

LL-37 upregulates neutrophil respiratory burst and extracellular trap formation in response to influenza A virus, but reduces cytokine generation triggered by the virus.
Keywords: innate immunity, antimicrobial peptides, FPRL-1, defensin
Abstract
Recent studies have shown that the human cathelicidin, LL-37, has antiviral activity against IAV in vitro and in vivo. Neutrophils are important cellular components of the initial innate response to IAV infection. In addition to its direct antimicrobial activities, LL-37 has important immunomodulatory effects. In this study, we explore how LL-37 affects interactions of IAV with human neutrophils. LL-37 did not alter neutrophil uptake of IAV but significantly increased neutrophil H2O2 responses to the virus. IAV stimulated production of NETs in vitro, and this response was increased by preincubating the virus with LL-37. NADPH-oxidase blockade did not reduce IAV-induced NET formation or the increased NET response stimulated by LL-37 + IAV. The increased respiratory burst and NET responses were, however, inhibited by preincubating cells with a formyl peptide receptor blocker, indicating that LL-37 engages these receptors when complexed with IAV. Responses to IAV alone were not inhibited by formyl peptide receptor blockade. It has been reported that LL-37 reduces proinflammatory cytokine responses during IAV infection in vivo. We now show that IAV alone potentiated release of IL-8 from neutrophils, and preincubation with LL-37 reduced IAV-stimulated IL-8 release. These results confirm that LL-37 modulates human neutrophil responses to IAV in a distinctive manner and could have important bearing on the protective effects of LL-37 during IAV infection in vivo.
Introduction
LL-37 is a member of a large family of cationic antimicrobial peptides called cathelicidins expressed in many species and has broad-spectrum antimicrobial activity [1]. The cathelicidins contain a signal peptide, a cathelin-like domain, and antimicrobial domain. The antimicrobial domain is released by cleavage by proteases. There is only one human cathelicidin, and its antimicrobial domain is called LL-37. Like the defensins, LL-37 is produced by neutrophils and macrophages. LL-37 is distinguished from HNPs in that it is not only produced by phagocytes but also by epithelial cells in response to various activating stimuli. β-Defensins are another group of antimicrobial peptides produced by epithelial cells. LL-37 and β-defensins serve as part of an initial barrier against infection, play a role in repair of damaged epithelium, and also act as “alarmins” to trigger recruitment of inflammatory cells [2]. LL-37 expression is regulated by distinct mechanisms from the β-defensins, and it has distinct receptors on immune cells from those of the defensins.
Most studies of LL-37 have focused on its antibacterial activity; however, more recently, antiviral activities of LL-37 have been described as well. For instance, LL-37 inhibits HIV in vitro and improves outcome of IAV infection in mice through inhibition of viral replication and reduction of virus-induced proinflammatory cytokine generation [3]. Up-regulation of LL-37 expression by stimulation with leukotriene B4 correlated with improved outcome of IAV infection in mice [4]. We have partially characterized the mechanism of anti-IAV activity of LL-37 [5]. LL-37 does not block hemagglutination activity, cause viral aggregation, or reduce viral uptake by epithelial cells; rather, it inhibits viral replication at a postentry step before viral RNA or protein synthesis in the cell. There are two likely sources of LL-37 in the IAV-infected respiratory tract. Neutrophils are an abundant source of LL-37, and they are the predominant recruited inflammatory cell during the first phase of IAV infection [6]. LL-37 is released from specific granules of neutrophils upon cell activation. Respiratory epithelial cells also produce LL-37 in response to inflammatory stimuli. LL-37 expression by the respiratory epithelium is also up-regulated by active metabolites of vitamin D. There is some evidence that the propensity for epidemic influenza to occur in winter months is related, in part, to lower levels of vitamin D in the population [7]. Alveolar macrophages also play a key role in the innate response to IAV infection [6], and these cells are another important source of LL-37.
Most research on the antimicrobial activity of LL-37 has focused on binding and killing of various pathogens; however, LL-37 also has strong immunomodulatory effects that could be important in vivo. LL-37 stimulates chemotaxis of neutrophils, monocytes, and T cells [8]. There is evidence for several specific receptors for LL-37 on these cells, including FPRL-1 on neutrophils, monocytes, and lymphocytes [9]; GAPDH in the cytoplasm of monocytes [10]; and CXCR2 on neutrophils [11]. LL-37 has complex effects on monocyte and macrophage activation. LL-37 can promote inflammatory responses of macrophages in concert with IL-1β [12] and can direct macrophage differentiation in a proinflammatory direction [13]; however, LL-37 also inhibits some macrophage responses [14] and binds to and inhibits monocyte responses to LPS [15]. Finally, LL-37 can activate respiratory epithelial cells by interacting with epidermal growth factor receptors [16].
In this paper, we examine how LL-37 modulates the interactions of IAV with human neutrophils. There is extensive evidence that neutrophils play an important role in host defense against IAV. As noted, neutrophils are the most abundant cell recruited to the airway in the first phase of IAV infection. Neutrophils have been found to play a protective role during IAV infection, although results have varied depending on the model used [17–20]. Neutrophils also appear to modulate the subsequent adaptive immune response [21, 22]. We demonstrate that LL-37 substantially modulates the response of human neutrophils to IAV.
MATERIALS AND METHODS
Virus preparations
The Phil82 strain was kindly provided by Dr. E. Margot Anders (University of Melbourne, Australia) and grown in the chorioallantoic fluid of 10-day-old chicken eggs and purified on a discontinuous sucrose gradient, as described previously [23]. The virus was dialyzed against PBS to remove sucrose, aliquoted, and stored at −80°C until needed. Post-thawing, the viral stocks contained ∼5 × 108 infectious focus-forming units/ml.
LL-37 and other reagents
LL-37 was purchased from Phoenix Pharmaceuticals (Burlingame, CA, USA). sLL-37 was purchased from Abgent (San Diego, CA, USA).HNP-2 was purchased from Bachem (Torrance, CA, USA). The FPRL-1 agonist peptide WKYMVM and WRW4-blocking peptide were purchased from Phoenix Pharmaceuticals. PMA, DPI, fMLP, and LPS were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Human neutrophil preparation
Neutrophils from healthy volunteers were isolated to >95% purity by using Dextran precipitation, followed by Ficoll-Paque gradient separation for the separation of mononuclear cells (layering above the Ficoll-Paque) and neutrophils (below the Ficoll-Paque). The neutrophils were purified further by hypotonic lysis to eliminate any contaminating erythrocytes. Cell viability was determined to be >98% by trypan blue staining. The isolated neutrophils were resuspended at the appropriate concentrations in PBS and used within 2 h. Neutrophil collection was done with informed consent, as approved by the Institutional Review Board of Boston University School of Medicine (Boston, MA, USA).
Measurement of IAV uptake by neutrophils
FITC-labeled IAV (Phil82 strain) was prepared, and uptake of virus by neutrophils was measured by flow cytometry, as described [23]. In brief, IAV was treated with various doses of LL-37 for 30 min at 37°C. Then, it was incubated with neutrophils for 45 min at 37°C in the presence of control buffer. The concentrations of LL-37, shown in Fig. 1, are those present in the final incubation with neutrophils. Trypan blue (0.2 mg/ml) was added to these samples to quench extracellular fluorescence. Following washing, the neutrophils were fixed with 1% paraformaldehyde, and neutrophil-associated fluorescence was measured using flow cytometry. The mean cell fluorescence (>1000 cells counted/sample) was measured.
Figure 1. Effects of LL-37 on neutrophil uptake of IAV.
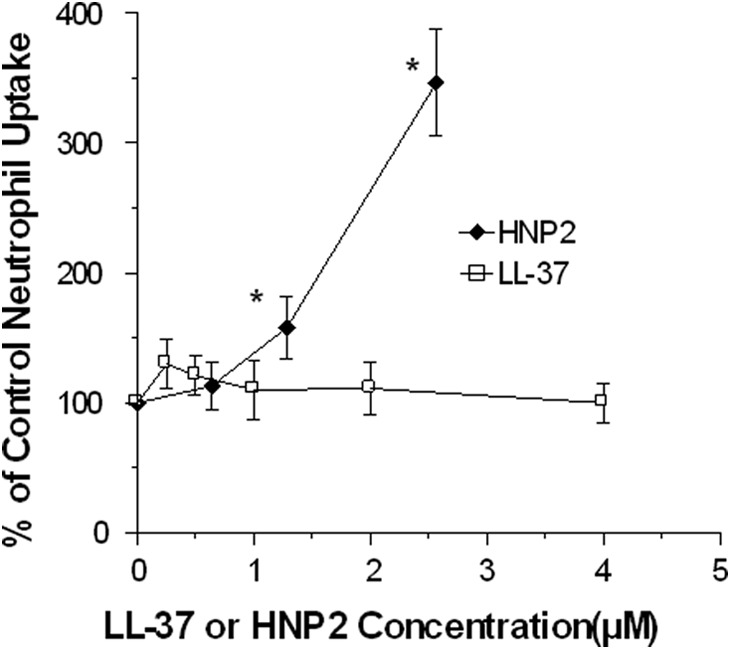
The virus was first preincubated with LL-37 or HNP-2 for 30 min at 37°C, and aliquots of these samples were then added to the neutrophils. The concentrations of LL-37 shown are those present in the final incubation with neutrophils, and they are given as molar concentrations to allow comparison of the effective concentrations of HNP2. The multiplicity of infection (MOI) for these experiments was ∼12 (or 12 infectious virions/neutrophil). HNP-2 caused significant increases in viral uptake, but LL-37 did not. Results are mean ± sem of five experiments using separate blood donors. *P < 0.05 compared with control using Student's t-test.
Measurement of neutrophil H2O2 production
H2O2 production was measured by assessing reduction in scopoletin fluorescence, as described previously [24]. Virus alone or virus that had been pretreated with various concentrations of LL-37 for 30 min at 37°C was added at Time 0 in the figure. The concentrations of LL-37 or sLL-37 shown are the final concentrations upon incubation with neutrophils. Measurements were made using a POLARstar OPTIMA fluorescent plate reader (BMG Labtech, Durham, NC, USA). WKYMVM peptide was used at a concentration of 10 nM. For WRW4 treatment, neutrophils were incubated with 20 μM WRW4 for 5 min at room temperature and then used for experiment.
Assessment of NET formation
For NET experiments, neutrophils were resuspended in PBS, supplemented with Ca2+ and Mg2+, and allowed to adhere on poly-L-lysine-coated, 96-well plates or on glass-bottom culture dishes (MatTek, Ashland, MA, USA) for 1 h in a CO2 incubator. In some experiments, neutrophils were pretreated with DPI (10 μM/5×106 cells) for 15 min at room temperature before adding to poly-L- lysine-coated plates. After 1 h, unadhered cells were removed, and adhered cells were incubated for 3 h in a CO2 incubator. Phil82 virus, which had been preincubated for 30 min, 37°C, with various concentrations of LL-37 or sLL-37, was included in this 3-h incubation with the neutrophils. For quantitative assessment of NETs, 5 μM Sytox Green (Invitrogen, Life Technologies, Grand Island, NY, USA) was added to samples on 96-well plates after the 3-h incubation period, and the plate was read on a POLARstar OPTIMA fluorescent plate reader (BMG Labtech). Immediately after reading, the plate was photographed on a fluorescent microscope. We chose the 3-h time-point for NET measurement based on preliminary experiments, showing that maximal NET formation in response to IAV or PMA (400 ng/ml) had occurred by this time. Repeated readings on the fluorescent plate reader were done over 30 additional min, and these showed stable relative values.
Neutrophil NET formation was also assessed using confocal microscopy. For confocal experiments, PMNs were adhered on poly-L-lysine-coated, glass-bottom culture dishes and incubated for 3 h with Phil82 virus and LL-37, followed by fixation with 1% paraformaldehyde. WGA-Oregon Green 488 (4 μg/ml) and DAPI 350 (Invitrogen) were used to stain the cell membrane and nucleus, respectively. The virus was Alexa Flour 594 labeled using a kit (Invitrogen), per the manufacturer's instructions. Confocal pictures were taken at Zeiss LSM510 on 100× magnification.
Measurement of IL-8 production by neutrophils
Phil82 virus was preincubated (30 min, 37°C) with control buffer (PBS) or various concentrations of LL-37 or sLL-37. For testing the effect of LL-37 on IL-8 production, neutrophils were treated with control buffer or the various virus preparations (e.g., virus alone, heat-treated virus, UV-inactivated virus, or live virus, which had been preincubated with LL-37 or sLL-37). This incubation was carried out for 45 min in a CO2 incubator in PBS buffer. No serum was present during the infection with virus. The concentrations of LL-37 or sLL-37 are the final concentrations added to the neutrophils. After this, the cells were pelleted and then resuspended and cultured in RPMI with 10% heat-inactivated autologous serum for 20 h. After 20 h, the supernatant was collected and assayed for IL-8 using a commercially available ELISA kit (BD Biosciences, San Diego, CA, USA), according to the manufacturer's instructions. Cells without any stimulus or with LPS as stimulus (100 ng/5×105 cells; Sigma-Aldrich) were used as NC and PC in the experiment. As additional controls, other cultures of the cells were treated with heat-inactivated IAV or UV-irradiated IAV.
LDH assay
The LDH assay was performed using the LDH cytotoxicity detection kit (Clontech, Mountain View, CA, USA), according to the manufacturer's instructions. In brief, human neutrophils were incubated with PBS, LL-37, or Phil82 IAV (with or without LL-37). The incubation time (20 h) and assay conditions were exactly the same as in the IL-8 assays. The LDH activity was measured after the 20-h incubation of cells with IAV and/or peptides. Controls included uninfected/untreated cells as NC and cells treated with lysis solution as PC. Results are expressed as mean absorbance values.
Apoptosis detection
Apoptosis was detected using the Annexin V-FITC apoptosis detection kit (Sigma-Aldrich), per the manufacturer's instructions. Before apoptosis detection, neutrophils were incubated with virus and/or LL-37 for 20 h. Culture conditions were the same as in IL-8 ELISA. After incubation, cells were washed in PBS and then resuspended in binding buffer provided with the kit. Then, the cells were incubated with Annexin V-FITC and/or propidium iodide for 10 min at room temperature, followed by washing and fixation with 1% paraformaldehyde. Fluorescence was measured by flow cytometry (BD Biosciences).
Statistics
Statistical comparisons were made using Student's paired, two-tailed t-test or ANOVA with post hoc test (Tukey's). ANOVA was used for multiple comparisons with a single control.
RESULTS
LL-37 does not increase neutrophil uptake of IAV
We have reported that preincubation of IAV with SP-D, HNPs, and retrocyclins increases viral uptake by neutrophils [25–27]. In contrast, LL-37 did not alter uptake of IAV by neutrophils (Fig. 1). The results shown involve preincubation of FITC-labeled virus with various doses of LL-37 for 30 min, followed by infection of neutrophils for 45 min. Results with HNP-2 are shown for comparison. For this assay, the molar concentrations of the peptides are given to allow direct comparison of the activity of LL-37 with HNP-2.
LL-37 potentiates neutrophil respiratory burst response to IAV
We have shown previously that IAV triggers an unusual respiratory burst response in neutrophils, in which H2O2 is generated without measurable production of superoxide [24]. Although LL-37 did not alter viral uptake by neutrophils, it did increase neutrophil H2O2 generation substantially in response to IAV (Fig. 2A). This effect was more pronounced when the virus was preincubated with LL-37 before addition to neutrophils; however, preincubation of neutrophils with LL-37 also increased the response to subsequent addition of IAV at the highest concentration of LL-37 tested (Fig. 2B). LL-37 alone (in the absence of IAV), at concentrations of 0.5–4 μg/ml, did not trigger increased H2O2 production compared with control buffer alone (results for the highest tested dose are shown in Fig. 2B). In addition, preincubation of IAV with sLL-37 at the same range of concentrations did not increase H2O2 production compared with IAV alone (data not shown).
Figure 2. Neutrophil H2O2 production in response to IAV, LL-37, or IAV + LL-37.
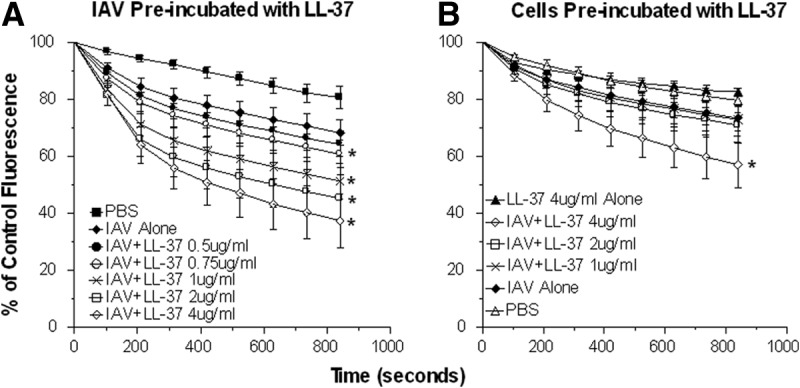
Neutrophils were treated with control buffer, IAV, LL-37, or combinations of IAV and LL-37, and H2O2 responses were measured by the fluorescent scopoletin assay. Decrease in scopoletin fluorescence corresponds to H2O2 production. (A) Results of treating neutrophils with IAV alone or combinations of IAV and LL-37. In these experiments, the virus and LL-37 were preincubated for 30 min at 37°C before addition to the neutrophils. These samples were diluted eightfold upon addition to the neutrophils (e.g., the sample noted as 4 μg/ml LL-37 was originally 32 μg/ml during the initial incubation with IAV). The concentrations of LL-37 shown are those present in the final incubation with neutrophils. The MOI for these experiments was 40. IAV alone elicited increased H2O2 generation compared with control buffer. Preincubation of IAV with LL-37 caused significant further increases in H2O2 generation in a dose-related manner compared with IAV alone. Doses of LL-37 ≥ 0.75 μg/ml caused significantly greater H2O2 generation than IAV alone (P<0.05). (B) Effects of preincubating neutrophils with LL-37, followed by adding IAV. In these experiments, LL-37 was added to the neutrophils first, and then, virus was added without washing off the LL-37. In this case, only the highest concentration of LL-37 (4 μg/ml) caused an increase in H2O2 generation compared with IAV alone (P<0.05 compared with IAV alone). For comparison, the results with LL-37 (4 μg/ml) alone (i.e., with no virus) are shown. The results shown are mean ± sem of nine experiments using separate neutrophil donors for each experiment. *Significant increase in H2O2 generation (P<0.05) compared with IAV alone, as measured by Student's t-test.
WKYMVM peptide is a strong inducer of neutrophil activation through formyl peptide receptors. As expected, WKYMVM strongly induced neutrophil H2O2 production (Fig. 3A). Preincubation of neutrophils with WRW4 (a FPRL-1 blocker) inhibited H2O2 production in response to the WKYMVM peptide (Fig. 3A). Unexpectedly, WRW4 significantly increased the response to IAV alone for unclear reasons. In contrast, WRW4 significantly blocked the ability of LL-37 to increase the H2O2 response triggered by IAV (Fig. 3B). The response to IAV + LL-37 + WRW4 was not significantly different from IAV + WRW4.
Figure 3. Effect of FPRL-1 blockade on H2O2 generation in response to IAV or IAV + LL-37.
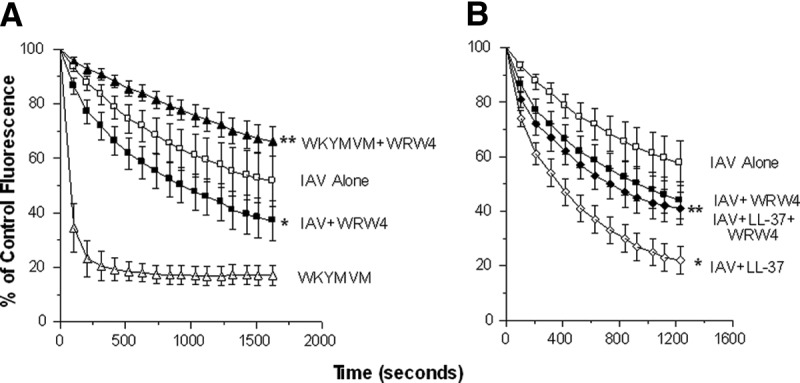
Experiments were performed as in Fig. 2. (A) Neutrophils were stimulated with IAV or the WKYMVM peptide alone or combined with the WRW4-blocking peptide. WRW4 markedly reduced the response to WKYMVM (P<0.001) but significantly increased the response to IAV (P<0.01). (B) IAV was pretreated with LL-37 (4 μg/ml), as in Fig. 2, and this again resulted in a significantly increased response compared with IAV alone (P<0.001). This response was reduced significantly by WRW4 (P<0.05), such that it was nearly the same at the response to IAV + WRW4 without LL-37. Results are mean ± sem of six experiments with separate neutrophil donors. *Significant increase in H2O2 generation compared with IAV alone (P<0.05) using Student's t-test; **WRW4 significantly reduced responses (P<0.05), as measured using Student's t-test.
IAV induces NET formation, and this response is increased by preincubation of the virus with LL-37
As shown in Fig. 4A, incubation of neutrophils with IAV significantly increased NET formation by neutrophils in vitro. The NETs were identified using Sytox Green to detect extracellular DNA. PMA (400 ng/ml) caused a greater increase in NET formation than IAV. We showed similar results with the Aichi68 H3N2 strain of IAV (i.e., Sytox Green fluorescence was increased significantly to 164±14% of control; P<0.01; n=5 using separate neutrophil donors). Figure 4B shows representative fluorescent micrographs from these experiments. A phase-contrast image of the control slide is included to indicate how many neutrophils were present in the field.
Figure 4. Induction of NETs by IAV or PMA.
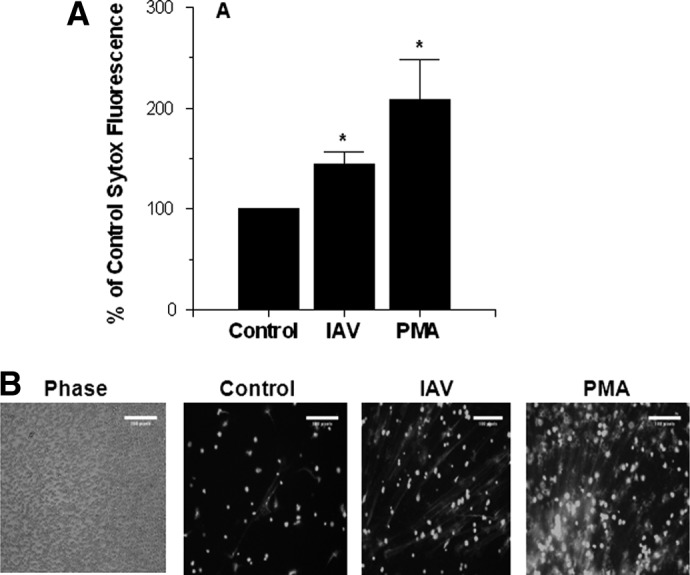
Neutrophils were exposed to control buffer, Phil82 IAV, or PMA (400 ng/ml) for 3 h, and Sytox Green fluorescence was measured by a plate-reading fluorometer (A) or microscopy (B). The MOI for these experiments was 50. Results in the graph are mean ± sem of six experiments, and separate neutrophil donors and microscopy results are representative of samples treated with control buffer, IAV, or PMA from these experiments. (B, left) Phase-contrast image to indicate how many neutrophils were present in a representative field. The three other panels were taken using fluorescent microscopy. The images were taken at 10× original magnification. *P < 0.05 compared with control buffer using Student's t-test.
We next preincubated IAV with LL-37 before infection of the neutrophils. As shown in Fig. 5A, preincubation with LL-37 increased NET formation significantly, triggered by IAV. LL-37 alone, at a range of concentrations, did not stimulate NET formation (results for 32 μg/ml of LL-37 alone are shown). We also preincubated the neutrophils with the NADPH-oxidase inhibitor DPI (10μM) to determine if this would prevent NET formation. Unexpectedly, this treatment did not alter NET formation in response to IAV or IAV + LL-37, even though it did strongly inhibit the PMA-induced response. NET formation in response to IAV or PMA alone was not inhibited by WRW4; however, the NET response to the combination of IAV and LL-37 was inhibited, indicating that the enhanced NET response caused by LL-37 is partially mediated through FPRL-1 (Fig. 5B). WKYMVM alone did not induce NET formation. We also found in separate experiments (not shown) that fMLP did not increase the NET response. Figure 5C shows representative fluorescent micrographs from these experiments.
Figure 5. Effects of LL-37 on neutrophil NET formation.
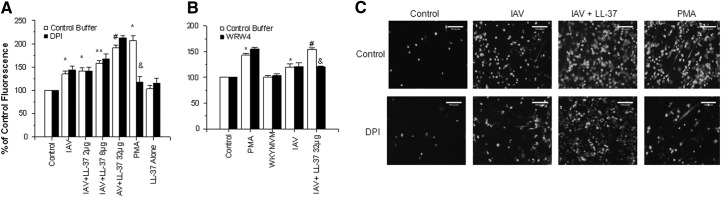
(A) Sytox Green fluorescence measurements of neutrophils treated with control buffer, PMA, IAV, IAV pretreated with the indicated concentrations (μg/ml) of LL-37, or LL-37 alone. In this set of experiments, the IAV samples preincubated with LL-37 were not diluted further upon addition to neutrophils. Black bars show results obtained with neutrophils preincubated with DPI, and DPI did not significantly reduce fluorescence of IAV- or IAV + LL-37-treated samples but did reduce that of samples treated with PMA. (B) Effects of WRW4 on NET formation in response to IAV, PMA, WKYMVM peptide, or IAV preincubated with LL-37. Results are mean ± sem of six experiments with separate neutrophil donors. (C) Fluorescent micrographs on NET formation in response to control buffer, IAV, IAV + LL-37 (32 μg/ml), or PMA. (Upper) Images show control neutrophils; (lower) neutrophils preincubated with DPI (10μM). Results are representative of three experiments with different neutrophil donors. The images were taken at 10× original magnification. *Fluorescence was increased significantly (P<0.01) compared with control cells alone, as measured by Student's t-test; **LL-37 caused significant increases (P<0.05) in NET response compared with IAV alone, as assessed by paired t-test; #LL-37 caused significant increases (P<0.05) in NET formation compared with IAV alone, as measured by ANOVA; &DPI or WRW4 significantly reduced (P<0.05) NET formation, as assessed by ANOVA.
We also wanted to determine the location of IAV with respect to NETs. Figure 6 shows representative confocal micrographs from two experiments of NET formation in response to IAV or IAV + LL-37 (32 μg/ml). In these experiments, LL-37 appeared to direct IAV to associate with the NETs rather than being internalized by the neutrophils themselves.
Figure 6. Location of IAV with respect to NETs using confocal microscopy.

Confocal microscopic images from two representative experiments of neutrophils treated with control buffer, IAV, or IAV + LL-37 (32 μg/ml). DNA was stained blue with DAPI, cell membranes green with WGA-Oregon Green, and IAV red with Alexa Fluor 594. NET formation was present in many fields of IAV-exposed cells but was more evident in cells treated with IAV + LL-37. The images shown were taken at 100× original magnification.
LL-37 reduces neutrophil IL-8 generation
An important finding in the report by Barlow et al. [3] was that LL-37 reduces cytokine generation, resulting from IAV infection in vivo. In preliminary experiments, we found that maximal IL-8 generation occurred after treatment of neutrophils with IAV or LPS for 20 h. As shown in Fig. 7, similar levels of increased IL-8 production were found at 20 h after treatment of neutrophils with infectious IAV or LPS. Heat-inactivated IAV did not cause any increase in IL-8 production. However, UV-inactivated IAV did elicit some increase in IL-8 generation above control levels. LL-37 or sLL-37 alone did not stimulate IL-8 production. LL-37 did, however, significantly reduce LPS- or IAV-stimulated IL-8 production in a dose-related manner. Of note, sLL-37 did not significantly alter the virus-induced response.
Figure 7. Effects of IAV or IAV plus LL-37 on neutrophil IL-8 generation.
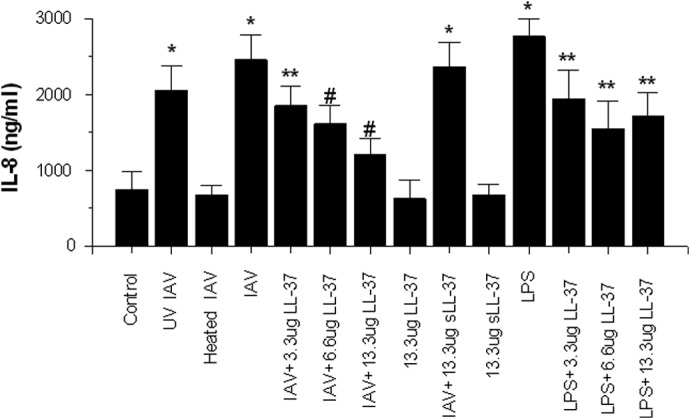
Neutrophils were stimulated with LPS, infectious IAV, or heat- or UV-inactivated IAV, and IL-8 production in the cell supernatants was measured 20 h later. LPS, infectious IAV, and UV-inactivated IAV all caused increased IL-8 production compared with control (untreated neutrophils). Before adding to neutrophils, the virus was preincubated with control buffer, LL-37, or sLL-37 for 30 min at 37°C. These samples were then further diluted 2.4-fold in addition to neutrophils. The amounts of LL-37 or sLL-37 were those present in the final incubation with neutrophils. The MOI for these experiments was 10. LL-37 reduced IL-8 generation in response to LPS or infectious IAV in a dose-related manner. sLL-37 did not reduce the IL-8 response to IAV. LL-37 or sLL-37 alone (in the absence of other stimuli) did not increase IL-8 generation compared with control. Results are mean ± sem of six experiments with separate neutrophil donors. *Significant increase at P < 0.05 compared with control, as measured by Student's t-test; **significant decreases in LPS induced IL-8 generation caused by LL-37, as assessed by Student's t-test; #significant decrease compared with IAV alone at P < 0.05, as measured by ANOVA.
We used LDH and apoptosis assays to be sure that the decrease in IAV-stimulated IL-8 production was not the result of altered neutrophil survival caused by LL-37 [28–30]. The experiments were carried out in the same manner as the IL-8 assay (e.g., incubation for 20 h). There was no significant change in LDH release into the supernatant induced by LL-37, alone or in combination with IAV (data not shown). The percent of cells in late apoptosis (measured by colabeling with Annexin 5 and propidium iodide and flow cytometry) was also not increased by LL-37 (mean±sem percent of cells in late apoptosis was 61±7 for IAV alone vs. 56±8.5 for IAV+LL-37; n=4 experiments with separate neutrophil donors).
DISCUSSION
It is likely that IAV encounters LL-37 in the respiratory tract in vivo coming from neutrophils, macrophages, and/or epithelial cells, all of which participate actively in the innate response to the virus. In our assays, we have used a range of LL-37 concentrations similar to those found to be active in viral or bacterial neutralization [3, 31, 32] or in inhibition of macrophage cytokine responses [33]. These concentrations of LL-37 exceed those generally measured in BALF in healthy individuals or patients with chronic obstructive pulmonary disease [34]. In contrast, a study of BALF from patients with cystic fibrosis showed significantly higher concentrations (e.g., up to 15 μg/ml) in some individuals [35]. The local concentrations of LL-37 at mucosal surfaces or at sites of neutrophil infiltration are unknown, although it is plausible to suspect that these concentrations would exceed those in BALF and would approach those used in this study.
LL-37 is present in NETs, along with a variety of other host defense proteins, including HNPs. The association of these cationic peptides with NETs appears to be mediated by electrostatic interactions. LL-37 has been demonstrated to improve outcome of IAV infection in vivo by Barlow et al. [3]. In their study, a reduction in viral load and also in inflammatory cytokine production was noted. We also have shown direct antiviral effects of LL-37 in epithelial cells [5]. We now demonstrate reduced virus-induced cytokine production in vitro by human neutrophils. Overall, these results suggest that LL-37 participates in host defense against IAV through direct antiviral effects and also through modulating inflammation.
Based on our results, LL-37 does not appear to cause increased ingestion of IAV by neutrophils. In this regard, LL-37 contrasts with HNPs and other innate immune proteins, such as collectins, as those proteins cause significantly increased IAV uptake by neutrophils [30–32]. This may relate to the inability of LL-37 to induce viral aggregation, in part [5], as collectins and defensins do induce viral aggregation [26, 27].
There is abundant evidence supporting the concept that neutrophils are an integral part of the innate immune response to IAV (see ref. [6] for review). In mouse models, neutrophils appear to play a largely protective role in IAV infection [17, 19, 22]; however, murine studies of severe lethal influenza (e.g., that caused by highly pathogenic H5N1) indicate that neutrophils may be harmful [36, 37]. We now show LL-37 significantly increases aspects of neutrophil activation in the presence of IAV, and IAV itself generates a respiratory burst response, and this response was markedly up-regulated by preincubation of IAV with LL-37. Here, again, the effects of LL-37 were distinct from those of HNPs, which reduce neutrophil respiratory burst responses to IAV [26]. We demonstrate that the increased respiratory burst response to IAV caused by LL-37 is mediated, to a significant extent, by signaling through the FPRL-1. Of note, LL-37 on its own did not stimulate any respiratory burst response, and optimal enhancement of the viral response was achieved when the virus was preincubated with LL-37. This suggests that LL-37 is more effective at triggering responses through FPRL-1 when combined with an additional ligand (e.g., the virus).
NET formation is now recognized to be an important mechanism through which neutrophils entrap and kill bacteria [38], but there is growing evidence that they play a role in viral infections. For instance, NETs have been implicated in host defense against HIV [39]. Recent papers have provided evidence of NET formation in vivo in the lungs of IAV-infected mice [18]. Li et al. [40] generated mice, with a conditional PAD4 gene deletion, which do not form NETs in vivo in response to LPS, PMA, or H2O2, as they lack the ability deiminate histones H3 and H4, which are felt to be a necessary step in the formation of NETs in response to these stimuli [40, 41]. Hemmers et al. [41] infected these mice with influenza, and although they had less weight loss than control mice after infection, there were no differences in viral titers or mortality.
We now demonstrate that IAV induces formation of NETs by human neutrophils in vitro through a distinctive pathway. A common pathway of NET formation involves generation of reactive oxygen species. As LL–37 increased production of H2O2 by IAV–treated neutrophils, we predicted that LL–37 would also increase NET formation in response. This was indeed the case; however, we could not show a direct link between the respiratory burst response and NET formation induced by IAV or the combination of IAV and LL–37. We have shown previously that DPI reduces neutrophil H2O2 production and apoptosis induced by IAV [42]. We confirm here that it reduces NET formation induced by PMA; however, DPI did not reduce IAV- or IAV + LL-37-mediated NET formation. Some other stimuli have also been reported to induce NETs without a requirement for oxidant production [43, 44]. IAV alone or IAV + LL-37 appears to represent additional instances of such an alternative pathway of NET formation. The ability of LL-37 to increase IAV-induced NET formation could also, in part, reflect its ability to protect the virus-induced NETs from degradation, as observed by Lande et al. [45]. The WRW4 peptide inhibited NET formation in response to the combination of LL-37 and IAV. Hence, the increased respiratory burst and NET response to IAV caused by LL-37 are mediated by FPRL-1 receptors engaged by LL-37; however, the two responses are not dependent on each other. It is unclear why FPRL-1 receptors trigger NET formation when LL-37 is complexed with IAV but not when they are triggered by soluble stimuli, such as WKYMVM or fMLP. Our current hypothesis is that these receptors trigger NET formation mainly when they are engaged by a particulate stimulus, such as IAV or bacteria. In any case, our finding that IAV (or IAV combined with LL-37) induces NET formation through a pathway that is not dependent on the respiratory burst per se raises the question of whether PAD4 or citrullinated histones are required in this process. Further studies will be needed to sort this out.
Although LL-37 increased respiratory burst and NET responses of neutrophils to IAV, it reduced IL-8 production triggered by the virus. The ability of IAV to induce cytokine production in neutrophils was not completely dependent on viral replication in the cells. In fact, UV-inactivated virus did induce significant IL-8 generation. UV inactivation destroys viral RNA but leaves the viral proteins and particles intact. In contrast, heat inactivation degrades viral particles and proteins, and this did ablate virus-induced cytokine generation. It appears, therefore, that intact but noninfectious viral particles are able to induce cytokine generation to some extent. Further studies will be needed to understand the mechanism through which LL-37 down-regulates IAV-induced IL-8 generation.
Conclusion
LL-37 significantly modulates the response of neutrophils to IAV. Coupled with our prior studies, we conclude that LL-37 has a distinct mode of antiviral activity and of interaction with neutrophils compared with HNPs and collectins. On the one hand, LL-37 has an anti-inflammatory effect by reducing IL-8 production by these cells in response to IAV. On the other hand, it increases neutrophil respiratory burst and NET responses to the virus. The ability of LL-37 to increase the respiratory burst and NET response of neutrophils is mediated, to a large extent, by its ability to engage FPRL-1 receptors. Although LL-37 had proinflammatory and anti-inflammatory effects in the context of IAV infection of neutrophils in our studies, overall, LL-37 appears to a beneficial component of the host response to IAV in vivo [3, 4].
NETs have been shown to mediate protection against bacterial infection but to be pathogenic in acute lung injury, cystic fibrosis, and autoimmune diseases (e.g., systemic lupus erythematosus) [46, 47]. Overall, neutrophils appear to play a protective role in IAV infection, although the contribution of NETs is unclear and may depend on the severity of infection or the presence of other factors, such as SP-D, which can bind to and clear NETs [48]. One possibility is that the ability of LL-37 to increase NET formation in response to IAV may promote viral clearance in vivo, as we demonstrate binding of IAV to NETs in vitro. The role of LL-37 and NETs in bacterial superinfection, resulting from IAV infection, is an area worthy of further study. Cardiovascular events are another source of morbidity and mortality during IAV epidemics. NETs have been shown to contribute to thrombosis in some settings [49], and it will be interesting to evaluate if they could contribute to thrombosis during severe IAV infection.
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health Grants AI-83222 and HL069031 (to K.L.H.).
Footnotes
- BALF
- bronchoalveolar lavage fluid
- DAPI
- 4′,6-diamidino-2-phenylindole
- DPI
- diphenyleneiodonium
- FPRL-1
- formyl peptide receptor-like 1
- HNP
- human neutrophil defensin
- IAV
- influenza A virus
- LDH
- lactate dehydrogenase
- NC
- negative control
- NET
- neutrophil extracellular trap
- PAD4
- peptidylarginine deiminase 4
- PC
- positive control
- Phil82
- Philippines 82/H3N2
- sLL-37
- scrambled LL-37
- SP-D
- surfactant protein D
- WGA
- wheat germ agglutinin
- WKYMVM
- Trp-Lys-Tyr-Met-Val-D-Met-NH2
- WRW4
- Trp-Arg-Trp-Trp-Trp-Trp-CONH2 (WRWWWW)
AUTHORSHIP
S.T. contributed to the design, performance of experiments, overall experimental plan, and writing of the manuscript. A.V. designed and performed experiments. E-j.K. did the initial design and performed some experiments with NETs. M.R.W. and K.L.H. designed and performed various experiments and contributed to the overall review of the research plan and manuscript.
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1. Gwyer Findlay E., Currie S. M., Davidson D. J. (2013) Cationic host defence peptides: potential as antiviral therapeutics. BioDrugs 27, 479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oppenheim J. J., Yang D. (2005) Alarmins: chemotactic activators of immune responses. Curr. Opin. Immunol. 17, 359–365. [DOI] [PubMed] [Google Scholar]
- 3. Barlow P. G., Svoboda P., Mackellar A., Nash A. A., York I. A., Pohl J., Davidson D. J., Donis R. O. (2011) Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS One 6, e25333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gaudreault E., Gosselin J. (2008) Leukotriene B4 induces release of antimicrobial peptides in lungs of virally infected mice. J. Immunol. 180, 6211–6221. [DOI] [PubMed] [Google Scholar]
- 5. Tripathi S., Tecle T., Verma A., Crouch E., White M., Hartshorn K. L. (2013) The human cathelicidin LL-37 inhibits influenza A viruses through a mechanism distinct from that of surfactant protein D or defensins. J. Gen. Virol. 94, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tripathi S., White M. R., Hartshorn K. L. (2013) The amazing innate immune response to influenza A virus infection. Innate Immun. 10.1177/1753425913508992 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 7. Beard J. A., Bearden A., Striker R. (2011) Vitamin D and the anti-viral state. J. Clin. Virol. 50, 194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doss M., White M. R., Tecle T., Hartshorn K. L. (2010) Human defensins and LL-37 in mucosal immunity. J. Leukoc. Biol. 87, 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Y., Chen Q., Schmidt A. P., Anderson G. M., Wang J. M., Wooters J., Oppenheim J. J., Chertov O. (2000) LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J. Exp. Med. 192, 1069–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mookherjee N., Lippert D. N., Hamill P., Falsafi R., Nijnik A., Kindrachuk J., Pistolic J., Gardy J., Miri P., Naseer M., Foster L. J., Hancock R. E. (2009) Intracellular receptor for human host defense peptide LL-37 in monocytes. J. Immunol. 183, 2688–2696. [DOI] [PubMed] [Google Scholar]
- 11. Zhang Z., Cherryholmes G., Chang F., Rose D. M., Schraufstatter I., Shively J. E. (2009) Evidence that cathelicidin peptide LL-37 may act as a functional ligand for CXCR2 on human neutrophils. Eur. J. Immunol. 39, 3181–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu J., Mookherjee N., Wee K., Bowdish D. M., Pistolic J., Li Y., Rehaume L., Hancock R. E. (2007) Host defense peptide LL-37, in synergy with inflammatory mediator IL-1beta, augments immune responses by multiple pathways. J. Immunol. 179, 7684–7691. [DOI] [PubMed] [Google Scholar]
- 13. Van der Does A. M., Beekhuizen H., Ravensbergen B., Vos T., Ottenhoff T. H., van Dissel J. T., Drijfhout J. W., Hiemstra P. S., Nibbering P. H. (2010) LL-37 directs macrophage differentiation toward macrophages with a proinflammatory signature. J. Immunol. 185, 1442–1449. [DOI] [PubMed] [Google Scholar]
- 14. Nijnik A., Pistolic J., Wyatt A., Tam S., Hancock R. E. (2009) Human cathelicidin peptide LL-37 modulates the effects of IFN-gamma on APCs. J. Immunol. 183, 5788–5798. [DOI] [PubMed] [Google Scholar]
- 15. Scott M. G., Davidson D. J., Gold M. R., Bowdish D., Hancock R. E. (2002) The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J. Immunol. 169, 3883–3891. [DOI] [PubMed] [Google Scholar]
- 16. Tjabringa G. S., Aarbiou J., Ninaber D. K., Drijfhout J. W., Sorensen O. E., Borregaard N., Rabe K. F., Hiemstra P. S. (2003) The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J. Immunol. 171, 6690–6696. [DOI] [PubMed] [Google Scholar]
- 17. Tate M. D., Ioannidis L. J., Croker B., Brown L. E., Brooks A. G., Reading P. C. (2011) The role of neutrophils during mild and severe influenza virus infections of mice. PLoS One 6, e17618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Narasaraju T., Yang E., Samy R. P., Ng H. H., Poh W. P., Liew A. A., Phoon M. C., van Rooijen N., Chow V. T. (2011) Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 179, 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tate M. D., Deng Y. M., Jones J. E., Anderson G. P., Brooks A. G., Reading P. C. (2009) Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J. Immunol. 183, 7441–7450. [DOI] [PubMed] [Google Scholar]
- 20. Tate M. D., Brooks A. G., Reading P. C. (2008) The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir. Res. 9, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hufford M. M., Richardson G., Zhou H., Manicassamy B., Garcia-Sastre A., Enelow R. I., Braciale T. J. (2012) Influenza-infected neutrophils within the infected lungs act as antigen presenting cells for anti-viral CD8(+) T cells. PLoS One 7, e46581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tate M. D., Brooks A. G., Reading P. C., Mintern J. D. (2012) Neutrophils sustain effective CD8(+) T-cell responses in the respiratory tract following influenza infection. Immunol. Cell Biol. 90, 197–205. [DOI] [PubMed] [Google Scholar]
- 23. Hartshorn K. L., Collamer M., Auerbach M., Myers J. B., Pavlotsky N., Tauber A. I. (1988) Effects of influenza A virus on human neutrophil calcium metabolism. J. Immunol. 141, 1295–1301. [PubMed] [Google Scholar]
- 24. Hartshorn K. L., Collamer M., White M. R., Schwartz J. H., Tauber A. I. (1990) Characterization of influenza A virus activation of the human neutrophil. Blood 75, 218–226. [PubMed] [Google Scholar]
- 25. Doss M., White M. R., Tecle T., Gantz D., Crouch E. C., Jung G., Ruchala P., Waring A. J., Lehrer R. I., Hartshorn K. L. (2009) Interactions of alpha-, beta-, and theta-defensins with influenza A virus and surfactant protein D. J. Immunol. 182, 7878–7887. [DOI] [PubMed] [Google Scholar]
- 26. Tecle T., White M. R., Gantz D., Crouch E. C., Hartshorn K. L. (2007) Human neutrophil defensins increase neutrophil uptake of influenza A virus and bacteria and modify virus-induced respiratory burst responses. J. Immunol. 178, 8046–8052. [DOI] [PubMed] [Google Scholar]
- 27. Hartshorn K. L., Crouch E. C., White M. R., Eggleton P., Tauber A. I., Chang D., Sastry K. (1994) Evidence for a protective role of pulmonary surfactant protein D (SP-D) against influenza A viruses. J. Clin. Invest. 94, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nagaoka I., Tamura H., Hirata M. (2006) An antimicrobial cathelicidin peptide, human CAP18/LL-37, suppresses neutrophil apoptosis via the activation of formyl-peptide receptor-like 1 and P2X7. J. Immunol. 176, 3044–3052. [DOI] [PubMed] [Google Scholar]
- 29. Li H. N., Barlow P. G., Bylund J., Mackellar A., Bjorstad A., Conlon J., Hiemstra P. S., Haslett C., Gray M., Simpson A. J., Rossi A. G., Davidson D. J. (2009) Secondary necrosis of apoptotic neutrophils induced by the human cathelicidin LL-37 is not proinflammatory to phagocytosing macrophages. J. Leukoc. Biol. 86, 891–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barlow P. G., Beaumont P. E., Cosseau C., Mackellar A., Wilkinson T. S., Hancock R. E., Haslett C., Govan J. R., Simpson A. J., Davidson D. J. (2010) The human cathelicidin LL-37 preferentially promotes apoptosis of infected airway epithelium. Am. J. Respir. Cell Mol. Biol. 43, 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Currie S. M., Findlay E. G., McHugh B. J., Mackellar A., Man T., Macmillan D., Wang H., Fitch P. M., Schwarze J., Davidson D. J. (2013) The human cathelicidin LL-37 has antiviral activity against respiratory syncytial virus. PLoS One 8, e73659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Feng X., Sambanthamoorthy K., Palys T., Paranavitana C. (2013) The human antimicrobial peptide LL-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant Acinetobacter baumannii. Peptides 49, 131–137. [DOI] [PubMed] [Google Scholar]
- 33. Brown K. L., Poon G. F., Birkenhead D., Pena O. M., Falsafi R., Dahlgren C., Karlsson A., Bylund J., Hancock R. E., Johnson P. (2011) Host defense peptide LL-37 selectively reduces proinflammatory macrophage responses. J. Immunol. 186, 5497–5505. [DOI] [PubMed] [Google Scholar]
- 34. Golec M., Reichel C., Lemieszek M., Mackiewicz B., Buczkowski J., Sitkowska J., Skorska C., Dutkiewicz J., Milanowski J., Ziesche R. (2012) Cathelicidin LL-37 in bronchoalveolar lavage and epithelial lining fluids from COPD patients and healthy individuals. J. Biol. Regul. Homeost. Agents 26, 617–625. [PubMed] [Google Scholar]
- 35. Chen C. I., Schaller-Bals S., Paul K. P., Wahn U., Bals R. (2004) beta-Defensins and LL-37 in bronchoalveolar lavage fluid of patients with cystic fibrosis. J. Cyst. Fibros. 3, 45–50. [DOI] [PubMed] [Google Scholar]
- 36. Ichikawa A., Kuba K., Morita M., Chida S., Tezuka H., Hara H., Sasaki T., Ohteki T., Ranieri V. M., dos Santos C. C., Kawaoka Y., Akira S., Luster A. D., Lu B., Penninger J. M., Uhlig S., Slutsky A. S., Imai Y. (2013) CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am. J. Respir. Crit. Care Med. 187, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brandes M., Klauschen F., Kuchen S., Germain R. N. (2013) A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 154, 197–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brinkmann V., Reichard U., Goosmann C., Fauler B., Uhlemann Y., Weiss D. S., Weinrauch Y., Zychlinsky A. (2004) Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. [DOI] [PubMed] [Google Scholar]
- 39. Saitoh T., Komano J., Saitoh Y., Misawa T., Takahama M., Kozaki T., Uehata T., Iwasaki H., Omori H., Yamaoka S., Yamamoto N., Akira S. (2012) Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 12, 109–116. [DOI] [PubMed] [Google Scholar]
- 40. Li P., Li M., Lindberg M. R., Kennett M. J., Xiong N., Wang Y. (2010) PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 207, 1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hemmers S., Teijaro J. R., Arandjelovic S., Mowen K. A. (2011) PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One 6, e22043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Engelich G., White M., Hartshorn K. L. (2001) Neutrophil survival is markedly reduced by incubation with influenza virus and Streptococcus pneumoniae: role of respiratory burst. J. Leukoc. Biol. 69, 50–56. [PubMed] [Google Scholar]
- 43. Pilsczek F. H., Salina D., Poon K. K., Fahey C., Yipp B. G., Sibley C. D., Robbins S. M., Green F. H., Surette M. G., Sugai M., Bowden M. G., Hussain M., Zhang K., Kubes P. (2010) A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 185, 7413–7425. [DOI] [PubMed] [Google Scholar]
- 44. Marcos V., Zhou Z., Yildirim A. O., Bohla A., Hector A., Vitkov L., Wiedenbauer E. M., Krautgartner W. D., Stoiber W., Belohradsky B. H., Rieber N., Kormann M., Koller B., Roscher A., Roos D., Griese M., Eickelberg O., Doring G., Mall M. A., Hartl D. (2010) CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat. Med. 16, 1018–1023. [DOI] [PubMed] [Google Scholar]
- 45. Lande R., Ganguly D., Facchinetti V., Frasca L., Conrad C., Gregorio J., Meller S., Chamilos G., Sebasigari R., Riccieri V., Bassett R., Amuro H., Fukuhara S., Ito T., Liu Y. J., Gilliet M. (2011) Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 3, 73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cheng O. Z., Palaniyar N. (2013) NET balancing: a problem in inflammatory lung diseases. Front. Immunol. 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kahlenberg J. M., Carmona-Rivera C., Smith C. K., Kaplan M. J. (2013) Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 190, 1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Douda D. N., Jackson R., Grasemann H., Palaniyar N. (2010) Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. J. Immunol. 187, 1856–1865. [DOI] [PubMed] [Google Scholar]
- 49. Gardiner E. E., Andrews R. K. (2012) Neutrophil extracellular traps (NETs) and infection-related vascular dysfunction. Blood Rev. 26, 255–259. [DOI] [PubMed] [Google Scholar]