

Abstract
The clinical syndrome of Huntington’s disease is notable for a triad of motor, cognitive and emotional features. All HD patients eventually become occupationally disabled; however the factors that render HD patients unable to maintain employment have not been extensively studied. This review begins by discussing the clinical triad of HD, highlighting the distinction in the motor disorder between involuntary movements such as chorea, and voluntary movement impairment, with the latter contributing more to functional disability. Cognitive disorder clearly contributes to disability, though the relative contribution compared to motor is difficult to unravel, especially since many of the tests used to asses “cognition” have a strong motor component. The role of emotional changes in disability needs more study. The literature on contributions to functional disability, driving impairment and nursing home placement is reviewed. Relevant experience is presented from the longstanding JHU HD observational study on motor vs cognitive onset, and on cognitive and motor features at the time when individuals discontinued working. Finally, we briefly review government policies in several countries on disability determination. We interpret the data from our own studies and from the literature to indicate that there is usually a close relationship between cognitive and motor dysfunction, and that it is critical to take both into consideration in determining disability.
Introduction: The Triad of Signs and Symptoms in HD
The clinical syndrome of Huntington’s disease is notable for a triad of motor, cognitive and emotional features1–5. Although all HD patients eventually become occupationally disabled, the factors that render HD patients unable to maintain employment have not been extensively studied. One difficulty in this line of research lies in the fact that different occupations require different cognitive and motor capabilities. Thus, for example, the factors that render an accountant, a jeweler, or a roofer disabled are likely to be dissimilar. A second difficulty is that many patients are able to maintain employment with accommodations, including marked reductions in work duties. A third issue is that self-employed individuals (e.g., craftspeople, consultants, free-lance accountants, real-estate brokers) may slowly curtail their activities so that it is difficult to determine when disability begins. This article will summarize some of the issues for consideration, in the context of the HD clinical triad, including some of the experiences at the HD Center at Johns Hopkins, which has been in existence for over thirty years.
The progressive motor disorder of HD is a major source of functional disability. The motor disorder is commonly assessed via the brief semiquantitative clinical motor exam in the Unified Huntington’s Disease Rating Scale6. The motor disorder can be divided into two prominent components—involuntary and voluntary movement disorders. Chorea and related involuntary movements (such as athetosis or, occasionally, tic-like movements) tend to be an early feature of HD (except in the relatively rare juvenile onset form with long CAG repeat lengths). A second component of the motor disorder termed “motor impairment” consists of voluntary movement deficits (including incoordination, bradykinesia, motor sequencing difficulties and ideomotor apraxia). Factor analysis of the Quantitative Neurological Exam (QNE), which was the precursor of the UHDRS, identified chorea and motor impairment as two separate factors7). A factor analysis of the UHDRS has resulted in two comparable factors8.
Clinicopathologic studies have found that loss of medium spiny neurons in the striatum and overall severity of striatal pathology correlate with motor impairment score, but not with chorea9, 10. MIS progresses more steadily than chorea and MIS correlates with impairment of activities of daily living, while chorea does not11, 12. Thus, while chorea is very distinctive and often useful in making the diagnosis, functional impairment tends to arise more frequently from MIS.
All patients with HD also have progressive cognitive impairment. This is often characterized by executive dysfunction, especially early on in the disease, but other cognitive realms are also affected, including learning and memory, motor planning and working memory (eg the ability to perform tasks such serial 7s, which involves computation based on information held in memory). This is sometimes termed a “subcortical” dementia syndrome, characterized by slowed cognitive processing and impairments in sustained attention, memory retrieval, and executive control13–15. Cognitive impairment is a major contributor to disability among patients with HD16, 17, and can be an early feature of the illness16, 18. The relative contribution of cognitive and motor impairment to disability is difficult to unravel, especially since many of the “cognitive” tests whose results are used to highlight the importance of cognition actually have major motor aspects.
The emotional manifestations of HD can also be very disruptive to social and occupational functioning, though they are less predictable in the natural history. These frequently include apathy, irritability and major or minor depression, and less frequently delusions or other psychosis, and sometimes sexual or other disinhibited behavior. It is becoming increasingly apparent that apathy begins relatively early and this can be a debilitating symptom. However there have been relatively few studies of these features in relation to disability, and there is a great need for future studies.
Functional Disability
Several studies have examined the elements of functional disability in HD. In an early study, Rothlind et al assessed eighty patients with HD using the Huntington’s Disease-Activities of Daily Living Questionnaire (HD-ADL)12, 16 Sixty-seven patients also completed a comprehensive assessment of cognitive and voluntary motor functioning and chorea. Both cognitive and motor items were correlated with HD-ADL total score. Psychomotor speed and the ability to regulate attention appeared to be particularly important determinants of everyday functioning in mild HD, even after accounting for individual differences in the severity of chorea and voluntary motor impairment.
Marder et al. assessed factors affecting the annual rate of functional decline using the Total Functional Capacity Scale (TFC) and the Independence Scale (IS) in 960 patients with definite HD followed prospectively for a mean of 18.3 months8. Longer disease duration and better cognitive status at baseline (as assessed by verbal fluency, symbol digit, and Stroop interference items of the UHDRS) at baseline were associated with a slower rate of decline in TFC score. Depressive symptomatology was the only factor associated with more rapid decline on the IS score. Age at onset of HD, sex, weight, and education did not affect decline on either score. In the early stages of HD (duration 0 to 5 years), better neuropsychological performance was associated with a less rapid decline in TFC, and none of the assessed covariates affected the rate of IS decline. Aside from the issue of unadjusted multiple statistical comparisons, less impaired or more slowly progressing subjects may have been overrepresented in this cohort (especially if the most severely impaired patients dropped out due to institutionalization or other factors).
Mahant et al. analyzed data on 1,026 patients, followed for a median of 2.7 years, using a mixed effects model19. Chorea and dystonia were not major determinants of disability. However disability did correlate with the motor score--excluding chorea and dystonia—and with performance on the Symbol-Digit Modalities Test (SDMT), a task requiring rapid transcoding between numbers and arbitrary symbols.
Ho et al examined determinants of the scores on the health-related quality of life (HrQOL)20. Multiple regression analyses showed that greater impairments in HrQOL were associated with higher levels of depressive mood and lower functional capacity. Motor symptoms and cognitive function were not found to be as closely linked with HrQOL.
Hart et al. retrospectively examined the relationship between motor HD type and cognitive (assessed as in Marder et al. above) and functional capacity in the European Huntington’s Disease Network Registry cohort 8, 21. Patients were classified as predominantly choreatic (n=528) or hypokinetic-rigid (n=432) according to their scores on items of the total motor score (922 patients were of a mixed type, and were not included in the analysis). Motor subtype was found to contribute significantly to general functioning (TFC score; p<.001), accounting for group differences in age, sex, education, disease duration, CAG repeat number, and UHDRS total motor score (TMS). Hypokinetic-rigid subjects performed significantly worse on all cognitive tests and TFC than did choreatic subjects, and motor subtype was the second most contributing variable after TMS in all linear regression models (partial R2=0.078, p<.001). Because differences in disease duration were taken into account in the analyses, an increased probability of converting to a more hypokinetic-rigid motor type with advancing disease does not fully account for this study’s findings.
Driving
Because driving a motor vehicle is a highly complicated activity that is often relied upon for both work and leisure, giving up driving may be considered an important marker of increasing disability in HD. In an early study Rebock et al assessed neurological and cognitive impairments in relation to automobile driving22. In a group of 73 HD outpatients, 53 (72%) continued to drive after illness onset. Those no longer driving had more severe symptoms than those still driving. Twenty-nine HD patients who were still driving and 16 healthy control subjects underwent a clinical examination, a cognitive examination, and a driving-simulator assessment. HD patients performed significantly worse than control subjects on the driving-simulator tasks and were more likely to have been involved in a collision in the preceding 2 years (58% of HD vs. 11% of control subjects). Patients with collisions were less functionally impaired but had slower simple reaction time scores than did those without collisions. The study concluded that HD patients are at increased risk for accidents, but patients who have accidents are not easily distinguished from those who do not.
In a retrospective review of UHDRS and supplemental neuropsychological testing from 74 charts, Beglinger et al. found that lower global cognitive performance (as assessed by the Repeatable Battery for the Assessment of Neuropsychological Status) and UHDRS TFC scores were the best predictors of driving cessation (R2=0.65; p<0.0001)23. Measures of learning (p=0.006) and psychomotor speed/attention (p=0.003) appeared to account for the overall cognitive finding, and no motor variables (including chorea and oculomotor function) reached statistical significance as predictors of driving cessation in multivariate analyses. This retrospective cross-sectional study was limited by its reliance of clinicians’ assessments of driving ability, and lack of formal driving assessments.
Devos et al. compared 30 active drivers with HD (disease stages 1–2, corresponding to mild functional disability) with 30 age and gender-matched healthy control drivers, and identified poor performance on the SDMT, Stroop Word Reading subtest, and Trail Making Test part B as the screening measures that best predict failure on a comprehensive fitness to drive evaluation24. The combination of these 3 tests provided a model (R2=0.49) to predict fitness to drive, correctly classifying 26/30 (87%) patients. 15 out of 30 assessed patients failed the evaluation, but 11 of those 15 were unaware of their decreased driving ability. The UHDRS motor score was not retained in the prediction model. The outcome on this fitness to drive evaluation was based on detailed visual and cognitive testing, as well as on a standardized on-road test. However, the authors did not conduct follow-up assessments to evaluate the predictive accuracy of the clinical assessment battery.
The same group recently compared results of on-road driving assessments (13 specific skills divided into 4 clusters) between 30 patients with HD and 30 age-matched healthy control drivers25. In addition to tests from their prior study, they administered several visual and cognitive tests performed at the fitness-to-drive center to model road driving behavior in HD. The high driving test failure rate (47% of HD patients, 0% of controls) was similar to the authors’ prior study. TFC scores discriminated significantly between pass and fail groups. However, 2 of 6 subjects with HD who had maximal TFC scores failed the driving evaluation, suggesting presence of driving impairment even in some individuals with very early stages of HD. Total on-road score and performance in operational, tactical, and visuo-integrative clusters correlated strongly (Spearman rho>0.50) with the pass/fail decision. Strong correlations were found between the total score of the on-road test and motor functions (UHDRS–motor section), information processing (Stroop Color and Word subtests), executive control, divided attention, and visual tracking and working memory (SDMT). Additionally, selective attention, (measured with the Useful Field of View Test (which requires localization of a car embedded in an array of triangles while focusing on a central fixation task at different speeds of presentation) was strongly associated (Spearman rho>0.50) with the total on-road score and all driving clusters.
Nursing Home Placement
Rosenblatt et al. investigated factors contributing to institutionalization in 799 HD patients followed at the Baltimore Huntington’s Disease Center28. 88 of the patients were relocated to care facilities were institutionalized in a number of different facilities during the 9.2 (±6.0) years of follow up. Scores on the Quantified Neurologic Examination (QNE; R2=0.203, p<.001), Huntington’s disease Activities of Daily Living Scale (ADL scale; R2=0.259, p<.001), and the MISMotor Impairment Score (MIS; R2=0.173, p<.001) were found to have the strongest correlations with time until institutionalization. In a stepwise Cox model of ADL, QNE, and MMSE, only ADL was left in the equation, suggesting that the variation in time to institutionalization explained by QNE and MMSE is also explained by ADL; this was in line with previous findings10 (Rosenblatt et al. 2003). Psychiatric symptoms did not significantly predict institutionalization.
Neither age at onset nor CAG repeat length number alone were significantly correlated with disease duration at time of institutionalization. However, when controlling for age at onset, CAG number was significantly associated with disease duration at the time of admission to a facility. It should be noted that none of these studies included information on caregiver stress, which is an important predictor of institutionalization In other dementing conditions, and may have been associated with high patient loss to follow up 28.
Features of Patients Who Stop Working: JHU Experience
Thirty-five patients with HD who were followed in the longitudinal cohort study at the Johns Hopkins University Center Without Walls for Huntington’s Disease discontinued gainful employment between annual visits. The 20 women and 15 men had an average of 14.7 years of education (i.e., 2½ years of college). Their average age of clinical onset of HD was 38.1 years (SD=11.3), and their average age of retirement or disability was 47.0 years (SD=11.6).
All patients in the longitudinal cohort study were evaluated neurologically, psychiatrically, and neuropsychologically at annual visits to the Center. The clinical and neuropsychological test scores of the patients who stopped working are shown in Table 1 for the visit immediately prior to their cessation of employment and for their first unemployed visit. Performance on tests measuring speed of mental processing changed the most over these two visits, with effect sizes approaching 0.80 (the conventional cut-off for a large effect). It is noteworthy that manual speed and dexterity (Grooved Pegboard Test) did not differ significantly between the working and disabled visits, suggesting that declining motor control per se is not strongly associated with occupational disability in HD. The WAIS-R and Wisconsin Card Sorting Test were also not different between visits (data not shown). Cognitive and motor tests both showed strong correlations with loss of ability to work. These data should be interpreted cautiously, since the sample sizes are relatively small and do not allow a multivariate analysis. However, they do suggest the closely intertwined contributions of motor and cognitive difficulties to functional disability.
Table 1.
Clinical scales and neuropsychological test scores (means =/−SD) from annual assessment at visit just prior to retiring or becoming disabled and visit just after stopping work. Visits are nominally one year apart.
| Last Visit Still Working | First Visit Not Working | p | Cohen’s d | |
|---|---|---|---|---|
|
| ||||
| Quantified Neurological Exam | ||||
| Chorea scale | 7.15 (4.55) | 8.56 (4.23) | .021 | 0.41 |
| Motor Impairment scale | 6.32 (4.02) | 7.68 (4.39) | .010 | 0.47 |
| Eye Movement scale | 4.56 (2.45) | 5.12 (2.84) | NS | |
| Total | 29.03 (16.21) | 35.82 (18.29) | <.001 | 0.75 |
|
| ||||
| Mini-Mental State Exam | 26.57 (2.87) | 26.17 (3.68) | NS | |
|
| ||||
| HD-ADL Scale | 7.58 (7.23) | 14.42 (10.19) | .002 | 0.73 |
|
| ||||
| Irritability Scale | 12.14 (9.73) | 14.32 (10.29) | NS | |
|
| ||||
| Apathy Scale | 13.91 (7.68) | 16.22 (7.79) | .027 | 0.49 |
|
| ||||
| Hamilton Depression Scale | 4.03 (3.85) | 5.45 (3.44) | NS | |
|
| ||||
| Grooved Pegboard Test | ||||
| Dominant hand sec. | 88.95 (23.11) | 93.74 (22.59) | NS | |
| Nondominant hand sec. | 104.89 (27.88) | 110.83 (32.20) | NS | |
|
| ||||
| Symbol Digit Modalities Test (written) | 38.71 (12.36) | 34.24 (12.74) | .002 | 0.79 |
|
| ||||
| Brief Test of Attention | 11.45 (4.08) | 11.61 (4.78) | NS | |
|
| ||||
| Controlled Oral Word Association Test | 25.84 (12.27) | 25.78 (15.27) | NS | |
|
| ||||
| Developmental Test of Visual-Motor Integration, raw | 16.28 (4.14) | 14.72 (4.97) | .007 | 0.51 |
|
| ||||
| Hopkins Verbal Learning Test | ||||
| Total of Recall Trials | 22.80 (6.59) | 20.74 (6.12) | .018 | 0.42 |
| Delayed Recall | 8.31 (2.44) | 9.06 (2.70) | NS | |
| Recognition Discrimination | 9.44 (2.06) | 9.76 (1.81) | NS | |
|
| ||||
| Hopkins Board | ||||
| Errors to Criterion | 6.05 (6.19) | 7.95 (7.38) | NS | |
| Trials to Criterion | 6.05 (3.12) | 6.45 (3.25) | NS | |
| Delayed Recall of Items | 8.30 (1.03) | 8.45 (0.76) | NS | |
| Delayed Recall of Locations | 8.10 (1.62) | 8.20 (1.15) | NS | |
|
| ||||
| Trail Making Test | ||||
| Part A time | 43.03 (17.53) | 59.83 (47.56) | .024 | 0.40 |
| Part B time | 125.14 (86.81) | 184.31 (161.13) | .007 | 0.48 |
|
| ||||
| Stroop Color-Word Test | ||||
| Word T-score | 35.49 (8.91) | 31.49 (10.73) | <.001 | 0.76 |
| Color T-score | 33.14 (9.06) | 28.89 (10.62) | <.001 | 0.72 |
| Color-Word T-score | 38.23 (11.05) | 36.97 (9.68) | NS | |
| Interference T-score | 51.11 (7.09) | 53.17 (4.33) | NS | |
Cognitive vs Motor Early Signs and Symptoms: JHU Experience
A related issue that often arises is to what extent there is a pure motor or pure cognitive “onset” of HD. This issue is reviewed in the Pausen et al article in this issue and also discussed in Biglan et al and Vinther-Jensen et al, so will not be discussed in detail here18, 29. Our impression is that while different individuals and families notice different aspects of the disorder, and different aspects of the disorder may be more pronounced at one time or another, it is very difficult to separate motor and cognitive features. Many of the “cognitive” tests whose results are used to highlight the importance of cognition actually have major motor aspects (eg SDMT, Trail-Making and certainly “tapping” tests), so this may be a function of measurement, but motor and cognitive ability do tend to decline together. Nevertheless, there is a minority of individuals for whom onset of cognitive signs and symptoms precedes onset of motor signs and symptoms.
This is corroborated by a review of cognitive versus motor onset in patients in our clinic at JHU. For many years we have used a semi-structured interview to gather information on the onset of signs and symptoms of motor disorder versus signs and symptoms of cognitive disorder. Information is recorded when the informants can give a clear estimate of onset of signs and symptoms noticeable to subjects or family members or both. Because onset is so gradual, onset is assessed to the nearest year. There were 208 cases with information on both cognitive and motor onset. As shown in the graph in Figure 1, the time of onset of noticeable and troubling motor versus cognitive signs and symptoms were quite contiguous for most subjects, ie within a year or two of each other for the majority of subjects. Overall, of the 208 patients studied, 55% had motor onset first; 32% had the same year of cognitive and motor onset; and 28 cases (13%) had cognitive onset first. On average the motor onset preceded cognitive onset by 4.1 years when they were different. Five out of the 208 individuals (or less than 3%) had cognitive onset 5 or more years prior to motor onset. It would be interesting to see if there was anything else unusual about these cases. There was no relationship between repeat length and timing of cognitive vs motor onset
Figure 1.
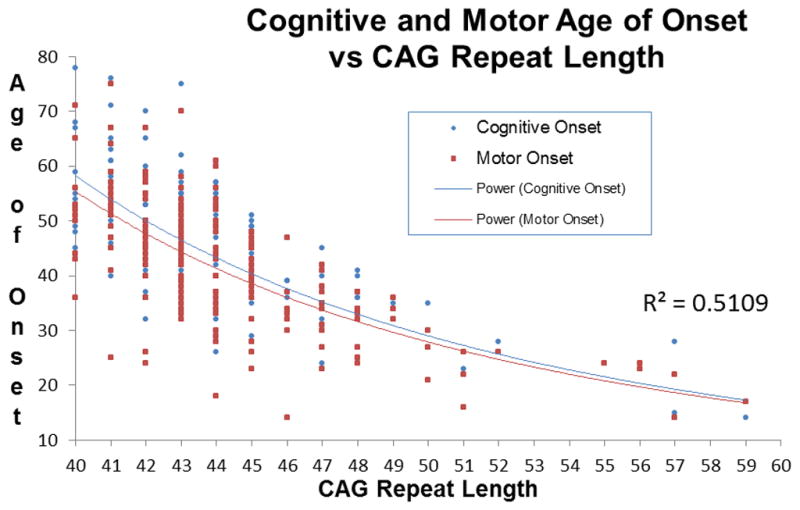
CAG repeat length vs motor and cognitive onset. Motor onset is on average 4.1 years earlier than cognitive onset, with little variation over this range of repeat lengths. “Power” refers to trendlines for cognitive and motor onset respectively.
Conclusions--Cognitive and Motor Signs and Symptoms in HD
Overall, we believe the data from our Center and from the literature support the idea that cognitive and motor signs and symptoms are closely linked. This is consistent with the idea of cortical loops subserving the coordination of motor, cognitive and emotional function, integrated in the basal ganglia. While the projections from different cortical regions have topographic representation within the basal ganglia circuit (see Figure 2), there are mechanisms for convergence and functional integration30–33. Thus cognitive and motor dysfuntion together appear to contribute to disability. The role of emotional dysfunction is much less well-understood. From the standpoint of basal ganglia circuitry, it may not be surprising that emotional signs and symptoms generally are less well correlated with motor dysfunction that cognitive signs and symptoms are—the pathways from the emotional areas of cortex project via the ventral striatum, which is further separated from the motor areas of striatum.
Figure 2.

Schematic diagram of basal ganglia circuits, emphasizing the close relationship among motor, cognitive and emotional circuits. Cortical region A indicates motor areas such as supplemental motor cortex, B indicates cortical areas especially involved with cognitive function such as dorsolateral prefrontal or parietal association cortex, and C represents cortical areas especially involved with emotion such as orbito-frontal, temporal or cingulate cortex. The circuits are parallel, but projections from these regions interdigitate, and recurrent projections enable the motor, cognitive and emotional information to be integrated before the efferent streams return back to the cortex. However emotional information may remain relatively distinct, via the ventral striatum. Adapted from Alexander et al 198630.
Disability Determination by Government Agencies
United States
Disability Framework
In the United States, the designation of occupational disability is managed by the Social Security Administration (SSA), and is separated into two programs; Social Security Income (SSI) is for people with substantially limited income, work history and assets; and Social Security Disability Income (SSDI) is for people for individuals who have a work history. The medical criteria for SSI and SSDI are identical. An individual must be permanently disabled to qualify.
The SSA uses a five step sequential evaluation process to determine disability (http://www.ssa.gov/OP_Home/rulings/di/01/SSR86-08-di-01.html). Each step can be expressed in the form of a question.
Can the individual engage in Substantial Gainful Activity? This refers to a maximal earnings threshold.
Is the individual’s condition severe? An individual must be severely limited in his or her ability to perform work activities, for a continuous period of one year.
Does the individual meet or equal one of SSA’s Medical Listings? SSA organizes their listings into 14 sections, by body system, and there are separate listings for adults and children. These listings are referred to colloquially as the Blue Book.
Can the individual perform at his or her current job, or any past job that he/she has held in the last 15 years? If the individual cannot perform past work, the process proceeds to the fifth step.
Can the individual do any type of work? Social Security considers a claimant’s age, education, work experience and physical and mental condition to make this determination.
An individual can either be judged to be disabled at Step 3 (having or meeting a listing, such as blindness), or at Step 5 (inability to perform any job). Based upon the most recent data from the Social Security (FY 2007–FY 2011), most individuals with Huntington’s disease were allowed at Step 3. Thus an individual with a collection of symptoms that are not singularly disabling can qualify for disability based upon the combined impact of the symptoms.
Compassionate Allowances (CAL)
In 2012, Adult Onset HD and Juvenile Onset HD were added to the Social Security Administration’s Compassionate Allowances List (CAL), an initiative used by SSA to fast-track conditions for disability. Cases are still evaluated using the sequential evaluation process. Individuals with juvenile onset HD are automatically flagged for fast-tracked processing, whereas individuals with adult-onset HD must allege a certain level of severity of symptoms to qualify. Examples of allegations that trigger CAL are mid-stage HD, late-stage HD, advanced HD, and Huntington’s chorea.
SSA Criteria for HD
Huntington’s disease is currently listed in Section 11.00 of the Blue Book (Adult Neurological Listings). There is currently no listing for Juvenile Onset HD in the Childhood Neurological Listings section. SSA does not have a separate listing for HD, and instead lists it with other neurodegenerative disorders, in Section 11.17. An individual can either meet the listing with disorganization of motor function, or, via ‘mixed brain syndrome’, which is found in 12.02, in the Adult Mental listings.
The genetic test is not currently a pre-requisite of receiving disability for HD, nor is showing a positive test for a gene expansion sufficient to be considered disabled.
At the time of writing, the Social Security Administration had just released a revision of their neurological listings, and childhood neurological listings (20 CFR Pt. 404, 2014). SSA is currently in the process of taking public comments, so it may be several years until they are finalized. The Mental listings process of being revised (20 CFR Pt. 404 & 416, 2010).
Worldwide
Worldwide, there is a wide variability in the disability evaluation process. Australia, Canada, and the U.K. will be briefly compared.
Australia
Australia is in the process of transitioning from a State-run system, with varying framework and criteria, to a Federally-run system. The National Disability Insurance Agency (NDIA) is in place in some parts of Australia. Individuals will also be evaluated based upon their ability to work. According to NDIA staff, many of the planners, professionals evaluating individuals for disability purposes will themselves have lived the experience of disability either personally or in a professional capacity.
Canada
Canada’s disability system assesses an individual’s functional capacity. Unlike the U.S. and Australia, Canada does not have specific impairment listings. Instead, medical adjudicators, usually trained nurses, evaluate an individual’s condition. Like the United States, Canada also uses a sequential evaluation process. In order to be disabled, an individual must prove that his or her condition is severe and prolonged, and prevents him or her from performing substantially gainful occupation. Severity is defined as a mental or physical disability that regularly prevents an individual from doing the work that they do, as well as any other work. Prolonged is defined either as resulting in death, or likely to continue indefinitely. Substantially Gainful Occupation is measured in terms of profitability (amount earned), productivity and motivation (working despite medical advice to the contrary). Canada has a fast-track system, but does not include HD in this list.
United Kingdom
The United Kingdom also does not use specific medical criteria. The disability payment system in the U.K, called the Personal Independence Payment (PIP), is not based on medical diagnoses, but rather upon holistic, functional criteria deemed to be the most essential for everyday life.
PIP is measured in terms of activities of daily living (ADLs) and mobility. The daily living criteria of PIP include such items as: Preparing and cooking food, Washing and bathing, Dressing and undressing, Communicating verbally, and Making budgeting decisions. The mobility criteria of PIP are based on the ability to plan and follow a journey, as well as the ability to ambulate.
A complete inability to do perform an activity in any of these spheres would qualify a person for the PIP. Similarly to disability under Step 5 in the United States, an individual can also be found disabled if he or she is limited in a combination of these areas.
International Comparisons
There is no consensus on how best to evaluate disability for Huntington’s disease. Although disability can be determined based either upon specific symptom criteria (U.S. & Australia) or on functional criteria describing the inability to work (Canada & U.K), there is a great deal of variability in both the medical and the functional framework.
Overall Conclusions and Future Directions
It is clear that both cognitive and motor features of HD contribute to functional disability. We believe that the timing and relationship of these two features, and their respective contributions, may vary from individual to individual, based both on the individual natural history of the disease, and also on the individual’s occupation. In most cases, cognitive and motor contributions are closely linked. Emotional features may also be important contributors, though the data on this are relatively scant. Depression may be especially important, though depression is also the most treatable of the psychiatric features of HD, so it may not contribute as much to long-term disability. Apathy is under-appreciated as a feature of early disease, and may be an important factor in disability. As the biology of HD becomes better understood, and new treatments are developed, improving our understanding of the features of the disease that cause functional disability becomes imperative. It will be important to study the roles of physical therapy, exercise and cognitive remediation, to see if they might be able to mitigate the effects of the disease on functional capacity. A better understanding of the determinants of functional disability in HD may also guide choice of outcome measures for clinical trials.
Acknowledgments
We thank our patients and families and research subjects, without whom these studies would not be possible. We thank Susan and Marshal Folstein for the initial organization of the HD Center, many ideas about HD and in particular for the semi-structured onset interview. Figure 1 is modified from Figure 1 of Alexander GE, Ann. Rev. Neurosci. 1986. 9: 357–81. We thank Greg Churchill and Maryjane Ong for data analysis and figure preparation for Figure 2. Supported by NINDS HD Center Grant NS16375, and HDSA Center of Excellence grant. We thank Alice Wexler for reading and commenting on the ms. Part of paragraph 2 is adapted from Ross, C. A. et al. Nat. Rev. Neurol. 10, 204–216 (2014).
References
- 1.Walker FO. Huntington’s disease. Lancet. 2007;369(9557):218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 2.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet neurology. 2011;10(1):83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 3.Ross CA, Aylward EH, Wild EJ, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nature reviews Neurology. 2014;10(4):204–216. doi: 10.1038/nrneurol.2014.24. [DOI] [PubMed] [Google Scholar]
- 4.Folstein SE. The psychopathology of Huntington’s disease. Research publications - Association for Research in Nervous and Mental Disease. 1991;69:181–191. [PubMed] [Google Scholar]
- 5.Folstein SE. Huntington’s disease: a disorder of families. Baltimore: Johns Hopkins University Press; 1989. [Google Scholar]
- 6.Unified Huntington’s Disease Rating Scale: reliability and consistency. Huntington Study Group. Movement disorders: official journal of the Movement Disorder Society. 1996;11(2):136–142. doi: 10.1002/mds.870110204. [DOI] [PubMed] [Google Scholar]
- 7.Folstein SE, Jensen B, Leigh RJ, Folstein MF. The measurement of abnormal movement: methods developed for Huntington’s disease. Neurobehavioral toxicology and teratology. 1983;5(6):605–609. [PubMed] [Google Scholar]
- 8.Marder K, Zhao H, Myers RH, et al. Rate of functional decline in Huntington’s disease. Huntington Study Group. Neurology. 2000;54(2):452–458. doi: 10.1212/wnl.54.2.452. [DOI] [PubMed] [Google Scholar]
- 9.Guo Z, Rudow G, Pletnikova O, et al. Striatal neuronal loss correlates with clinical motor impairment in Huntington’s disease. Movement disorders: official journal of the Movement Disorder Society. 2012;27(11):1379–1386. doi: 10.1002/mds.25159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenblatt A, Abbott MH, Gourley LM, et al. Predictors of neuropathological severity in 100 patients with Huntington’s disease. Annals of neurology. 2003;54(4):488–493. doi: 10.1002/ana.10691. [DOI] [PubMed] [Google Scholar]
- 11.Rosenblatt A, Liang KY, Zhou H, et al. The association of CAG repeat length with clinical progression in Huntington disease. Neurology. 2006;66(7):1016–1020. doi: 10.1212/01.wnl.0000204230.16619.d9. [DOI] [PubMed] [Google Scholar]
- 12.Bylsma FW, Rothlind J, Hall MR, Folstein SE, Brandt J. Assessment of adaptive functioning in Huntington’s disease. Movement disorders: official journal of the Movement Disorder Society. 1993;8(2):183–190. doi: 10.1002/mds.870080212. [DOI] [PubMed] [Google Scholar]
- 13.Brandt J, Folstein SE, Folstein MF. Differential cognitive impairment in Alzheimer’s disease and Huntington’s disease. Annals of neurology. 1988;23(6):555–561. doi: 10.1002/ana.410230605. [DOI] [PubMed] [Google Scholar]
- 14.Brandt J, Inscore AB, Ward J, et al. Neuropsychological deficits in Huntington’s disease gene carriers and correlates of early “conversion”. The Journal of neuropsychiatry and clinical neurosciences. 2008;20(4):466–472. doi: 10.1176/appi.neuropsych.20.4.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brandt J, Strauss ME, Larus J, Jensen B, Folstein SE, Folstein MF. Clinical correlates of dementia and disability in Huntington’s disease. Journal of clinical neuropsychology. 1984;6(4):401–412. doi: 10.1080/01688638408401231. [DOI] [PubMed] [Google Scholar]
- 16.Rothlind JC, Bylsma FW, Peyser C, Folstein SE, Brandt J. Cognitive and motor correlates of everyday functioning in early Huntington’s disease. The Journal of nervous and mental disease. 1993;181(3):194–199. doi: 10.1097/00005053-199303000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Mayeux R, Stern Y, Herman A, Greenbaum L, Fahn S. Correlates of early disability in Huntington’s disease. Annals of neurology. 1986;20(6):727–731. doi: 10.1002/ana.410200613. [DOI] [PubMed] [Google Scholar]
- 18.Biglan KM, Zhang Y, Long JD, et al. Refining the diagnosis of Huntington disease: the PREDICT-HD study. Frontiers in aging neuroscience. 2013;5:12. doi: 10.3389/fnagi.2013.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahant N, McCusker EA, Byth K, Graham S Huntington Study G. Huntington’s disease: clinical correlates of disability and progression. Neurology. 2003;61(8):1085–1092. doi: 10.1212/01.wnl.0000086373.32347.16. [DOI] [PubMed] [Google Scholar]
- 20.Ho AK, Gilbert AS, Mason SL, Goodman AO, Barker RA. Health-related quality of life in Huntington’s disease: Which factors matter most? Movement disorders: official journal of the Movement Disorder Society. 2009;24(4):574–578. doi: 10.1002/mds.22412. [DOI] [PubMed] [Google Scholar]
- 21.Hart EP, Marinus J, Burgunder JM, et al. Better global and cognitive functioning in choreatic versus hypokinetic-rigid Huntington’s disease. Movement disorders: official journal of the Movement Disorder Society. 2013;28(8):1142–1145. doi: 10.1002/mds.25422. [DOI] [PubMed] [Google Scholar]
- 22.Rebok GW, Bylsma FW, Keyl PM, Brandt J, Folstein SE. Automobile driving in Huntington’s disease. Movement disorders: official journal of the Movement Disorder Society. 1995;10(6):778–787. doi: 10.1002/mds.870100611. [DOI] [PubMed] [Google Scholar]
- 23.Beglinger LJ, Prest L, Mills JA, et al. Clinical predictors of driving status in Huntington’s disease. Movement disorders: official journal of the Movement Disorder Society. 2012;27(9):1146–1152. doi: 10.1002/mds.25101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Devos H, Nieuwboer A, Tant M, De Weerdt W, Vandenberghe W. Determinants of fitness to drive in Huntington disease. Neurology. 2012;79(19):1975–1982. doi: 10.1212/WNL.0b013e3182735d11. [DOI] [PubMed] [Google Scholar]
- 25.Devos H, Nieuwboer A, Vandenberghe W, Tant M, De Weerdt W, Uc EY. On-road driving impairments in Huntington disease. Neurology. 2014;82(11):956–962. doi: 10.1212/WNL.0000000000000220. [DOI] [PubMed] [Google Scholar]
- 26.Nance MA, Sanders G. Characteristics of individuals with Huntington disease in long-term care. Movement disorders: official journal of the Movement Disorder Society. 1996;11(5):542–548. doi: 10.1002/mds.870110509. [DOI] [PubMed] [Google Scholar]
- 27.Wheelock VL, Tempkin T, Marder K, et al. Predictors of nursing home placement in Huntington disease. Neurology. 2003;60(6):998–1001. doi: 10.1212/01.wnl.0000052992.58107.67. [DOI] [PubMed] [Google Scholar]
- 28.Rosenblatt A, Kumar BV, Margolis RL, Welsh CS, Ross CA. Factors contributing to institutionalization in patients with Huntington’s disease. Movement disorders: official journal of the Movement Disorder Society. 2011;26(9):1711–1716. doi: 10.1002/mds.23716. [DOI] [PubMed] [Google Scholar]
- 29.Vinther-Jensen T, Larsen IU, Hjermind LE, et al. A clinical classification acknowledging neuropsychiatric and cognitive impairment in Huntington inverted question marks disease. Orphanet journal of rare diseases. 2014;9(1):114. doi: 10.1186/s13023-014-0114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annual review of neuroscience. 1986;9:357–381. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- 31.Utter AA, Basso MA. The basal ganglia: an overview of circuits and function. Neuroscience and biobehavioral reviews. 2008;32(3):333–342. doi: 10.1016/j.neubiorev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Draganski B, Kherif F, Kloppel S, et al. Evidence for segregated and integrative connectivity patterns in the human Basal Ganglia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28(28):7143–7152. doi: 10.1523/JNEUROSCI.1486-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haber SN. The primate basal ganglia: parallel and integrative networks. Journal of chemical neuroanatomy. 2003;26(4):317–330. doi: 10.1016/j.jchemneu.2003.10.003. [DOI] [PubMed] [Google Scholar]