

Abstract
Although the association of common genetic variation in the extended MHC region with schizophrenia is the most significant yet discovered, the MHC region is one of the more complex regions of the human genome, with unusually high gene density and long-range linkage disequilibrium. The statistical test on which the MHC association is based is a relatively simple, additive model which uses logistic regression of SNP genotypes to predict case-control status. However, it is plausible that more complex models underlie this association. Using a well-characterized sample of trios, we evaluated more complex models by looking for evidence for: (a) non-random mating for HLA alleles, schizophrenia risk profiles, and ancestry; (b) parent-of-origin effects for HLA alleles; and (c) maternal-fetal genotype incompatibility in the HLA. We found no evidence for non-random mating in the parents of individuals with schizophrenia in terms of MHC genotypes or schizophrenia risk profile scores. However, there was evidence of non-random mating that appeared mostly to be driven by ancestry. We did not detect over-transmission of HLA alleles to affected offspring via the general TDT test (without regard to parent of origin) or preferential transmission via paternal or maternal inheritance. We evaluated the hypothesis that maternal-fetal HLA incompatibility may increase risk for schizophrenia using eight classical HLA loci. The most significant alleles were in HLA-B, HLA-C, HLA-DQB1, and HLA-DRB1 but none was significant after accounting for multiple comparisons. We did not find evidence to support more complex models of gene action, but statistical power may have been limiting.
Keywords: Schizophrenia, human leukocyte antigens, non-random mating, maternal-fetal incompatibility
1. Introduction
Common genetic variation in the major histocompatibility complex (MHC) on 6p22.1 is a risk factor for many complex human diseases. The association of common genetic variation in the extended MHC region with schizophrenia is the most significant yet discovered (P ~10−12) (Ripke et al., 2011) and meets community standards in human genetics for replication (Chanock SJ, 2007). However, the MHC region is one of the more complex regions of the human genome, with unusually high gene density and long-range linkage disequilibrium. As a result, the genome-wide significant evidence for association involves more than 100 SNPs, extends a very large distance (26–33 Mb), and encompasses around 300 genes.
The statistical test on which the MHC association is based is simple, using logistic regression of SNP genotypes to predict case-control status under an additive model. It is plausible that more complex models underlie this association. First, non-random mating (i.e., the tendency for mating partners to have greater phenotypic similarity than expected by chance) occurs for many physiological traits (Merikangas, 1982) as well as schizophrenia (Lichtenstein et al., 2006). Non-random mating can lead to complex biases in genomic studies (Redden and Allison, 2006) and may even be driven by genetic variation in the MHC region (Havlicek and Roberts, 2009). Second, parent-of-origin effects (variable genetic risk depending on the parent from which an allele is inherited) can occur in the MHC (Bassett, 2011; Chao et al., 2010). If this mechanism is operative, statistical models explicitly including such effects could assist in refining the currently broad and ill-defined MHC-schizophrenia association. Finally, maternal-fetal genotype incompatibility occurs when specific combinations of maternal and fetal genotypes yield an adverse prenatal environment (Childs et al., 2011). During pregnancy, maternal antibodies to paternal HLAs can be detected (Palmer, 2010). Since maternal antibodies to fetal antigens have been observed in a large proportion of healthy pregnancies, it is possible that maternal recognition or sensitization of paternally-derived fetal HLAs dissimilar to maternal HLAs may be beneficial for implantation and maintenance of pregnancy (Palmer et al., 2006). If paternally-derived fetal HLAs are similar to the maternal HLAs, maternal sensitization can fail to occur and lead to adverse fetal outcomes. Maternal-fetal genotype incompatibility may increase the risk of prenatal/obstetric complications (Cowan et al., 1994; Ober et al., 1998; Schneider et al., 1994; Verp et al., 1993), and there is some evidence that risk of schizophrenia may also be elevated (Palmer, 2010; Palmer et al., 2006).
Evaluation of these potentially more complex, MHC-themed models is difficult or impossible to do in case-control studies. Using a well-characterized sample of parent-affected offspring trios, we evaluated the evidence for: (a) non-random mating for HLA alleles and MHC SNPs, schizophrenia risk profiles, and ancestry; (b) parent-of-origin effects for HLA alleles and MHC SNPs; and (c) maternal-fetal genotype incompatibility in the HLA.
2. Materials and Methods
2.1. Subjects and genotyping
The study sample comprised 698 parent-offspring trio families from Bulgaria with 727 affected offspring (50.2% male). All subjects were genotyped with Affymetrix 6.0 chips at the Broad Institute(Ruderfer et al., 2011). We performed quality control (QC) steps in which we removed subjects with high genotype missing rates (> 2%) or high Mendelian errors per individual (> 2000 SNPs) along with SNPs with high missing rates (> 2%), strong deviation from Hardy-Weinberg Equilibrium (p < 1×10−6 in parents although there were none in the MHC region), frequency difference to HapMap3 CEU (> 0.15)(Altshuler et al., 2010), or high Mendelian errors per SNP (> 4). After QC, there were 642 probands in 624 complete trios (607 families with 1 proband, 16 families with 2 probands, and 1 family with 3 probands). Of the 642 probands, 544 (85%) had a diagnosis of schizophrenia and 98 (15%) schizoaffective disorder (Table S1), and 12 (1.0%) fathers and 36 (2.9%) mothers were diagnosed with schizophrenia or schizoaffective disorder. We kept 657,466 successfully genotyped SNPs which contain 1,704 SNPs in the extended MHC region (chr6: 26–33Mb).
In order to impute classical HLA alleles, we used a dataset created by the MHC Working Group of the Type 1 Diabetes Genetics Consortium (Pereyra et al., 2010). This dataset contains genotypes (MHC tag SNPs and direct determination of alleles for eight MHC genes at four digit resolution) for 2,767 unrelated individuals of European ancestry. The eight MHC genes are HLA-A, HLA-B, HLA-C, HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DPA1, and HLA-DPB1 (Brown et al., 2009), and the dataset enables imputation of 377 HLA alleles, 3,852 SNPs, and 372 amino acid changing polymorphisms. We used Beagle (Browning and Browning, 2009) to phase and impute classical HLA alleles (Robinson et al., 2011) for these subjects. We selected 189 HLA alleles imputed with high confidence (imputation quality > 0.8) and no Mendelian inheritance errors for further analysis.
2.2. Non-Random mating
We evaluated non-random mating in three ways. First, to evaluate non-random mating for HLA alleles and MHC SNPs, we used methods described in Chaix et al. (Chaix et al., 2008). The genetic similarity of a mating pair c at a genetic marker in the HLA region was estimated as R, the ratio of probabilities of identity in state: R=(Qc – Qm) / (1-Qm), where Qc is the proportion of identical variants in the mating pair (0 if different alleles, 0.5 if one allele identical, or 1 if both alleles were the same) and Qm is the mean proportion of identical variants over all possible pairs of individuals in the sample. We summarized R by computing its mean for mating pairs across the imputed HLA alleles or across SNPs with R > 0 indicating genetic similarity and R < 0 indicating genetic dissimilarity between mating pairs relative to random mates in the sample. The significance of R was assessed using permutation and random shuffling of mating pairs 1,000 times.
Second, we evaluated evidence for non-random mating that might be driven by sub-clinical phenotypes genetically related to schizophrenia by using risk profile scores (Purcell et al., 2009) genome-wide, genome-wide excluding MHC, and MHC only. There are robust and replicable findings that liability to schizophrenia is distributed across the genome and can be assessed by a weighted sum of the number of associated risk alleles (Purcell et al., 2009; Ripke et al., 2011; Ruderfer et al., 2011). We used the PGC schizophrenia sample (Ripke et al., 2011) as the discovery sample (excluding the subjects from this study) to generate a set of markers for generating risk profiles in these trios. The risk profile set contained 64,254 SNPs and was a subset of the full GWAS results file after filtering for allele frequency 0.02–0.98, imputation INFO score > 0.9, approximate linkage equilibrium (r2 ≤ 0.25 within 500kb windows), and association threshold PT < 0.2 (see Figure S6 in reference (Ripke et al., 2011) for justification for selection of PT < 0.2). Using this risk profile, we calculated the schizophrenia risk profile scores for each of the parents in these Bulgarian trios, and assessed non-random mating via the correlation between mothers and fathers. As a comparison, we performed the same analysis for the HapMap3 CEU founders. Third, we evaluated evidence for non-random mating based on ancestry as determined using multidimensional scaling (MDS) dimensions (Purcell et al., 2007; Sebro et al., 2010) and again used HapMap3 CEU founders for comparison.
2.3. Parent-of-origin effects
We used the transmission disequilibrium test (Spielman and Ewens, 1993) implemented in PLINK (Purcell et al., 2007) to determine whether any HLA allele was more frequently transmitted to the affected offspring than the other allele without regard to the gender of the transmitting parent. We then refined this general analysis by separately considering transmissions from heterozygous fathers versus heterozygous mothers to affected offspring to assess whether the transmission of risk alleles was biased towards one parent.
2.4. Maternal-fetal genotype incompatibility
To evaluate the presence of maternal-fetal genotype incompatibility on risk for schizophrenia, we used “MFG” option in Mendel (Childs EJ, 2010; Sinsheimer et al., 2003). The MFG option implements an affected-only likelihood ratio test relying on the joint estimation of offspring allelic effects, maternal allelic effects, and interactions between maternal and offspring genotypes. As the Mendel MFG implementation can handle only biallelic loci, we converted multi-allelic HLA types into binary format and tested for MFG incompatibility at each biallelic HLA marker. Over all alleles at each HLA locus, we designated an index allele as “G” (e.g. HLA-A*0101), and “A” for all the other alleles at the locus. For example, an individual with two HLA-A*0101 alleles would be assigned “G/G” at marker HLA-A*0101, and an individual with genotype HLA-A*0301/0302 would be assigned “A/A” at the same marker. With our genotype notation of biallelic HLA marker, there are 7 possible maternal-child genotype combinations between mother and child (Table S2). Childs et al. (Childs et al., 2011) and Palmer et al. (Palmer et al., 2006) defined offspring as being MFG incompatible for a polymorphism if mother and child have identical genotypes or if the mother is heterozygous and the offspring is homozygous. Following their definition, we defined the following two combinations as MFG incompatible: (gm= G/G, gc=G/G), (gm= A/G, gc=G/G) where gm=maternal genotype and gc=child genotype. We did not consider (gm= A/G, gc=A/A), (gm= A/A, gc=A/A), or (gm= A/G, gc=A/G) as incompatible since “A” denotes other alleles at the index allele being considered. Mendel maximizes the log-likelihoods under our MFG incompatibility model and a null model in which all mother-offspring genotype combinations confer the same risk of disease. For each biallelic HLA marker, Mendel calculates a likelihood ratio test statistic and reports an asymptotic p-value.
3. Results
3.1. Non-random mating
First, we tested for genetic similarity in founders of HapMap2 CEU, HapMap3 CEU, and our trio sample using imputed HLA alleles (Table S3) and SNPs (Table S4). To calibrate the method described in (Chaix et al., 2008) and replicate their finding, we repeated the non-random mating analysis of 5,708 MHC SNPs (chr6: 29.6 −33.3 Mb) with MAF ≥5% in HapMap2 CEU founders and confirmed the previously reported results (a slight but statistically significant dissimilarity in the MHC region, R=−0.064, p=0.016). We applied these procedures to 1,175 MHC SNPs (chr6: 29.6 −33.3 Mb) with MAF ≥1% and to imputed 4-digit HLA alleles across class I and II genes in 42 mating pairs from HapMap3 CEU. However, unlike the HapMap2 CEU results, there was no evidence for genetic similarity in HapMap3 CEU based neither on SNPs (R=−0.047, p=0.175) nor on imputed HLA alleles (six HLA genes, R=0.009, p=0.589; eight HLA genes, R=0.003, p=0.521). Derti et al. (Derti et al., 2010) suggest that the HapMap2-HapMap3 discrepancy could be due to influential outliers. Next, we applied similar approaches to 1,082 MHC SNPs (chr6: 29.6 −33.3 Mb) with MAF≥1% and class I and II HLA genes in our sample. Although we observed marginally significant similarity among parents of schizophrenic offspring using SNPs (R=0.0082, p=0.052), there was no evidence for similarity using HLA genes (six HLA genes, R=0.002, p=0.534; eight HLA genes, R=0.004, p=0.804). In summary, we failed to find evidence to support the hypothesis that parents of schizophrenic offspring are more likely to be genetically similar in MHC than parents of healthy offspring.
Second, we evaluated non-random mating in regard to risk profile scores derived from PGC schizophrenia results. As higher risk profile scores have been highly significantly but weakly correlated with schizophrenia, parents of offspring with schizophrenia are, in general, likely to have higher risk profile scores than parents of healthy children (Figure S1). Moreover, if parental mating was influenced by similarity for phenotypic traits related to risk profile scores, an appreciable positive correlation between parents might be observed. The genome-wide correlation in risk profile scores between parents was highly significant (Figure 1, Pearson’s r=0.174, asymptotic p=1.3×10−5, and permutation p < 1×10−4, Figure S2). However, the paternal-maternal risk profile non-parametric correlation was not significant (Spearman’s ρ=0.065, p=0.105) (Table 1), suggesting that the Pearson correlation could be unduly influenced by outliers. After removing 137 ancestral outliers detected by smartpca in EigenSoft (Price et al., 2006), we found that Pearson’s correlation was no longer significant (Pearson’s r=−0.07, p=0.11, Figure 1, Table 1). Of the 137 ancestral outliers, 130 individuals were control and 7 were diagnosed with schizophrenia. Correlations excluding MHC (Pearson’s r=0.16, p=6.1×10−5 ; Spearman’s ρ=0.05, p=0.24) were quite similar to those including MHC. Correlations in MHC only were null (Pearson’s r=0.01, p=0.76 ; Spearman’s ρ=−0.03, p=0.52). We applied the same risk score profile to the HapMap3 CEU founders, and observed non-significant results genome-wide (Pearson’s r=−0.04, p=0.80; Spearman’s ρ=−0.03, p=0.81; Figure 1, Table 1). Correlations excluding MHC (Pearson’s r=−0.02, p=0.90; Spearman’s ρ=0.02, p=0.91) were insignificant, similar to genome-wide results. Although non-parametric correlations in MHC only were significant (Spearman’s ρ=−0.32, p=0.01), parametric correlations in MHC were insignificant (Pearson’s r=−0.23, p=0.08). Taken together, we conclude that there is no strong evidence to support non-random mating based on schizophrenia risk profile scores calculated across genome-wide or across MHC in either Bulgarian sample or HapMap3 CEU.
Figure 1. Spousal correlation in Bulgarian sample and HapMap3 CEU using polygene scores.
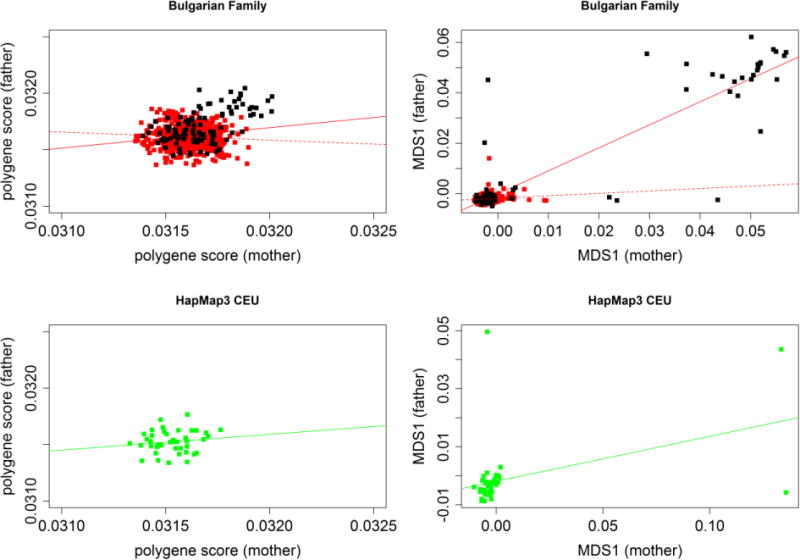
Upper panels depict polygene risk scores and MDS1 of Bulgarian founders. Black dots indicate genetic outliers detected by Eigenstrat. Solid red line represents a regression line when including all pairs. Dotted line represents a regression line after removing the genetic outliers. Lower panels show polygene risk scores and MDS1 of HapMap 3 CEU founders.
Table 1.
Summary of non-random mating using risk profile scores and multidimensional scaling (MDS).
| Bulgarian Families | HapMap3 CEU | |||
|---|---|---|---|---|
| Pearson correlation (p-value) | Spearman correlation (p-value) | Pearson correlation (p-value) | Spearman correlation (p-value) | |
| (a) Risk score profile | ||||
| Genome-wide | 0.17 (1.3E-5) | 0.07 (0.11) | −0.04 (0.80) | −0.03 (0.81) |
| Genome-wide (After removing outliers) | −0.07 (0.11) | −0.05 (0.28) | NA | NA |
| Excluding MHC | 0.16 (6.1E-5) | 0.05 (0.24) | −0.02 (0.90) | 0.02 (0.91) |
| MHC only | 0.01 (0.76) | −0.03 (0.52) | −0.23 (0.08) | −0.32 (0.01) |
| (b) MDS | ||||
| MDS1 | 0.92 (< 2.2E-16) | 0.23 (7.6E-9) | 0.41 (0.006) | 0.45 (0.003) |
| MDS1 (After removing outliers) | 0.11 (0.01) | 0.12 (0.006) | 0.54 (0.001) | 0.52 (0.002) |
| MDS2 | −0.02 (0.69) | 0.08 (0.05) | 0.90 (8.9E-16) | 0.86 (< 2.2E-16) |
| MDS3 | 0.01 (0.73) | −0.004 (0.92) | 0.98 (< 2.2E-16) | 0.95 (< 2.2E-16) |
Finally, we assessed non-random mating in regard to genetic ancestry. The first multidimensional scale score was positively correlated between the parents of probands with schizophrenia (Pearson’s r=0.92, p<2.2×10−16, and Spearman’s ρ=0.23, p=7.6×10−9), suggesting ancestry-related non-random mating. After removing 137 ancestry outliers detected by smartpca, the maternal-paternal correlation was greatly attenuated (r=0.11, p=0.01, Figure 1, Table 1). There were no significant correlations between intra-parental scores on the second or third multidimensional scale scores. We also applied the same procedure to the HapMap3 CEU founders, and observed significant correlations in the first three MDS values, indicating that there is ancestral non-random mating among the HapMap3 CEU founders. Even after removing 7 outliers detected by EigenSoft (Price et al., 2006), correlations in the first three MDS values remained strongly significant. We conclude that there is supportive evidence for non-random mating with respect to genetic ancestry in these Bulgarian trios as well as HapMap 3 CEU.
3.2. Association testing
We performed TDT and parent-of-origin analyses on 1704 SNPs in the extended MHC region (chr6: 26–33Mb) and on 189 HLA alleles with imputation quality > 0.8. TDT analysis revealed that 4 SNPs (rs2894249, rs2107191, rs3129932, rs3117194), HLA-DPB1*0201, and HLA-DPB1*1101 were significant (Table 2, Figure S3 for 2-digit HLA alleles; Figure S4 for 4-digit HLA alleles). However, after multiple testing adjustment using false discovery rate method (Benjamini, 1995), all the adjusted p-values were greater than 0.6 and hence we concluded that no SNP or HLA allele was significantly over-transmitted to affected offspring without regard to parents. Parent-of-origin analysis, which separately considered paternal and maternal over-transmission of risk allele to affected offspring, suggested that 2 SNPs (rs2107191, rs3117194), HLA-A*0301, HLA-C*1203, and HLA-DPB1*0201 were over-transmitted paternally whereas 3 SNPs (rs886403, rs1632857, rs1634717), HLA-A*1101, and HLA-C*0401 were over-transmitted maternally (Table 3, Figure S3 for 2-digit HLA alleles; Figure S4 for 4-digit HLA alleles). Again, no SNP or HLA alleles survived after multiple testing adjustments.
Table 2.
TDT results for 189 imputed HLA alleles with imputation quality > 0.8 and 1704 SNPs in extended MHC region.
| Marker | Transmitted counts | Untransmitted counts | Odds ratio | Chisq | P-value | Empirical P-value | FDR adjusted P-value |
|---|---|---|---|---|---|---|---|
| (a) HLA alleles | |||||||
| HLA-DPB1*02 | 200 | 163 | 1.23 | 3.77 | 0.052 | 0.076 | 0.96 |
| HLA-DPB1*0201 | 197 | 161 | 1.22 | 3.62 | 0.057 | 0.090 | 0.96 |
| HLA-DPB1*11 | 6 | 1 | 6.00 | 3.57 | 0.059 | 0.079 | 0.96 |
| HLA-DPB1*1101 | 6 | 1 | 6.00 | 3.57 | 0.059 | 0.079 | 0.96 |
| (b) SNPs | |||||||
| rs2894249 | 161 | 114 | 1.41 | 8.03 | 0.005 | 0.005 | 0.67 |
| rs2107191 | 347 | 277 | 1.25 | 7.85 | 0.005 | 0.006 | 0.67 |
| rs3129932 | 161 | 117 | 1.38 | 6.96 | 0.008 | 0.010 | 0.71 |
| rs3117194 | 344 | 279 | 1.23 | 6.78 | 0.009 | 0.008 | 0.71 |
Over-transmitted HLA alleles with p-value < 0.06 and over-transmitted SNPs with p-value < 0.01 are shown in this table. Empirical p-values were obtained via permutation in which transmitted/untransmitted status constantly was flipped for all SNPs for a given family, preserving the LD and linkage information between markers and siblings. FDR adjusted p-value was calculated using the method of Benjamini and Hochberg (1995).
Table 3.
Parent-of-origin test results for 189 imputed HLA alleles with imputation quality > 0.8 and 1704 SNPs in extended MHC region.
| HLA allele | T:U (PAT) | P (PAT) | T:U (MAT) | P (MAT) | Z (POO) | P (POO) | FDR adjusted P-value |
|---|---|---|---|---|---|---|---|
| (a) HLA alleles | |||||||
| HLA-A*03 | 78:47 | 0.005 | 42:73 | 0.004 | 3.975 | 7.1E-5 | 0.28 |
| HLA-A*0301 | 75:45 | 0.006 | 39:72 | 0.002 | 4.102 | 4.1E-5 | 0.28 |
| HLA-C*1203 | 76:53 | 0.042 | 66:71 | 0.668 | 1.758 | 0.079 | 0.82 |
| HLA-DPB1*02 | 104:77 | 0.045 | 96:86 | 0.459 | 0.902 | 0.367 | 0.82 |
| HLA-DPB1*0201 | 104:76 | 0.036 | 94:86 | 0.549 | 1.062 | 0.288 | 0.82 |
| (b) SNPs | |||||||
| rs2107191 | 191:136 | 0.003 | 157:140 | 0.353 | 1.396 | 0.163 | 0.97 |
| rs3117194 | 188:137 | 0.005 | 156:142 | 0.417 | 1.378 | 0.168 | 0.97 |
| (c) HLA alleles | |||||||
| HLA-A*11 | 47:55 | 0.426 | 58:34 | 0.012 | −2.368 | 0.018 | 0.51 |
| HLA-A*1101 | 47:55 | 0.426 | 58:34 | 0.012 | −2.368 | 0.018 | 0.51 |
| HLA-C*04 | 102:100 | 0.944 | 113:85 | 0.047 | −1.37 | 0.167 | 0.75 |
| HLA-C*0401 | 102:100 | 0.944 | 113:85 | 0.047 | −1.37 | 0.167 | 0.75 |
| (d) SNPs | |||||||
| rs886403 | 139:144 | 0.767 | 157:110 | 0.004 | −2.273 | 0.023 | 0.96 |
| rs1632857 | 149:155 | 0.688 | 169:114 | 0.001 | −2.592 | 0.009 | 0.96 |
| rs1634717 | 156:154 | 0.909 | 174:129 | 0.012 | −1.762 | 0.078 | 0.96 |
HLA alleles or SNPs with significantly more transmission to affected offspring paternally (highlighted with blue) or maternally (highlighted with pink) are listed. For HLA alleles, P(PAT) or P(MAT) <0.06 are shown in this table. For SNPs, P(PAT) or P(MAT) <0.01 are shown in this table. This test separately considers transmission of risk alleles from heterozygous fathers versus heterozygous mothers to affected offspring. Under the null, there is no differential transmission paternally or maternally. P(PAT) represents p-value for paternal over-transmission. T:U (PAT) means paternal transmitted counts versus untransmitted counts. Z (POO) represents Z score for difference in paternal versus maternal odds ratios to contrast parent-of-origin effects. P (POO) is the asymptotic two-sided p-value for Z (POO). FDR adjusted p-values for P(PAT) or P(MAT) were calculated using the method of Benjamini and Hochberg (1995).
3.3. Maternal-fetal genotype incompatibility
We tested the hypothesis of maternal-fetal HLA incompatibility effects on schizophrenia. Of the 189 HLA alleles tested, 7 alleles in HLA-C, HLA-B, HLA-DRB1, HLA-DQB1 loci appeared to be associated with schizophrenia (Table 4, Figure S3 for 2-digit HLA alleles; Figure S4 for 4-digit HLA alleles). Due to previous findings that HLA-B matching effect on schizophrenia was exclusive to female offspring (Childs et al., 2011; Palmer et al., 2006), we tested for the MFG incompatibility effects on female probands by performing analyses on families with female probands. Although we found significant MFG incompatibility effects at HLA-B*56 and HLA-B*5601 with all probands, we failed to detect such effects at HLA-B in female probands (p =0.2 for HLA-B*56 and HLA-B*5601). The MFG incompatibility effect at HLA-DRB1*01 and HLA-DRB1*0101 remained significant in both all and female probands. However, after multiple testing correction using false discovery rate method (Benjamini, 1995), all the adjusted p-values were 1.0.
Table 4.
Summary of MFG incompatibility test.
| HLA allele | P-value1 (all probands) | P-value2 (female probands) |
|---|---|---|
| HLA-B*56 | 0.047 | 0.201 |
| HLA-B*5601 | 0.047 | 0.202 |
| HLA-C*03 | 0.038 | 0.141 |
| HLA-C*0303 | 0.029 | 0.153 |
| HLA-DRB1*01 | 0.004 | 0.051 |
| HLA-DRB1*0101 | 0.003 | 0.033 |
| HLA-DQB1*0301 | 0.037 | 0.185 |
: Families with all probands were included in analysis.
: Families with female probands were included in analysis.
4. Discussion
Using a relatively large and well-characterized trio sample, our study looked for evidence of non-random mating, parent-of-origin effects for HLA alleles, and maternal-fetal genotype incompatibility in the HLA. The results are consistent with three conclusions.
First, there was evidence of non-random mating by ancestry that appeared mostly to be driven by a subset of subjects who were ancestry outliers. We speculate that this could reflect within-group mating by minority groups within Bulgaria (e.g., Turks or Roma). Without data from additional trios sampled without respect to affection status, we cannot know whether this is a risk factor for schizophrenia or the result of chance, geographic propinquity, or some other process mechanistically unrelated to schizophrenia. However, we found no compelling evidence for non-random mating in the parents of individuals with schizophrenia in terms of MHC genotypes or schizophrenia risk profile scores.
Second, after correcting for multiple testing, we did not detect over-transmission of HLA alleles to affected offspring via the general TDT test (without regard to parent of origin) or preferential transmission via paternal or maternal inheritance.
Third, we evaluated the hypothesis that maternal-fetal HLA incompatibility may increase risk for schizophrenia by examining the effect of maternal-fetal HLA matching in all probands, female probands using imputed alleles of eight classical HLA loci. The most significant alleles were in HLA-B, HLA-C, HLA-DQB1, HLA-DRB1 but none was significant after accounting for multiple comparisons. In contrast to the more general and less assumption-laden approach we chose, Palmer et al. (Palmer, 2010; Palmer et al., 2006) and Childs et al. (Childs et al., 2011) applied a modified MFG test to three multi-allelic loci of HLA-A, HLA-B, and HLA-DRB1. They observed an association between HLA-B matching and schizophrenia in female offspring. Although we found significant HLA-B matching effect using families with all probands, we failed to replicate HLA-B matching effect using families with female probands only. Failure of replication could be due to methodological differences or different population (Finland versus Bulgaria) or HLA typing (imputed versus genotyped) or different sample sizes (274 families versus 624 families). Regarding methodological differences, they combined alleles to reduce the number of allele frequencies estimated at each locus and tested for HLA matching effect on schizophrenia at each HLA locus as a whole whereas we tested for HLA matching effect at each allele of each HLA locus. As discussed in their paper, however, combining alleles may result in binning low-risk mother-offspring combinations with high-risk ones. Different results may be obtained depending on how to combine the alleles.
These analyses must be viewed in context of several limitations. First, although the present sample is, to our knowledge, the largest trio sample with GWAS data available for schizophrenia, it is possible that true effects could have gone undetected due to limited power. As an illustration, for one of our hypotheses involving the general TDT, we had 80% power to detect genotypic relative risks of 1.37 and 1.79 for minor allele frequencies of 0.30 and 0.05 (624 case-parent trios, α=0.00625 (8 tests), log-additive model, and 0.4% disease risk in the general population) (Gauderman, 2006). These effect sizes are quite large (Ripke et al., 2011) and thus power was probably insufficient, although we have no strong precedents with which to derive effect size expectations under the more complex models. Second, other models such as a model including gene by environment interaction are possible. Third, this Bulgarian sample is atypical in terms of frequency and effect size of the ancestral haplotype across European samples as the ISC study (Purcell et al., 2009) originally documented.
Supplementary Material
References
- Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Bonnen PE, de Bakker PI, Deloukas P, Gabriel SB, Gwilliam R, Hunt S, Inouye M, Jia X, Palotie A, Parkin M, Whittaker P, Chang K, Hawes A, Lewis LR, Ren Y, Wheeler D, Muzny DM, Barnes C, Darvishi K, Hurles M, Korn JM, Kristiansson K, Lee C, McCarrol SA, Nemesh J, Keinan A, Montgomery SB, Pollack S, Price AL, Soranzo N, Gonzaga-Jauregui C, Anttila V, Brodeur W, Daly MJ, Leslie S, McVean G, Moutsianas L, Nguyen H, Zhang Q, Ghori MJ, McGinnis R, McLaren W, Takeuchi F, Grossman SR, Shlyakhter I, Hostetter EB, Sabeti PC, Adebamowo CA, Foster MW, Gordon DR, Licinio J, Manca MC, Marshall PA, Matsuda I, Ngare D, Wang VO, Reddy D, Rotimi CN, Royal CD, Sharp RR, Zeng C, Brooks LD, McEwen JE. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467(7311):52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS. Parental origin, DNA structure, and the schizophrenia spectrum. Am J Psychiatry. 2011;168(4):350–353. doi: 10.1176/appi.ajp.2011.11010173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Ya, H Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995;57:289–300. [Google Scholar]
- Brown WM, Pierce J, Hilner JE, Perdue LH, Lohman K, Li L, Venkatesh RB, Hunt S, Mychaleckyj JC, Deloukas P. Overview of the MHC fine mapping data. Diabetes Obes Metab. 2009;11(Suppl 1):2–7. doi: 10.1111/j.1463-1326.2008.00997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84(2):210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaix R, Cao C, Donnelly P. Is mate choice in humans MHC-dependent? PLoS Genet. 2008;4(9):e1000184. doi: 10.1371/journal.pgen.1000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanock SJ, M T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G, Hirschhorn JN, Abecasis G, Altshuler D, Bailey-Wilson JE, Brooks LD, Cardon LR, Daly M, Donnelly P, Fraumeni JF, Jr, Freimer NB, Gerhard DS, Gunter C, Guttmacher AE, Guyer MS, Harris EL, Hoh J, Hoover R, Kong CA, Merikangas KR, Morton CC, Palmer LJ, Phimister EG, Rice JP, Roberts J, Rotimi C, Tucker MA, Vogan KJ, Wacholder S, Wijsman EM, Winn DM, Collins FS. Replicating genotype-phenotype associations. Nature. 2007 Jun 7;447(7145):665–660. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- Chao MJ, Herrera BM, Ramagopalan SV, Deluca G, Handunetthi L, Orton SM, Lincoln MR, Sadovnick AD, Ebers GC. Parent-of-origin effects at the major histocompatibility complex in multiple sclerosis. Hum Mol Genet. 2010;19(18):3679–3689. doi: 10.1093/hmg/ddq282. [DOI] [PubMed] [Google Scholar]
- Childs EJ, P C, Lange K, Sinsheimer JS. Modeling maternal-offspring gene-gene interactions: the extended-MFG test. Genetic Epidemiology. 2010;34:512–521. doi: 10.1002/gepi.20508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs EJ, Sobel EM, Palmer CG, Sinsheimer JS. Detection of intergenerational genetic effects with application to hla-B matching as a risk factor for schizophrenia. Hum Hered. 2011;72(3):161–172. doi: 10.1159/000332051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan LD, Hudson L, Bobele G, Chancellor I, Baker J. Maternal-Fetal Hla Sharing and Risk of Newborn Encephalopathy and Seizures – a Pilot-Study. J Child Neurol. 1994;9(2):173–177. doi: 10.1177/088307389400900214. [DOI] [PubMed] [Google Scholar]
- Derti A, Cenik C, Kraft P, Roth FP. Absence of evidence for MHC-dependent mate selection within HapMap populations. PLoS Genet. 2010;6(4):e1000925. doi: 10.1371/journal.pgen.1000925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauderman WaJM. QUANTO 1.1: A computer program for power and sample size calculations for genetic-epidemiology studies. 2006 http://hydra.usc.edu/gxe.
- Havlicek J, Roberts SC. MHC-correlated mate choice in humans: a review. Psychoneuroendocrinology. 2009;34(4):497–512. doi: 10.1016/j.psyneuen.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Lichtenstein P, Bjork C, Hultman CM, Scolnick E, Sklar P, Sullivan PF. Recurrence risks for schizophrenia in a Swedish national cohort. Psychol Med. 2006;36(10):1417–1425. doi: 10.1017/S0033291706008385. [DOI] [PubMed] [Google Scholar]
- Merikangas KR. Assortative mating for psychiatric disorders and psychological traits. Arch Gen Psychiatry. 1982;39(10):1173–1180. doi: 10.1001/archpsyc.1982.04290100043007. [DOI] [PubMed] [Google Scholar]
- Ober C, Hyslop T, Elias S, Weitkamp LR, Hauck WW. Human leukocyte antigen matching and fetal loss: results of a 10 year prospective study. Hum Reprod. 1998;13(1):33–38. doi: 10.1093/humrep/13.1.33. [DOI] [PubMed] [Google Scholar]
- Palmer CGS. Evidence for Maternal-Fetal Genotype Incompatibility as a Risk Factor for Schizophrenia. J Biomed Biotechnol. 2010 doi: 10.1155/2010/576318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CGS, Hsieh HJ, Reed EF, Lonnqvist J, Peltonen L, Woodward JA, Sinsheimer JS. HLA-B maternal-fetal genotype matching increases risk of schizophrenia. Am J Hum Genet. 2006;79(4):710–715. doi: 10.1086/507829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereyra FP, Jia XM, McLaren PJ, Telenti A, de Bakker PIW, Walker BD, Ripke S, Brumme CJ, Pulit SL, Carrington M, Kadie CM, Carlson JM, Heckerman D, Graham RR, Plenge RM, Deeks SG, Gianniny L, Crawford G, Sullivan J, Gonzalez E, Davies L, Camargo A, Moore JM, Beattie N, Gupta S, Crenshaw A, Burtt NP, Guiducci C, Gupta N, Carrington M, Gao XJ, Qi Y, Yuki Y, Piechocka-Trocha A, Cutrell E, Rosenberg R, Moss KL, Lemay P, O’Leary J, Schaefer T, Verma P, Toth I, Block B, Baker B, Rothchild A, Lian J, Proudfoot J, Alvino DML, Vine S, Addo MM, Allen TM, Altfeld M, Henn MR, Le Gall S, Streeck H, Haas DW, Kuritzkes DR, Robbins GK, Shafer RW, Gulick RM, Shikuma CM, Haubrich R, Riddler S, Sax PE, Daar ES, Ribaudo HJ, Agan B, Agarwal S, Ahern RL, Allen BL, Altidor S, Altschuler EL, Ambardar S, Anastos K, Anderson B, Anderson V, Andrady U, Antoniskis D, Bangsberg D, Barbaro D, Barrie W, Bartczak J, Barton S, Basden P, Basgoz N, Bazner S, Bellos NC, Benson AM, Berger J, Bernard NF, Bernard AM, Birch C, Bodner SJ, Bolan RK, Boudreaux ET, Bradley M, Braun JF, Brndjar JE, Brown SJ, Brown K, Brown ST, Burack J, Bush LM, Cafaro V, Campbell O, Campbell J, Carlson RH, Carmichael JK, Casey KK, Cavacuiti C, Celestin G, Chambers ST, Chez N, Chirch LM, Cimoch PJ, Cohen D, Cohn LE, Conway B, Cooper DA, Cornelson B, Cox DT, Cristofano MV, Cuchural G, Czartoski JL, Dahman JM, Daly JS, Davis BT, Davis K, Davod SM, Deeks SG, DeJesus E, Dietz CA, Dunham E, Dunn ME, Ellerin TB, Eron JJ, Fangman JJW, Farel CE, Ferlazzo H, Fidler S, Fleenor-Ford A, Frankel R, Freedberg KA, French NK, Fuchs JD, Fuller JD, Gaberman J, Gallant JE, Gandhi RT, Garcia E, Garmon D, Gathe JC, Gaultier CR, Gebre W, Gilman FD, Gilson I, Goepfert PA, Gottlieb MS, Goulston C, Groger RK, Gurley TD, Haber S, Hardwicke R, Hardy WD, Harrigan PR, Hawkins TN, Heath S, Hecht FM, Henry WK, Hladek M, Hoffman RP, Horton JM, Hsu RK, Huhn GD, Hunt P, Hupert MJ, Illeman ML, Jaeger H, Jellinger RM, John M, Johnson JA, Johnson KL, Johnson H, Johnson K, Joly J, Jordan WC, Kauffman CA, Khanlou H, Killian RK, Kim AY, Kim DD, Kinder CA, Kirchner JT, Kogelman L, Kojic EM, Korthuis T, Kurisu W, Kwon DS, LaMar M, Lampiris H, Lanzafame M, Lederman MM, Lee DM, Lee JML, Lee MJ, Lee ETY, Lemoine J, Levy JA, Llibre JM, Liguori MA, Little SJ, Liu AY, Lopez AJ, Loutfy MR, Loy D, Mohammed DY, Man A, Mansour MK, Marconi VC, Markowitz M, Marques R, Martin JN, Martin HL, Mayer KH, McElrath MJ, McGhee TA, McGovern BH, McGowan K, McIntyre D, Mcleod GX, Menezes P, Mesa G, Metroka CE, Meyer-Olson D, Miller AO, Montgomery K, Mounzer KC, Nagami EH, Nagin I, Nahass RG, Nelson MO, Nielsen C, Norene DL, O’Connor DH, Ojikutu BO, Okulicz J, Oladehin OO, Oldfield EC, Olender SA, Ostrowski M, Owen WF, Pae E, Parsonnet J, Pavlatos AM, Perlmutter AM, Pierce MN, Pincus JM, Pisani L, Price LJ, Proia L, Prokesch RC, Pujet HC, Ramgopal M, Rathod A, Rausch M, Ravishankar J, Rhame FS, Richards CS, Richman DD, Robbins GK, Rodes B, Rodriguez M, Rose RC, Rosenberg ES, Rosenthal D, Ross PE, Rubin DS, Rumbaugh E, Saenz L, Salvaggio MR, Sanchez WC, Sanjana VM, Santiago S, Schmidt W, Schuitemaker H, Sestak PM, Shalit P, Shay W, Shirvani VN, Silebi VI, Sizemore JM, Skolnik PR, Sokol-Anderson M, Sosman JM, Stabile P, Stapleton JT, Starrett S, Stein F, Stellbrink HJ, Sterman FL, Stone VE, Stone DR, Tambussi G, Taplitz RA, Tedaldi EM, Telenti A, Theisen W, Torres R, Tosiello L, Tremblay C, Tribble MA, Trinh PD, Tsao A, Ueda P, Vaccaro A, Valadas E, Vanig TJ, Vecino I, Vega VM, Veikley W, Wade BH, Walworth C, Wanidworanun C, Ward DJ, Warner DA, Weber RD, Webster D, Weis S, Wheeler DA, White DJ, Wilkins E, Winston A, Wlodaver CG, van’t Wout A, Wright DP, Yang OO, Yurdin DL, Zabukovic BW, Zachary KC, Zeeman B, Zhao M, Study IHC. The Major Genetic Determinants of HIV-1 Control Affect HLA Class I Peptide Presentation. Science. 2010;330(6010):1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redden DT, Allison DB. The effect of assortative mating upon genetic association studies: Spurious associations and population substructure in the absence of admixture. Behav Genet. 2006;36(5):678–686. doi: 10.1007/s10519-006-9060-0. [DOI] [PubMed] [Google Scholar]
- Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, Lin DY, Duan J, Ophoff RA, Andreassen OA, Scolnick E, Cichon S, St Clair D, Corvin A, Gurling H, Werge T, Rujescu D, Blackwood DH, Pato CN, Malhotra AK, Purcell S, Dudbridge F, Neale BM, Rossin L, Visscher PM, Posthuma D, Ruderfer DM, Fanous A, Stefansson H, Steinberg S, Mowry BJ, Golimbet V, De Hert M, Jonsson EG, Bitter I, Pietilainen OP, Collier DA, Tosato S, Agartz I, Albus M, Alexander M, Amdur RL, Amin F, Bass N, Bergen SE, Black DW, Borglum AD, Brown MA, Bruggeman R, Buccola NG, Byerley WF, Cahn W, Cantor RM, Carr VJ, Catts SV, Choudhury K, Cloninger CR, Cormican P, Craddock N, Danoy PA, Datta S, de Haan L, Demontis D, Dikeos D, Djurovic S, Donnelly P, Donohoe G, Duong L, Dwyer S, Fink-Jensen A, Freedman R, Freimer NB, Friedl M, Georgieva L, Giegling I, Gill M, Glenthoj B, Godard S, Hamshere M, Hansen M, Hansen T, Hartmann AM, Henskens FA, Hougaard DM, Hultman CM, Ingason A, Jablensky AV, Jakobsen KD, Jay M, Jurgens G, Kahn RS, Keller MC, Kenis G, Kenny E, Kim Y, Kirov GK, Konnerth H, Konte B, Krabbendam L, Krasucki R, Lasseter VK, Laurent C, Lawrence J, Lencz T, Lerer FB, Liang KY, Lichtenstein P, Lieberman JA, Linszen DH, Lonnqvist J, Loughland CM, Maclean AW, Maher BS, Maier W, Mallet J, Malloy P, Mattheisen M, Mattingsdal M, McGhee KA, McGrath JJ, McIntosh A, McLean DE, McQuillin A, Melle I, Michie PT, Milanova V, Morris DW, Mors O, Mortensen PB, Moskvina V, Muglia P, Myin-Germeys I, Nertney DA, Nestadt G, Nielsen J, Nikolov I, Nordentoft M, Norton N, Nothen MM, O’Dushlaine CT, Olincy A, Olsen L, O’Neill FA, Orntoft TF, Owen MJ, Pantelis C, Papadimitriou G, Pato MT, Peltonen L, Petursson H, Pickard B, Pimm J, Pulver AE, Puri V, Quested D, Quinn EM, Rasmussen HB, Rethelyi JM, Ribble R, Rietschel M, Riley BP, Ruggeri M, Schall U, Schulze TG, Schwab SG, Scott RJ, Shi J, Sigurdsson E, Silverman JM, Spencer CC, Stefansson K, Strange A, Strengman E, Stroup TS, Suvisaari J, Terenius L, Thirumalai S, Thygesen JH, Timm S, Toncheva D, van den Oord E, van Os J, van Winkel R, Veldink J, Walsh D, Wang AG, Wiersma D, Wildenauer DB, Williams HJ, Williams NM, Wormley B, Zammit S, Sullivan PF, O’Donovan MC, Daly MJ, Gejman PV. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43(10):969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J, Mistry K, McWilliam H, Lopez R, Parham P, Marsh SG. The IMGT/HLA database. Nucleic Acids Res. 2011;39(Database issue):D1171–1176. doi: 10.1093/nar/gkq998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderfer DM, Kirov G, Chambert K, Moran JL, Owen MJ, O’Donovan MC, Sklar P, Purcell SM. A family-based study of common polygenic variation and risk of schizophrenia. Mol Psychiatry. 2011;16(9):887–888. doi: 10.1038/mp.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider K, Knutson F, Tamsen L, Sjoberg O. HLA antigen sharing in preeclampsia. Gynecol Obstet Invest. 1994;37(2):87–90. doi: 10.1159/000292531. [DOI] [PubMed] [Google Scholar]
- Sebro R, Hoffman TJ, Lange C, Rogus JJ, Risch NJ. Testing for non-random mating: evidence for ancestry-related assortative mating in the Framingham heart study. Genet Epidemiol. 2010;34(7):674–679. doi: 10.1002/gepi.20528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinsheimer JS, Palmer CG, Woodward JA. Detecting genotype combinations that increase risk for disease: maternal-fetal genotype incompatibility test. Genet Epidemiol. 2003;24(1):1–13. doi: 10.1002/gepi.10211. [DOI] [PubMed] [Google Scholar]
- Spielman RS, Ewens WJ. Transmission Disequilibrium Test (Tdt) for Linkage and Linkage Disequilibrium between Disease and Marker. Am J Hum Genet. 1993;53(3):863–863. [Google Scholar]
- Verp MS, Sibul M, Billstrand C, Bellen G, Hsu M, Ober C. Maternal-fetal histocompatibility in intrauterine growth retarded and normal weight babies. Am J Reprod Immunol. 1993;29(4):195–198. doi: 10.1111/j.1600-0897.1993.tb00586.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.