

Abstract
The development of clinical vectors to correct genetic mutations that cause inherited myopathies and related disorders of skeletal muscle is advancing at an impressive rate. Adeno-associated virus (AAV) vectors are attractive for clinical use because (i) AAVs do not cause human disease, and (ii) these vectors are able to persist for years. New vectors are now becoming available as gene therapy delivery tools, and recent preclinical experiments have demonstrated the feasibility, safety and efficacy of gene therapy with AAV for long-term correction of muscle pathology and weakness in myotubularin-deficient canine and murine disease models. In this review, we present recent advances in the application of gene therapies to treat inherited muscle disorders including Duchenne Muscular Dystrophy and X-linked Myotubular Myopathy. Potential areas for therapeutic synergies between rehabilitation medicine and genetics are also discussed.
What is gene therapy and when will it be used in clinical practice?
Gene therapy - the process of introducing foreign genomic materials into host cells to elicit therapeutic benefit - became available for clinical practice on November 2, 2012, when Glybera (alipogene tiparvovec) became the first gene therapy in the Western world to receive market approval for patients with lipoprotein lipase (LPL) deficiency, a rare genetic disease previously without effective treatment(1, 2). Since 1989, gene therapy clinical trials have been undertaken in 31 countries with more than 1800 human trials ongoing, completed or approved worldwide(3). Many of these trials target rare “orphan diseases”. The Orphan Drug Act of 1983 defined an orphan product as a drug intended to treat a condition affecting fewer than 200,000 persons in the United States or a drug that would not be expected to be profitable within seven years following FDA approval(4). Orphan disease designation allows a sponsor to apply for market protection for the product following approval. 2,700 orphan drug designations and more than 400 approvals associated with these designations were approved as of 2012(4). While most gene therapy trials have addressed cancer or cardiovascular disease, a significant number of gene therapy trials have targeted rare monogenic (single gene) diseases (Table 1)(3). These groundbreaking therapies involve the insertion of DNA sequences that encode functional, therapeutic genes into patients to replace mutated dysfunctional genes causing disease. In the case of Glybera, a DNA sequence encodes a therapeutic LPL gene packaged within a vector, in this case an adeno-associated virus (AAV). The recombinant AAV is injected into a patient harboring a mutant disease-causing LPL gene. The injected AAV is capable of shuttling the replacement DNA sequence from inside the vector to the cells of the targeted tissue. Once inside, the patient’s own cellular machinery works to transcribe the new replacement DNA sequence to produce the therapeutic protein to treat the patient’s disease (Figure 1). Besides the success of Glybera, other notable examples of gene therapy successes are highlighted in clinical trials undertaken in rare genetic childhood diseases such as X-linked severe combined immunodeficiency (SCID-X1) and Leber congenital amaurosis (LCA). In the first example, SCID-X1 results in recurrent and often fatal infections caused by genetic deficiency in cellular and humoral immunity. Long-term follow-up of nine SCID-X1 boys treated in a French adenovirus-mediated gene therapy trial reported eight survivors after nearly ten years(5). In the second example, LCA leads to congenital blindness caused by mutations in a retinal gene that causes progressive loss of vision; young patients become completely blind by adulthood. Three independent gene therapy trials for LCA patients have been initiated, and follow-up results indicate improvement in vision for up to 2 years with no serious adverse events(6–8). These and other gene therapy successes have been offset by serious, and rarely fatal, adverse events that lead to early clinical trial “holds”. The most famous case was the death of Jesse Gelsinger in 2000, who was the 19th patient enrolled in a gene therapy trial of a deficiency of ornithine transcarbamylase (OTCD)(9). Despite these early setbacks, the field of gene therapy continues to move forward at an extraordinary pace.
Table 1.
Monogenic disorders reported in gene therapy clinical trials
| Adrenoleukodystrophy |
| α-1 antitrypsin deficiency |
| Becker muscular dystrophy |
| β-thalassaemia |
| Canavan disease |
| Chronic granulomatous disease |
| Cystic fibrosis |
| Duchenne muscular dystrophy |
| Fabry disease |
| Familial adenomatous polyposis |
| Familial hypercholesterolaemia |
| Fanconi anaemia |
| Galactosialidosis |
| Gaucher’s disease |
| Gyrate atrophy |
| Hemophilia A and B |
| Hurler syndrome |
| Hunter syndrome |
| Huntington’s chorea |
| Junctional epidermolysis bullosa |
| Late infantile neuronal ceroid lipofuscinosis |
| Leukocyte adherence deficiency |
| Limb girdle muscular dystrophy |
| Lipoprotein lipase deficiency* |
| Mucopolysaccharidosis type VII |
| Ornithine transcarbamylase deficiency |
| Pompe disease |
| Purine nucleoside phosphorylase deficiency |
| Recessive dystrophic epidermolysis bullosa |
| Sickle cell disease |
| Severe combined immunodeficiency |
| Tay Sachs disease |
| Wiskott–Aldrich syndrome |
Glybera approved in Europe
Figure 1.

AAV mediated gene therapy. A, Genome structure of wild type AAV, a linear single-stranded DNA. ITR, inverted terminal repeats, act in cis and important for AAV replication, genome packaging and transcription; Rep, rep gene produces 4 Rep proteins act in trans in all phases of AAV life cycle; Cap, capsid gene, produces 3 viral capsid proteins; Genome structure of recombinant AAV vector, AAV rep and cap genes are replaced by transgene of interest under the control of a promoter of interest. B, AAV vector production, delivery and assessments of clinical outcome. Co-transfection of AAV production cells such as HEK293 cells with 3 plasmids: (1) an AAV ITR-containing plasmid carrying the gene of interest; (2) a plasmid carrying the AAV rep and cap genes; and (3) a plasmid providing the helper genes isolated from adenovirus (Ad). Recombinant AAV vectors will be administrated via intramuscular or intra-vascular or other routes into subjects, up-taken by target cells, transgene will be expressed and clinical outcomes will be measured as a result of functions of the transgene product.
Will gene replacement therapy be useful for inherited muscle diseases like muscular dystrophy?
Most gene therapy clinical trials have targeted genes involved in cancer(3) with fewer trials initiated in monogenic diseases, such as Duchenne muscular dystrophy (DMD). Muscular dystrophies make up only a small percentage of the monogenic diseases (Table 1), notably Becker, Duchenne and Limb Girdle muscular dystrophy. Pompe disease, amongst others listed in Table 1, is not considered a muscular dystrophy even though skeletal muscles of affected patients grow progressively weaker due to accumulation of abnormal proteins within the muscle’s contractile tissue. Therapeutic approaches at replacing defective genes in monogenic diseases of muscle, like DMD, include the use of recombinant viral vectors engineered to target specific tissues. In the 1960s, the discovery of naturally occurring adeno-associated virus (AAV) isolates led to the clinical application of recombinant AAV vectors with early successes in clinical trials (10). AAV vectors are attractive for clinical use because AAVs are not associated with human disease(2), the virus persists in the infected host for years and a large “toolkit” of AAV vectors is available as clinical gene therapy delivery tools(11). In addition to muscle diseases, AAV vectors have been used in clinical gene therapy trials targeted to the liver for the treatment of hemophilia B, to the lung for treatment of cystic fibrosis, to the brain for treatment of Parkinson’s, Batten’s, and Canavan’s disease, to joints for treatment of rheumatoid arthritis and to the eye for treatment of LCA(10).
In recent years, AAV–mediated gene replacement has rapidly moved from preclinical studies to clinical trials due to encouraging results from animal models. Major challenges surrounding this strategy include: (1) effective delivery methods to target muscles throughout the body, including diaphragm and cardiac muscle, and (2) host immune responses to the therapeutic vector(12). The latter challenge was encountered in a double-blinded, randomized, controlled phase I trial of limb-girdle muscular dystrophy(13, 14), AAV1-MCK.human-alpha-Sarcoglycan (SGCA) was injected locally into the extensor digitorum brevis muscle in 3 patients, and sustained transgene expression was observed in 2 out 3 patients after 6 months. Humoral and cellular immune responses to the AAV capsid proteins were detected in the patient who failed to show expression. Another phase I trial on DMD from Mendell et al (15) also raised the potential of cellular immune responses to either self or non-self dystrophin epitopes. In the study, AAV vectors carrying a truncated but functional dystrophin gene under the control of a CMV promoter were injected into the bicep of 6 DMD patients. None of the patients displayed transgene expression with 4 out 6 patients showing detectable T cell responses against the transgene product. Two of the patients had dystrophin-specific T cell responses before the treatment. Taken together, these trials are informative to emphasize the importance of pre-screening patients for pre-existing immune responses to both AAV capsid proteins and transgene product, and also to develop strategies to circumvent immune responses, such as using a transient course of immunosuppression, shown to be effective in a dog model of DMD(16, 17).
Can genes be repaired?
While AAV-mediated gene therapy is regarded as a gene replacement strategy, another exciting development, termed exon skipping, focuses on gene repair. Approximately 70% of all DMD mutations are due to single or multiple exon deletions. Such deletions disrupt the open reading frame of dystrophin and hence, result in a premature truncated protein. In most cases, selective removal of specific exons can restore the reading frame and produce a partially functional dystrophin protein for clinical benefit(18–21). Antisense oligonucleotides (AOs) targeting pre-mRNA to modulate splicing have been used to induce exon skipping, and the first human phase I trials(22, 23) focused on skipping exon 51, which, if successful, would be able to correct ~13% of DMD patients with specific deletions within exons 42 to 50. A therapeutic exon skipping AO, 2-O-methyl AO termed “PRO051”(23), and AVI-4658, a morpholino conjugated AO(22) that targets an internal sequence of exon 51 was injected intramuscularly into DMD patients. AOs used in these trials were well tolerated, demonstrated successful exon skipping, and all patients demonstrated dystrophin expression to levels between 3–12% and 22–35% by PRO051 and AVI-4658, respectively. Following these initial clinical trials of intramuscular administration, phase I/II trials using systemic delivery of the two drugs were tested for dose, safety and efficacy(24, 25). Dose-dependent restoration of dystrophin expression was observed following weekly administration of the experimental compounds through abdominal subcutaneous injections in two cohorts. While no significant differences was detected in patient’s walking ability following a 12-week long treatment(24), patients treated with weekly PMO-AO (AVI-4658)(25) for 48 weeks demonstrated improvement in stabilization of the muscle and in the six-minute walking distance test (6MWT) (26). A larger confirmatory phase III trial is in the planning for 2014. Clinical trials for skipping other exons are also underway or in planning(18). These “personalized gene therapy medicines” generate hope for DMD patients, with the caveat that effective therapy will need to restore dystrophin expression in both skeletal and cardiac muscle, and that treatment will need to be persistent. The future challenge for clinical development with AOs is that current forms have a short half-life, and more than 85% of AOs are cleared from the circulation within 24 hours. This short half-life requires weekly injections to maintain a therapeutic level. Long-term outcomes of these AOs are also unknown. Intramuscular administration of these agents may be limited in treating skeletal muscle, and treating the cardiac muscle will need to be addressed for this strategy to be an effective treatment for DMD associated cardiomyopathy.
Non-sense stop codon read-through is another method of gene editing, and potentially benefits ~ 13% of DMD boys with premature termination codon (PTC) mutations. Aminoglycoside antibiotics initially demonstrated the capacity to induce ribosomal read through of premature stop codon(27), but not efficient and too toxic to be used for long term treatment. Ataluren (PTC124) was identified to be effective in this matter in mdx mice, and subsequently tested in human DMD trials(28–30). The drug was well tolerated in general and the dystrophin protein was detected with treated patients showing improvement in the 6MWT. However, correlation between the level of dystrophin expression and the 6MWT remains unclear. More trials are currently under way and the same treatment strategy could be potentially applied to other muscle diseases including spinal muscular atrophy. Progress in developing other approaches targeting repair of the muscle membrane due to lack of dystrophin is also under development. For example, increasing the level of the compensatory protein utrophin (31, 32), and upregulation of glycosylation of a-dystroglycan to improve extracellular matrix attachment (33).
Can defective genes be completely replaced? Surprising lessons learned from dogs
In 2008, a case report of a 5-month-old Labrador retriever was published in a Canadian veterinary medical journal(34). The dog presented with weakness, muscle atrophy and histopathological changes in skeletal muscle consistent with a centronuclear myopathy. Because male littermates were similarly affected, the authors postulated that the disease was X-linked, giving rise to the possibility that the disorder could be analogous to X-linked myotubular myopathy (XLMTM). It was through the tireless and extraordinary efforts of Alison Rockett Frase, that our research group was able to acquire a first-degree relative, a dog named “Nibs”, a Labrador retriever coming from a line of dogs with a history suspicious for XLMTM. We later discovered that Nibs harbored a canine MTM1 mutation(34), the same gene known to cause myotubular myopathy in patients [reviewed in “The Miracle of Nibs” (35)]. Mrs. Frase recalls the story of her odyssey locating and retrieving the founding carrier dog from a farmer in Canada. The following excerpt describes how a determined mother of an affected child can help shape the future of research for a disorder like myotubular myopathy: “In the fall of 2008, a female Labrador Retriever was discovered to carry the same gene as I do for my son’s muscle disorder called mytubular myopathy (MTM). To date, this was the first MTM large animal ever discovered by researchers anywhere in the world….Nibs was a beautiful Labrador Retriever that was instrumental in giving us puppies that carry the myotubular myopathy (MTM1) gene that affects our son Joshua. I am so grateful to Nibs for the initial 12 puppy litter…Our second litter of MTM pups has been born…Knowledge gained from these animals may one day lead to treatments not only for MTM, but other neuromuscular diseases. It will be a miracle for our son Josh and thousands of children like him if our goals are achieved.” Joshua Frase passed away on December 24, 2010, less than 2 years after this was written. His legacy, the Joshua Frase Foundation, set into motion research that will, hopefully, develop the first effective treatment for this devastating disorder.
XLMTM is an orphan disease, affecting 1/50,000 live male births worldwide(36) with only supportive, palliative care available for patients(37). This inherited muscle disease results from loss-of-function mutations in the Myotubularin 1 gene (MTM1) (38) that encodes the founder of a family of 3-phosphoinositide phosphatases acting on the second messengers phosphatidylinositol 3-monophosphate [PI(3)P] and phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2] (39, 40). Although myotubularin is expressed ubiquitously, loss of this enzyme profoundly affects skeletal muscles causing hypotrophic myofibers and structural abnormalities, with associated weakness (41). No effective therapy exists for XLMTM. Management of the disease generally consists of mechanical ventilation, gastrostomy feeding tubes, antibiotics (for respiratory infections), orthotics to prevent skeletal limb contractures and surgical treatment to alleviate severe spinal deformities. In spite of aggressive medical care, the average life expectancy is only about two years and most who survive beyond this age require mechanical ventilation.
Animal models of the disease currently exist in zebrafish, mice and notably, in dogs (41–43). The murine phenotype resembles human XLMTM, with similar pathology and early mortality. In a mouse knockout model of XLMTM, local delivery of the wild-type myotubularin gene (MTM1) via an AAV vector reversed characteristic pathological features and rescued the function of injected limb muscles(44). Buj-Bello et al were the first to report a gene therapy success in the Mtm1 knockout mouse in 2008. That same year, the Joshua Frase Foundation provided our research group access to a female Labrador retriever harboring an MTM1 gene mutation that was later proven by Beggs et al to cause a canine version of the human disease(45). From this single founding female our group established a canine breeding colony to study effects of the disease in dogs. Initial data revealed that affected males display a phenotype directly analogous to human XLMTM: progressive and severe muscular weakness(46) leading to the inability to walk, weak ventilatory muscles leading to respiratory impairment(47) and early death. Based upon the early experience with gene replacement in the Mtm1 knockout mouse(44), a similar gene replacement strategy was initiated in the XLMTM dog in 2011.
For eventual gene therapy of XLMTM patients, our goal was to use a predictive large animal model (the XLMTM dog) to refine the delivery system, to assess critical safety parameters such as the potential host immune response to vector and transgene, and to optimize efficacy measurements. In collaboration with the French non-profit institute, Généthon(48), cohorts of Mtm1 knockout mice were first tested for response to systemic Mtm1 gene replacement via tail vein injection. Results indicated that a single systemic treatment with AAV-Mtm1 sufficed for long-term (at least one year) survival and essentially complete amelioration of symptoms of mice with myotubularin-deficient muscles(49). Using the same AAV vectors produced by Généthon scientists and tested in mice, our collaborative research group confirmed that local gene replacement therapy, delivered intramuscularly into the hind limb of young XLMTM dogs, reversed pathological changes in myotubularin-deficient skeletal muscles. Remarkably, the treated muscles also showed nearly normal strength at six weeks post-injection, compared to very weak muscles (only 20 percent of normal strength) in saline-injected contralateral limbs. In subsequent experiments, intravascular administration of AAV8-MTM1 at the same dose used in mice was well tolerated in dogs, rescued the skeletal muscle pathology and respiratory function, and prolonged life for over one year (Figure 2). Together these initial studies demonstrated the feasibility, safety and efficacy of gene therapy with AAV for long-term correction of muscle pathology and weakness observed in myotubularin-deficient mouse and dog models, and paved the way to clinical trials aimed at correcting this devastating disease in patients.
Figure 2.

The first XLMTM dog treated with intravascular gene replacement therapy (AAV8-MTM1) given at 9 weeks-of-age. Untreated XLMTM dogs do not survive beyond 6 months of age, and succumb to severe muscle weakness and respiratory insufficiency. Shown here, is an affected dog one year post-infusion with AAV8-MTM1. Systemic treatment resulted in amelioration of the severe muscle pathology and weakness observed in untreated dogs.
Rehabilitation and gene therapy
As highlighted in the discussion above, research on the role of gene therapy in the treatment of muscular disorders has undergone impressive growth over the course of the past decade, with a considerable portion of this research being focused on inherited myopathies such as DMD and XLMTM. Gene replacement therapy in such disorders, however, represents only a fraction of the spectrum of muscular disorders addressed in rehabilitation medicine that might benefit from application of a gene therapy approach. Equally significant are inquiries into the genetic regulation of age-related muscle atrophy, muscle breakdown in hypercatabolic states (as seen with malignancy or infection), as well as recovery from muscle injuries associated with mechanical trauma, ischemia or denervation.
Investigation of potential gene therapy strategies to enhance muscle regeneration and/or counteract the effects of muscle atrophy and degeneration is underway to elucidate (and experimentally manipulate) molecular events at the transcriptional, translational and post-translational stages. Examples include research on the bioavailability of growth factors such as TGF1-3(50, 51), IGF-1(52, 53) and growth-differentiation factors such as GDF8/myostatin(54, 55) that play a role in the proliferation and differentiation of muscle stem cells (reviewed in this supplement by Jasuja and LeBrasseur). The ability to therapeutically regulate expression of these factors has the potential to prevent muscle loss and enhance muscle recovery, and thus is aligned with key concerns in the area of rehabilitation medicine. To date, however, much of the research in these areas has remained within the molecular and cellular biology domains, with limited dissemination to the rehabilitation research community. Translational research on the rehabilitation applications of these therapies remains in the nascent stages, though advocacy for advancement of research to define the cellular and molecular mechanisms that are induced and/or enhanced by rehabilitation therapies continues to grow(56).
Increasingly demonstrated in rehabilitation medicine literature is research on how mechanical stimuli such as stretch or compression affect intracellular signaling and gene expression (i.e. “mechanotransduction”)(57–59). Studies on up-regulation of growth factors in response to tensile and/or compressive loading have been undertaken in tendon (showing up-regulation of IGF-1)(60, 61), as well as muscle (showing up-regulation of MGF)(58, 62). The mechanisms underlying mechanotransduction are still being elucidated, and appear to involve not only chemical signaling pathways, but also direct physical coupling via cytoskeletal elements between membrane structures and nuclear structures. Less understood are the effects of mechanical stimulus on gene therapy delivery and efficacy; Prior studies have shown that cyclical mechanical stretch enhances AAV2 mediated gene transfer to cultured vascular smooth muscle cells(63), but to our knowledge, effects of mechanical stretch on AAV gene transduction have not been reported in skeletal muscle. Our research group is investigating effects of passive stretch in cultured skeletal muscle on the uptake and expression of various AAV serotypes, a process thought to occur via clathrin-dependent endocytosis(64). A series of follow-on studies will examine the effect of exercise on gene therapy uptake/efficacy in vivo. Rehabilitation research is needed using similar paradigms along with measures of motor recovery in human subjects undergoing gene transfer.
In other emerging areas of research on gene therapy for treatment of muscular disorders, the interplay of genetic factors and cellular factors is central (e.g. up-regulating expression of growth factors and the availability of responsive precursor cells that retain the ability to differentiate into functionally mature excitable tissues such as nerve and muscle). Indeed in some cases, the means for delivery of gene therapy relies directly upon transplant of cells that have been transfected with the genetic elements of interest. The importance of this synergistic interplay is underscored in a 2008 study by Haastert et al(65) in rats with sciatic nerve injury to determine whether voluntary exercise enhances the effects of gene therapy delivered via Schwann cells over-expressing FGF-2. These authors found that rats treated with FGF-2 gene therapy plus exercise showed enhanced regeneration of myelinated axons in comparison to sedentary animals, and furthermore that mRNA levels of regeneration associated proteins in lumbar spinal cord were significantly higher in exercised vs. sedentary animals. Gene therapies and cellular therapies are each important tools in the emerging era of biologic therapeutics. While cellular and gene therapies each have independent potential as treatments to combat muscular dystrophy, congenital myopathies, and muscle damage, the potential for synergistic therapies is also an important aspect of future treatment strategies. Moreover, as demonstrated by the work of Hasstert et al above, further potentiation of cellular and gene therapies may be effected by the strategic use of exercise interventions, thus positioning rehabilitation researchers and clinicians to offer significant contributions in the development of these important therapeutic advances.
Areas for future research: Where Rehabilitation meets Genetics
As we continue to explore how rehabilitation medicine and genetic medicine complement each other, we can begin to define potential therapeutic synergies by combining rehabilitation and genetic approaches. Areas for further research that emerge at the intersection of these two clinical/research domains are presented in Figures 3 and 4. In this review, we have already discussed advances in the application of gene therapies to treat inherited muscle disorders such as Duchenne Muscular Dystrophy and X-linked Myotubular Myopathy. Specific inquires that should be addressed in the future include whether a dose-response effect for therapeutic exercise in the rehabilitation of inherited muscle disorders exists, and if so whether a minimal effective exercise “dose” (versus toxic “dose”) can be defined. Similarly for gene therapy, dose-response curves need to be established and examined with an eye toward minimizing both potential costs and side effects associated with the delivery of large inocula. An intriguing possibility, built on the principles of mechanotransduction discussed earlier, is that by targeting gene therapy delivery to elements over-expressed in muscle after mechanical loading, we may be able to facilitate an activity-dependent up-regulation of therapeutic gene products. Rehabilitation researchers and clinicians thus stand to offer important insights on how gene therapies may be potentiated by the strategic manipulation of exercise timing and intensity. This could have implications, for example, in myopathic patients weaning from chronic ventilator support, where gene replacement therapy might be coupled with a pulmonary rehabilitation/exercise program. Ongoing dialogue about the interplay of biological and behavioral interventions will be essential in moving gene therapy for muscle disorders forward in the coming decade, and will constitute a central focus in the evolving field of Regenerative Rehabilitation.
Figure 3.
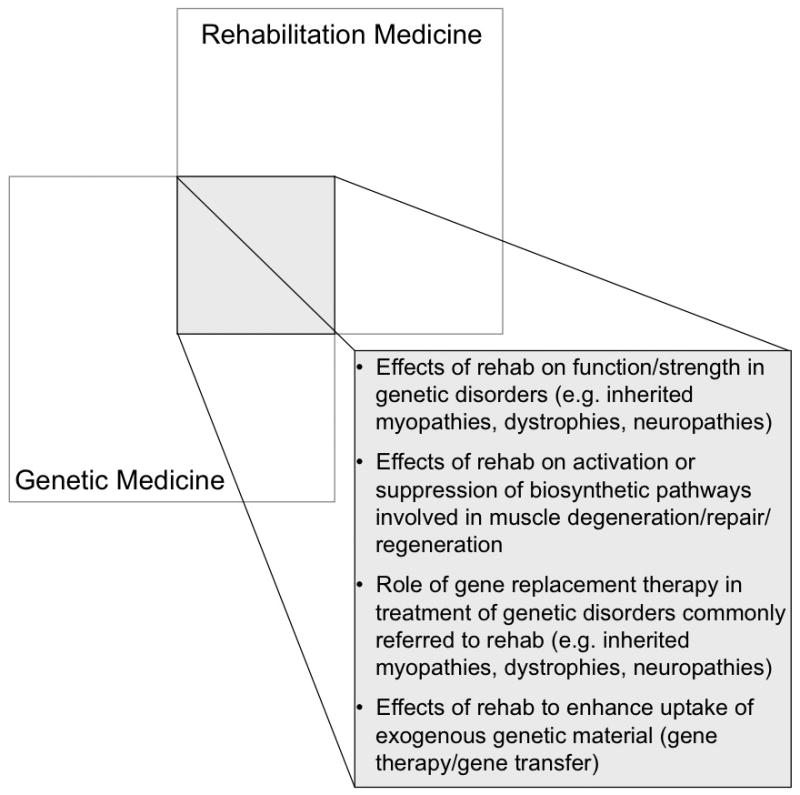
Areas for future research at the interface of rehabilitation medicine and genetic medicine.
Figure 4.

Conceptual framework for areas of overlap between rehabilitation and genetic medicine.
Table 2.
Transcription factors in skeletal muscle potentially responsive to rehabilitation strategies
| Factor | Description |
|---|---|
| Myo D | Transcriptional regulatory protein, induced during muscle regeneration(66 ) |
| Myogenic factor 5 (Myf5) | Transcriptional regulatory protein induced during muscle regeneration(67–69) |
| Mechano growth factor (MGF) | A splice variant of IGF-I that leads to muscle hypertrophy via activation of satellite cells(70) |
| Myogenic regulatory factor 4 (Mrf4) | Myogenic regulatory factor induced during muscle regeneration(71–73) |
| Myogenin | Myogenic regulatory factor induced during muscle regeneration(74–76) |
| Transforming growth factor beta (TGF-beta) | Considered a key signaling molecule in cardiac hypertrophy and failure, among others (77–79) |
| IGF-1 (Insulin-like growth Factor-1) | Implicated in the control of skeletal muscle growth, differentiation, survival, and regeneration, and considered a promising therapeutic agent in staving off the advance of muscle weakness (80–84) |
References
- 1.Kastelein JJ, Ross CJ, Hayden MR. From mutation identification to therapy: discovery and origins of the first approved gene therapy in the Western world. Human gene therapy. 2013;24:472–478. doi: 10.1089/hum.2013.063. [DOI] [PubMed] [Google Scholar]
- 2.Nayerossadat N, Maedeh T, Ali PA. Viral and nonviral delivery systems for gene delivery. Adv Biomed Res. 2012;1:27. doi: 10.4103/2277-9175.98152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2012 - an update. J Gene Med. 2013;15:65–77. doi: 10.1002/jgm.2698. [DOI] [PubMed] [Google Scholar]
- 4.Byrne BJ. Pathway for approval of a gene therapy orphan product: treading new ground. Molecular therapy: the journal of the American Society of Gene Therapy. 2013;21:1465–1466. doi: 10.1038/mt.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, Martinache C, Rieux-Laucat F, Latour S, Belohradsky BH, Leiva L, Sorensen R, Debre M, Casanova JL, Blanche S, Durandy A, Bushman FD, Fischer A, Cavazzana-Calvo M. Efficacy of gene therapy for X-linked severe combined immunodeficiency. The New England journal of medicine. 2010;363:355–364. doi: 10.1056/NEJMoa1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simonelli F, Maguire AM, Testa F, Pierce EA, Mingozzi F, Bennicelli JL, Rossi S, Marshall K, Banfi S, Surace EM, Sun J, Redmond TM, Zhu X, Shindler KS, Ying GS, Ziviello C, Acerra C, Wright JF, McDonnell JW, High KA, Bennett J, Auricchio A. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Molecular therapy: the journal of the American Society of Gene Therapy. 2010;18:643–650. doi: 10.1038/mt.2009.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maguire AM, High KA, Auricchio A, Wright JF, Pierce EA, Testa F, Mingozzi F, Bennicelli JL, Ying GS, Rossi S, Fulton A, Marshall KA, Banfi S, Chung DC, Morgan JI, Hauck B, Zelenaia O, Zhu X, Raffini L, Coppieters F, De Baere E, Shindler KS, Volpe NJ, Surace EM, Acerra C, Lyubarsky A, Redmond TM, Stone E, Sun J, McDonnell JW, Leroy BP, Simonelli F, Bennett J. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374:1597–1605. doi: 10.1016/S0140-6736(09)61836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung DC, Lee V, Maguire AM. Recent advances in ocular gene therapy. Curr Opin Ophthalmol. 2009;20:377–381. doi: 10.1097/ICU.0b013e32832f802a. [DOI] [PubMed] [Google Scholar]
- 9.Wilson JM. Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol Genet Metab. 2009;96:151–157. doi: 10.1016/j.ymgme.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 10.Asokan A, Schaffer DV, Samulski RJ. The AAV vector toolkit: poised at the clinical crossroads. Molecular therapy: the journal of the American Society of Gene Therapy. 2012;20:699–708. doi: 10.1038/mt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi VW, McCarty DM, Samulski RJ. AAV hybrid serotypes: improved vectors for gene delivery. Curr Gene Ther. 2005;5:299–310. doi: 10.2174/1566523054064968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Tapscott SJ, Storb R. Local gene delivery and methods to control immune responses in muscles of normal and dystrophic dogs. Methods Mol Biol. 2011;709:265–275. doi: 10.1007/978-1-61737-982-6_17. [DOI] [PubMed] [Google Scholar]
- 13.Mendell JR, Rodino-Klapac LR, Rosales XQ, Coley BD, Galloway G, Lewis S, Malik V, Shilling C, Byrne BJ, Conlon T, Campbell KJ, Bremer WG, Taylor LE, Flanigan KM, Gastier-Foster JM, Astbury C, Kota J, Sahenk Z, Walker CM, Clark KR. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Ann Neurol. 2010;68:629–638. doi: 10.1002/ana.22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mendell JR, Rodino-Klapac LR, Rosales-Quintero X, Kota J, Coley BD, Galloway G, Craenen JM, Lewis S, Malik V, Shilling C, Byrne BJ, Conlon T, Campbell KJ, Bremer WG, Viollet L, Walker CM, Sahenk Z, Clark KR. Limb-girdle muscular dystrophy type 2D gene therapy restores alpha-sarcoglycan and associated proteins. Ann Neurol. 2009;66:290–297. doi: 10.1002/ana.21732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, Bowles D, Gray S, Li C, Galloway G, Malik V, Coley B, Clark KR, Li J, Xiao X, Samulski J, McPhee SW, Samulski RJ, Walker CM. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Kuhr CS, Allen JM, Blankinship M, Gregorevic P, Chamberlain JS, Tapscott SJ, Storb R. Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol Ther. 2007;15:1160–1166. doi: 10.1038/sj.mt.6300161. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, Storb R, Halbert CL, Banks GB, Butts TM, Finn EE, Allen JM, Miller AD, Chamberlain JS, Tapscott SJ. Successful regional delivery and long-term expression of a dystrophin gene in canine muscular dystrophy: a preclinical model for human therapies. Mol Ther. 2012;20:1501–1507. doi: 10.1038/mt.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jarmin S, Kymalainen H, Popplewell L, Dickson G. New developments in the use of gene therapy to treat Duchenne muscular dystrophy. Expert Opin Biol Ther. 2013 doi: 10.1517/14712598.2014.866087. [DOI] [PubMed] [Google Scholar]
- 19.Fairclough RJ, Wood MJ, Davies KE. Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nat Rev Genet. 2013;14:373–378. doi: 10.1038/nrg3460. [DOI] [PubMed] [Google Scholar]
- 20.Hoffman EP, Bronson A, Levin AA, Takeda S, Yokota T, Baudy AR, Connor EM. Restoring dystrophin expression in duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through. Am J Pathol. 2011;179:12–22. doi: 10.1016/j.ajpath.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson SF, Crosbie RH, Miceli MC, Spencer MJ. Emerging genetic therapies to treat Duchenne muscular dystrophy. Curr Opin Neurol. 2009;22:532–538. doi: 10.1097/WCO.0b013e32832fd487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C, Guglieri M, Ashton E, Abbs S, Nihoyannopoulos P, Garralda ME, Rutherford M, McCulley C, Popplewell L, Graham IR, Dickson G, Wood MJ, Wells DJ, Wilton SD, Kole R, Straub V, Bushby K, Sewry C, Morgan JE, Muntoni F. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–928. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, den Dunnen JT, Koop K, van der Kooi AJ, Goemans NM, de Kimpe SJ, Ekhart PF, Venneker EH, Platenburg GJ, Verschuuren JJ, van Ommen GJ. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357:2677–2686. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 24.Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, Holling T, Janson AA, Platenburg GJ, Sipkens JA, Sitsen JM, Aartsma-Rus A, van Ommen GJ, Buyse G, Darin N, Verschuuren JJ, Campion GV, de Kimpe SJ, van Deutekom JC. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- 25.Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJ, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McDonald CM, Henricson EK, Han JJ, Abresch RT, Nicorici A, Elfring GL, Atkinson L, Reha A, Hirawat S, Miller LL. The 6-minute walk test as a new outcome measure in Duchenne muscular dystrophy. Muscle Nerve. 2010;41:500–510. doi: 10.1002/mus.21544. [DOI] [PubMed] [Google Scholar]
- 27.Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. 1999;104:375–381. doi: 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 29.Finkel RS, Flanigan KM, Wong B, Bonnemann C, Sampson J, Sweeney HL, Reha A, Northcutt VJ, Elfring G, Barth J, Peltz SW. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation duchenne muscular dystrophy. PLoS One. 2013;8:e81302. doi: 10.1371/journal.pone.0081302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finkel RS. Read-through strategies for suppression of nonsense mutations in Duchenne/Becker muscular dystrophy: aminoglycosides and ataluren (PTC124) J Child Neurol. 2010;25:1158–1164. doi: 10.1177/0883073810371129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sonnemann KJ, Heun-Johnson H, Turner AJ, Baltgalvis KA, Lowe DA, Ervasti JM. Functional substitution by TAT-utrophin in dystrophin-deficient mice. PLoS Med. 2009;6:e1000083. doi: 10.1371/journal.pmed.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tinsley JM, Fairclough RJ, Storer R, Wilkes FJ, Potter AC, Squire SE, Powell DS, Cozzoli A, Capogrosso RF, Lambert A, Wilson FX, Wren SP, De Luca A, Davies KE. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One. 2011;6:e19189. doi: 10.1371/journal.pone.0019189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen HH, Jayasinha V, Xia B, Hoyte K, Martin PT. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc Natl Acad Sci U S A. 2002;99:5616–5621. doi: 10.1073/pnas.082613599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cosford KL, Taylor SM, Thompson L, Shelton GD. A possible new inherited myopathy in a young Labrador retriever. The Canadian veterinary journal La revue veterinaire canadienne. 2008;49:393–397. [PMC free article] [PubMed] [Google Scholar]
- 35.Frase AR. The miracle of Nibs. 2009 http://www.joshuafrase.org/uploads/JFF-The story of Nibs.pdf.
- 36.Heckmatt JZ, Sewry CA, Hodes D, Dubowitz V. Congenital centronuclear (myotubular) myopathy. A clinical, pathological and genetic study in eight children. Brain. 1985;108(Pt 4):941–964. doi: 10.1093/brain/108.4.941. [DOI] [PubMed] [Google Scholar]
- 37.Jungbluth H, Wallgren-Pettersson C, Laporte J. Centronuclear (myotubular) myopathy. Orphanet J Rare Dis. 2008;3:26. doi: 10.1186/1750-1172-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laporte J, Hu LJ, Kretz C, Mandel JL, Kioschis P, Coy JF, Klauck SM, Poustka A, Dahl N. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. 1996;13:175–182. doi: 10.1038/ng0696-175. [DOI] [PubMed] [Google Scholar]
- 39.Laporte J, Blondeau F, Buj-Bello A, Tentler D, Kretz C, Dahl N, Mandel JL. Characterization of the myotubularin dual specificity phosphatase gene family from yeast to human. Hum Mol Genet. 1998;7:1703–1712. doi: 10.1093/hmg/7.11.1703. [DOI] [PubMed] [Google Scholar]
- 40.Robinson FL, Dixon JE. Myotubularin phosphatases: policing 3- phosphoinositides. Trends Cell Biol. 2006;16:403–412. doi: 10.1016/j.tcb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 41.Buj-Bello A, Laugel V, Messaddeq N, Zahreddine H, Laporte J, Pellissier JF, Mandel JL. The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc Natl Acad Sci U S A. 2002;99:15060–15065. doi: 10.1073/pnas.212498399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dowling JJ, Vreede AP, Low SE, Gibbs EM, Kuwada JY, Bonnemann CG, Feldman EL. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. 2009;5:e1000372. doi: 10.1371/journal.pgen.1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beggs AH, Bohm J, Snead E, Kozlowski M, Maurer M, Minor K, Childers MK, Taylor SM, Hitte C, Mickelson JR, Guo LT, Mizisin AP, Buj-Bello A, Tiret L, Laporte J, Shelton GD. MTM1 mutation associated with X-linked myotubular myopathy in Labrador Retrievers. Proc Natl Acad Sci U S A. 2010;107:14697–14702. doi: 10.1073/pnas.1003677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buj-Bello A, Fougerousse F, Schwab Y, Messaddeq N, Spehner D, Pierson CR, Durand M, Kretz C, Danos O, Douar AM, Beggs AH, Schultz P, Montus M, Denefle P, Mandel JL. AAV-mediated intramuscular delivery of myotubularin corrects the myotubular myopathy phenotype in targeted murine muscle and suggests a function in plasma membrane homeostasis. Hum Mol Genet. 2008;17:2132–2143. doi: 10.1093/hmg/ddn112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beggs AH, Bohm J, Snead E, Kozlowski M, Maurer M, Minor K, Childers MK, Taylor SM, Hitte C, Mickelson JR, Guo LT, Mizisin AP, Buj-Bello A, Tiret L, Laporte J, Shelton GD. MTM1 mutation associated with X-linked myotubular myopathy in Labrador Retrievers. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14697–14702. doi: 10.1073/pnas.1003677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grange RW. Muscle function in a canine model of X-Linked Myotubular Myopathy. Muscle Nerve. 2012 doi: 10.1002/mus.23463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goddard MA, Mitchell EL, Smith BK, Childers MK. Establishing clinical end points of respiratory function in large animals for clinical translation. Physical medicine and rehabilitation clinics of North America. 2012;23:75–94. xi. doi: 10.1016/j.pmr.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 48.Butler D. French move past Genethon to gene-therapy research. Nature. 1993;361:671. doi: 10.1038/361671a0. [DOI] [PubMed] [Google Scholar]
- 49.Childers MK, Joubert R, Poulard K, Moal C, Grange RW, Doering JA, Lawlor MW, Rider BE, Jamet T, Daniele N, Martin S, Riviere C, Soker T, Hammer C, Van Wittenberghe L, Lockard M, Guan X, Goddard M, Mitchell E, Barber J, Williams JK, Mack DL, Furth ME, Vignaud A, Masurier C, Mavilio F, Moullier P, Beggs AH, Buj-Bello A. Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Sci Transl Med. 2014;6:220ra210. doi: 10.1126/scitranslmed.3007523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 51.Beggs ML, Nagarajan R, Taylor-Jones JM, Nolen G, Macnicol M, Peterson CA. Alterations in the TGFbeta signaling pathway in myogenic progenitors with age. Aging Cell. 2004;3:353–361. doi: 10.1111/j.1474-9728.2004.00135.x. [DOI] [PubMed] [Google Scholar]
- 52.Musaro A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001;27:195–200. doi: 10.1038/84839. [DOI] [PubMed] [Google Scholar]
- 53.Scicchitano BM, Rizzuto E, Musaro A. Counteracting muscle wasting in aging and neuromuscular diseases: the critical role of IGF-1. Aging (Albany NY) 2009;1:451–457. doi: 10.18632/aging.100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gomes AR, Soares AG, Peviani S, Nascimento RB, Moriscot AS, Salvini TF. The effect of 30 minutes of passive stretch of the rat soleus muscle on the myogenic differentiation, myostatin, and atrogin-1 gene expressions. Arch Phys Med Rehabil. 2006;87:241–246. doi: 10.1016/j.apmr.2005.08.126. [DOI] [PubMed] [Google Scholar]
- 55.LeBrasseur NK, Schelhorn TM, Bernardo BL, Cosgrove PG, Loria PM, Brown TA. Myostatin inhibition enhances the effects of exercise on performance and metabolic outcomes in aged mice. J Gerontol A Biol Sci Med Sci. 2009;64:940–948. doi: 10.1093/gerona/glp068. [DOI] [PubMed] [Google Scholar]
- 56.Ambrosio F, Tarabishy A, Kadi F, Brown EH, Sowa G. Biological basis of exercise-based treatments for musculoskeletal conditions. PMR. 2011;3:S59–63. doi: 10.1016/j.pmrj.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kjaer M. Role of extracellular matrix in adaptation of tendon and skeletal muscle to mechanical loading. Physiol Rev. 2004;84:649–698. doi: 10.1152/physrev.00031.2003. [DOI] [PubMed] [Google Scholar]
- 58.Goldspink G. Gene expression in muscle in response to exercise. J Muscle Res Cell Motil. 2003;24:121–126. doi: 10.1023/a:1026041228041. [DOI] [PubMed] [Google Scholar]
- 59.Khan KM, Scott A. Mechanotherapy: how physical therapists’ prescription of exercise promotes tissue repair. Br J Sports Med. 2009;43:247–252. doi: 10.1136/bjsm.2008.054239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Olesen JL, Heinemeier KM, Haddad F, Langberg H, Flyvbjerg A, Kjaer M, Baldwin KM. Expression of insulin-like growth factor I, insulin-like growth factor binding proteins, and collagen mRNA in mechanically loaded plantaris tendon. J Appl Physiol (1985) 2006;101:183–188. doi: 10.1152/japplphysiol.00636.2005. [DOI] [PubMed] [Google Scholar]
- 61.Heinemeier KM, Olesen JL, Schjerling P, Haddad F, Langberg H, Baldwin KM, Kjaer M. Short-term strength training and the expression of myostatin and IGF-I isoforms in rat muscle and tendon: differential effects of specific contraction types. J Appl Physiol (1985) 2007;102:573–581. doi: 10.1152/japplphysiol.00866.2006. [DOI] [PubMed] [Google Scholar]
- 62.Jarvinen TA, Jarvinen TL, Kaariainen M, Aarimaa V, Vaittinen S, Kalimo H, Jarvinen M. Muscle injuries: optimising recovery. Best Pract Res Clin Rheumatol. 2007;21:317–331. doi: 10.1016/j.berh.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 63.Sen S, Conroy S, Hynes SO, McMahon J, O’Doherty A, Bartlett JS, Akhtar Y, Adegbola T, Connolly CE, Sultan S, Barry F, Katusic ZS, O’Brien T. Gene delivery to the vasculature mediated by low-titre adeno-associated virus serotypes 1 and 5. J Gene Med. 2008;10:143–151. doi: 10.1002/jgm.1133. [DOI] [PubMed] [Google Scholar]
- 64.Bartlett JS, Wilcher R, Samulski RJ. Infectious entry pathway of adeno- associated virus and adeno-associated virus vectors. J Virol. 2000;74:2777–2785. doi: 10.1128/jvi.74.6.2777-2785.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haastert K, Ying Z, Grothe C, Gomez-Pinilla F. The effects of FGF-2 gene therapy combined with voluntary exercise on axonal regeneration across peripheral nerve gaps. Neurosci Lett. 2008;443:179–183. doi: 10.1016/j.neulet.2008.07.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Legerlotz K, Smith HK. Role of MyoD in denervated, disused, and exercised muscle. Muscle & nerve. 2008;38:1087–1100. doi: 10.1002/mus.21087. [DOI] [PubMed] [Google Scholar]
- 67.Ustanina S, Carvajal J, Rigby P, Braun T. The myogenic factor Myf5 supports efficient skeletal muscle regeneration by enabling transient myoblast amplification. Stem Cells. 2007;25:2006–2016. doi: 10.1634/stemcells.2006-0736. [DOI] [PubMed] [Google Scholar]
- 68.Gayraud-Morel B, Chretien F, Flamant P, Gomes D, Zammit PS, Tajbakhsh S. A role for the myogenic determination gene Myf5 in adult regenerative myogenesis. Dev Biol. 2007;312:13–28. doi: 10.1016/j.ydbio.2007.08.059. [DOI] [PubMed] [Google Scholar]
- 69.Anderson JE, Vargas C. Correlated NOS-Imu and myf5 expression by satellite cells in mdx mouse muscle regeneration during NOS manipulation and deflazacort treatment. Neuromuscular disorders: NMD. 2003;13:388–396. doi: 10.1016/s0960-8966(03)00029-4. [DOI] [PubMed] [Google Scholar]
- 70.Goldspink G. Impairment of IGF-I gene splicing and MGF expression associated with muscle wasting. Int J Biochem Cell Biol. 2006;38:481–489. doi: 10.1016/j.biocel.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 71.Sambasivan R, Comai G, Le Roux I, Gomes D, Konge J, Dumas G, Cimper C, Tajbakhsh S. Embryonic founders of adult muscle stem cells are primed by the determination gene Mrf4. Dev Biol. 2013;381:241–255. doi: 10.1016/j.ydbio.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 72.Becker C, Della Gaspera B, Guyot M, Donsez E, Armand AS, Charbonnier F, Launay T, Chanoine C. Expression of MRF4 protein in adult and in regenerating muscles in Xenopus. Dev Dyn. 2003;227:445–449. doi: 10.1002/dvdy.10318. [DOI] [PubMed] [Google Scholar]
- 73.Pavlath GK, Dominov JA, Kegley KM, Miller JB. Regeneration of transgenic skeletal muscles with altered timing of expression of the basic helix-loop-helix muscle regulatory factor MRF4. Am J Pathol. 2003;162:1685–1691. doi: 10.1016/S0002-9440(10)64303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu LN, Ren Y, Chen JQ, Wang YZ. Effects of myogenin on muscle fiber types and key metabolic enzymes in gene transfer mice and C2C12 myoblasts. Gene. 2013;532:246–252. doi: 10.1016/j.gene.2013.09.028. [DOI] [PubMed] [Google Scholar]
- 75.Naka A, Iida KT, Nakagawa Y, Iwasaki H, Takeuchi Y, Satoh A, Matsuzaka T, Ishii KA, Kobayashi K, Yatoh S, Shimada M, Yahagi N, Suzuki H, Sone H, Yamada N, Shimano H. TFE3 inhibits myoblast differentiation in C2C12 cells via down-regulating gene expression of myogenin. Biochemical and biophysical research communications. 2013;430:664–669. doi: 10.1016/j.bbrc.2012.11.094. [DOI] [PubMed] [Google Scholar]
- 76.Aguiar AF, Vechetti-Junior IJ, Alves de Souza RW, Castan EP, Milanezi-Aguiar RC, Padovani CR, Carvalho RF, Silva MD. Myogenin, MyoD and IGF-I regulate muscle mass but not fiber-type conversion during resistance training in rats. International journal of sports medicine. 2013;34:293–301. doi: 10.1055/s-0032-1321895. [DOI] [PubMed] [Google Scholar]
- 77.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–606. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ramos-Mondragon R, Galindo CA, Avila G. Role of TGF-beta on cardiac structural and electrical remodeling. Vasc Health Risk Manag. 2008;4:1289–1300. doi: 10.2147/vhrm.s3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tsuchida K. Activins, myostatin and related TGF-beta family members as novel therapeutic targets for endocrine, metabolic and immune disorders. Curr Drug Targets Immune Endocr Metabol Disord. 2004;4:157–166. doi: 10.2174/1568008043339901. [DOI] [PubMed] [Google Scholar]
- 80.Barberi L, Dobrowolny G, Pelosi L, Giacinti C, Musaro A. Muscle involvement and IGF-1 signaling in genetic disorders: new therapeutic approaches. Endocr Dev. 2009;14:29–37. doi: 10.1159/000207474. [DOI] [PubMed] [Google Scholar]
- 81.Giovannini S, Marzetti E, Borst SE, Leeuwenburgh C. Modulation of GH/IGF-1 axis: potential strategies to counteract sarcopenia in older adults. Mech Ageing Dev. 2008;129:593–601. doi: 10.1016/j.mad.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Philippou A, Maridaki M, Halapas A, Koutsilieris M. The role of the insulin-like growth factor 1 (IGF-1) in skeletal muscle physiology. In Vivo. 2007;21:45–54. [PubMed] [Google Scholar]
- 83.Musaro A, Dobrowolny G, Rosenthal N. The neuroprotective effects of a locally acting IGF-1 isoform. Exp Gerontol. 2007;42:76–80. doi: 10.1016/j.exger.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 84.Grounds MD. Reasons for the degeneration of ageing skeletal muscle: a central role for IGF-1 signalling. Biogerontology. 2002;3:19–24. doi: 10.1023/a:1015234709314. [DOI] [PubMed] [Google Scholar]