A, B) Western blot analysis of H3K4me3, H3K9me2, H3K27me3 and LSD1 after 48 hours of vehicle or HCI2509 treatment at varying concentrations in (A) AN3CA and (B) KLE cells. Images are representative of two repeat experiments performed in triplicate. (C, D) qRT-PCR analysis of LSD1 target genes, HMOX1 and CDH1, after treatment with 3X EC50 for (C) AN3CA and (D) KLE cells. Data represents the mean and standard deviation (n = 3) and all replicates were normalized to internal housekeeping gene RPL19.
In complex with the estrogen and androgen hormone receptors, LSD1 is shown to activate target gene expression through removal of repressive H3K9 methylation. H3K9me2 is also shown to be largely depleted during EMT through an LSD1-dependent mechanism and this loss of H3K9me2 is associated with transformation [41]. Thus, we evaluated the effects of HCI2509 on H3K9me2 and observed an increase in H3K9me2 in AN3CA cells (Figure 2A). Interestingly, treatment with HCI2509 showed no effect on H3K9me2 in KLE cells (Figure 2B). We also predicted that changes in global methylation status in either H3K4 or H3K9 would occur in synchrony with additional global changes to chromatin state, so we also blotted for H3K27me3, a mark typically associated with gene repression and heterochromatin [26]. HCI2509 treatment induced a dose-dependent increase in H3K27me3 in both cell lines. The observed elevation of histone methylation by HCI2509 occurred with no change observed for LSD1 protein levels (Figure 2A, B).
We also asked whether HCI2509 modulated expression of LSD1 target genes. Induction of HMOX1 has been shown to be a biological readout for LSD1 engagement by HCI2509 [35, 38]. We additionally evaluated the expression of CDH1 (E-cadherin). E-cadherin is a cell-surface adhesion molecule that is repressed during SNAIL-LSD1-mediated EMT and is often misregulated in Type II endometrial cancer [42, 43]. HCI2509 treatment induced increased transcription of both HMOX1 and CDH1 in both AN3CA and KLE cell lines (Figure 2C, D), suggesting LSD1 target engagement by HCI2509.
LSD1 inhibition disrupts normal cell cycle progression in human endometrial cancer cell lines
The observation of decreased proliferative rates prompted us to test the effect of HCI2509 treatment on cell cycle progression in both AN3CA and KLE cells. Cell cycle analysis was performed with either vehicle or HCI2509 exposure at 300 nM, 1 μM, or 3 μM for 48 hours. AN3CA cells showed a dose-dependent increase in the percentage of cells in S-phase (Figure 3A). This was accompanied by a decrease in the G0/G1 population. In a time-course experiment 3 μM HCI2509 shows an accumulation of cells in the G0/G1 population from 6–12 hours before developing the increased S-phase fraction at 24 and 48 hours (Additional file 2: Figure S3A). KLE cells show a similar accumulation in early S-phase with increasing concentrations of HCI2509 (Figure 3B). Unlike the AN3CA data, the increase in the S-phase fraction occurs at the expense of the G2/M population of cells. The time-course experiment with KLE cells in 3 μM HCI2509 interestingly never passed through the same distribution as observed for 1 μM HCI2509 at 48 hours, and failed to show any obvious change until 48 hours (Additional file 2: Figure S3B). These data suggest that LSD1 inhibition with HCI2509 perturbs cell cycle progression in both Type II endometrial carcinoma cell lines, most likely through an accumulation in early S-phase.
Figure 3.
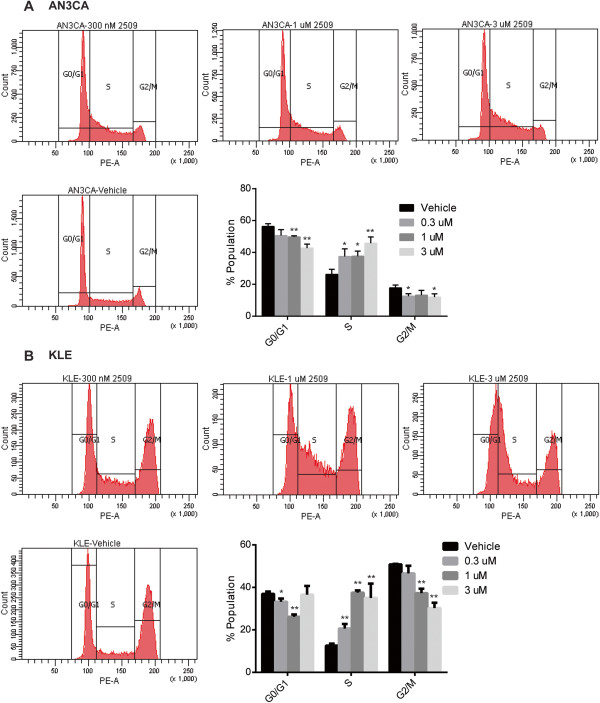
Dose-dependent cell cycle perturbation in Type II EC cell lines with HCI2509 treatment. (A, B) Cell cycle populations of (A) AN3CA and (B) KLE cell lines after exposure to vehicle, 300 nM, 1 μM, and 3 μM HCI2509 for 48 hours. 2 × 104 counts and 1 × 104 counts were used for AN3CA and KLE cells, respectively. Data is representative of four biological replicates. Mean and standard deviation are plotted (* p < 0.05, ** p < 0.01).
HCI2509 induces apoptosis in AN3CA and KLE cells
In addition to cell cycle disruption, we investigated the mechanism causing negative cell doubling in both AN3CA and KLE cells. We hypothesized that HCI2509 treatment may cause apoptotic cell death and therefore tested both cell lines for caspase 3/7 activation. Caspase activity was assayed in parallel with cell viability using 3X the EC50 and comparing to vehicle control. Viability and caspase activation were assessed over a time-course of 72 hours in both cell lines. Interestingly, in the context of HCI2509 treatment AN3CA showed decreased cell viability and caspase activity over the course of 48 hours with increased caspase activation occurring at 72 hours (Figure 4A). The decrease in caspase activation during the first 48 hours of treatment is likely due to a decreased number of cells/well due to cytostatsis relative to vehicle. HCI2509-treated KLE cells showed a concomitant increase in caspase activity and decrease in cell viability over 72 hours (Figure 4B). These data suggest an initial cytostasis which is followed by apoptotic cell death induced after 48 hours. We next confirmed apoptotic cell death using fluorescent TUNEL staining. AN3CA and KLE cells were treated with either vehicle or 3X EC50 HCI2509 for 72 hours and then assayed for TUNEL staining. Both cell lines showed decreased cell density and the presence of apoptotic cells with HCI2509 treatment, while vehicle treated cells appeared healthy and well spread on the coverslip (Figure 4C, D, Additional file 3: Figure S4A, S4B). Internal controls for the TUNEL assay are reported in Additional file 3: Figure S4C. These results confirmed apoptotic cell death induced by HCI2509 treatment.
Figure 4.
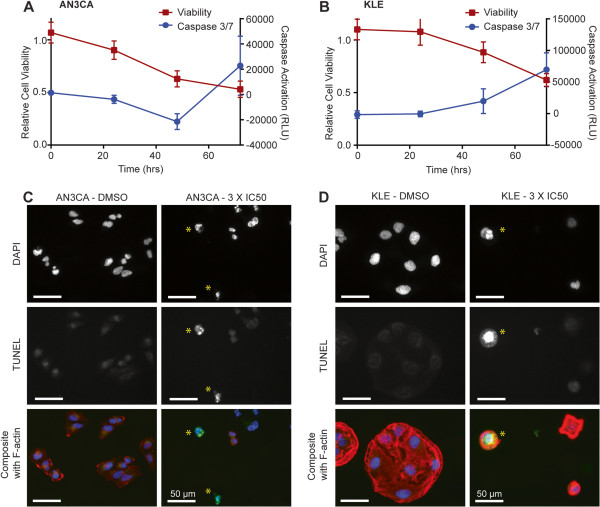
HCI2509 induces apoptotic cell death. (A, B) Cell viability and caspase activation at 0, 24, 48, and 72 hours in (A) AN3CA and (B) KLE cells treated with 3X EC50 HCI2509. Measurements were normalized to their respective vehicle (0.3% DMSO) sample at the appropriate time point. (C, D) Fluorescence microscopy images of (C) AN3CA and (D) KLE cell lines after exposure to either vehicle or 3X EC50 HCI2509 and then stained with TUNEL for apoptotic nuclei (green), DAPI for nuclei (blue), and phalloidin for actin (red). HCI2509 treatment induced apoptosis with apoptotic cells marked with (*).
HCI2509 leads to tumor regression in an orthotopic endometrial carcinoma mouse xenograft model
We further evaluated the efficacy of HCI2509 in an orthotopic xenograft model of endometrial carcinoma utilizing the KLE cell line stably transfected with luciferase to facilitate bioluminescence imaging. After implantation (day 0) and recovery, bioluminescence was measured weekly for the duration of the study (42 d). Total body weight was measured 3 times weekly, and weekly points were plotted (Figure 5A). At day 7, animals with detectable tumor were randomized into vehicle only and HCI2509 treatment groups (Additional file 4: Figure S5A). We observed the tumor luminescence values were better fit to a log-normal distribution than a normal distribution, which is common for various biological phenomena such as latency times for infections or survival times after a diagnosis of cancer (Additional file 4: Figure S5B) [44]. For this reason, the geometric mean of the tumor volumes for both conditions are plotted (Figure 5B). Values observed at day 7 were higher than those observed for the remainder of the study and therefore excluded from the graph. This initial burst of proliferation, and associated luminescence, followed by a drop off before later hitting exponential growth is commonly observed in xenograft studies. After 35 days of treatment (day 42 of the study) proliferating disease was observed in all of the vehicle treated animals, while 5 of the 9 drug treated animals showed no detectable luminescence (Figure 5C). Lack of luminescence is incorporated as the background reading of the instrument for each day of the experiment, as determined by an unimplanted, non-tumor bearing, healthy control. We used a Fisher’s exact test to evaluate the effect of treatment vs. vehicle on either tumor or regression and found HCI2509 significantly associated with tumor regression (p = 0.034). No difference in body weight was seen between the vehicle and treatment groups indicating tolerability of HCI2509. The luminescence readout for the untreated control group are plotted together with data from the vehicle and treatment groups in Additional file 4: Figure S5D as are the body weight measurements including the non-tumor bearing control. When considered with the in vitro data suggesting decreased proliferation, transformation and induced apoptosis in concert with increased global histone methylation and LSD1 engagement, these data support LSD1 inhibition with HCI2509 as a potential therapeutic strategy for Type II endometrial carcinoma.
Figure 5.
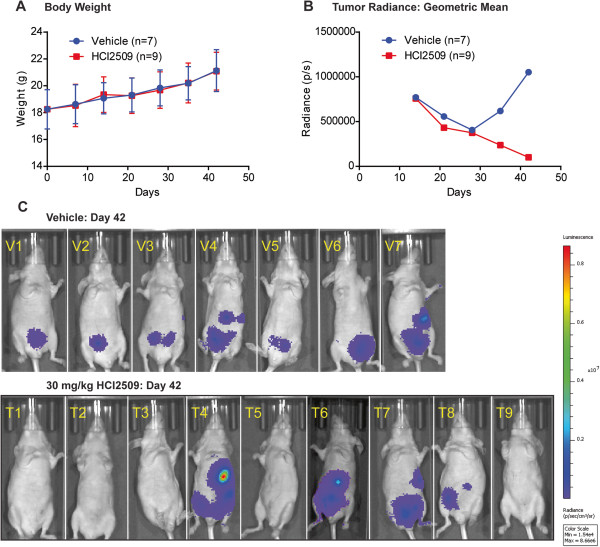
HCI2509 treatment causes tumor regression in vivo . (A) Average total body weight (g) of mice in both groups, vehicle and HCI2509 treatment, starting at implantation (day 0) through the course of the study. Data points shown represent the mean and standard deviation. (B) Quantified bioluminscence measurements of both the vehicle and HCI2509 treatment groups. Data is plotted as the geometric mean of total flux (photons/second). Daily treatment was initiated on day 7 (day 0 = implantation), such that day 14 represents the first day of imaging after the start of treatment. (C) Individual mouse images from study day 42 (day 35 of treatment). All images are on the same luminescence scale from 1.54 × 104 p/s to 8.66 × 106 p/s. Note: Animal #6 in the treatment group was sacrificed on study day 36, therefore the image is from study day 35 imaging.
Discussion
LSD1 is an emerging target for poorly differentiated and aggressive solid malignancies. Our findings suggest that LSD1 inhibition holds potential as a new therapeutic strategy for Type II endometrial cancer, which may accompany current state of the art treatment of EC in the future. Targeted LSD1 inhibition with HCI2509 showed potent anti-cancer activity both in vitro and in vivo with multiple tumor regressions observed in our orthotopic EC model. Type II EC constitutes an unmet medical need, with disproportionately high number of annual EC deaths relative to the proportion of Type II EC diagnoses as compared to Type I disease. While it is known that epigenetics, genetics, and the environment all contribute to the development of EC, recent studies demonstrating mutations in chromatin remodeling complexes as driver events in Type II EC [15–17] underscore the need for research to evaluate more effective and new therapeutic strategies targeting these mechanisms.
Chromatin modifiers or ubiquitin ligase complexes were recently implicated in 35% of clear cell endometrial and 50% of serous endometrial tumors [15]. One of the most commonly altered genes was CHD4, a member of the NuRD complex, along with the observation of frequent mutations in MBD3, another NuRD component [15]. CHD4 mutations were all predicted to disrupt normal function of the protein, suggesting a functional role in the development of EC [15]. LSD1 is bound by NuRD and has been shown to repress both tumor suppressor genes [45] and genes associated with metastasis and invasion [46] in complex with NuRD. It is possible that the role of LSD1 is altered in endometrial cancer through functional mutations in NuRD members, and this results in sensitivity to LSD1 inhibition. However, the role of NuRD mutations in endometrial cancer remain unstudied. Detailed studies addressing the role of NuRD and whether LSD1 and NuRD work in concert in endometrial cancer could lead to insight regarding which patients may benefit from LSD1 inhibition or other epigenetic intervention.
This is especially true in light of the results showing that not only are LSD1 substrates affected by HCI2509, H3K27me3 was elevated in both cell lines, suggesting LSD1 inhibition exhibited downstream epignomic regulatory effects. Interestingly, decreased H3K27me3 is associated with poorer survival in both breast and ovarian cancers [47], though this has not been studied in Type II EC. The H3K27 methyltransferase EZH2 is overexpressed in ~60% of Type II EC and has been linked to focal adhesion kinase (FAK) and deregulation of E-cadherin [13]. This presents another possible avenue to define the functional linkage of epigenetic misregulation with Type II EC biology. The differences observed between cell lines with respect to H3K9me2 are consistent with the highly contextual dependence of LSD1 function. In summary, the histone methylation data presented here contrasts with that shown for HCI2509 in Ewing sarcoma [38], and emphasizes the importance of additional mechanistic studies to be conducted in the future to better define LSD1 biology.
Corroborating other in vitro findings, results observed for the effects of HCI2509 on the cell cycle showed a dose-dependent increase in S-phase and decrease in the G2/M for both AN3CA and KLE cells. Time course analysis revealed what appeared to be a moderate G1/G0 arrest at 12 hours in AN3CA cells, though not in the KLE cells. The primary effect seen was an accumulation in early S-phase in both cell lines. LSD1 has been shown to play a critical role in maintaining the cell cycle in embryonic stem cells [48, 49], as well as promoting proliferation and cell cycle progression in cancer cells [50, 51], and the data here is consistent with this observation.
One of the biggest limitations in studying new epigenetic therapies in a given disease is the lack of mechanistic understanding to distinguish which molecular events are drivers and passengers in tumorigenesis. In the meantime, translational progress requires potent and specific tool compounds and to validate new therapeutic strategies. Because epigenetics represents the intersection between genes and the environment, it is likely that the phenomena observed in tissue culture will not represent the disease state in a mouse, and further, the difficulties translating from mouse studies to humans is well documented [52]. To mitigate these issues, we placed emphasis on early testing of whether epigenetic modulation with an LSD1 inhibitor would work in vivo in Type II EC. Further, we also wanted to recapitulate the tumor environment as reliably as possible in an orthotopic setting using relevant human cancer cells. In our KLE model, the generated tumors showed a dip in luminescent signal after the first week as is common and in the vehicle group signal rebounded in an exponential growth pattern by day 42. The doubling time for KLE cells in tissue culture is fairly slow, around 72 hours, indicating in vivo disease progression rate being consistent with the character of the cell line. We were encouraged to see signal present throughout the abdominal cavity in several mice throughout the study, as this suggested an invasive and disseminated disease. Based on the limited number of animals in this pilot in vivo study, we favored endpoints over additional tissue evaluation of responsive tumors to better understand molecular effects caused by HCI2509 treatment, rendering responsive tumors unavailable for additional experiments. Further dose finding, frequency, and survival studies are planned.
Ultimately, this is the first data including histone methylation changes, target gene elevation, and induced apoptosis in EC and is very encouraging. Additional studies should evaluate LSD1 inhibition in more translational and patient-derived models, both in vitro and in vivo. To do so will require expanding the mechanistic insight based on the recent implication of chromatin remodelers in Type II EC using more potent and specific tool compounds. Additional investigation of epigenetics, as well as the relationship between specific pharmacodynamic and pharmacokinetic markers of response, will be needed to gain an in depth understanding of these mechanisms in the development of EC. Furthermore, LSD1 inhibition with HCI2509 should be evaluated for synergistic effects with other targeted inhibitors of other pathways implicated in Type II EC, such as FAK [13] signaling, as well as conventional treatment modalities including hormone therapy currently applied in the treatment of EC.
Conclusions
In conclusion, we have demonstrated that the treatment of Type II endometrial carcinoma cell lines with the LSD1 inhibitor HCI2509 decreased proliferation and transformation, induced histone methylation and LSD1 target gene expression, perturbed cell cycle progression, and induced apoptotic cell death in vitro. Moreover, in an orthotopic endometrial carcinoma animal model with human KLE cells, HCI2509 treatment resulted in 5/9 tumor regressions over the course of 42 days. Taken together these findings support further investigation of the role of LSD1 in Type II endometrial carcinoma biology as well as LSD1 inhibition as a novel therapeutic strategy for this aggressive gynecologic malignancy.
Authors’ information
Sunil Sharma and Margit Janat-Amsbury are co-senior authors.
Electronic supplementary material
Additional file 1: Figure S2: Changes to histone H3 lysine 4 monomethyl and dimethyl marks with HCI2509 treatment. (A, B) Western blot analysis of H3K4me1 and H3K4me2 after 48 hours of vehicle or HCI2509 treatment at varying concentrations in (A) AN3CA and (B) KLE cells. Images are representative of two repeat experiments performed in triplicate. (PDF 49 KB)
Additional file 2: Figure S3: Time course evaluation of cell cycle perturbations caused by HCI2509 treatment. (A, B) Cell cycle populations of (A) AN3CA and (B) KLE cell lines after exposure to vehicle (0 and 48 hours) or 3 μM HCI2509 (6, 12, 24, and 48 hours). 2 × 104 counts and 1 × 104 counts were used for AN3CA and KLE cells, respectively. Data is representative of four biological replicates. (PDF 554 KB)
Additional file 3: Figure S4: TUNEL assay replicates and controls. (A, B) Fluorescence microscopy images of (A) AN3CA and (B) KLE cell lines after exposure to either vehicle or 3X EC50 HCI2509 and then stained with TUNEL for apoptotic nuclei (green), DAPI for nuclei (blue), and phalloidin for actin (red). HCI2509 treatment induced apoptosis with apoptotic cells marked with (*). (C) Fluorescence microscopy images of TUNEL negative and positive controls with untreated AN3CA and KLE cells. Negative controls were generated by adding labeled nucleotide with no enzyme and positive controls were generated by pretreating DNase before TUNEL labeling. Cells are stained with TUNEL (green), DAPI (blue), and phalloidin for actin (red). (PDF 194 KB)
Additional file 4: Figure S5: HCI2509 treatment causes tumor regression in vivo. (A) Individual mouse images from study day 7 (day 0 of treatment). All images are on the same luminescence scale from 1.54 × 104 p/s to 8.66 x 106 p/s. (B) Quantified bioluminscence measurements of both the vehicle and HCI2509 treatment groups pooled. Total flux (photons/second) was rank ordered and plotted on a semi-log plot. The linearity of the log-transformed data supports a log-normal distribution. (C) Fisher’s exact test shows significant association of HCI2509 treatment with tumor regression. Both the observed and expected contingency tables are shown with the reported p-value. (D) Tumor volume and body weight measurements including both the untreated and unimplanted control. Tumor volumes are plotted as the geometric mean of the observed luminescent signal and body weight is plotted as the average and SD. (PDF 150 KB)
Acknowledgments
We thank Jason Tanner and Jeffrey Theisen for their support, discussion, and thoughtful feedback. This work was supported by the American Foundation for Pharmaceutical Education 2013 Pre-Doctoral Fellowship (AFPE), NCI grant P30 CA042014 awarded to the Huntsman Cancer Institute, and the Department of Obstetrics and Gynecology.
Abbreviations
- APC
Adenomatous polyposis coli
- ARID1A
AT rich interactive domain 1A
- CD133
Cluster of differentiation 133
- CDH1
E-cadherin
- EC
Endometrial carcinoma
- EC50
Effective concentration at 50%
- EMT
Epithelial-to-mesenchymal transition
- EP300
E1A binding protein p300
- EZH2
Enhancer of zeste homolog 2
- FAK
Focal adhesion kinase
- FGFR
Fibroblast growth factor receptor
- H3
Histone H3
- H3K4 (me3)
Histone H3 lysine 4 (dimethylated)
- H3K9 (me2)
Histone H3 lysine 9 (trimethylated)
- H3K27 (me3)
Histone H3 lysine 27 (trimethylated)
- HMOX1
Heme oxygenase 1
- LSD1
Lysine-specific demethylase 1
- MGMT
O-6-methylguanine-DNA methyltransferase
- MLH1
mutL homolog 1
- MPA
Medroxyprogesterone 17-acetate
- NuRD
Nucleosome remodeling and deacetylase
- PTEN
Phosphatase and tensin homolog
- TAF1
TATA box binding protein (TBP)-associated factor
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling.
Footnotes
Competing interests
SS is a founder and Chief Medical Officer of Salarius Pharmaceuticals, a company focused on epigenetic therapies for cancer.
Authors’ contributions
ERT, SG and JB performed the experiments and wrote the manuscript. SS and MJA (corresponding author) designed and supervised the experiments and wrote the manuscript. VS and MJA contributed reagents, facilities and personnel. All authors read and approved the final manuscript.
Contributor Information
Emily R Theisen, Email: emily.theisen@hci.utah.edu.
Snehal Gajiwala, Email: u0724410@utah.edu.
Jared Bearss, Email: jared.bearss@hci.utah.edu.
Venkataswamy Sorna, Email: swamy@hci.utah.edu.
Sunil Sharma, Email: sunil.sharma@hci.utah.edu.
Margit Janat-Amsbury, Email: margit.janat-amsbury@hsc.utah.edu.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer Statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 3.National Cancer Institute: Endometrial Cancer Home Page. http://www.cancer.gov/cancertopics/types/endometrial
- 4.Ferlay J, Shin H-R, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2008;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 5.Sutton G, Axelrod JH, Bundy BN, Roy T, Homesley HD, Malfetano JH, Mychalczak BR, King ME. Whole abdominal radiotherapy in the adjuvant treatment of patients with stage III and IV endometrial cancer: a gynecologic oncology group study. Gynecol Oncol. 2005;97:755–763. doi: 10.1016/j.ygyno.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 6.Bokhman JV. Two pathogenic types of endometrial carcinoma. Gynecol Oncol. 1983;15:10–17. doi: 10.1016/0090-8258(83)90111-7. [DOI] [PubMed] [Google Scholar]
- 7.Mendivil A, Schulder KM, Gehrig PA. Non-endometriod adenocarcinoma of the uterine corpus: a review of selected histological subtypes. Cancer Control. 2009;16:46–52. doi: 10.1177/107327480901600107. [DOI] [PubMed] [Google Scholar]
- 8.Xiong Y, Dowdy SC, Bosquet JG, Zhao Y, Eberhardt NL, Podratz KC, Jiang S-W. Epigenetic-mediated upregulation of progesterone receptor B gene in endometrial cancer cell lines. Gynecol Oncol. 2005;99:135–141. doi: 10.1016/j.ygyno.2005.05.035. [DOI] [PubMed] [Google Scholar]
- 9.Tao MH, Freudenheim JL. DNA methylation in endometrial cancer. Epigenetics. 2010;5:491–498. doi: 10.4161/epi.5.6.12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friel AM, Zhang L, Curley MD, Therrien VA, Sergent PA, Belden SE, Borger DR, Mohapatra G, Zukerberg LR, Foster R, Rueda BR. Epigenetic regulation of CD133 and tumorgenicity of CD133 positive and negative endometrial cancer cells. Reprod Biol Endocrinol. 2010;8:147. doi: 10.1186/1477-7827-8-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fakhry H, Miyamoto T, Kashima H, Suzuki A, Ke H, Konishi I, Shiozawa T. Immunohistochemical detection of histone deacetylases in endometrial carcinoma: involvement of histone deacetylase 2 in the proliferation of endometrial carcinoma cells. Hum Pathol. 2010;41:848–858. doi: 10.1016/j.humpath.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 12.Jiang S, Dowdy SC, Meng XW, Wang Z, Jones MB, Podratz, Jiang S-W. Histone deacetylase inhibitors induce apoptosis in both Type I and Type II endometrical cancer cells. Gynecol Oncol. 2007;105:493–500. doi: 10.1016/j.ygyno.2007.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou J, Roh J-W, Bandyopadhyay S, Chen Z, Munkarah AR, Hussein Y, Alosh B, Jazaerly T, Hayek K, Semaan A, Sood AK, Ali-Fehmi R. Overexpression of enhancer of zeste homolog 2 (EZH2) and focal adhesion kinase (FAK) in high grade endometrial carcinoma. Gynecol Oncol. 2013;128:344–348. doi: 10.1016/j.ygyno.2012.07.128. [DOI] [PubMed] [Google Scholar]
- 14.Eskander RN, Ji T, Huynh B, Wardeh R, Randall LM, Hoang B. Inhibitor of enhancer of zeste homolog 2 (EZH2) expression is associated with decreased tumor cell proliferation, migration, and invasion in endometrial cancer cell lines. Int J Gynecol Cancer. 2013;23:997–1005. doi: 10.1097/IGC.0b013e318296a265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le Gallo M, O’Hara AJ, Rudd ML, Urick ME, Hansen NF, O’Neil NJ, Price JC, Zhang S, England BM, Godwin AK, Sgroi DC, Hieter P, Mullikin JC, Merino MJ, Bell DW, NISC Comparative Sequencing Program Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012;44:1310–1315. doi: 10.1038/ng.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E, Guzzo F, English DP, Varughese J, Gasparrini S, Bortolomai I, Buza N, Hui P, Abu-Khalaf M, Ravaggi A, Bignotti E, Bandiera E, Romani C, Todeschini P, Tassi R, Zanotti L, Carrara L, Pecorelli S, Silasi DA, Ratner E, Azodi M, Schwartz PE, Rutherford TJ, Stiegler AL, Mane S, et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A. 2013;110:2916–2921. doi: 10.1073/pnas.1222577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Gallo M, Bell DW. The emerging genomic landscape of endometrial cancer. Clin Chem. 2014;60:98–110. doi: 10.1373/clinchem.2013.205740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 19.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123:8–13. doi: 10.1002/ijc.23607. [DOI] [PubMed] [Google Scholar]
- 21.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 22.Islam AB, Richter WF, Lopez-Bigas N, Benevolenskaya EV. Selective targeting of histone methylation. Cell Cycle. 2011;10:413–424. doi: 10.4161/cc.10.3.14705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Popovic R, Licht JD. Emerging epigenetic targets and therapies in cancer medicine. Cancer Discov. 2012;2:405–413. doi: 10.1158/2159-8290.CD-12-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 25.Metzger E, Wissmann M, Yin N, Mueller JM, Schneider R, Peters AHFM, Guenther T, Buettner R, Schuele R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 26.Lachner M, O’Sullivan RJ, Jenuwein T. An epigenetic road map for histone lysine methylation. J Cell Sci. 2003;116:2117–2124. doi: 10.1242/jcs.00493. [DOI] [PubMed] [Google Scholar]
- 27.Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, Ora I, Pajtler K, Klein-Hitpass L, Kuhfittig-Kulle S, Metzger E, Schüle R, Eggert A, Buettner R, Kirfel J. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69:2065–2071. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- 28.Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, Miller CJ, Ogilvie DJ, Somervaille TC. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473–487. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 29.Lim S, Janzer A, Becker A, Zimmer A, Schüle R, Buettner R, Kirfel J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis. 2010;31:512–520. doi: 10.1093/carcin/bgp324. [DOI] [PubMed] [Google Scholar]
- 30.Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, Solleder G, Bastian PJ, Ellinger J, Metzger E, Schüle R, Buettner R. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006;66:11341–11347. doi: 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]
- 31.Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, Field HI, Neal DE, Yamaue H, Ponder BA, Nakamura Y, Hamamoto R. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer. 2011;128:574–586. doi: 10.1002/ijc.25349. [DOI] [PubMed] [Google Scholar]
- 32.Zhao ZK, Yu HF, Wang DR, Dong P, Chen L, Wu WG, Ding WJ, Liu YB. Overexpression of lysine specific demethylase 1 predicts worse prognosis in primary hepatocellular carcinoma patients. World J Gastroenterol. 2012;18:6651–6656. doi: 10.3748/wjg.v18.i45.6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schildhaus HU, Riegel R, Hartmann W, Steiner S, Wardelmann E, Merkelbach-Bruse S, Tanaka S, Sonobe H, Schüle R, Buettner R, Kirfel J. Lysine-specific demethylase 1 is highly expressed in solitary fibrous tumors, synovial sarcomas, rhabdomyosarcomas, desmoplastic small round cell tumors, and malignant peripheral nerve sheath tumors. Hum Pathol. 2011;42:1667–1675. doi: 10.1016/j.humpath.2010.12.025. [DOI] [PubMed] [Google Scholar]
- 34.Bennani-Baiti I-M, Machado I, Llombart-Bosch A, Kovar H. Lysine-specific demethylase 1 (LSD1/KDM1A/AOF2/BHC110) is expressed and is an epigenetic drug target in chondrosarcoma, Ewing’s sarcoma, osteosarcoma, and rhabdomyosarcoma. Hum Pathol. 2012;43:1300–1307. doi: 10.1016/j.humpath.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Sorna V, Theisen ER, Stephens B, Warner SL, Bearss DJ, Vankayalapati H, Sharma S. High-throughput virtual screening identifies novel N’-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible inhibitors of LSD1. J Med Chem. 2013;56:9496–9508. doi: 10.1021/jm400870h. [DOI] [PubMed] [Google Scholar]
- 36.Stephens BJ, Theisen ER, Warner SL, Sharma S, Bearss DJ. Proceedings of the American Association for Cancer Research 103rd Annual Meeting 2012: 31 March −4 April 2012. IL: Chicago; 2012. Abstract 1045: Activity of the LSD1 inhibitor HCI-2509 in ER-negative breast cancer cells. [Google Scholar]
- 37.Theisen ER, Bearss J, Sorna V, Bearss DJ, Sharma S. Proceedings of the American Association for Cancer Research 104th Annual Meeting 2013. DC: Washington; 2013. Abstract 3: Targeted inhibition of LSD1 in castration-resistant prostate cancer. [Google Scholar]
- 38.Sankar S, Theisen ER, Bearss J, Mulvihill T, Hoffman LM, Sorna V, Beckerle MC, Sharma S, Lessnick SL. LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin Cancer Res. 2014;20:4584–4597. doi: 10.1158/1078-0432.CCR-14-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lessnick SL, Dacwag CS, Golub TR. The Ewing’s sarcoma oncoprotein EWS/FLI induces a p53-dependent growth arrest in primary human fibroblasts. Cancer Cell. 2002;1:393–401. doi: 10.1016/S1535-6108(02)00056-9. [DOI] [PubMed] [Google Scholar]
- 40.Nakamura M, Kyo S, Zhang B, Zhang X, Mizumoto Y, Takakura M, Maida Y, Mori N, Hashimoto M, Ohno S, Inoue M. Prognostic impact of CD133 expression as a tumor-initiating cell marker in endometrial cancer. Hum Pathol. 2010;41:1516–1529. doi: 10.1016/j.humpath.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 41.McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-mesenchymal transition. Nat Struct Mol Biol. 2011;18:867–874. doi: 10.1038/nsmb.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin T, Ponn A, Hu X, Law BK, Lu J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene. 2010;29:4896–4904. doi: 10.1038/onc.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Llobet D, Pallares J, Yeramian A, Santacana M, Eritja N, Velasco A, Dolcet X, Matias-Guiu X. Molecular pathology of endometrial carcinoma: practical aspects from the diagnostic and therapeutic viewpoints. J Clin Pathol. 2009;62:777–785. doi: 10.1136/jcp.2008.056101. [DOI] [PubMed] [Google Scholar]
- 44.Limpert E, Stahel WA, Abbt M. Log-normal distributions across the sciences: keys and clues. Bioscience. 2001;51:341–352. doi: 10.1641/0006-3568(2001)051[0341:LNDATS]2.0.CO;2. [DOI] [Google Scholar]
- 45.Sankar S, Bell R, Stephens B, Zhuo R, Sharma S, Bearss DJ, Lessnick SL. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene. 2013;32:5089–5100. doi: 10.1038/onc.2012.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, Liang J, Sun L, Yang X, Shi L, Li R, Li Y, Zhang Y, Li Q, Yi X, Shang Y. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009;138:660–672. doi: 10.1016/j.cell.2009.05.050. [DOI] [PubMed] [Google Scholar]
- 47.Wei Y, Xia W, Zhang Z, Liu J, Wang H, Adsay NV, Albarracin C, Yu D, Abbruzzese JL, Mills GB, Bast RC, Jr, Hortobagyi GN, Hung MC. Loss of trimethylation at lysine 27 of histone H3 is a predictor of poor outcome in breast, ovarian, and pancreatic cancers. Mol Carcinog. 2008;47:701–706. doi: 10.1002/mc.20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nair VD, Ge Y, Balasubramaniyan N, Kim J, Okawa Y, Chikina M, Troyanskaya O, Sealfon SC. Involvement of histone demethylase LSD1 in short-time-scale gene expression changes during cell cycle progression in embryonic stem cells. Mol Cell Biol. 2012;32:4861–4876. doi: 10.1128/MCB.00816-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yin F, Lan R, Zhang X, Zhu L, Chen F, Xu Z, Liu Y, Ye T, Sun H, Lu F, Zhang H. LSD1 regulates pluripotency of embryonic stem/carcinoma cells through histone deacetylase 1-mediated deacetylation of histone H4 at lysine 16. Mol Cell Biol. 2014;34:158–179. doi: 10.1128/MCB.00631-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scoumanne A, Chen X. The lysine-specific demethylase 1 is required for cell proliferation in both p53-dependent and -independent manners. J Biol Chem. 2007;282:15471–15475. doi: 10.1074/jbc.M701023200. [DOI] [PubMed] [Google Scholar]
- 51.Cho HS, Suzuki T, Dohmae N, Hayami S, Unoki M, Yoshimatsu M, Toyokawa G, Takawa M, Chen T, Kurash JK, Field HI, Ponder BA, Nakamura Y, Hamamoto R. Demethylation of RB regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells. Cancer Res. 2011;71:655–660. doi: 10.1158/0008-5472.CAN-10-2446. [DOI] [PubMed] [Google Scholar]
- 52.Lum DH, Matsen C, Welm AL, Welm BE. Overview of human primary tumorgraft models: comparisons with traditional oncology preclinical models and the clinical relevance and utility of primary tumorgrafts in basic and translational oncology research. Curr Protoc Pharmacol. 2012;Chapter 14:Unit 14.22. doi: 10.1002/0471141755.ph1422s59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pre-publication history
- The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/14/752/prepub
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S2: Changes to histone H3 lysine 4 monomethyl and dimethyl marks with HCI2509 treatment. (A, B) Western blot analysis of H3K4me1 and H3K4me2 after 48 hours of vehicle or HCI2509 treatment at varying concentrations in (A) AN3CA and (B) KLE cells. Images are representative of two repeat experiments performed in triplicate. (PDF 49 KB)
Additional file 2: Figure S3: Time course evaluation of cell cycle perturbations caused by HCI2509 treatment. (A, B) Cell cycle populations of (A) AN3CA and (B) KLE cell lines after exposure to vehicle (0 and 48 hours) or 3 μM HCI2509 (6, 12, 24, and 48 hours). 2 × 104 counts and 1 × 104 counts were used for AN3CA and KLE cells, respectively. Data is representative of four biological replicates. (PDF 554 KB)
Additional file 3: Figure S4: TUNEL assay replicates and controls. (A, B) Fluorescence microscopy images of (A) AN3CA and (B) KLE cell lines after exposure to either vehicle or 3X EC50 HCI2509 and then stained with TUNEL for apoptotic nuclei (green), DAPI for nuclei (blue), and phalloidin for actin (red). HCI2509 treatment induced apoptosis with apoptotic cells marked with (*). (C) Fluorescence microscopy images of TUNEL negative and positive controls with untreated AN3CA and KLE cells. Negative controls were generated by adding labeled nucleotide with no enzyme and positive controls were generated by pretreating DNase before TUNEL labeling. Cells are stained with TUNEL (green), DAPI (blue), and phalloidin for actin (red). (PDF 194 KB)
Additional file 4: Figure S5: HCI2509 treatment causes tumor regression in vivo. (A) Individual mouse images from study day 7 (day 0 of treatment). All images are on the same luminescence scale from 1.54 × 104 p/s to 8.66 x 106 p/s. (B) Quantified bioluminscence measurements of both the vehicle and HCI2509 treatment groups pooled. Total flux (photons/second) was rank ordered and plotted on a semi-log plot. The linearity of the log-transformed data supports a log-normal distribution. (C) Fisher’s exact test shows significant association of HCI2509 treatment with tumor regression. Both the observed and expected contingency tables are shown with the reported p-value. (D) Tumor volume and body weight measurements including both the untreated and unimplanted control. Tumor volumes are plotted as the geometric mean of the observed luminescent signal and body weight is plotted as the average and SD. (PDF 150 KB)