

Abstract
Arteriovenous malformations (AVMs) are tortuous vessels characterized by arteriovenous (AV) shunts, which displace capillaries and shunt blood directly from artery to vein. Notch signaling regulates embryonic AV specification by promoting arterial, as opposed to venous, endothelial cell (EC) fate. To understand the essential role of endothelial Notch signaling in postnatal AV organization, we used inducible Cre-loxP recombination to delete Rbpj, a mediator of canonical Notch signaling, from postnatal ECs in mice. Deletion of endothelial Rbpj from birth resulted in features of AVMs by P14, including abnormal AV shunting and tortuous vessels in the brain, intestine and heart. We further analyzed brain AVMs, as they pose particular health risks. Consistent with AVM pathology, we found cerebral hemorrhage, hypoxia and necrosis, and neurological deficits. AV shunts originated from capillaries (and possibly venules), with the earliest detectable morphological abnormalities in AV connections by P8. Prior to AV shunt formation, alterations in EC gene expression were detected, including decreased Efnb2 and increased Pai1, which encodes a downstream effector of TGFβ signaling. After AV shunts had formed, whole-mount immunostaining showed decreased Efnb2 and increased Ephb4 expression within AV shunts, suggesting that ECs were reprogrammed from arterial to venous identity. Deletion of Rbpj from adult ECs led to tortuosities in gastrointestinal, uterine and skin vascular beds, but had mild effects in the brain. Our results demonstrate a temporal requirement for Rbpj in postnatal ECs to maintain proper artery, capillary and vein organization and to prevent abnormal AV shunting and AVM pathogenesis.
Keywords: Arteriovenous, Notch, Cerebrovascular, Endothelial cell, Rbpj, Mouse
INTRODUCTION
Arteriovenous malformations (AVMs) are vascular lesions that disrupt the typical artery-capillary-vein organization of blood vessels. AVMs pass arterial blood directly to vein via arteriovenous (AV) shunts and thus supplant normal capillary connections. Vessels in AVMs are enlarged, tortuous and may coalesce into a vessel entanglement or nidus (The Arteriovenous Malformation Study Group, 1999; Lawton, 2014; Young and Yang, 2004). High-flow AV shunts are prone to rupture and result in hypoperfusion of surrounding tissue (The Arteriovenous Malformation Study Group, 1999; Fleetwood and Steinberg, 2002; Friedlander, 2007). AVMs are currently managed by surgical resection, embolization and radiosurgery (Friedman, 2013; Lawton, 2014; Lee, 2013; Murray et al., 2013; Zacest et al., 2013); however, these procedures carry risk, and not all AVM cases qualify for these interventions. Thus, alternative treatment options are needed.
AVMs can form throughout the body but are particularly damaging in the brain, where they may lead to hemorrhage, seizures, ischemia, neurological impairment and stroke (Achrol et al., 2009; Fleetwood and Steinberg, 2002; Friedlander, 2007; Murray et al., 2013). Causal genetic lesions have been identified for very few brain AVM (BAVM) cases (Young and Yang, 2004). For example, mutations in either ACVRL1 or ENG result in the human disease hereditary hemorrhagic telangiectasia (HHT) (Johnson et al., 1996; McAllister et al., 1994); however, the molecular causes of BAVMs remain largely unknown.
AV organization within a vascular bed depends on the acquisition and maintenance of arterial versus venous endothelial cell (EC) identity (Murphy et al., 2012; Wang et al., 1998). Although hemodynamic influences are involved, genetic programs also determine arterial or venous EC fate, even in the absence of blood flow (Kim et al., 2008; Lawson et al., 2001, 2002; Quillien et al., 2014; Thurston and Yancopoulos, 2001; Wang et al., 1998; Zhong et al., 2001). Notch signaling has an established role in cell fate determination in many cell types. In the developing vasculature, Notch receptors and ligands are expressed by arterial ECs (Villa et al., 2001) and are involved in determining arterial EC fate (Lawson et al., 2001, 2002; Quillien et al., 2014; Thurston and Yancopoulos, 2001; Zhong et al., 2001); thus, perturbations to Notch signaling affect AV gene expression. During vertebrate embryogenesis, loss of Notch signaling leads to numerous vascular abnormalities and the altered expression of arterial and venous marker genes (Kim et al., 2008; Krebs et al., 2004, 2010, 2000; Lawson et al., 2001; Limbourg et al., 2005). In mouse embryos, loss of either Notch1 or Rbpj gene function results in direct anastomoses between arterial and venous circulations (Krebs et al., 2004, 2010). Rbpj encodes a bHLH transcription factor that mediates canonical Notch signaling. In Rbpj mutant mouse embryos, expression of the arterial marker Efnb2 is reduced in ECs, suggesting a loss of arterial EC identity (Krebs et al., 2004).
Forced activation of Notch results in vascular lesions that disrupt AV organization and alter AV marker expression. Activation of Notch1 in ECs results in dorsal aortae fusing with developing cardinal veins in the mouse embryo (Krebs et al., 2010). In postnatal mouse endothelium, expression of a constitutively active Notch4 mutation leads to and sustains features of AVMs, including AV shunts and tortuous vessels, in brain, liver, uterus and lung (Carlson et al., 2005; Miniati et al., 2010; Murphy et al., 2008, 2012). Vessels from these mice show increased Efnb2 expression at the expense of Ephb4, suggesting that mutant venous ECs acquire arterial gene expression (Carlson et al., 2005; Murphy et al., 2008). Interestingly, increased expression of Notch signaling molecules is seen in samples from human BAVM patients (Murphy et al., 2009; ZhuGe et al., 2009), raising the possibility that activating mutations in Notch might contribute to human BAVM formation.
Arterial expression of Notch signaling molecules is maintained after birth, suggesting a role for Notch in the postnatal vasculature. Human patients with mutations in RBPJ are affected by Adams–Oliver syndrome and may present with cardiovascular anomalies and/or skin telangiectasias (Hassed et al., 2012). Given that the expression of a Notch gain-of-function mutation in the endothelium just after birth reprograms the molecular identity of arterial and venous ECs and leads to abnormal AV shunts in immature mouse brains, and that loss of endothelial Notch during embryogenesis also reprograms the AV identity of ECs and results in AV anastomoses, we tested whether Notch signaling is required in postnatal endothelium to maintain AV organization and AV marker expression. We found that deletion of Rbpj from postnatal ECs alters AV gene expression and leads to features of AVMs in mice.
RESULTS
Endothelial deletion of Rbpj from birth results in AV shunts and hemorrhage in P14 mouse brains
To test whether loss of canonical Notch from postnatal endothelium leads to abnormal AV shunt formation in immature mice, we deleted Rbpj from ECs (supplementary material Fig. S1) using an inducible Cre-loxP system. We used the Cdh5(PAC)-CreERT2 transgene for CreERT2 expression in ECs (Sorensen et al., 2009; Wang et al., 2010). We bred Cdh5(PAC)-CreERT2; Rbpjflox/flox mice, administered Tamoxifen (TAM) at postnatal day (P) 1 and P2, and harvested tissue at P14 (hereafter referred to as immature RbpjiΔEC). We first examined fluorescent casts of superficial cerebrovasculature (schematized in supplementary material Fig. S2). In control casts, capillary beds lying at the AV interface were readily seen (Fig. 1A); however, in mutant casts, enlarged AV connections were found (Fig. 1B; supplementary material Movies 1 and 2). We next used a Cre-responsive membrane-localized GFP reporter allele (mT/mG) (Muzumdar et al., 2007) to visualize whole-mount cerebrovasculature and to gauge the extent of CreERT2 activity (CreERT2-mediated excision results in mGFP expression in ECs) (supplementary material Fig. S3A-D). In control brains, arteries and veins were connected via capillaries. In all RbpjiΔEC brains examined, however, enlarged AV connections directly joined arteries and veins. To determine the extent of AV connection enlargement, we measured the diameters of AV connections from control and mutant cerebral vessels (supplementary material Fig. S3C,D). Average AV connection diameter was larger in RbpjiΔEC mutants than in controls (Fig. 1G), and the average capillary diameter from our controls is consistent with published results (Murphy et al., 2012). This suggests that Rbpj is required to maintain the diameter of AV connections in immature brain vasculature.
Fig. 1.
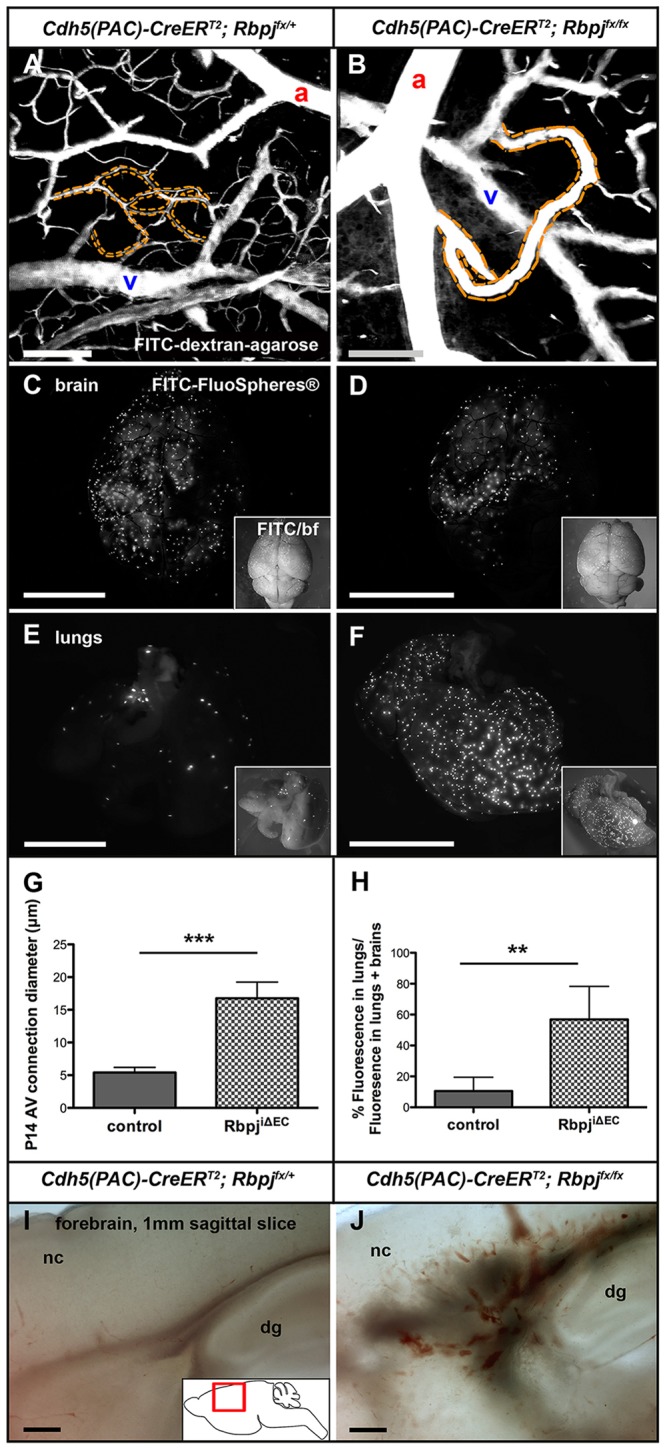
Endothelial deletion of Rbpj from birth results in abnormal AV shunts in P14 brains. (A,B) 2PEFM images of cerebrovascular casts. (A) Capillaries (dashed outline) lie between arteries (a) and veins (v) in controls. (B) In mutants, enlarged AV connections (dashed outline) directly connect arteries to veins. (C-F) Microspheres lodged in control brain capillaries (C) and very few circulated to the lungs (E). Some microspheres were retained in RbpjiΔEC brain (D), but most bypassed the brain and lodged in lung capillaries (F). Insets show merged fluorescence and brightfield (bf). (G) The diameter of AV connections was increased in RbpjiΔEC brains, as compared with controls. ***P=0.0001. N=6 controls (39 AV connections), N=6 mutants (40 AV connections). (H) Quantification of microsphere (FluoSphere) fluorescence, represented as percentage of lung/(lung+brain) fluorescence. **P=0.0066. N=5 controls, N=5 mutants. (I,J) Brightfield images of 1 mm sagittal brain slices show evidence of hemorrhage in mutants (J) as compared with control (I). N=3 controls, N=3 mutants. P-values were according to Student's t-test. nc, neocortex; dg, dentate gyrus. Scale bars: 100 μm in A,B,I,J; 5 mm in C-F.
To test whether enlarged AV connections in RbpjiΔEC mutants represented functional AV shunts, permitting flow of blood directly from arteries to veins, we performed a microsphere passage assay. Microspheres (15 μm diameter) injected into the left common carotid artery should become lodged in brain capillaries (∼5 μm diameter), as was observed in control mice (Fig. 1C,E). However, microspheres readily passed through the enlarged AV connections in RbpjiΔEC brains and lodged in the lungs (Fig. 1D,F, quantified in 1H), suggesting that these enlarged connections function as AV shunts. We also noted areas of hemorrhage within RbpjiΔEC brains, but not controls (Fig. 1I,J), consistent with high-flow AV shunts being prone to rupture. These data suggest that endothelial Rbpj is required for proper AV organization in the immature postnatal brain.
Endothelial deletion of Rbpj from birth results in irregular vessels and increased vascular density in P14 brains
To examine non-surface vessels, we generated 3D reconstructions of 2PEFM images from sagittal sections through immature mouse brains. In control forebrain, vessels dived into the brain perpendicular to the pial surface, creating parallel columns of vessels (Fig. 2A; supplementary material Movie 3). In some RbpjiΔEC brains, vascular organization was disrupted, and tortuous vessels formed focal tangles (Fig. 2B, bracket; supplementary material Movie 4), resembling niduses associated with BAVMs. In RbpjiΔEC cerebellum, blood vessels generally appeared wider in diameter than in controls, with select vessels greatly enlarged (Fig. 2C,D, arrow; supplementary material Movies 5 and 6). Some regions of RbpjiΔEC cerebellum showed sparse endothelium (Fig. 2D, asterisks), which is indicative of tissue necrosis caused by poor blood flow, a consequence of direct AV shunting.
Fig. 2.
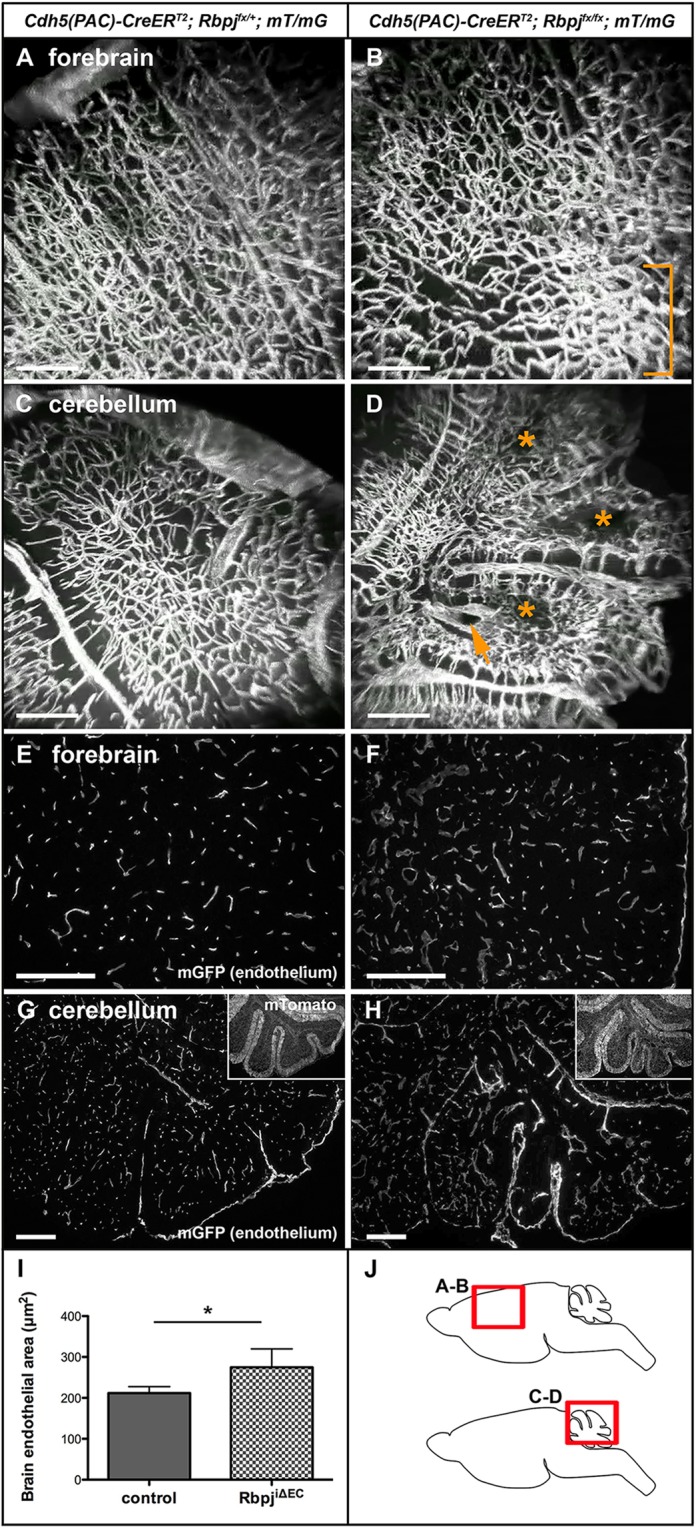
Endothelial deletion of Rbpj from birth results in large, irregular vessels and increased vascular density in P14 brains. (A,B) 3D reconstructions derived from 2PEFM for mGFP for control (A) and RbpjiΔEC (B) forebrain. Bracket indicates vessel entanglement in the mutant. (C,D) 2PEFM 3D reconstructions from control (C) and mutant (D) cerebellum. Asterisks highlight avascular regions; arrow indicates enlarged vessel. N=3 controls, N=3 mutants. (E-H) Sagittal sections through immature forebrain (E,F) and cerebellum (G,H) show increased vascular density in RbpjiΔEC brains (F,H) compared with controls (E,G). Insets show CreERT2-negative cells that expressed mTomato. (I) Area of mGFP-positive endothelium was increased in RbpjiΔEC brains. *P<0.0421, according to Student's t-test. N=5 controls, N=5 mutants. (J) Schematics indicating regions shown in A-D in sagittal brain slices. Scale bars: 200 μm.
To evaluate vascular density, we quantified mGFP-positive endothelium in control and RbpjiΔEC mice (Fig. 2E-H). We found increased vascular density in RbpjiΔEC versus control brains (Fig. 2I), suggesting that deletion of endothelial Rbpj leads to increased endothelial density in the immature brain.
Abnormalities in P14 RbpjiΔEC brain parenchyma are consistent with poor tissue perfusion
Because AV shunts are poor conduits for nutrient and oxygen exchange, we reasoned that the surrounding brain parenchymal tissue would be affected in RbpjiΔEC mice. We examined the cerebellum, as it undergoes a dynamic and substantial morphogenetic phase during the early postnatal period (P1-P14) (Acker et al., 2001). Gross analysis of immature RbpjiΔEC brains revealed consistently misshapen folia in all cerebella examined (Fig. 3B,C). Histopathological analysis revealed multiple regions of incomplete infarcts, characterized by areas of tissue necrosis (Fig. 3D,E) as well as loss of Purkinje neurons and internal granular cell neurons, with a marked thinning of the molecular layer (Fig. 3F,G). Together, these cerebellar changes are consistent with impaired blood flow and tissue perfusion.
Fig. 3.
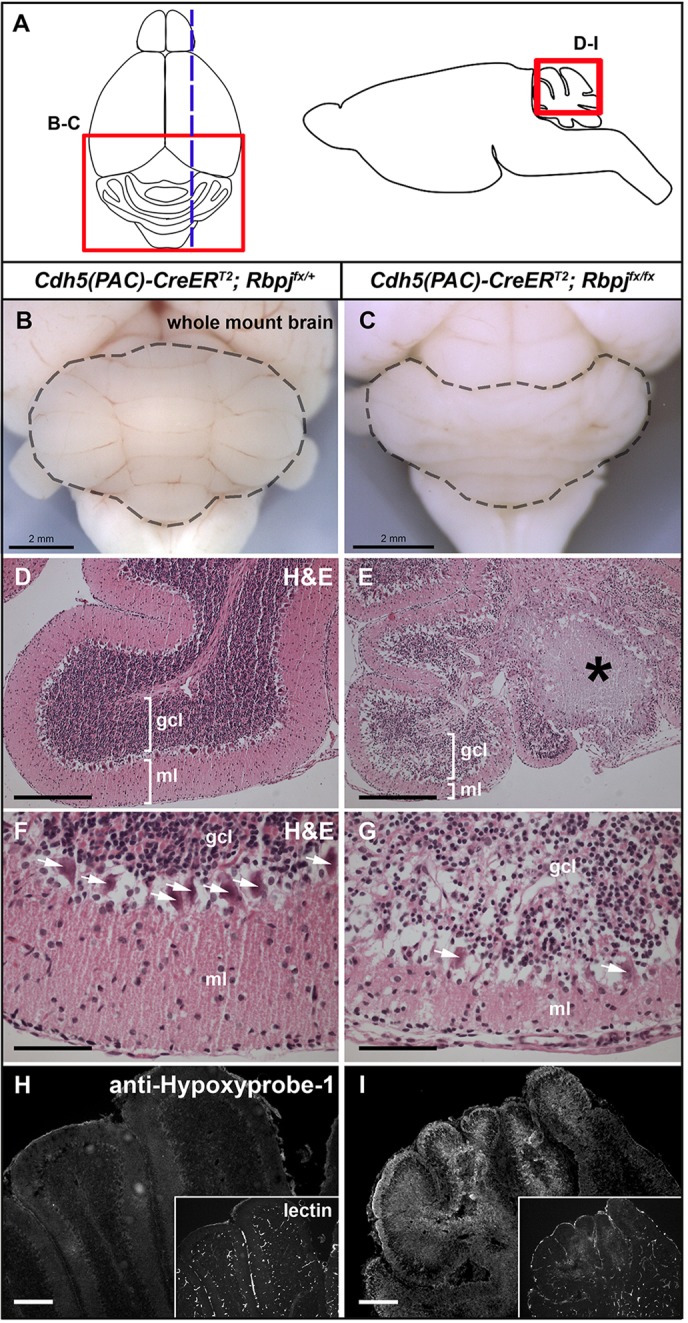
Endothelial deletion of Rbpj from birth results in gross and histopathological abnormalities in P14 brains. (A) Dorsal view of brain (left) indicating region shown in B,C; the dashed line indicates the plane of section in D-I. The sagittal view (right) indicates regions of cerebellum shown in D-I (at different magnifications). (B,C) Whole-mount brain in dorsal view. The cerebellum (outlined) is abnormal in the mutant (C; 40/40 mice), as compared with the control (B, 0/40 mice). (D-G) H&E staining indicates area of necrosis (asterisk in E), disrupted Purkinje cells (arrows in G) and reduced molecular layer (E,G) in RbpjiΔEC cerebellum. F,G are magnified regions from D,E. N=8 controls, N=6 mutants. (H,I) Immunostaining against pimonidazole HCl (anti-Hypoxyprobe-1) indicates hypoxic cells in RbpjiΔEC cerebellum. Insets show perfused vasculature highlighted by biotinylated tomato lectin. N=3 controls, N=3 mutants. ml, molecular layer; gcl, granule cell layer. Scale bars: 2 mm in B,C; 200 μm in D,E,H,I; 50 μm in F,G.
We hypothesized that necrosis and other pathological defects result from hypoxia caused by AV shunting. We assayed for tissue hypoxia by immunostaining against pimonidazole. Swathes of hypoxia were observed in mutant cerebella (Fig. 3I). No evidence for hypoxia was observed in controls (Fig. 3H). Furthermore, hypoxic regions of the mutant cerebellum showed reduced FITC-lectin (tomato) fluorescence, suggesting poor vascular perfusion of these regions. Notably, the regions of mutant cerebellum most affected upon pathological examination (medial folia) corresponded to areas with marked hypoxia. These data suggest that RbpjiΔEC brain tissue was insufficiently perfused to allow proper tissue oxygenation.
Together, these findings demonstrate that endothelial deletion of Rbpj from birth affects aspects of the surrounding immature brain parenchyma. These results, which are consistent with the perfusion defects that we have described and with consequences of AV shunting, suggest a role for endothelial Rbpj in promoting and/or maintaining proper vascular morphology and tissue perfusion in the postnatal mouse brain.
Endothelial deletion of Rbpj from birth results in vascular abnormalities resembling AVMs in select P14 organs
To determine whether deletion of endothelial Rbpj from birth results in vascular abnormalities in vascular beds other than the brain, we examined multiple organs in immature RbpjiΔEC mice. Gross examination revealed enlarged vessels in the intestinal mesentery (Fig. 4A,B) and enlarged coronary vessels on the ventral and dorsal surfaces of the heart (Fig. 4G,H). Histopathology of the intestine showed expanded vascular lumens in mesentery and submucosa (Fig. 4C-F, quantified in 4K). In addition, the smooth muscle layer of mesenteric arteries was reduced in thickness (supplementary material Fig. S4A,B, quantified in S4E), while immunostaining against CD13 (Anpep – Mouse Genome Informatics) suggested increased submucosal pericyte coverage (supplementary material Fig. S4C,D, quantified in S4E). Histopathology of the heart (the four-chamber view) revealed that blood vessels in the myocardium of the lateral ventricle were drastically enlarged in lumen (Fig. 4I,J, quantified in 4K), and many vessels in the mutants exhibited tortuous and complex morphology. Smooth muscle cell thickness (supplementary material Fig. S5A,B, quantified in S5E) and pericyte coverage (supplementary material Fig. S5C,D, quantified in S5F) were unaltered in RbpjiΔEC myocardium. These results suggest that Rbpj is required in postnatal endothelium to maintain vessel caliber and peri-endothelial support cell coverage in select organs.
Fig. 4.

Endothelial deletion of Rbpj from birth results in vascular abnormalities resembling AVMs in the intestine and heart at P14. (A,B) Live imaging of intestine. Mesentery arteries (red arrowheads) and veins (blue arrows) appear enlarged in the mutant (B) compared with the control (A). N=3 controls, N=4 mutants. (C-F) H&E staining of intestine show a complex network of enlarged, tortuous vessels in the mutant mesentery (C,D; red arrowheads, arteries; blue arrows, veins) and submucosa (E,F; yellow arrows, capillaries). (G,H) Ventral view of heart. Vessels on the ventricle surface appear enlarged in RbpjiΔEC mice (H). N=12 controls, N=8 mutants. (I,J) H&E staining of ventricular myocardium shows enlarged vessels (arrows) in the mutant (J). (K) Quantification of vessel size in intestine mesentery, submucosa and heart. The inner (lumenal) perimeter of vessels was measured by tissue section. Mesentery: **P=0.0061; N=5 controls (78 vessels), N=5 mutants (77 vessels). Submucosa: *P=0.0301; N=5 controls (112 vessels), N=5 mutants (92 vessels). Myocardium: *P=0.0305; N=5 controls (97 vessels), N=5 mutants (94 vessels). P-values were according to Student's t-test. Scale bars: 5 mm in A,B; 100 μm in C,D; 50 μm in E,F; 2 mm in G,H; 200 μm in I,J.
Endothelial deletion of Rbpj from birth leads to illness and death in immature mice
To determine whether overall health and survival are affected in RbpjiΔEC mice, we performed Kaplan–Meier survival analysis. Deletion of endothelial Rbpj from birth resulted in no lethality by P7, ∼50% lethality by P14 and ∼90% lethality by P20 (Fig. 5A). P14 (but not P7) RbpjiΔEC mutants were smaller than littermate controls and had lower body weight (Fig. 5B,C). Mutant mice displayed impaired movement by P14; they were slow to initiate movement and walked with splayed limbs (supplementary material Movie 7). These impairments were progressive, as differences in body weight and gait/movement among control and RbpjiΔEC mice were not observed prior to ∼P11. These results suggest that RbpjiΔEC mice develop both cerebral and systemic AV shunting that may lead to impaired brain function and compromised overall health and survival.
Fig. 5.
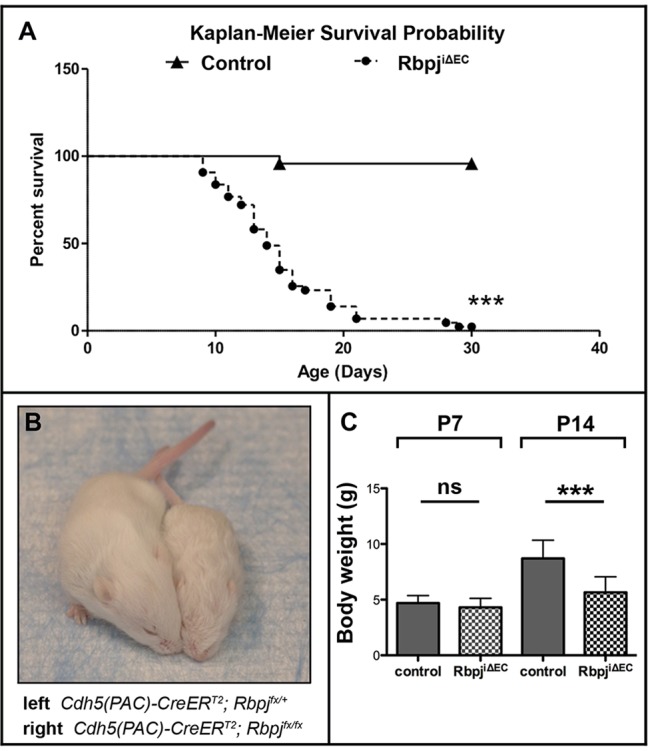
Endothelial deletion of Rbpj from birth leads to illness and death in P14 mice. (A) Kaplan–Meier survival curve indicating 50% probability of survival for P14 RbpjiΔEC mice. ***P<0.0001. N=24 controls, N=43 mutants. (B) Overall body size was compromised in RbpjiΔEC mice. (C) Total body weight was reduced in P14 RbpjiΔEC mice, but not in P7 RbpjiΔEC mice. P7: P=0.217 (not significant); N=42 controls, N=29 mutants. P14: ***P<0.0001; N=48 controls, N=49 mutants. P-values were according to Student's t-test.
Endothelial deletion of Rbpj at 6 weeks results in mild defects in the mature brain at 12 weeks
The mouse brain undergoes extensive vascular and parenchymal morphogenesis in the early postnatal period (Wang et al., 1992) and may be particularly susceptible, at this stage, to endothelial-specific genetic lesions. Thus, we hypothesized that deletion of Rbpj from adult ECs would not lead to brain AV shunting. TAM was administered to 6-week-old Cdh5(PAC)-CreERT2; Rbpjflox/flox; mT/mG (mature RbpjiΔEC) and Cdh5(PAC)-CreERT2; Rbpjflox/+; mT/mG (mature control) mice. Tissue was harvested and analyzed at 12 weeks of age (6 weeks post-TAM). No obvious defects were observed in 12-week RbpjiΔEC whole-mount brains in terms of size and gross morphology (Fig. 6A,B). Analysis of surface cerebrovasculature showed no difference between AV connection diameter in control and RbpjiΔEC brains (Fig. 6C,D, quantified in 6O). Histopathological analysis showed mild pathology, characterized by scattered dilated blood vessels in the cerebellum (Fig. 6E,F). Interestingly, the striatum of the 12-week mutants showed evidence of vascular injury, which was characterized by the presence of scattered hypoxic-ischemic neurons in the caudate nucleus (Fig. 6G,H). The regional differences in pathological features between immature and mature mouse brains suggests that the vascular defects and their neurological outcomes might be age dependent.
Fig. 6.

Endothelial deletion of Rbpj from 6 weeks leads to mild brain abnormalities and to vascular abnormalities resembling AVMs in gastrointestinal tract, uterus and skin at 12 weeks. TAM administered at 6 weeks; tissue harvested at 12 weeks. (A,B) Whole brain, dorsal view (ant, anterior; post, posterior). Gross abnormalities were not observed in mutant brain (B) as compared with control (A). N=4 controls, N=5 mutants. (C,D) Whole-mount mGFP fluorescence, produced by mT/mG. Capillaries (dashed outline) lie at the artery-to-vein (a-v) interface in control (C) and mutant (D) brains. (E-H) H&E staining shows scattered dilated blood vessels in mutant cerebellum (F, arrows) and hypoxic-ischemic neurons in mutant striatum (H, arrows). N=5 controls, N=6 mutants. (I,J) An entanglement of vessels (outlined) connects the IVC to the gastrointestinal system in 0/4 controls and 6/6 mutants. (K-N) Vessels associated with non-gravid/non-post-partum uterus and skin were enlarged and tortuous in mutants (L,N). (O) Quantification of vessel diameter from brain, uterus and skin. Brain AV connections: P=0.4476; N=3 controls (30 AV connections), N=8 mutants (77 AV connections). Uterus: *P=0.0177; N=3 controls (27 vessels), N=4 mutants (40 vessels). Skin: **P=0.0026; N=4 controls (4 vessels), N=6 mutants (6 vessels). P-values were according to Student's t-test. Scale bars: 5 mm in A,B,M,N; 100 μm in C,D; 200 μm in E,F; 50 μm in G,H; 2 mm in I-L.
Endothelial deletion of Rbpj at 6 weeks results in vascular abnormalities resembling AVMs in select organs at 12 weeks
Expression of endothelial Notch4* during adulthood leads to hallmarks of AVMs (but not BAVMs) in mature mice. We speculated that deletion of Rbpj from adult ECs would lead to vascular abnormalities resembling AVMs in vascular beds other than the brain, and we analyzed 12-week RbpjiΔEC mice for gross vascular abnormalities. A tangle of tortuous vessels – resembling a characteristic nidus of AVMs – was seen joining the inferior vena cava (IVC) and the gastrointestinal tract in RbpjiΔEC mice (N=6/6; Fig. 6I,J). Enlarged vessels were observed in non-gravid/non-post-partum uterus and skin (Fig. 6K-N, quantified in 6O). Motor skills and body size appeared unchanged in 12-week RbpjiΔEC mice, and no significant difference in body weight was seen [control, 33.25±6.03 g; RbpjiΔEC, 30.7±6.56 g (mean±s.d.); P=0.25 by Student's t-test; N=16 controls, N=19 mutants]. Similar defects were observed in mice analyzed at 6 months of age (TAM at 6 weeks). These data show that deletion of Rbpj from adult ECs results in vascular lesions characteristic of AVMs in some organs, suggesting that endothelial Rbpj is required for the prevention of AVMs in certain mature vascular beds.
AV connections in RbpjiΔEC brains appear abnormal by P8, following TAM delivery at P1 and P2
To understand the changes to immature brain vasculature when Rbpj was deleted from endothelium at birth, we analyzed AV connections from P5 to P10. Prior to P8, control and RbpjiΔEC AV connections were not distinguishable (Fig. 7A-D); however, at P8 and P10 the AV connections appeared abnormally large, as compared with controls (Fig. 7E-H). Next, we analyzed alpha-smooth muscle actin (αSMA) expression. In both control and mutant P7 brains, αSMA was present in arteries and first-branch arterioles but absent from AV connections (Fig. 7I,J). Similarly, at P14, αSMA was restricted to arteries/arterioles and spared from venules, capillaries (control, Fig. 7K) and AV shunts (RbpjiΔEC, Fig. 7L). These data suggest that AV shunts in RbpjiΔEC mice originate from αSMA-negative vessels. To determine whether increased EC proliferation was associated with AV shunt formation, we analyzed EC BrdU incorporation from P5-P7. We observed no evidence for increased EC proliferation in P7 RbpjiΔEC cerebrovasculature (Fig. 7M,N, quantified in 7O). Together, these results suggest that, by the eighth day after TAM delivery, AV connections originate from αSMA-negative capillaries and/or venules without increased EC proliferation.
Fig. 7.

Following endothelial Rbpj deletion from birth, brain AV connections are abnormal by P8, lack αSMA and do not proliferate. (A-H) Whole-mount timecourse images showing brain AV connections (arrows). At P8, RbpjiΔEC AV connections (F) appear different than in controls (E). For each time point, N=3 controls, N=3 mutants, with at least seven fields imaged from each brain. (I-L) Whole-mount αSMA immunostaining. Arrows point to AV connections, which lacked αSMA staining in both control and mutant. For P7, N=4 controls, N=3 mutants; for P14, N=3 controls, N=3 mutants. (M-O) BrdU incorporation in ECs from P5-P7 was unchanged in mutant (N) versus control (M) brain (as quantified in O). Arrow indicates EC nucleus. P=0.9752, according to Student's t-test. N=4 controls (29 fields), N=5 mutants (33 fields). a, artery; v, vein. Scale bars: 100 μm in A-L; 25 μm in M,N.
Decreased Efnb2 and increased Pai1 expression in P7 brain ECs following TAM administration at P1 and P2
We next used quantitative RT-PCR to analyze changes in EC gene expression at P7, a stage immediately before detectable vascular abnormalities and therefore without the confounding effects of high flow-driven gene expression changes elicited by AV shunts. We examined the Notch downstream positive effectors Hey1, Hey2 and Efnb2. Decreases were close to significant for Hey1 and significant for Hey2 and Efnb2. Ephb4, a negative Notch downstream gene, was insignificantly increased. Expression of Vegfa, Smad4, Tgfb1 and Rasa1 was not significantly changed; however, Pai1 (Serpine1 – Mouse Genome Informatics), a TGFβ target gene, was increased ∼3-fold. These data demonstrate that at P7, prior to AV shunt formation in RbpjiΔEC brains, ECs already exhibit changes in gene expression.
Reduced Efnb2 and increased Ephb4 expression in P14 brain AV shunts following endothelial deletion of Rbpj from birth
The role of Notch in AV fate specification is well described (Kim et al., 2008; Lawson et al., 2001, 2002; Thurston and Yancopoulos, 2001; Zhong et al., 2001), and a gain-of-function Notch mutation leads to AV shunt formation associated with increased arterial marker and decreased venous marker expression (Murphy et al., 2012). To determine whether AV specification was affected in AV shunts following endothelial loss of Rbpj, we examined the expression of AV marker genes in P14 mouse brains. Coup-TFII (Nr2f2 – Mouse Genome Informatics), an orphan nuclear receptor, promotes venous specification by suppressing Notch (You et al., 2005); we hypothesized that Coup-TFII expression would not be altered following endothelial loss of Rbpj. β-gal expression in RbpjiΔEC; Coup-TFIIflox-nlacZ mutants was restricted to venous ECs and unchanged, compared with controls (Fig. 8B,C).
Fig. 8.
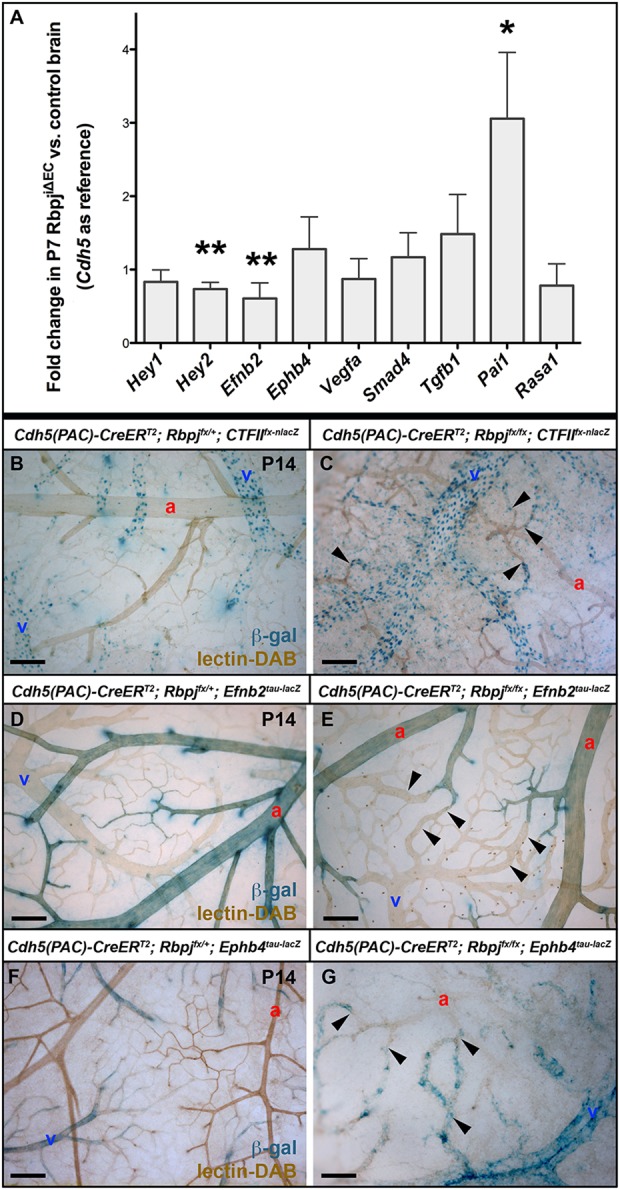
Altered gene expression in brains following endothelial deletion of Rbpj from birth. (A) Quantitative RT-PCR analysis on purified P7 brain ECs. Hey1 (P=0.0538), Hey2 (**P=0.0029) and Efnb2 (**P=0.0063) decreased; Ephb4 (P=0.1795), Vegfa (P=0.3118), Smad4 (P=0.2686), Tgfb1 (P=0.0806) and Rasa1 (P=0.2372) did not change significantly; and Pai1 (*P=0.0198) increased in the mutant. For each gene, N=6 mutant and control pairs; except Pai1 and Rasa1, N=4 pairs. Quantitative PCRs were run in triplicate. P-values were according to Student's t-test. (B-G) Whole-mount β-gal staining on P14 brains. (B,C) Coup-TFIIfx-nlacZ was expressed in veins (v), venules and the venous portion of AV connections in control (B) and mutant (C) brains. Arrowheads indicate AV shunts. N=4 controls, N=6 mutants. (D) In controls, Efnb2tau-lacZ was expressed by arterial (a) ECs, including the arterial portion of AV connections. (E) In mutants, Efnb2tau-lacZ expression was retained to a certain level in arteries and arterioles but was absent from AV shunts (arrowheads). N=7 controls, N=8 mutants. (F) In controls, Ephb4tau-lacZ was expressed by veins but not arteries. (G) In mutants, Ephb4tau-lacZ was misexpressed in AV shunts and arterioles (arrowheads). N=10 controls, N=5 mutants. Scale bars: 100 μm.
We analyzed the expression of Efnb2 and Ephb4, which encode downstream Notch effectors that are molecular markers for arterial and venous endothelial identity, respectively (Davy and Soriano, 2007). We hypothesized that ECs lacking Rbpj would have reduced Efnb2 expression and increased Ephb4 expression in P14 brains. In controls, β-gal produced by Efnb2tau-lacZ was expressed by ECs in arteries, arterioles and the arterial segment of capillaries. In RbpjiΔEC; Efnb2tau-lacZ brains, β-gal expression was retained in arteries and arterioles but was absent from AV shunts (Fig. 8D,E). Because Efnb2tau-lacZ expression provides a clear demarcation between β-gal-positive arterioles and β-gal-negative AV shunts, we measured the AV connection diameter and found it to be increased in RbpjiΔEC versus control brains expressing Efnb2tau-lacZ (supplementary material Fig. S3E-G).
In control and mutant brains, β-gal produced by Ephb4tau-lacZ was expressed by venous ECs, including the venous segment of capillaries (Fig. 8F,G). In RbpjiΔEC; Ephb4tau-lacZ brains, β-gal expression extended throughout the AV shunts and arterioles. Loss of endothelial Rbpj thus permitted misexpression of the venous marker Ephb4 in the arterial compartment and resulted in AV shunts devoid of Efnb2 arterial marker expression. Together, these results suggest that endothelial Rbpj is required to maintain the proper expression of genes specifying AV identity in the immature brain.
DISCUSSION
Here, we show that postnatal deletion of the transcription factor Rbpj, and thus Notch signaling, from the endothelium leads to features associated with AVMs. We detected AV shunts and enlarged, tortuous vessels in multiple vascular beds following endothelial deletion of Rbpj. Furthermore, we found that these effects are temporally regulated, with extensive AV shunting in the immature mouse brain, whereas the mature mouse brain is only mildly affected. Increased Pai1 expression, which was already significant in P7 brain ECs, suggests that TGFβ signaling is augmented prior to AV shunt formation. Downregulation of Efnb2 arterial marker expression and upregulation of Ephb4 venous marker expression indicated that the ECs in AV shunts are genetically reprogrammed to express venous and not arterial endothelial markers. Together, these results suggest that Rbpj is required in postnatal endothelium to maintain AV identity and to prevent AV shunting in mice. Our data demonstrate a novel role for endothelial Rbpj in the postnatal vasculature.
Rbpj is required in postnatal endothelium to prevent AVMs
We have shown that postnatal deletion of endothelial Rbpj leads to abnormal AV shunting and tortuous vessels, as features associated with AVMs, in multiple organs in immature and mature mice. Using both high-resolution imaging and a microsphere passage assay, we have shown that enlarged AV connections in the brain function as direct AV shunts in RbpjiΔEC mice. Consistent with our results, deletion of endothelial Rbpj from mouse embryos leads to abnormal vascular connections, permitting blood to flow from the cardiac outflow tract directly to the venous circulation (Krebs et al., 2004). However, there are important distinctions between the previously reported embryonic defects and the current postnatal findings. First, the AV connections and fusions in Rbpj mutant embryos are not bona fide AV shunts that displace a normal capillary bed. Second, the mutant embryos carry one Rbpjnull allele, which confounds interpretations of endothelial specificity. More recently, endothelial deletion of Rbpj has been shown to impair vascular remodeling both in the yolk sac (Copeland et al., 2011) and in the postnatal retina (Ehling et al., 2013), resulting in large diameter vessels; however, it is not known whether these vessels are direct AV shunts.
Our data also show that endothelial deletion of Rbpj from the adult vasculature results in abnormalities in select vascular beds. We report vessel enlargement or entanglement in adult uterus and gastrointestinal tract, but not in adult brain. Previous studies have shown that global deletion of Rbpj from adult mice leads to increased EC proliferation (Dou et al., 2008; Li et al., 2012; Wang et al., 2009) and vascular anomalies resembling veno-occlusive disease of the liver (Wang et al., 2009) and aortic valve disease (Li et al., 2012). Despite effects on the endothelium and vasculature, these studies did not report similar morphological defects to those of our study, such as vessel enlargement or entanglement. Importantly, the earlier studies are based on global deletion of Rbpj in the adult, whereas our study examines endothelial Rbpj deletion. Together, our results demonstrate specific requirements for Rbpj in postnatal endothelium to maintain AV identity and organization and to prevent AVMs.
Distinct roles for endothelial Rbpj to prevent AV shunt formation and to regulate vascular density
Avascular areas together with large, tortuous vessels within a tissue typically suggest AV shunting, as shunts may selectively steal blood flow from, and cause the regression of, smaller neighboring vessels (Murphy et al., 2012). However, we observed an overall increase in endothelial density in both forebrain and cerebellum, along with AV shunts. Because immature RbpjiΔEC mice do not survive past P21, the AV shunts might not advance enough to lead to blood flow-induced vessel enlargement and regression. Consistent with our results, increased vessel density and impaired vascular remodeling have been observed in yolk sac and in postnatal retina following deletion of endothelial Rbpj (Copeland et al., 2011; Ehling et al., 2013; Pitulescu et al., 2010), and increased vessel area was reported in the cerebrovasculature of another mouse model of BAVMs (Yao et al., 2013).
Conversely, a gain-of-function Notch4* mutation results in BAVMs that are accompanied by decreased vessel density in immature mice (Murphy et al., 2008). The disparity in the relationship between vessel density and AV shunts in Notch loss-of-function (RbpjiΔEC) and Notch gain-of-function (Notch4*) mutants suggests distinct, independent functions for Notch signaling in the endothelium. These distinctions also raise the possibility that brain AV shunts observed in immature RbpjiΔEC mice versus Notch4* mice may arise from different mechanisms, perhaps initially involving increased angiogenesis or impaired vessel remodeling; however, both lesions may eventually lead to vessel expansion via blood flow-mediated effects.
Deletion of endothelial Rbpj results in vascular abnormalities similar to those associated with Notch mutations in humans
Our results suggest that deletion of Rbpj from postnatal endothelium leads to hallmarks of clinically defined AVMs in mice affecting the brain, skin, uterus and intestine. Consistent with our data, it is reported that mutations in human RBPJ may lead to skin telangiectasias, which are small vascular malformations directly under the surface of the skin (Hassed et al., 2012). Currently, known genetic lesions account for very few human AVM cases (Young and Yang, 2004), and it is of upmost clinical interest to describe and investigate novel molecular mechanisms of AVM formation and progression. Our results support the possibility that RBPJ may underlie AVM pathology in human patients. Our RbpjiΔEC mice provide an elegant animal model with which to study the role of Rbpj in AVMs and to explore therapeutic avenues for AVM treatment.
Rbpj differentially regulates immature versus mature postnatal brain endothelium
Our data demonstrate that the mature brain endothelium is less susceptible to Rbpj deletion than the immature brain endothelium, which requires intact Rbpj. Deletion of endothelial Rbpj from birth results in AV shunts and hypoxic injury in the immature brain, whereas deletion during adulthood results in milder vascular abnormalities and ischemia. Adult endothelial Rbpj deletion leads to enlarged, tortuous vessels in mature vascular beds associated with the gastrointestinal tract, uterus and skin. Similarly, activation of the Notch4* mutation from birth results in brain AV shunts (Murphy et al., 2008), whereas Notch4* activation post-weaning leads to AV shunts in the gastrointestinal tract, uterus, skin and lung (Carlson et al., 2005; Miniati et al., 2010) but not in the brain (Carlson et al., 2005). Our results suggest that Rbpj is required in postnatal endothelium during two distinct temporal windows, but selectively in organs such as the brain. This differential regulation provides insight into AVM pathogenesis and the potential treatment of AVMs in select organs. Furthermore, as BAVM prevalence is highest among young adults (Hofmeister et al., 2000; Meyer-Heim and Boltshauser, 2003), animal models, such as RbpjiΔEC, that develop features of BAVMs in immature mice are most relevant to investigating mechanisms of BAVM pathogenesis.
Endothelial deletion of Rbpj from birth may interfere with capillary remodeling in the immature cerebrovasculature
Our timecourse data show that, by P8, abnormal AV connections are apparent in RbpjiΔEC brains. At this age, brain capillaries undergo progressive remodeling, including narrowing (Wang et al., 1992), and this process seems to be impaired in RbpjiΔEC brains, as a possible effect of endothelial deletion of Rbpj and a potential contributing factor in the initiation of AV shunting in the mutant brains. We did not detect capillary regression in RbpjiΔEC brains in this timecourse analysis. We did find that mutant AV shunts lack αSMA, suggesting that AV shunting originates from capillaries and/or venules. Defects in capillaries and/or venules might not be the only defects in RbpjiΔEC vasculature nor the only cause for AV shunt formation; however, our data are consistent with a primary abnormality at the capillary or venule level. Further investigation, including the identification of capillary-specific and venule-specific markers, will be required to further resolve the RbpjiΔEC vascular abnormalities.
Loss of endothelial Rbpj alters a downstream TGFβ effector in brain ECs prior to AV shunt formation
Our results show that endothelial expression of Pai1 was increased in RbpjiΔEC brains 7 days post-TAM injection at birth. Pai1 is regulated by TGFβ via Smad proteins (Blokzijl et al., 2003; Dong et al., 2002). Increased Pai1 suggests increased TGFβ signaling following endothelial deletion of Rbpj. Mutations in TGFβ pathway members are associated with AVMs in humans, mice and zebrafish (Corti et al., 2011; Johnson et al., 1996; Kim et al., 2012; McAllister et al., 1994; Park et al., 2008, 2009; Urness et al., 2000). Recently, TGFβ and Notch were shown to synergistically regulate retinal vascular morphogenesis (Larrivee et al., 2012) and BAVM formation (Yao et al., 2013) in mice. Our data suggest that Notch negatively regulates TGFβ signaling in postnatal brain endothelium.
Loss of endothelial Rbpj alters AV specification in the immature brain vasculature
The molecular identity of AVMs is unknown and, given that different genetic lesions can lead to AVMs, the expression of AV markers is likely to differ among AVMs. Notch is a crucial regulator of AV specification and is both necessary and sufficient to promote arterial and repress venous EC identity (Krebs et al., 2004; Murphy et al., 2012, 2008). Notch activity positively regulates arterial Efnb2 and negatively regulates venous Ephb4 in zebrafish (Lawson et al., 2001; Thurston and Yancopoulos, 2001; Zhong et al., 2001) and mouse (Kim et al., 2008) embryos. Consistent with this, we found that loss of endogenous Rbpj (and thus Notch signaling) from the endothelium results in reduced Efnb2 expression at P7 and P14 in mice. The increase in Ephb4 expression in isolated ECs at P7 was not significant, probably owing to mixed populations of isolated ECs. Ephb4 expression was increased beyond the veins and in AV shunts at P14, as assessed by whole-mount staining. Taken together, these data suggest that endothelial deletion of Rbpj, or loss of canonical Notch signaling, permits venous identity at the expense of arterial identity.
AV specification is also altered in AV shunts of the Notch4 gain-of-function brains; however, the identity of these gain-of-function Notch4 shunts is the converse of that of RbpjiΔEC shunts, as Efnb2 is increased and Ephb4 is decreased (Murphy et al., 2012). Given that both loss and gain of Notch signaling result in AV shunts, despite contrasting molecular identities, we propose that failure to maintain AV identity may underlie AV shunt formation in these mutant mice.
Tightly controlled Notch signaling is crucial for the maintenance of AV organization
Our results indicate temporal and spatial requirements for endothelial Rbpj in the postnatal vascular system. Deletion of Rbpj from the endothelium at birth leads to AV shunts in the brain and other organs, whereas deletion of Rbpj from adult endothelium does not lead to AV shunts in the brain, but does so in other organs. A similar temporal susceptibility in the brain vasculature is observed following endothelial expression of Notch4* (Murphy et al., 2012). As either loss or gain of Notch gene function leads to AV shunt formation, fine-tuned modulation of Notch signaling would be crucial to ensure safe and effective treatments for Notch-associated AVMs. Thus, our work defining the temporal requirements for Notch in brain endothelium might inform potential therapeutic strategies.
MATERIALS AND METHODS
Mice
Mouse lines: Cdh5(PAC)-CreERT2 (Sorensen et al., 2009), Rbpjflox (Tanigaki et al., 2002), Rosa26mT/mG (Muzumdar et al., 2007), Coup-TFIIflox-nlacZ (Takamoto et al., 2005), Efnb2tau-lacZ (Wang et al., 1998) and Ephb4tau-lacZ (Gerety et al., 1999) were kindly provided by Ralf Adams (Max Planck Institute for Molecular Biomedicine, Münster, Germany), Tasuku Honjo (Kyoto University, Japan), Liqun Luo (Stanford University School of Medicine, Stanford, CA, USA), Sophia Tsai (Baylor College of Medicine, Houston, TX, USA), and, the latter two, by David Anderson (California Institute of Technology, Pasadena, CA, USA), respectively. 100 μg TAM (Sigma) in 50 μl peanut oil (Planters) was injected into stomach at P1 and P2. Two milligram TAM in 100 μl peanut oil was injected intraperitoneally (IP) into adults once daily for five consecutive days. Animals were maintained and treated in accordance with UCSF IACUC guidelines.
Vascular staining and imaging
mGFP (produced by mT/mG) was imaged from whole-mount cortex, 8 μm cryosections and 1 mm cleared slices (Selever et al., 2011). For casting, 1% FITC-dextran (2000 kDa; Sigma)/1% low-melting Sea Plaque agarose (Cambrex)/PBS was transcardially perfused. Two-photon excitation fluorescence microscopy (2PEFM) was performed on casts for mGFP (mT/mG). 3D reconstructions were generated using ImageJ (NIH) and Imaris (Bitplane). BrdU imaging used Yokogawa spinning disk confocal microscopy and ImageJ at UCSF Biological Imaging Development Center. See also supplementary methods.
Immunostaining and β-gal detection
BrdU (Fisher; 100 mg/kg body weight in 0.9% saline) was injected IP at P5, P6 and P7. Immunostaining for BrdU followed Pitulescu et al. (2010). Whole-mount immunostaining with αSMA-Cy3 antibody (Sigma, C6198, Lot 079K4834) was according to Murphy et al. (2012), and with Rbpj antibody (1:100; Cell Signaling, 5313, Lot 1) followed the manufacturer's recommendations. Section immunostaining with αSMA-Cy3 (1:500; Sigma) and CD13-Alexa488 (1:1000; Serotec, MCA1270A488, Lot 0304) antibodies followed Nielsen and Dymecki (2010). Immunostaining against pimonidazole (HPI) followed the manufacturer's recommendations. β-gal detection followed Murphy et al. (2012). For lectin perfusion, 50 μg biotinylated or FITC-Lycopersicon esculentum (tomato) lectin (Vector Labs) in 125 μl PBS was injected into the IVC; detection was with Alexa 647-streptavidin or HRP-streptavidin (1:1000; Jackson ImmunoResearch) and DAB Peroxidase Substrate (Vector Labs).
Quantitative RT-PCR
P7 brain dissociation followed Zhou et al. (2014). Myelin was removed using Myelin Removal Beads (Miltenyi Biotec) and LS+ Positive Selection Columns (Miltenyi Biotec). ECs were sorted for mGFP (mT/mG) on a MoFlo cytometer (Beckman-Coulter) at UCSF Parnassus Flow Cytometry Core. Purity was >98%. Total RNA was isolated using the RNeasy Plus Micro Kit (Qiagen); cDNA was generated using the Transcriptor First Strand cDNA Synthesis Kit (Roche). Quantitative PCR was carried out using iQ SYBR Green (Bio-Rad) and DNA Engine Opticon 2 machinery (MJ Research). The comparative Ct method was used for data analysis (with Cdh5 as reference gene). Primers are listed in supplementary material Table S1.
Microsphere passage and hypoxia assays
Seventy-five microliters of 15 μm FluoSpheres (Invitrogen) were injected into the left common carotid artery. Hypoxia assay was performed as directed by the manufacturer (HPi-100, HPI). Image capture used Image-Pro Plus (Media Cybernetics) or SlideBook (Intelligent Imaging Innovations) software. For further details see supplementary methods.
Histology
Hematoxylin and Eosin (H&E) staining was performed by the Gladstone Histology and Light Microscopy Core.
Quantification analysis
ImageJ was used to measure smooth muscle cell layer thickness in mesentery, AV connection diameters, endothelium area (mGFP fluorescence), FITC-FluoSphere fluorescence and CD13 immunofluorescence. Photoshop Extended (Adobe) was used to measure the perimeter length of vessel lumens and the area of smooth muscle cell coverage in myocardial arteries.
Statistical analysis
Statistical analysis used Prism software (GraphPad). Unpaired Student's t-test with Welch's correction was used to compare data among groups. P<0.05 was considered significant. Error bars in figures represent s.d. from the mean.
Supplementary Material
Acknowledgements
We thank Lawrence Huang, Karyn Joanne Catalano, Luda Urisman and Allison Soung for technical assistance.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
C.M.N., R.A.W. and H.C. conceptualized and designed experiments; C.M.N., H.C., V.W.D. and Y.K. performed experiments; C.M.N., R.A.W., H.C., E.J.H. and V.W.D. analyzed data; C.M.N. and R.A.W. wrote the manuscript.
Funding
This research was supported by the National Institutes of Health [NIH R01 NS067420, NIH R56NS06742, NIH R01 HL075033], Vascular Cures (formerly the Pacific Vascular Research Foundation), Frank A. Campini Foundation, Mildred V. Strouss Trust, American Heart Association [grant-in-aid 10GRNT4170146 and GRNT 16850032] to R.A.W.; by Tobacco-Related Disease Research Grants Program (TRDRP) Office of the University of California [grant 20FT-0069 and NIH F32 HL110724] to C.M.N.; and by TRDRP [grant 20FT-0081] to H.C. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.108951/-/DC1
References
- Achrol, A. S., Guzman, R., Varga, M., Adler, J. R., Steinberg, G. K. and Chang, S. D. (2009). Pathogenesis and radiobiology of brain arteriovenous malformations: implications for risk stratification in natural history and posttreatment course. Neurosurg. Focus 26, E9 10.3171/2009.2.FOCUS0926 [DOI] [PubMed] [Google Scholar]
- Acker, T., Beck, H. and Plate, K. H. (2001). Cell type specific expression of vascular endothelial growth factor and angiopoietin-1 and -2 suggests an important role of astrocytes in cerebellar vascularization. Mech. Dev. 108, 45-57 10.1016/S0925-4773(01)00471-3 [DOI] [PubMed] [Google Scholar]
- Blokzijl, A., Dahlqvist, C., Reissmann, E., Falk, A., Moliner, A., Lendahl, U. and Ibanez, C. F. (2003). Cross-talk between the Notch and TGF-β signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J. Cell Biol. 163, 723-728 10.1083/jcb.200305112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, T. R., Yan, Y., Wu, X., Lam, M. T., Tang, G. L., Beverly, L. J., Messina, L. M., Capobianco, A. J., Werb, Z. and Wang, R. (2005). Endothelial expression of constitutively active Notch4 elicits reversible arteriovenous malformations in adult mice. Proc. Natl. Acad. Sci. USA 102, 9884-9889 10.1073/pnas.0504391102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland, J. N., Feng, Y., Neradugomma, N. K., Fields, P. E. and Vivian, J. L. (2011). Notch signaling regulates remodeling and vessel diameter in the extraembryonic yolk sac. BMC Dev. Biol. 11, 12 10.1186/1471-213X-11-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti, P., Young, S., Chen, C.-Y., Patrick, M. J., Rochon, E. R., Pekkan, K. and Roman, B. L. (2011). Interaction between alk1 and blood flow in the development of arteriovenous malformations. Development 138, 1573-1582 10.1242/dev.060467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davy, A. and Soriano, P. (2007). Ephrin-B2 forward signaling regulates somite patterning and neural crest cell development. Dev. Biol. 304, 182-193 10.1016/j.ydbio.2006.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, C., Zhu, S., Wang, T., Yoon, W. and Goldschmidt-Clermont, P. J. (2002). Upregulation of PAI-1 is mediated through TGF-β/Smad pathway in transplant arteriopathy. J. Heart Lung Transplant. 21, 999-1008 10.1016/S1053-2498(02)00403-5 [DOI] [PubMed] [Google Scholar]
- Dou, G.-R., Wang, Y.-C., Hu, X.-B., Hou, L.-H., Wang, C.-M., Xu, J.-F., Wang, Y.-S., Liang, Y.-M., Yao, L.-B., Yang, A.-G.et al. (2008). RBP-J, the transcription factor downstream of Notch receptors, is essential for the maintenance of vascular homeostasis in adult mice. FASEB J. 22, 1606-1617 10.1096/fj.07-9998com [DOI] [PubMed] [Google Scholar]
- Ehling, M., Adams, S., Benedito, R. and Adams, R. H. (2013). Notch controls retinal blood vessel maturation and quiescence. Development 140, 3051-3061 10.1242/dev.093351 [DOI] [PubMed] [Google Scholar]
- Fleetwood, I. G. and Steinberg, G. K. (2002). Arteriovenous malformations. Lancet 359, 863-873 10.1016/S0140-6736(02)07946-1 [DOI] [PubMed] [Google Scholar]
- Friedlander, R. M. (2007). Arteriovenous malformations of the brain. N. Engl. J. Med. 356, 2704-2712 10.1056/NEJMcp067192 [DOI] [PubMed] [Google Scholar]
- Friedman, W. A. (2013). Stereotactic radiosurgery of intracranial arteriovenous malformations. Neurosurg. Clin. N. Am. 24, 561-574 10.1016/j.nec.2013.05.002 [DOI] [PubMed] [Google Scholar]
- Gerety, S. S., Wang, H. U., Chen, Z.-F. and Anderson, D. J. (1999). Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol. Cell 4, 403-414 10.1016/S1097-2765(00)80342-1 [DOI] [PubMed] [Google Scholar]
- Hassed, S. J., Wiley, G. B., Wang, S., Lee, J.-Y., Li, S., Xu, W., Zhao, Z. J., Mulvihill, J. J., Robertson, J., Warner, J.et al. (2012). RBPJ mutations identified in two families affected by Adams-Oliver syndrome. Am. J. Hum. Genet. 91, 391-395 10.1016/j.ajhg.2012.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmeister, C., Stapf, C., Hartmann, A., Sciacca, R. R., Mansmann, U., terBrugge, K., Lasjaunias, P., Mohr, J. P., Mast, H. and Meisel, J. (2000). Demographic, morphological, and clinical characteristics of 1289 patients with brain arteriovenous malformation. Stroke 31, 1307-1310 10.1161/01.STR.31.6.1307 [DOI] [PubMed] [Google Scholar]
- Johnson, D. W., Berg, J. N., Baldwin, M. A., Gallione, C. J., Marondel, I., Yoon, S.-J., Stenzel, T. T., Speer, M., Pericak-Vance, M. A., Diamond, A.et al. (1996). Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 13, 189-195 10.1038/ng0696-189 [DOI] [PubMed] [Google Scholar]
- Kim, Y. H., Hu, H., Guevara-Gallardo, S., Lam, M. T. Y., Fong, S.-Y. and Wang, R. A. (2008). Artery and vein size is balanced by Notch and ephrin B2/EphB4 during angiogenesis. Development 135, 3755-3764 10.1242/dev.022475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J.-H., Peacock, M. R., George, S. C. and Hughes, C. C. W. (2012). BMP9 induces EphrinB2 expression in endothelial cells through an Alk1-BMPRII/ActRII-ID1/ID3-dependent pathway: implications for hereditary hemorrhagic telangiectasia type II. Angiogenesis 15, 497-509 10.1007/s10456-012-9277-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs, L. T., Xue, Y., Norton, C. R., Shutter, J. R., Maguire, M., Sundberg, J. P., Gallahan, D., Closson, V., Kitajewski, J., Callahan, R.et al. (2000). Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 14, 1343-1352. [PMC free article] [PubMed] [Google Scholar]
- Krebs, L. T., Shutter, J. R., Tanigaki, K., Honjo, T., Stark, K. L. and Gridley, T. (2004). Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 18, 2469-2473 10.1101/gad.1239204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs, L. T., Starling, C., Chervonsky, A. V. and Gridley, T. (2010). Notch1 activation in mice causes arteriovenous malformations phenocopied by ephrinB2 and EphB4 mutants. Genesis 48, 146-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrivée, B., Prahst, C., Gordon, E., del Toro, R., Mathivet, T., Duarte, A., Simons, M. and Eichmann, A. (2012). ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell 22, 489-500 10.1016/j.devcel.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson, N. D., Scheer, N., Pham, V. N., Kim, C. H., Chitnis, A. B., Campos-Ortega, J. A. and Weinstein, B. M. (2001). Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 128, 3675-3683. [DOI] [PubMed] [Google Scholar]
- Lawson, N. D., Vogel, A. M. and Weinstein, B. M. (2002). sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev. Cell 3, 127-136 10.1016/S1534-5807(02)00198-3 [DOI] [PubMed] [Google Scholar]
- Lawton, M. T. (2014). Seven AVMs: Tenets and Techniques for Resection, p. 352 New York: Thieme Medical Publishers. [Google Scholar]
- Lee, J. H. (2013). Complete reduction with traction of the infraorbital neurovascular bundle in a delayed case of white-eyed blow-out fracture. J. Craniofac. Surg. 25, e54-55. [DOI] [PubMed] [Google Scholar]
- Li, Z., Feng, L., Wang, C.-M., Zheng, Q.-J., Zhao, B.-J., Yi, W., Zhang, J.-Z., Wang, Y.-M., Guo, H.-T., Yi, D.-H.et al. (2012). Deletion of RBP-J in adult mice leads to the onset of aortic valve degenerative diseases. Mol. Biol. Rep. 39, 3837-3845 10.1007/s11033-011-1162-y [DOI] [PubMed] [Google Scholar]
- Limbourg, F. P., Takeshita, K., Radtke, F., Bronson, R. T., Chin, M. T. and Liao, J. K. (2005). Essential role of endothelial Notch1 in angiogenesis. Circulation 111, 1826-1832 10.1161/01.CIR.0000160870.93058.DD [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister, K. A., Grogg, K. M., Johnson, D. W., Gallione, C. J., Baldwin, M. A., Jackson, C. E., Helmbold, E. A., Markel, D. S., McKinnon, W. C., Murrell, J.et al. (1994). Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 8, 345-351 10.1038/ng1294-345 [DOI] [PubMed] [Google Scholar]
- Meyer-Heim, A. D. and Boltshauser, E. (2003). Spontaneous intracranial haemorrhage in children: aetiology, presentation and outcome. Brain Dev. 25, 416-421 10.1016/S0387-7604(03)00029-9 [DOI] [PubMed] [Google Scholar]
- Miniati, D., Jelin, E. B., Ng, J., Wu, J., Carlson, T. R., Wu, X., Looney, M. R. and Wang, R. A. (2010). Constitutively active endothelial Notch4 causes lung arteriovenous shunts in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 298, L169-L177 10.1152/ajplung.00188.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, P. A., Lam, M. T. Y., Wu, X., Kim, T. N., Vartanian, S. M., Bollen, A. W., Carlson, T. R. and Wang, R. A. (2008). Endothelial Notch4 signaling induces hallmarks of brain arteriovenous malformations in mice. Proc. Natl. Acad. Sci. USA 105, 10901-10906 10.1073/pnas.0802743105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, P. A., Lu, G., Shiah, S., Bollen, A. W. and Wang, R. A. (2009). Endothelial Notch signaling is upregulated in human brain arteriovenous malformations and a mouse model of the disease. Lab. Invest. 89, 971-982 10.1038/labinvest.2009.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, P. A., Kim, T. N., Lu, G., Bollen, A. W., Schaffer, C. B. and Wang, R. A. (2012). Notch4 normalization reduces blood vessel size in arteriovenous malformations. Sci. Transl. Med. 4, 117ra8 10.1126/scitranslmed.3002670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, A. L., Dally, M., Jeffreys, A., Hwang, P. and Anderson, J. F. (2013). Neuropsychological outcomes of stereotactic radiotherapy for cerebral arteriovenous malformations. J. Clin. Neurosci. 21, 601-606. 10.1016/j.jocn.2013.08.007 [DOI] [PubMed] [Google Scholar]
- Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L. and Luo, L. (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593-605 10.1002/dvg.20335 [DOI] [PubMed] [Google Scholar]
- Nielsen, C. M. and Dymecki, S. M. (2010). Sonic hedgehog is required for vascular outgrowth in the hindbrain choroid plexus. Dev. Biol. 340, 430-437 10.1016/j.ydbio.2010.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. O., Lee, Y. J., Seki, T., Hong, K.-H., Fliess, N., Jiang, Z., Park, A., Wu, X., Kaartinen, V., Roman, B. L.et al. (2008). ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood 111, 633-642 10.1182/blood-2007-08-107359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. O., Wankhede, M., Lee, Y. J., Choi, E. J., Fliess, N., Choe, S. W., Oh, S. H., Walter, G., Raizada, M. K., Sorg, B. S.et al. (2009). Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J. Clin. Invest. 119, 3487-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitulescu, M. E., Schmidt, I., Benedito, R. and Adams, R. H. (2010). Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat. Protoc. 5, 1518-1534 10.1038/nprot.2010.113 [DOI] [PubMed] [Google Scholar]
- Quillien, A., Moore, J. C., Shin, M., Siekmann, A. F., Smith, T., Pan, L., Moens, C. B., Parson, M. J. and Lawson, N. D. (2014). Distinct Notch signaling outputs pattern the developing arterial system. Development 141, 1544-1552 10.1242/dev.099986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selever, J., Kong, J.-Q. and Arenkiel, B. R. (2011). A rapid approach to high-resolution fluorescence imaging in semi-thick brain slices. J. Vis. Exp. pe2807. 10.3791/2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen, I., Adams, R. H. and Gossler, A. (2009). DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood 113, 5680-5688 10.1182/blood-2008-08-174508 [DOI] [PubMed] [Google Scholar]
- Takamoto, N., You, L.-R., Moses, K., Chiang, C., Zimmer, W. E., Schwartz, R. J., DeMayo, F. J., Tsai, M.-J. and Tsai, S. Y. (2005). COUP-TFII is essential for radial and anteroposterior patterning of the stomach. Development 132, 2179-2189 10.1242/dev.01808 [DOI] [PubMed] [Google Scholar]
- Tanigaki, K., Han, H., Yamamoto, N., Tashiro, K., Ikegawa, M., Kuroda, K., Suzuki, A., Nakano, T. and Honjo, T. (2002). Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat. Immunol. 3, 443-450 10.1038/ni793 [DOI] [PubMed] [Google Scholar]
- The Arteriovenous Malformation Study Group. (1999). Arteriovenous malformations of the brain in adults. N. Engl. J. Med. 340, 1812-1818 10.1056/NEJM199906103402307 [DOI] [PubMed] [Google Scholar]
- Thurston, G. and Yancopoulos, G. D. (2001). Developmental biology: gridlock in the blood. Nature 414, 163-164 10.1038/35102664 [DOI] [PubMed] [Google Scholar]
- Urness, L. D., Sorensen, L. K. and Li, D. Y. (2000). Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat. Genet. 26, 328-331 10.1038/81634 [DOI] [PubMed] [Google Scholar]
- Villa, N., Walker, L., Lindsell, C. E., Gasson, J., Iruela-Arispe, M. L. and Weinmaster, G. (2001). Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech. Dev. 108, 161-164 10.1016/S0925-4773(01)00469-5 [DOI] [PubMed] [Google Scholar]
- Wang, D.-B., Blocher, N. C., Spence, M. E., Rovainen, C. M. and Woolsey, T. A. (1992). Development and remodeling of cerebral blood vessels and their flow in postnatal mice observed with in vivo videomicroscopy. J. Cereb. Blood Flow Metab. 12, 935-946 10.1038/jcbfm.1992.130 [DOI] [PubMed] [Google Scholar]
- Wang, H. U., Chen, Z.-F. and Anderson, D. J. (1998). Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93, 741-753 10.1016/S0092-8674(00)81436-1 [DOI] [PubMed] [Google Scholar]
- Wang, L., Wang, C.-M., Hou, L.-H., Dou, G.-R., Wang, Y.-C., Hu, X.-B., He, F., Feng, F., Zhang, H.-W., Liang, Y.-M.et al. (2009). Disruption of the transcription factor recombination signal-binding protein-Jkappa (RBP-J) leads to veno-occlusive disease and interfered liver regeneration in mice. Hepatology 49, 268-277 10.1002/hep.22579 [DOI] [PubMed] [Google Scholar]
- Wang, Y., Nakayama, M., Pitulescu, M. E., Schmidt, T. S., Bochenek, M. L., Sakakibara, A., Adams, S., Davy, A., Deutsch, U., Lüthi, U.et al. (2010). Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483-486 10.1038/nature09002 [DOI] [PubMed] [Google Scholar]
- Yao, Y., Yao, J., Radparvar, M., Blazquez-Medela, A. M., Guihard, P. J., Jumabay, M. and Bostrom, K. I. (2013). Reducing Jagged 1 and 2 levels prevents cerebral arteriovenous malformations in matrix Gla protein deficiency. Proc. Natl. Acad. Sci. USA 110, 19071-19076 10.1073/pnas.1310905110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You, L.-R., Lin, F.-J., Lee, C. T., DeMayo, F. J., Tsai, M.-J. and Tsai, S. Y. (2005). Suppression of Notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature 435, 98-104 10.1038/nature03511 [DOI] [PubMed] [Google Scholar]
- Young, W. L. and Yang, G.-Y. (2004). Are there genetic influences on sporadic brain arteriovenous malformations? Stroke 35, 2740-2745 10.1161/01.STR.0000145054.35083.32 [DOI] [PubMed] [Google Scholar]
- Zacest, A. C., Caon, J., Roos, D. E., Potter, A. E. and Sullivan, T. (2013). LINAC radiosurgery for cerebral arteriovenous malformations: a single centre prospective analysis and review of the literature. J. Clin. Neurosci. 21, 241-245. 10.1016/j.jocn.2013.03.040 [DOI] [PubMed] [Google Scholar]
- Zhong, T. P., Childs, S., Leu, J. P. and Fishman, M. C. (2001). Gridlock signalling pathway fashions the first embryonic artery. Nature 414, 216-220 10.1038/35102599 [DOI] [PubMed] [Google Scholar]
- Zhou, L., Sohet, F. and Daneman, R. (2014). Purification of endothelial cells from rodent brain by immunopanning. Cold Spring Harb. Protoc. 2014, 65-77 10.1101/pdb.prot074963 [DOI] [PubMed] [Google Scholar]
- ZhuGe, Q., Zhong, M., Zheng, W., Yang, G.-Y., Mao, X., Xie, L., Chen, G., Chen, Y., Lawton, M. T., Young, W. L.et al. (2009). Notch-1 signalling is activated in brain arteriovenous malformations in humans. Brain 132, 3231-3241 10.1093/brain/awp246 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.