

Gr-1+CD11b+ myeloid cells drive CD80 downregulation on hepatic B cells, in the context of liver metastases.
Keywords: B cells, immunosuppression, liver, STAT3
Abstract
LM escape immune surveillance, in part, as a result of the expansion of CD11b+MC, which alter the intrahepatic microenvironment to promote tumor tolerance. HBC make up a significant proportion of liver lymphocytes and appear to delay tumor progression; however, their significance in the setting of LM is poorly defined. Therefore, we characterized HBC and HBC/CD11b+MC interactions using a murine model of LM. Tumor-bearing livers showed a trend toward elevated absolute numbers of CD19+ HBC. A significant increase in the frequency of IgMloIgDhi mature HBC was observed in mice with LM compared with normal mice. HBC derived from tumor-bearing mice demonstrated increased proliferation in response to TLR and BCR stimulation ex vivo compared with HBC from normal livers. HBC from tumor-bearing livers exhibited significant down-regulation of CD80 and were impaired in inducing CD4+ T cell proliferation ex vivo. We implicated hepatic CD11b+MC as mediators of CD80 down-modulation on HBC ex vivo via a CD11b-dependent mechanism that required cell-to-cell contact and STAT3 activity. Therefore, CD11b+MC may compromise the ability of HBC to promote T cell activation in the setting of LM as a result of diminished expression of CD80. Cross-talk between CD11b+MC and HBC may be an important component of LM-induced immunosuppression.
Introduction
A significant proportion of colorectal cancer patients will develop LM over the course of their disease, and modern multimodality management will not be curative for the majority of patients. An important factor contributing to the biologic aggressiveness of LM is the tolerogenic nature of the intrahepatic space [1–3]. The liver contains an abundance of cells capable of suppressing the immune response, including Tregs, MDSC, and tumor-associated macrophages, all of which expand in response to inflammatory or neoplastic stimuli [4–7]. The importance and nature of tumor-induced immunosuppression have been demonstrated in numerous studies [8–10].
Although HBC represent a large proportion of liver lymphocytes and interact with hepatic T cells [11, 12], their role in the LM environment remains virtually unexplored. Murine data regarding the role of B cells in cancer is complex, demonstrating that B cells inhibit anti-tumor responses in some models, while enhancing tumor protection in others [13–19]. In humans, tumor-infiltrating B cells have been studied most extensively in breast carcinoma, where B cells have been linked to favorable survival [20, 21]. To date, we have a limited understanding of how HBC may impact the biology of liver tumors. The function of intrahepatic immune cells compared with their counterparts in other sites is often distinct [3, 11, 12, 22]. Therefore, an investigation of how HBC function is affected by liver tumors is warranted.
As immunotherapy for solid tumors becomes increasingly integrated into clinical practice, we must enhance our understanding of the immunologic context within which liver tumors develop and progress. Whether HBC promote T cell suppression or activation in the context of LM is unknown. We speculated that HBC immune function is impaired by the abundance of suppressive immune cells in the liver, of which CD11b+MC comprise a major fraction in the setting of abdominal malignancy [23]. Therefore, we focused on how CD11b+MC may modulate HBC phenotype and function. In addition to the well-known role of MDSC as negative regulators of T cell function [24, 25], a recent study demonstrated that MDSC can inhibit B cell mitogenic responses in the setting of a retroviral infection [26]. We hypothesized that liver CD11b+MC suppress HBC function and the ability of HBC to stimulate T cells in the setting of LM.
In this report, we describe HBC phenotypic and functional changes induced by LM. HBC from tumor-bearing livers showed a more mature phenotype and high proliferative capacity but exhibited CD80 down-regulation that was reversible upon removal from the tumor environment. CD11b+MC were responsible for CD80 down-regulation on HBC and resulted in impaired T cell stimulation by HBC. CD80 loss on HBC was dependent on CD11b+MC CD11b expression and STAT3 activity. The results described herein further our understanding of the role of HBC in LM. Rescue of HBC immune function through prevention of CD80 down-regulation may be a rational therapeutic objective.
MATERIALS AND METHODS
Mice and tumor cell injections
C57BL/6, BALB/c, and μMT B6.129S2-Ighm/J male, 6- to 8-week-old mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Animals were maintained at the Roger Williams Medical Center animal facility under specific pathogen-free conditions. To generate LM, mice were anesthetized and injected via spleen with 2.5 × 106 MC38CEA cells. Two minutes after injection, spleens were removed to confine metastases to the liver. “Normal” control animals were splenectomized without tumor injection. All surgical procedures and animal handling were carried out according to Institutional Animal Care and Use Committee guidelines at Roger Williams Medical Center.
Generation of luciferase-positive MC38CEA-luc cell line
MC38CEA murine colorectal carcinoma cells, a generous gift of Dr. Jeffrey Schlom at the U.S. National Institutes of Health, were grown in DMEM (Cellgro, Mediatech, Manassas, VA, USA), supplemented with 10% FBS (Sigma Life Science, St. Louis, MO, USA) and penicillin/streptomycin (Cellgro, Mediatech). MC38CEA cells were transduced with a lentivirus luciferase construct (pLenti-UbC-Luc2; Applied Biological Materials, Richmond, BC, Canada). Briefly, diluted viral stock (1×106 CFU/mL) was added to subconfluent MC38CEA cells in the presence of polybrene. Transduced cells were selected by puromycin (0.3 μg/mL; Sigma-Aldrich, St. Louis, MO, USA) resistance. To isolate pure cell lines derived from single-cell clones, two rounds of cloning by limiting dilution were performed. Clones were screened for luciferase activity using the IVIS 100 bioimaging system (Xenogen/PerkinElmer Life Sciences, Waltham, MA, USA). The clones were tested further in vivo to confirm the activity in live animals.
In vivo bioimaging of tumors
Abdominal walls of mice with LM were shaved to remove fur that absorbs light and reduces the sensitivity of the assay. Each anesthetized animal was injected i.p. with 200 μl potassium luciferin (15 mg/mL; Gold Biotechnology, St. Louis, MO, USA). The following settings were used to detect luciferase activity of the tumor cells in vivo, 15 min after injection: field of view = D, f-stop = 1, exposure = 0.5 s. Visual data were analyzed further using PerkinElmer Living Image software and luminescence quantified in total photon counts.
ELISAs
Serum and supernatant levels of anti-CEA IgG were determined using an in-house ELISA. Costar 96-well ELISA plates were coated overnight with 250 ng/well rCEA (Fitzgerald Industries International, Acton, MA, USA). The solution was removed after the incubation and plates were blocked with 1% BSA in PBS for 1 h at room temperature. Serum was diluted in 0.05% Tween-20, 1% BSA in PBS from 1:20 to 1:160, and added to plates for 2 h at room temperature. The plates were washed with 0.05% Tween-20, 1% BSA in PBS, and AP-conjugated secondary antibody, diluted 1:1500, was added (goat anti-mouse IgG-AP; Southern Biotechnology, Birmingham, AL, USA). Plates were washed after 2 h and AP Yellow (p-nitrophenyl phosphate) substrate (Sigma-Aldrich) added. Fifty μL 2N H2SO4 was added after 4 h to stop the reaction. Plates were read at 405 nm using BioTek Eon microplate spectrophotometer (BioTek Instruments, Winooski, VT, USA).
Liver leukocyte isolation and purification
Liver NPC were isolated from tumor-bearing mice, as described previously with modifications [3]. Red blood cell lysis was performed using 2 mL Ammonium-chloride-potassium lysing buffer (Gibco, Life Technologies, Grand Island, NY, USA) for 5 min at room temperature. Total hepatic leukocytes were Fc blocked using anti-FcγRII/III mAb 2.4G2 (AbD Secotec, Raleigh, NC, USA). Immunomagnetic beads against CD45, CD19, Thy 1.2, and CD11b were used to purify total hepatic leukocytes, B cells, T cells, and CD11b+MC, respectively (Miltenyi Biotec, Auburn, CA, USA). Typical purity of beaded B cells and T cells was 90–95%. Following 10–14 days of LM growth, the CD11b+ fraction is predominantly a CD11b+MC based on phenotype (>80% CD11b+Gr-1+) and potent in vitro T cell-suppressive function (data not shown).
Flow cytometry
Antibodies specific for the following surface markers were used: B220 (RA3-6B2), CD19 (1D3), IgM (II/41), IgD (11-26c.2a), CD80 (16-10A1), CD86 (GL1), CD45 (30-F11), I-A (AF6-120.1), Gr-1 (RB6-8C5), and CD11b (M1/17; BD Biosciences, San Jose, CA, USA) and PD-L1 (10F.9G2; BioLegend, San Diego, CA, USA). A CyAn ADP flow cytometer (Beckman Coulter, Indianapolis, IN, USA) was used to collect cells for analysis. Unstained cells and single-stained controls were used to determine laser voltages and calculate compensations. Postacquisition analysis was carried out using FlowJo software (Tree Star, Ashland, OR, USA).
Adoptive transfers
Splenic B cells from C57BL/6 mice were isolated using anti-CD19 immunomagnetic beads and were loaded with 5μM CellTrace CFSE (Invitrogen, Eugene, OR, USA), according to the manufacturer's instructions. CFSE-labeled B cells (2.5×106 cells/mouse) were injected via portal vein into normal or tumor-bearing livers of C57BL/6 mice. After 48 h, the livers were harvested, and the CD45+ fraction was analyzed by flow cytometry to evaluate CD80 expression on CFSE+B220+ cells.
B cell proliferation assay
HBC were loaded with 5 μM CFSE, and were stimulated ex vivo with LPS (10 μg/mL; L5293; Sigma-Aldrich), CpG 1826 (25 μg/mL; InvivoGen, San Diego, CA, USA), or AffiniPure F(ab′)2 fragment goat anti-mouse IgM (15 μg/mL; Jackson ImmunoResearch, West Grove, PA, USA) and anti-mouse CD40 (10 μg/mL; Miltenyi Biotec). After 4 days of culture, the cells were harvested and proliferation assessed by CFSE dilution.
B cell coculture assays
B and T cells or B cells and CD11b+MC were cocultured at a 1:2 ratio in round- bottom, 96-well tissue-culture plates. For Transwell experiments, 24-well cell-culture inserts with 1.0 μm pore size membranes (BD Biosciences) were used. B Cell surface-marker expression was tested at 24 and 48 h of culture. To block CD11b on CD11b+MC, 5 μg/well LEAF-purified anti-mouse/human CD11b antibody (clone M1/70; BioLegend) was used. The following drugs were used to inhibit the immunosuppressive products of CD11b+MC: L-NMMA (300 μM; Calbiochem, San Diego, CA, USA), anti-PD-L1 antibody (29F1.A12; 3 μg/mL; BioLegend), 1-MT (0.5 mM; Sigma-Aldrich), nor-NOHA (2 μM; Calbiochem), JSI-124 (1.5 μM; Sigma-Aldrich), and Stattic (100 μM; Sigma-Aldrich).
Allogeneic T cell proliferation assay
BALB/c Thy 1.2+ splenocytes were loaded with 0.5μM CFSE. HBC were purified from C57BL/6 mice, as described above. BD Fixation Buffer (BD Biosciences) containing paraformaldehyde was used to fix HBC, which were incubated for 30 min at 4°C in fixative and washed four times. B and T cells were mixed at 1:2, 1:1, and 2:1 ratios and cultured for 4 days on round-bottom 96-well plates. The cells were washed and immunostained with anti-CD4 antibody conjugated to Pacific Blue to measure CD4+ T cell CFSE dilution. CD80/86 mAb [LEAF-purified anti-mouse CD86 (GL-1) and CD80 (16-10A1); BioLegend] at 50 μg/mL were used to block allogeneic T cell costimulation by HBC.
CAR-T generation and cytotoxicity assay
C57BL/6J splenocytes were activated in anti-CD3-coated (10 μg/mL; eBioscience, San Diego, CA, USA), 750-mL flasks (BD Falcon; BD Biosciences) with 20 μg/mL anti-CD28 (eBioscience) and 100 μg/mL murine rIL-2 (R&D Systems, Minneapolis, MN, USA) in RPMI with L-glutamine (Corning: Cellgro), supplemented with 10% FBS (Sigma-Aldrich) and antibiotic/antimycotic (Corning: Cellgro). Retroviral supernatant, containing tandem molecules of hMN14 sFv-CD8α fused to a hybrid CD28/CD3ζ CAR, was used to transduce activated splenocytes, as described previously [27], to create second-generation anti-CEA CAR-T. Anti-CEA CAR-Ts were maintained in RPMI, supplemented, as above, with 100 μg/mL IL-2. MC38CEA-luc cells were irradiated (500 rad) before culture. CD11b+MC (CD11b+Gr-1+) were isolated by FACS. CAR-Ts, MC38CEA-luc, and CD11b+MC were cocultured at a 1:2:1 ratio for 4 h in optical 96-well plates. At the end of incubation, the plate was washed and 150 μg/mL luciferin added for 15 min at 37°C. Luminescence was measured using the IVIS 100 bioimaging system (Xenogen/PerkinElmer Life Sciences).
Statistical analysis
Data were analyzed and graphed using Microsoft Excel (Microsoft, Redmond, WA, USA) and Prism V5 (GraphPad Software, La Jolla, CA, USA) software. Student's t-test was calculated using Microsoft Excel software. P < 0.05 was considered statistically significant.
RESULTS
B cells delay but do not prevent progression of LM
To gain insight into the potential impact of HBC on LM progression, we compared tumor growth in WT and μMT mice. Tumor growth was enhanced in μMT compared with WT mice, which was most apparent at Days 4 and 8 following tumor injections (Fig. 1A and B). Sera of tumor-bearing mice were tested at 2 weeks following tumor administration for the presence of anti-CEA IgG to determine whether antibody responses against the tumor were generated. The majority of mice with tumors had detectable titers of anti-CEA IgG (Fig. 1C), confirming that tumor-specific antibodies are produced in mice with LM. However, the humoral response was not sufficiently protective against LM progression, as all mice with intact HBC ultimately died with large tumor burdens.
Figure 1. Tumor growth is accelerated in μMT mice.
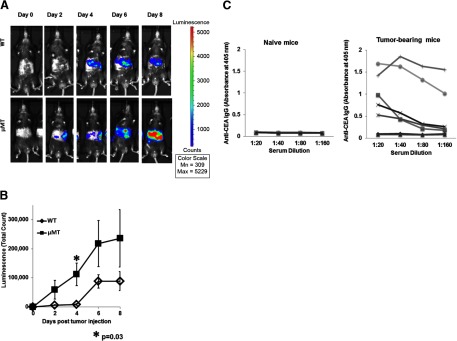
WT (n=5) and μMT (n=8) mice were injected via portal circulation with luciferase-positive MC38CEA cells and were imaged every 2 days after i.p. injection with luciferin. The images show one representative animal from each group and were taken with uniform settings and normalized to the same scale for comparison (A). The average luminescence for the two groups was determined using Living Image software (B). Error bars are ± sem of the group. P values are based on Student's t-test. Serum from normal (n=4) and tumor-bearing (n=7) mice was diluted and tested by ELISA for anti-CEA IgG (C). Antibody levels were determined by measuring optical density at 405 nm. Lines represent individual animals.
LM promote B cell maturation and increase B cell-proliferative capacity
Before assessing HBC immunologic function, we wished to determine whether LM induced HBC dysfunction at a broad level. The effect of LM on HBC phenotype was assessed in C57BL/6 mice injected with MC38CEA cells. Flow cytometric analysis revealed that in normal livers, B cells coexpressed CD19 and B220 (Fig. 2A) and comprised a significant proportion of CD45+ hepatic lymphocytes (54±3%) based on CD19 expression (Fig. 2B and C, left). Similar frequencies of HBC were obtained using B220 (CD45R) as a B cell marker (data not shown). The overall frequency of B cells in tumor-bearing livers was reduced twofold (P=0.005) compared with normal livers (Fig. 2C, left). However, the difference in the absolute number of B cells/liver was not statistically significant (Fig. 2C, right; P=0.1). Marked expansion of other intrahepatic cellular compartments, including Gr-1+CD11b+ cells (4.3±0.5% in normal liver vs. 17.6±1.1% in tumor-bearing liver; P=0.0005), occurred in response to LM (data not shown), which reduced the HBC frequency.
Figure 2. Maturation and proliferation of HBC are increased in tumor-bearing livers.

NPC were stained with CD19 and B220 to demonstrate that HBC are CD19/B220 double-positive. SSC, Side-scatter; FSC, forward-scatter (A). HBC cell frequency and absolute numbers (as a proportion of CD45+ liver leukocytes) in normal and tumor-bearing livers were determined using flow cytometry (B and C). The frequencies of transitional and mature B cells were determined in normal and tumor-bearing livers at 2 weeks after tumor cell administration (D) and at 7 and 17 days post-tumor cell administration (E). HBC loaded with 5 μM CFSE were stimulated ex vivo with LPS (10 μg/mL), CpG (25 μg/mL), and IgM/CD40 (15 μg/mL/10 μg/mL) for 4 days before proliferation was measured by flow cytometry (F and G). Representative average results of an experiment with three animals/group (Sham and tumor) are shown. Bar charts are averages of at least two experiments with three animals/group. Error bars are sem, and P values based on Student's t-test.
The impact of LM on B cell maturation was determined by measuring IgM and IgD expression on CD19+ cells. LM skewed HBC toward the mature phenotype, characterized by low IgM and high IgD expression (1.9-fold increase; P=0.04; Fig. 2D). HBC maturation required more than 1 week of LM growth (Fig. 2E).
The proliferative potential of HBC from normal and tumor-bearing livers was examined to evaluate the impact of LM on HBC function. B Cells from normal and tumor-bearing livers were loaded with CFSE and activated using several mitogenic stimuli. B cells from normal livers were generally refractory to TLR and BCR stimulation (Fig. 2F and G). In contrast, B cells from tumor-bearing livers divided vigorously in response to LPS, stimulatory CpG, and anti-IgM/anti-CD40 antibody cocktail, suggesting that LM do not induce global HBC dysfunction (Fig. 2F and G).
HBC from tumor-bearing livers show potent induction of T cell proliferation ex vivo despite CD80/CD86 down-regulation
Our examination of HBC immune function began with an assessment of costimulatory molecule expression. We observed a substantial down-regulation of the costimulatory CD80 and CD86 molecules (fivefold change, P=0.0005 for CD80; twofold change, P=0.04 for CD86) among HBC harvested from tumor-bearing animals compared with normal HBC (Fig. 3A and B). CD40 expression was high on HBC and was not affected by LM (data not shown). Further experiments focused on CD80, as CD86 expression is relatively low on HBC (11.7±2%).
Figure 3. CD80 and CD86 are down-regulated on HBC in the presence of LM.

The frequencies of CD80+ and CD86+ B cells (CD19+) were determined for normal and tumor-bearing livers (A and C). Results are representative of four experiments with three animals/group. CD80 expression was determined on mature (IgMloIgDhi) and immature (IgMhiIgDlo) HBC (B). Two and one-half million splenic B cells labeled with CFSE were adoptively transferred into normal and tumor-bearing livers via portal vein. CD80 expression was measured on B220+CFSE+ cells after 48 h (D). The results are representative of two experiments that used two to four animals/group.
As the expanded HBC population was largely mature (65%) in phenotype (Fig. 2D), we examined CD80 expression on HBC subsets. CD80 expression was reduced significantly on mature (IgMloIgDhi) HBC in tumor-bearing livers compared with normal livers (Fig. 3C; P=0.01), whereas immature (IgMhiIgDlo) HBC expressed low CD80 and were not affected by LM. To confirm LM-induced HBC CD80 loss in vivo, CFSE-labeled splenic B cells were adoptively transferred into normal and tumor-bearing livers via portal vein injection and analyzed 48 h post-transfer. Normal and tumor-bearing livers contained similar absolute numbers of CFSE+B220+ adoptively transferred cells. However, a 2.3-fold down-regulation of CD80 expression (P=0.04) was seen in adoptively transferred splenic B cells in tumor-bearing livers compared with normal livers (Fig. 3D).
CD80/86 costimulation is well established as a necessary component of T cell activation by APCs, including B cells [28, 29]. Based on CD80/86 down-regulation, seen on B cells from tumor-bearing livers, we hypothesized that LM would render HBC poor stimulators of T cell proliferation. Indeed, the blocking of CD80/86 with mAb abolished allogeneic T cell proliferation when CFSE-loaded T cells from BALB/c were cocultured with HBC from C57BL/6 mice with established LM (Fig. 4A). It was, therefore, surprising that B cells from tumor-bearing livers were not impaired in their ability to induce proliferation of CD4+ allogeneic T cells ex vivo (Fig. 4B). T cell proliferation rates were similar at 1:2, 1:1, and 2:1 B:T cell ratios using B cells from normal and tumor-bearing livers (Fig. 4C). As such, we sought to determine whether the LM-induced CD80 down-regulation was reversed ex vivo.
Figure 4. HBC CD80 down-regulation recovers spontaneously in culture but is prevented by CD19− NPC.

CFSE-loaded T cells derived from the spleen of a BALB/c mouse were cocultured with HBC from normal C57BL/6 livers in the presence of blocking CD80/CD86 antibodies (both at 50 μg/mL) for 4 days (A), and CFSE dilution, indicative of T cell proliferation, was determined using flow cytometry. HBC from normal and tumor-bearing livers were cocultured with allogeneic, CFSE-loaded T cells for 4 days, and T cell proliferation was determined based on CFSE dilution (B). (C) Division of T cells at various B:T cell ratios (1:2, 1:1, and 2:1) are shown. Results are representative of two experiments with three mice/group. CD80 expression levels on HBC isolated from tumor-bearing livers cultured ex vivo were determined at 24 and 48 h of culture (D, upper). In addition, CD80 expression was determined on HBC from tumor-bearing livers cultured ex vivo in the presence of the CD19− liver leukocyte fraction (D, lower). Allogeneic T cell proliferation in the presence of HBC was measured using HBC from normal and tumor-bearing livers live or fixed with a paraformaldehyde fixative to maintain CD80 and CD86 expression (E). The bar charts show a representative experiment of two experiments with averaged three normal and three tumor-bearing mice. Error bars are sem for the group, and P values are based on Student's t-test.
Fixed HBC from tumor-bearing livers are less-effective stimulators of allogeneic T cell proliferation than normal HBC
We hypothesized that CD80 down-regulation on HBC is reversible and dependent on interaction with other cells within the tumor microenvironment. HBC were cultured alone or with CD19− NPC from tumor-bearing livers. When cultured alone, HBC CD80 expression recovered spontaneously in culture (8.4-fold increase; Fig. 4D, upper). Interestingly, coculture of B cells from tumor-bearing livers with the CD19− fraction from the same liver significantly diminished HBC CD80 recovery (Fig. 4D, lower), suggesting that the non-B cell fraction contained cells that prevented CD80 re-expression. B Cells derived from normal livers had significant levels of CD80 on Day 0 and remained high, with culture ex vivo inducing little change in CD80 expression (data not shown).
To investigate the function of B cells from tumor-bearing livers in their native state, HBC were fixed using paraformaldehyde before coculture with allogeneic T cells to prevent CD80 re-expression. B Cells from healthy livers were treated in the same fashion to demonstrate the ability of normal fixed B cells to induce T cell proliferation. Normal live and fixed B cells induced similar rates of T cell proliferation, which were not statistically different from those induced by the live B cells from tumor-bearing livers (Fig. 4E). In contrast, fixed B cells from tumor-bearing livers were impaired in inducing T cell proliferation compared with fixed B cells from healthy livers or live B cells from tumor-bearing livers (0.4-fold decrease, P=0.018; 0.6-fold decrease, P=0.02, respectively; Fig. 4B). The impairment of T cell activation by HBC from tumor-bearing livers was confirmed in an antigen-specific assay. HBC from tumor-bearing livers loaded with ovalbumin 329–337 induced significantly less OT-II splenic T cell proliferation compared with HBC from normal livers (data not shown).
CD11b+ myeloid cells mediate CD80 down-regulation on HBC ex vivo
We next sought to define which component of the CD19− cell population was responsible for suppressing CD80 expression on HBC. To exclude a tumor-derived soluble factor, we exposed HBC to medium from MC38CEA cell cultures and did not detect a down-regulation of CD80 (data not shown). The ability of CD19− cells from tumor-bearing livers to inhibit expression of CD80 by HBC (Fig. 4D, bottom) prompted us to investigate whether CD11b+MC that expand in tumor-bearing livers were responsible for this phenomenon. The demonstration of immunosuppressive phenotype and function heightened our interest in CD11B+ MC. The frequency of CD11b+MC increased from 4 ± 0.5% in normal livers to 18 ± 1% in tumor-bearing livers (P=0.0005; Supplemental Fig. 1A). CD11b+ cells (82±2% Gr-1+CD11b+; Supplemental Fig. 1B) were isolated from tumor-bearing livers, 2 weeks after MC38CEA cell injection to allow sufficient CD11b+MC expansion. CD11b+MC expressed significant levels of PD-L1 (65±7%; Supplemental Fig. 1C), and suppressed anti-CEA CAR-T killing of MC38CEA-luc cells (Fig. 5A). Having confirmed the suppressive nature of CD11b+ MC, we cocultured these cells with HBC from tumor-bearing livers, and CD80 expression was determined. Whereas CD80 expression was recovered on HBC cultured alone, the re-expression was inhibited in the presence of CD11b+MC (P=0.03 at 24 h, and P=0.03 at 48 h; Fig. 5B). In contrast, coculture with T cells (Thy1.2+ cells), which included Tregs, had no significant effect on CD80 re-expression.
Figure 5. CD80 expression on HBC is inhibited by CD11b+MC.

CD11b+MC, anti-CEA CAR-T and irradiated MC38CEA-luc cells were cocultured for 4 h. Cytotoxicity was measured by determining the loss of luminescence among viable MC38CEA-luc cells when 150 μg/mL luciferin was added to wells (A). HBC from normal (B) and tumor-bearing (C) livers were cocultured with CD11b+MC and T cells derived from tumor-bearing livers. CD80 levels were determined using flow cytometry before culture and at 24 and 48 h of culture. Graph bars are averages of three animals/group and are representative of three experiments. To determine the requirement for cell contact between HBC and CD11b+MC for CD80 down-regulation, HBC were cultured alone, with CD11b+MC derived from tumor-bearing livers or with CD11b+MC contained in Transwells (D and E). CD11b-mediated cell adhesion was blocked with 5 μg/well anti-CD11b mAb for 48 h in CD11b+MC/HBC coculture (F). Error bars are sem, and P values are based on Student's t-test.
The effect of CD11b+MC on HBC derived from normal livers was also investigated. HBC from normal livers were cocultured with CD11b+MC from tumor-bearing livers, and CD80 levels were measured at 24 and 48 h. CD80 expression levels were reduced on HBC derived from normal livers and cultured in the presence of CD11b+MC from tumor-bearing livers (P=0.02 at 24 h, and P=0.01 at 48 h; Fig. 5C). On the other hand, T cells had no effect on CD80 expression. CD11b+MC had no effect of HBC IgG production in vitro (data not shown).
CD11b+MC mediate CD80 down-regulation of HBC via direct contact and STAT3
CD11b+MC exert their immunosuppressive effects through a variety of secreted molecules, as well as membrane-bound proteins dependent on direct cellular contact. HBC, cultured with CD11b+MC, down-regulated CD80, whereas CD11b+MC, prevented from contacting HBC by Transwell inserts, did not affect HBC CD80 levels (P=0.003; Fig. 5D and E). CD11b+MC/HBC physical interactions may be mediated by adhesion molecules that bring the two cell types into close proximity. CD11b is an integrin highly expressed by CD11b+MC and may bind to ICAM-1 on B cells [30]. CD11b blockade with a mAb in CD11b+MC/HBC coculture assays partially restored CD80 expression on HBC compared with HBC/CD11b+MC without the antibody (twofold increase, P=0.006; Fig. 5F).
We also studied several compounds known to block immunosuppressive factors produced by MDSC. Inhibitors of reactive nitrogen and ROS, arginase, IDO, and PD-L1 were used to identify which CD11b+MC factors might mediate CD80 down-regulation on HBC. JSI-124, a plant-derived STAT3/ROS inhibitor, completely restored CD80 expression (Fig. 6A), suggesting that STAT3 signaling in CD11b+MC plays a role in CD80 down-regulation on HBC. JSI-124 had no effect on CD80 expression on HBC cultured alone (data not shown). In addition to inhibiting STAT3 phosphorylation, JSI-124 possesses a number of other biological activities, including cytokine transcription activation, ROS inhibition, and MDSC maturation induction [31–33]. To interrogate more precisely the STAT3 pathway in CD11b+MC, a highly selective STAT3 inhibitor, Stattic, was used in HBC/CD11b+MC cocultures. CD11b+MC pretreated for 1 h at 37°C with 100 μM Stattic were unable to affect CD80 down-regulation on HBC (Fig. 6B). In contrast, similarly pretreated HBC were fully susceptible to CD80 down-regulation mediated by CD11b+MC.
Figure 6. CD11b+MC mediate CD80 down-regulation on HBC in a STAT3-dependent manner.

HBC and CD11b+MC were cultured in the presence of several modulators of CD11b+MC-suppressive function [JSI-124, L-NMMA, nor-NOHA, anti-PD-L1 (aPD-L1), and 1-MT]. HBC CD80 expression levels were determined after 48 h of culture (A). The results are representative of at least two experiments, with bar charts showing the averages of three separate animals. CD11b+MC or HBC were pretreated with Stattic for 1 h at 37°C before being cocultured for 48 h (B). CD80 expression on HBC was determined and compared between groups.
DISCUSSION
The role of HBC in the progression of LM is poorly characterized despite a substantial presence of B cells in the liver. Despite LM promoting HBC maturation and increased HBC-proliferative potential, liver CD11b+MC induced loss of HBC costimulatory molecule expression leading to impaired T cell-stimulatory capacity ex vivo. The potent effects of liver CD11b+MC on HBC immune function were demonstrated by the similar levels of CD80 loss among normal HBC in vitro and splenic B cells in vivo, in addition to HBC from tumor-bearing mice. HBC loss of CD80 was dependent, in part, on CD11b+MC STAT3 activity and CD11b expression and was reversible upon removal from the suppressive intrahepatic milieu. Our data support the hypothesis that HBC immune dysfunction induced by the tumor microenvironment may limit the ability of HBC to activate T cells in vivo.
B Cells appear to promote tumor progression in some models [13, 14, 34, 35], but play a protective role in others [16, 17, 36]. We observed increased tumor growth in animals that lacked tumor-specific, IgG-producing B cells (Fig. 1A and B), which suggested that HBC may have a protective role in early LM progression. IL-10 expression was undetectable in freshly isolated HBC or in cultured HBC stimulated with LPS (data not shown). The absence of IL-10 production by HBC, shown by us as well as others [37], suggests that HBC in this model may not be regulatory B cells, as classically described [38]. Despite a reduced tumor burden in WT animals compared with μMT mice, all animals in both groups eventually succumbed to progressive LM. This suggested that although HBC may afford a modest degree of protection against tumor progression, the anti-tumor activity of HBC is far from sufficient to protect the host. These findings prompted us to investigate how LM impaired the ability of HBC to mediate effective anti-tumor immunity.
In seeking mechanisms through which the tumor microenvironment may limit HBC immune function, we found that HBC CD80 levels are markedly diminished in response to LM. The importance of CD80 expression on APCs (including B cells) in stimulating T cell activation is well established [28, 29, 39, 40]. Our finding that HBC loss of CD80 led to impaired T cell activation is consistent with prior reports [41–43]. Down-regulation of CD80 (and CD86) suggested that B cells in tumor-bearing livers may be compromised in their ability to function as APCs. Importantly, HBC spontaneously recovered CD80 expression in culture. The reversible nature of HBC dysfunction warrants further inquiry as to whether CD80 re-expression can be induced pharmacologically through prevention of specific suppressive mechanisms exploited by CD11b+MC.
The important role of CD11b+MC in mediating HBC immune dysfunction in our model is consistent with the broad array of immunosuppressive functions attributed to cells with this phenotype, including MDSC [5, 6, 44, 45]. The demonstration of in vivo CD80 loss among adoptively transferred splenic B cells, in addition to HBC isolated from normal and tumor-bearing hosts, further substantiated our novel finding that CD11b+MC potently inhibit B cell CD80. As such, liver CD11b+MC are able to inhibit the expression of costimulatory molecules on B cells that have not been exposed previously to the suppressive intrahepatic space or tumor microenvironment. The immunosuppressive effect of CD11b+MC, resulting in CD80 down-regulation, conforms well with the overall impact of MDSC on immune cells [46, 47]. Our data offer a novel mechanism through which CD11b+MC may impair antigen presentation in the setting of cancer [48–50].
By delving into the mechanisms of CD11b+MC suppression of HBC CD80 expression, we identified potential targets for reversing this effect. We established the importance of direct contact between HBC and CD11b+MC and implicated CD11b and STAT3 as mediators of HBC CD80 loss. We speculate that CD11b on CD11b+MC interacts with ICAM-1 (CD54) expressed by B cells [30, 51, 52]. Interestingly, CD11b blockade has been shown to reverse MDSC-mediated suppression of T cells [25], although it is not clear whether CD11b+MC use similar mechanisms for T and B cell suppression. As anti-CD11b treatment did not fully restore CD80 expression on HBC, it is likely that STAT3 activity cooperates in mediating HBC CD80 suppression. The ability of JSI-124 to reverse MDSC-mediated immunosuppression has been demonstrated in T cells, and we, therefore, suspected STAT3 may play a role in HBC CD80 loss [25]. We have also found that JSI-124 treatment leads to CD11b down-regulation on CD11b+MC (unpublished observation), which would abrogate some of the CD11b+MC/HBC interactions. JSI-124 appeared to have no direct effect on CD80 expression on HBC alone. JSI-124 has shown anti-tumor activity against cancer cells in vitro and was effective in suppressing tumor progression in mice [31, 32]. As JSI-124 has diverse biological effects, we specifically blocked STAT3 phosphorylation in CD11b+MC using Stattic to confirm that CD80 down-regulation on HBC relied on STAT3 signaling in CD11b+MC. Further inquiry is needed to define the precise roles of STAT3 signaling in promoting HBC CD80 down-regulation. It also remains to be demonstrated whether CD80 down-regulation can be reversed in vivo through STAT3 inhibition in our model.
An important limitation of our study is that in our examination of in vivo tumor progression, we were unable to use mice that were specifically deficient in HBC but had normal numbers of B cells in other organs. Although B cells can be depleted using antibodies, such as anti-IgM, this treatment results in a global B cell loss [14, 53]. Thus, we were unable to distinguish the specific effect of HBC on tumor progression from the overall impact of all B cells. It is possible that peripheral B cells that are present in blood and secondary lymphoid organs have anti-tumor properties, and this is the reason why μMT animals show accelerated tumor progression compared with WT mice. Moreover, other B cell surface molecules may be affected by the LM microenvironment. In addition to CD80 down-regulation, MHCII expression was reduced on HBC in the setting of LM, which may further curtail the ability of HBC to stimulate T cell activation in an antigen-specific manner (unpublished observation). We are currently investigating the mechanisms underlying MHCII down-regulation on HBC in our model. Finally, we did not explore the effects of LM progression on CD80 expression by other APC subsets, which may impact intrahepatic anti-tumor immunity.
The data described in this report may have direct relevance to understanding the role of HBC in the progression of LM. Impaired HBC CD80 expression through cross-talk with CD11b+MC may impair the immune response to LM. The reversible nature of HBC CD80 loss and identification of the roles of CD11b and STAT3 suggest therapeutic opportunities. It remains to be explored whether blocking CD11b+MC interactions with HBC or CD11b+MC signaling pathways to restore HBC CD80 expression will confer a clinical benefit in the setting of LM.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health Grant 1K08CA160662-01A1.
We thank Dr. Linda A. Spatz (Sophie Davis School of Biomedical Education, The City College of New York, The City University of New York, NY, USA) for critical review of the manuscript.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- 1-MT
- 1-methyl-L-tryptophan
- AP
- alkaline phosphatase
- APC
- antigen presenting cells
- CAR
- chimeric antigen receptor
- CAR-T
- chimeric antigen receptor-enhanced T cell
- CD11b+MC
- Gr-1+CD11b+ myeloid cell(s)
- CEA
- carcinoembryonic antigen
- HBC
- hepatic B cell(s)
- L-NMMA
- NG-monomethyl-L-arginine, monoacetate salt
- LM
- liver metastases
- MC38CEA-luc
- MC38 carcinoembryonic antigen luciferase
- MDSC
- myeloid-derived suppressor cell(s)
- μMT
- B cell-deficient
- nor-NOHA
- N-hydroxy-nor-L-arginine
- NPC
- nonparenchymal cell(s)
- PD-L1
- programmed cell death 1 ligand
- ROS
- reactive oxygen species
- Tregs
- regulatory T cell
- WT
- wild-type
AUTHORSHIP
M.T. designed the research, performed the experiments, analyzed data, and wrote the manuscript. G.R.P., R.A.B., and C.T.N. performed the experiments. N.J.E. wrote the manuscript. S.C.K. designed the research, analyzed data, and wrote the manuscript.
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1. Burghardt S., Erhardt A., Claass B., Huber S., Adler G., Jacobs T., Chalaris A., Schmidt-Arras D., Rose-John S., Karimi K., Tiegs G. (2013) Hepatocytes contribute to immune regulation in the liver by activation of the notch signaling pathway in T cells. J. Immunol. 191, 5574–5582. [DOI] [PubMed] [Google Scholar]
- 2. Connolly M. K., Bedrosian A. S., Malhotra A., Henning J. R., Ibrahim J., Vera V., Cieza-Rubio N. E., Hassan B. U., Pachter H. L., Cohen S., Frey A. B., Miller G. (2010) In hepatic fibrosis, liver sinusoidal endothelial cells acquire enhanced immunogenicity. J. Immunol. 185, 2200–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Katz S. C., Pillarisetty V. G., Bleier J. I., Shah A. B., DeMatteo R. P. (2004) Liver sinusoidal endothelial cells are insufficient to activate T cells. J. Immunol. 173, 230–235. [DOI] [PubMed] [Google Scholar]
- 4. Valzasina B., Piconese S., Guiducci C., Colombo M. P. (2006) Tumor-induced expansion of regulatory T cells by conversion of CD4+CD25− lymphocytes is thymus and proliferation independent. Cancer Res. 66, 4488–4495. [DOI] [PubMed] [Google Scholar]
- 5. Huang B., Pan P. Y., Li Q., Sato A. I., Levy D. E., Bromberg J., Divino C. M., Chen S. H. (2006) Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 66, 1123–1131. [DOI] [PubMed] [Google Scholar]
- 6. Connolly M. K., Mallen-St Clair J., Bedrosian A. S., Malhotra A., Vera V., Ibrahim J., Henning J., Pachter H. L., Bar-Sagi D., Frey A. B., Miller G. (2010) Distinct populations of metastases-enabling myeloid cells expand in the liver of mice harboring invasive and preinvasive intra-abdominal tumor. J. Leukoc. Biol. 87, 713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Palma M. (2012) Partners in crime: VEGF and IL-4 conscript tumour-promoting macrophages. J. Pathol. 227, 4–7. [DOI] [PubMed] [Google Scholar]
- 8. Rosenberg S. A., Yang J. C., Restifo N. P. (2004) Cancer immunotherapy: moving beyond current vaccines. Nat. Med. 10, 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Visser K. E., Schumacher T. N., Kruisbeek A. M. (2003) CD8+ T cell tolerance and cancer immunotherapy. J. Immunother. 26, 1–11. [DOI] [PubMed] [Google Scholar]
- 10. Siegel C. T., Schreiber K., Meredith S. C., Beck-Engeser G. B., Lancki D. W., Lazarski C. A., Fu Y. X., Rowley D. A., Schreiber H. (2000) Enhanced growth of primary tumors in cancer-prone mice after immunization against the mutant region of an inherited oncoprotein. J. Exp. Med. 191, 1945–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Novobrantseva T. I., Majeau G. R., Amatucci A., Kogan S., Brenner I., Casola S., Shlomchik M. J., Koteliansky V., Hochman P. S., Ibraghimov A. (2005) Attenuated liver fibrosis in the absence of B cells. J. Clin. Invest. 115, 3072–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moore J. W., Beattie L., Dalton J. E., Owens B. M., Maroof A., Coles M. C., Kaye P. M. (2012) B cell: T cell interactions occur within hepatic granulomas during experimental visceral leishmaniasis. PLoS One 7, e34143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shah S., Divekar A. A., Hilchey S. P., Cho H. M., Newman C. L., Shin S. U., Nechustan H., Challita-Eid P. M., Segal B. M., Yi K. H., Rosenblatt J. D. (2005) Increased rejection of primary tumors in mice lacking B cells: inhibition of anti-tumor CTL and TH1 cytokine responses by B cells. Int. J. Cancer 117, 574–586. [DOI] [PubMed] [Google Scholar]
- 14. Barbera-Guillem E., Nelson M. B., Barr B., Nyhus J. K., May K. F., Jr., Feng L., Sampsel J. W. (2000) B lymphocyte pathology in human colorectal cancer experimental and clinical therapeutic effects of partial B cell depletion. Cancer Immunol. Immunother. 48, 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qin Z., Richter G., Schuler T., Ibe S., Cao X., Blankenstein T. (1998) B Cells inhibit induction of T cell-dependent tumor immunity. Nat. Med. 4, 627–630. [DOI] [PubMed] [Google Scholar]
- 16. Schultz K. R., Klarnet J. P., Gieni R. S., HayGlass K. T., Greenberg P. D. (1990) The role of B cells for in vivo T cell responses to a Friend virus-induced leukemia. Science 249, 921–923. [DOI] [PubMed] [Google Scholar]
- 17. DiLillo D. J., Yanaba K., Tedder T. F. (2010) B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J. Immunol. 184, 4006–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rodriguez-Pinto D. (2005) B Cells as antigen presenting cells. Cell. Immunol. 238, 67–75. [DOI] [PubMed] [Google Scholar]
- 19. Watt V., Ronchese F., Ritchie D. (2007) Resting B cells suppress tumor immunity via an MHC class-II dependent mechanism. J. Immunother. 30, 323–332. [DOI] [PubMed] [Google Scholar]
- 20. Schmidt M., Bohm D., von Torne C., Steiner E., Puhl A., Pilch H., Lehr H. A., Hengstler J. G., Kolbl H., Gehrmann M. (2008) The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 68, 5405–5413. [DOI] [PubMed] [Google Scholar]
- 21. Milne K., Kobel M., Kalloger S. E., Barnes R. O., Gao D., Gilks C. B., Watson P. H., Nelson B. H. (2009) Systematic analysis of immune infiltrates in high-grade serous ovarian cancer reveals CD20, FoxP3 and TIA-1 as positive prognostic factors. PLoS One 4, e6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katz S. C., Pillarisetty V. G., Bleier J. I., Kingham T. P., Chaudhry U. I., Shah A. B., DeMatteo R. P. (2005) Conventional liver CD4 T cells are functionally distinct and suppressed by environmental factors. Hepatology 42, 293–300. [DOI] [PubMed] [Google Scholar]
- 23. Ma C., Kapanadze T., Gamrekelashvili J., Manns M. P., Korangy F., Greten T. F. (2012) Anti-Gr-1 antibody depletion fails to eliminate hepatic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol. 92, 1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Danna E. A., Sinha P., Gilbert M., Clements V. K., Pulaski B. A., Ostrand-Rosenberg S. (2004) Surgical removal of primary tumor reverses tumor-induced immunosuppression despite the presence of metastatic disease. Cancer Res. 64, 2205–2211. [DOI] [PubMed] [Google Scholar]
- 25. Kusmartsev S., Nefedova Y., Yoder D., Gabrilovich D. I. (2004) Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 172, 989–999. [DOI] [PubMed] [Google Scholar]
- 26. Green K. A., Cook W. J., Green W. R. (2013) Myeloid-derived suppressor cells in murine retrovirus-induced AIDS inhibit T- and B-cell responses in vitro that are used to define the immunodeficiency. J. Virol. 87, 2058–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emtage P. C., Lo A. S., Gomes E. M., Liu D. L., Gonzalo-Daganzo R. M., Junghans R. P. (2008) Second-generation anti-carcinoembryonic antigen designer T cells resist activation-induced cell death, proliferate on tumor contact, secrete cytokines, and exhibit superior antitumor activity in vivo: a preclinical evaluation. Clin. Cancer Res. 14, 8112–8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jenkins M. K., Taylor P. S., Norton S. D., Urdahl K. B. (1991) CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J. Immunol. 147, 2461–2466. [PubMed] [Google Scholar]
- 29. Kosco-Vilbois M. H., Gray D., Scheidegger D., Julius M. (1993) Follicular dendritic cells help resting B cells to become effective antigen-presenting cells: induction of B7/BB1 and upregulation of major histocompatibility complex class II molecules. J. Exp. Med. 178, 2055–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boyd A. W., Wawryk S. O., Burns G. F., Fecondo J. V. (1988) Intercellular adhesion molecule 1 (ICAM-1) has a central role in cell-cell contact-mediated immune mechanisms. Proc. Natl. Acad. Sci. USA 85, 3095–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McFarland B. C., Gray G. K., Nozell S. E., Hong S. W., Benveniste E. N. (2013) Activation of the NF-kappaB pathway by the STAT3 inhibitor JSI-124 in human glioblastoma cells. Mol. Cancer Res. 11, 494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Corzo C. A., Cotter M. J., Cheng P., Cheng F., Kusmartsev S., Sotomayor E., Padhya T., McCaffrey T. V., McCaffrey J. C., Gabrilovich D. I. (2009) Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 182, 5693–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nefedova Y., Nagaraj S., Rosenbauer A., Muro-Cacho C., Sebti S. M., Gabrilovich D. I. (2005) Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the Janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 65, 9525–9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Houghton A. N., Uchi H., Wolchok J. D. (2005) The role of the immune system in early epithelial carcinogenesis: B-ware the double-edged sword. Cancer Cell 7, 403–405. [DOI] [PubMed] [Google Scholar]
- 35. Schreiber H., Wu T. H., Nachman J., Rowley D. A. (2000) Immunological enhancement of primary tumor development and its prevention. Semin. Cancer Biol. 10, 351–357. [DOI] [PubMed] [Google Scholar]
- 36. Brodt P., Gordon J. (1978) Anti-tumor immunity in B lymphocyte-deprived mice. I. Immunity to a chemically induced tumor. J. Immunol. 121, 359–362. [PubMed] [Google Scholar]
- 37. Zhang H., Stolz D. B., Chalasani G., Thomson A. W. (2013) Hepatic B cells are readily activated by Toll-like receptor-4 ligation and secrete less interleukin-10 than lymphoid tissue B cells. Clin. Exp. Immunol. 173, 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. DiLillo D. J., Matsushita T., Tedder T. F. (2010) B10 Cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer Ann. N. Y. Acad. Sci. 1183, 38–57. [DOI] [PubMed] [Google Scholar]
- 39. Damle N. K., Klussman K., Linsley P. S., Aruffo A., Ledbetter J. A. (1992) Differential regulatory effects of intercellular adhesion molecule-1 on costimulation by the CD28 counter-receptor B7. J. Immunol. 149, 2541–2548. [PubMed] [Google Scholar]
- 40. Jomantaite I., Dikopoulos N., Kroger A., Leithauser F., Hauser H., Schirmbeck R., Reimann J. (2004) Hepatic dendritic cell subsets in the mouse. Eur. J. Immunol. 34, 355–365. [DOI] [PubMed] [Google Scholar]
- 41. Constant S., Schweitzer N., West J., Ranney P., Bottomly K. (1995) B Lymphocytes can be competent antigen-presenting cells for priming CD4+ T cells to protein antigens in vivo. J. Immunol. 155, 3734–3741. [PubMed] [Google Scholar]
- 42. O'Neill S. K., Cao Y., Hamel K. M., Doodes P. D., Hutas G., Finnegan A. (2007) Expression of CD80/86 on B cells is essential for autoreactive T cell activation and the development of arthritis. J. Immunol. 179, 5109–5116. [DOI] [PubMed] [Google Scholar]
- 43. Lim T. S., Goh J. K., Mortellaro A., Lim C. T., Hammerling G. J., Ricciardi-Castagnoli P. (2012) CD80 and CD86 differentially regulate mechanical interactions of T-cells with antigen-presenting dendritic cells and B-cells. PLoS One 7, e45185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yu J., Du W., Yan F., Wang Y., Li H., Cao S., Yu W., Shen C., Liu J., Ren X. (2013) Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 190, 3783–3797. [DOI] [PubMed] [Google Scholar]
- 45. Toh B., Wang X., Keeble J., Sim W. J., Khoo K., Wong W. C., Kato M., Prevost-Blondel A., Thiery J. P., Abastado J. P. (2011) Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 9, e1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Serafini P., Mgebroff S., Noonan K., Borrello I. (2008) Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 68, 5439–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen S., Akbar S. M., Abe M., Hiasa Y., Onji M. (2011) Immunosuppressive functions of hepatic myeloid-derived suppressor cells of normal mice and in a murine model of chronic hepatitis B virus. Clin. Exp. Immunol. 166, 134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gabrilovich D. I., Corak J., Ciernik I. F., Kavanaugh D., Carbone D. P. (1997) Decreased antigen presentation by dendritic cells in patients with breast cancer. Clin. Cancer Res. 3, 483–490. [PubMed] [Google Scholar]
- 49. Ostrand-Rosenberg S., Sinha P., Beury D. W., Clements V. K. (2012) Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 22, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Malmberg K. J. (2004) Effective immunotherapy against cancer: a question of overcoming immune suppression and immune escape? Cancer Immunol. Immunother. 53, 879–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang X., Moore D. J., Mohiuddin M., Lian M. M., Kim J. I., Sonawane S., Wang J., Gu Y., Yeh H., Markmann J. F., Deng S. (2008) Inhibition of ICAM-1/LFA-1 interactions prevents B-cell-dependent anti-CD45RB-induced transplantation tolerance. Transplantation 85, 675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maltzman J. S., Carmen J. A., Monroe J. G. (1996) Transcriptional regulation of the ICAM-1 gene in antigen receptor- and phorbol ester-stimulated B lymphocytes: role for transcription factor EGR1. J. Exp. Med. 183, 1747–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cerny A., Kimoto M., Hugin A. W., Merino R., Izui S. (1989) Anti-IgM treatment of C57BL/6-1pr/1pr mice: depletion of B cells reduces 1pr gene-induced lymphoproliferation and mononuclear cell vasculitis. Clin. Exp. Immunol. 77, 124–129. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.