

Abstract
We used a single cell RNA-seq strategy to create an atlas of gene expression patterns in the developing kidney. At several stages of kidney development, histologically uniform populations of cells give rise to multiple distinct lineages. We performed single cell RNA-seq analysis of total mouse kidneys at E11.5 and E12.5, as well as the renal vesicles at P4. We define an early stage of progenitor cell induction driven primarily by gene repression. Surprising stochastic expression of marker genes associated with differentiated cell types was observed in E11.5 progenitors. We provide a global view of the polarized gene expression already present in the renal vesicle, the first epithelial precursor of the nephron. We show that Hox gene read-through transcripts can be spliced to produce intergenic homeobox swaps. We also identify a surprising number of genes with partially degraded noncoding RNA. Perhaps most interesting, at early developmental times single cells often expressed genes related to several developmental pathways. This provides powerful evidence that initial organogenesis involves a process of multilineage priming. This is followed by a combination of gene repression, which turns off the genes associated with most possible lineages, and the activation of increasing numbers of genes driving the chosen developmental direction.
Keywords: Kidney development, Single cell analysis, Multilineage priming, Mouse
INTRODUCTION
The kidney provides a powerful model system for the study of mechanisms of development (Costantini and Kopan, 2010). The kidney forms in the intermediate mesoderm, with mutual inductive interactions between the cap mesenchyme (CM) and the branching ureteric bud (UB) directing formation of the nephrons and collecting duct system. We have previously described a laser capture/microarray atlas of the compartment-specific gene expression patterns that drive kidney development (Brunskill et al., 2008). We then refined this atlas using a series of cell type-specific transgenic GFP mice. For example, by using Meis1-GFP, Tie2-GFP and Mafb-GFP mice, coupled with fluorescence-activated cell sorting (FACS), the glomerulus was subdivided into the mesangial, endothelial and podoctye constituent cell types (Brunskill et al., 2011a; Brunskill and Potter, 2010, 2012a). Additional transgenic GFP mice allowed the analysis of CM, renal vesicles and renin-producing cells (Brunskill and Potter, 2012b; Brunskill et al., 2011b).
This work began to delineate the genetic blueprint of kidney development, defining the changing waves of gene expression driving nephrogenesis. Novel compartment-specific molecular markers were identified (Georgas et al., 2009). The resulting data helped to direct the generation of additional transgenic mice to further facilitate the study of kidney development, and provided a useful resource for the kidney research community (Harding et al., 2011; McMahon et al., 2008).
In this report, we extend the kidney development atlas to single cell resolution. At several stages of nephrogenesis there are interesting cellular level heterogeneities within a histologically uniform compartment. For example, at E11.5, the metanephric mesenchyme consists of cells that will contribute to the stromal, nephron and vascular components of the kidney. Although in situ hybridization studies have provided some definition of the distinct gene expression patterns of these cells (Mugford et al., 2009), a single cell level analysis of their global gene expression profiles could result in deeper insight. Similarly, the CM consists of both uninduced progenitors and induced cells that have begun to differentiate into nephrons. A single cell study could separate these cells and characterize the early molecular mechanisms of the induction process. Furthermore, the renal vesicle is the initial epithelial precursor of the nephron. Although in situ hybridizations have begun to define the polarized gene expression of the renal vesicle (Georgas et al., 2009), a single cell analysis could provide a more global view of the early patterning of this nephron precursor.
We primarily used microfluidic/robotic tools, coupled with RNA-seq, to generate single cell gene expression profiles of the developing kidney. We defined the global gene expression patterns for a total of 235 cells taken from the E11.5 metanephric mesenchyme, E12.5 total kidney and P4 renal vesicles. We observed unexpected gene expression combinations, including cells transcribing both Six2 and Foxd1. The results also showed surprising RNA splicing patterns, including intergenic splicing of Hox gene transcripts that resulted in homeobox swaps. The data also identified an unexpected number of genes with partially degraded/incomplete transcripts, reminding us that one must exercise caution in drawing transcription/translation conclusions. Furthermore, the process of nephron progenitor induction was examined and an early phase driven primarily by gene repression was found. In addition, the results provide global gene expression blueprints, at single cell resolution, for the E11.5 metanephric mesenchyme, for several compartments of the E12.5 kidney, and for the proximal and distal cells of the renal vesicle. Finally, we found abundant evidence for multilineage priming in kidney progenitor cells.
RESULTS
E11.5 metanephric mesenchyme single cell analysis
At E11.5 the metanephric kidney consists of the UB, an outgrowth of the nephric duct, and the surrounding metanephric mesenchyme (MM) (Costantini and Kopan, 2010). At E11.5, the MM includes Six2-expressing cells, closest to the UB, that will give rise to all of the epithelial cells of the nephron (Boyle et al., 2008; Kobayashi et al., 2008a), and Foxd1-expressing cells, surrounding the Six2-positive cells, that will produce the stromal cells located between the nephrons, as well as the mesangial cells of the glomerulus (Humphreys et al., 2010). The Six2 compartment can be further subdivided, with Cited1-expressing cells representing the uninduced fraction of the nephron progenitors (Mugford et al., 2009).
We carried out single cell resolution gene expression profiling of the E11.5 MM in order to create an atlas of the gene expression states of the multiple cell types, to define single cell gene expression heterogeneities present within types, and to better understand the molecular mechanisms of the early lineage decisions taking place. Our first experiments were carried out manually. Approximately 100 cells were selected and cDNA was generated and amplified using the SCAMP protocol (Brunskill et al., 2011b). The products were screened by PCR and the cells divided into bins based on expression of compartment specific markers, including Six2, Cited1 and Foxd1. A total of 33 cells, representing different categories, were then used for gene expression profiling using Affymetrix Gene 1.0 St arrays.
The relationships of the resulting E11.5 MM single cell gene expression patterns are illustrated in the heatmap of Fig. 1. The cells are labeled according to their expression of Six2 (S), Foxd1 (F) and Cited1 (C). It is interesting that many genes show reduced expression in the induced Six2-only cells (S) compared with the uninduced Six2 plus Cited1 cells (SC). The induction process at E11.5 includes repression of Cited1, Nmd3, Sumo1, Exosc8, Bud31, Rbm25, Sec11a, Ube2l3, Ppp1r7, Ube2n and many other genes (supplementary material Table S1).
Fig. 1.

Heatmap of differentially expressed genes for 33 single cells from E11.5 metanephric mesenchyme. Cells are labeled according to Foxd1 (F), Six2 (S) and Cited1 (C), expression. The induced Six2-only cells (S) show a large number of genes with reduced expression compared with the uninduced Six2 plus Cited1 (SC) cells. The cells with both Six2 and Foxd1 (SF) most closely resemble the Six2 only (S) cells in gene expression profile. It is also interesting to note that the Foxd1 (F) cells are distinct from the other cells in their high level expression of many genes. U indicates universal standard RNA made from entire newborn mice. Cells that did not express Six2, Foxd1 or Cited1 are marked N. Red, blue and yellow indicate high, low and intermediate expression levels, respectively. Clustering was based on the entire gene expression pattern and was not based on just Six2, Foxd1 and Cited1 expression.
The most striking feature of the Foxd1-expressing cells (F), at the far left of the heatmap (Fig. 1), was the high number of genes with relatively strong expression compared with the Six2-only expressing cells, or even compared with the uninduced Six2 plus Cited1-expressing cells. Genes with relatively high expression in this compartment included Foxd1, Lgals1, Isy1, Anxa2, Actr10, Snrnp27, Tmco1, Snrnp48 and Emp3 (supplementary material Table S1). The utility of Anxa2 as a stromal compartment marker was validated by immunostaining (supplementary material Fig. S1).
Cells co-expressing both Six2 and Foxd1, markers of nephron and stromal lineages, respectively, are generally not thought to exist during kidney development (Costantini and Kopan, 2010). A total of six such cells were identified, however, with two of them also expressing Cited1 (Fig. 1). Both the Six2 and Foxd1 expression levels in these cells were robust, showing raw signal values of 500-6500, well above background, which was ∼30-50 for these experiments. Furthermore, the gene expression profiles of these cells argue that the results cannot be explained by contamination, with two cells mistakenly profiled as one. Four out of the Six2/Foxd1 cells showed gene expression patterns that closely resembled cells expressing Six2 only. These cells did not show the strong gene activation signature seen in the Foxd1-only positive cells.
To validate the existence of Six2/Foxd1 dual-expressing cells, we used the Foxd1-Cre transgenic mouse to permanently label cells with the floxed stop Rosa26-lacZ reporter. Although most of the labeled cells were indeed stromal, as expected, we did observe a significant number of renal epithelial cells that were lacZ positive (Fig. 2A). This suggests that at some point in their developmental history a few nephron epithelial cells do show Foxd1 expression. We also performed immunostaining, monitoring Six2 and GFP expression using a Foxd1GFP-CRE knock-in transgenic. Multiple double-positive cells at the periphery of the Six2-positive domain were found at E11.5 (Fig. 2B-D). We observed only extremely rare Six2 and Foxd1(GFP) double-positive cells at E15.5 (Fig. 2E-G). In summary, the single cell transcriptional profiles showed several percent of cells with dual Six2-Foxd1 expression, whereas the immunostaining results suggested significantly fewer Six2-Foxd1 double-positive cells.
Fig. 2.
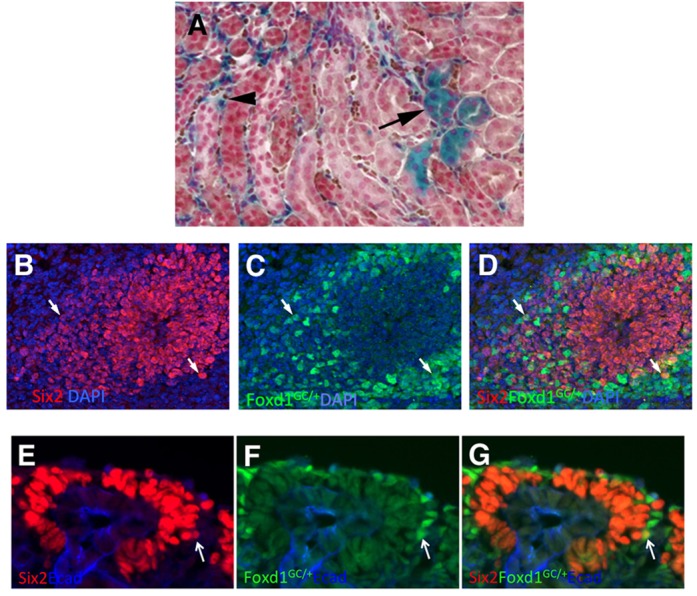
Cells expressing both Foxd1 and Six2. Cells expressing both Six2 and Foxd1 protein were present, but rarer than predicted by the transcriptional profiling. (A) A Foxd1-Cre transgenic mouse was mated to a floxed stop Rosa26-lacZ reporter mouse, with the resulting kidneys primarily showing stromal cell labeling, as expected (arrowhead), but with some tubular nephron epithelial cells also labeled (arrow). The counterstain used was nuclear Fast Red. (B-D) E11.5 kidney immunostained for Six2 (red) and Foxd1. The expression of Foxd1 was detected using a Foxd1-GFP-Cre knock-in mouse and GFP antibody. Two positive cells near the periphery of the Six2-positive population are marked with arrows. (E-G) E15.5 Foxd1-GFP-Cre knock-in embryos were immunostained for Six2 and GFP. A rare double-labeled cell, again at the periphery of the Six2-positive cells, is marked with an arrow.
We were also surprised to observe that the E11.5 MM single cells sometimes expressed a marker of a differentiated nephron cell type. For example, we have shown that Mafb is an excellent early podocyte marker (Brunskill et al., 2011a). At E11.5, there are no podocytes present, with the earliest sign of presumptive podocytes appearing in S-shaped bodies that first form at E12.5. Nevertheless, at E11.5 we observed two cells of the 33 total profiled that showed very robust (>1500 raw signal) expression of Mafb. The presence of such cells was confirmed by immunostaining (Fig. 3). Could these represent very early podocyte precursors, already lineage determined to make podocytes? Two lines of evidence suggest that this is not the case. First, these two cells did not express other podocyte markers. Indeed, another podocyte marker, podoplanin (Pdpn), also showed robust expression in two cells, but two different cells. This scattered pattern of expression was also observed for other podocyte markers. Second, the two cells shown expressing Mafb by microarray actually also expressed Foxd1, and not Six2, showing that they are probably in the stromal lineage and will not contribute to nephrons, which is where podocytes are found. The results suggest that progenitor cells can show occasional stochastic robust expression of genes associated with possible (or recently excluded) future developmental directions.
Fig. 3.
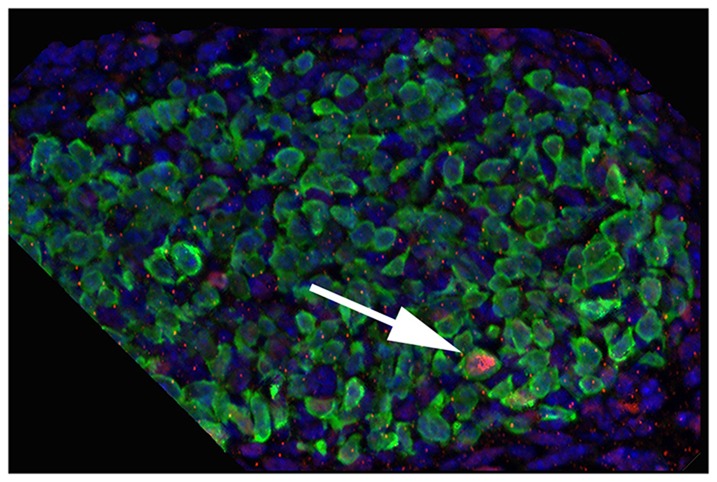
E11.5 MM cells show stochastic expression of markers of differentiated kidney cell types. The arrow shows a single cell with a strong immunostaining signal for Mafb (pink), a marker of glomerular podocytes. Blue, DAPI; green, Six2.
E11.5 single cell RNA-seq
We then re-examined the gene expression patterns of the E11.5 kidney using the microfluidics based Fluidigm C1 to define the RNA-seq gene expression profiles of an additional 58 single cells. We paid particular attention to the surprising microarray-based indication that E11.5 cells expressed unexpected combinations of genes. The RNA-seq data confirmed and extended these observations. For example, the RNA-seq data showed 22 of the 58 cells with significant (over 3 nRPKM) expression for Foxd1. Surprisingly, six of these cells also expressed significant levels of Six2.
Noncoding RNA
We observed that for many of the cells with Foxd1 transcription, the RNA-seq reads corresponded to only the 3′ UTR noncoding region of the gene (Fig. 4A). This suggests that Foxd1 transcripts present do not always encode protein. The single cell amplification technology used dT priming, which would detect only the 3′ ends of partially degraded RNAs. It is important to note that the RNA-seq reads for Gapdh showed quite uniform distribution across the length of the gene in almost all cells, arguing that the cell lysis/amplification/RNA-seq methods did not result in significant artifact RNA degradation. Instead, the restricted 3′ RNA-seq reads for some genes likely reflect differential turnover rates.
Fig. 4.

RNA-seq defined transcription and RNA processing patterns. (A) Foxd1. Cell 25 showed unexpected splicing (arrow) for this ‘intronless’ gene. Cell 50 showed transcripts restricted to the 3′ UTR (arrow), suggesting an absence of coding function. (B) Six2. Each cell shows a unique pattern of transcription/processing for the opposite strand (OS) LincRNA located 5′ of the Six2 gene. Cells 12 and 13 only showed transcripts from the 3′ exon. (C) Hox genes. Cell 4 showed intergenic splicing. Sequences within the second Hoxd12 exon served as a cryptic splice donor, providing an in-frame connection to the Hoxd11 second exon, resulting in a homeobox swap. Cell 29 used two novel exons, which were spliced with the second exon of Hoxd11. The positions of the genes, as per the UCSC genome browser are shown in blue, with introns marked by the lightest shade, and UTR sequences shown in the darkest shade. Arrows show unexpected splice events and the 3′ UTR transcripts of Foxd1.
It is also interesting to note that in two cells we observed a novel spliced form of the Foxd1 transcript. This removed a region including most of the 5′ UTR and part of the coding region, resulting in a 135 amino acid amino truncation of any possible resulting protein.
We also observed interesting single cell-specific variations in the patterns of transcription/processing of the Six2 opposite strand (OS) and canonical Six2 genes (Fig. 4B). For some cells, the Six2 RNA-seq reads detected were exclusively derived from the 3′ exon (cells 12, 13), and presumably noncoding. Single cells also showed unique patterns of transcription/processing of the Six2 OS region that would not be detected in ensemble averages defined by studies of population pools of cells.
Of interest, none of the single cells showed clear evidence of transcript-coding potential for both the Foxd1 and Six2 proteins. This then explains much of the disparity between the observed transcription patterns, with both microarrays and RNA-seq showing about 5% of cells with dual expression of Foxd1 and Six2 RNA, whereas the immunostaining and Foxd1-Cre results showed a lower frequency of expressing both proteins.
RNA splicing patterns
The single cell RNA-seq results provide a rich resource for the analysis of RNA splicing patterns. As an example consider the Hox genes. For cell 14, the Hoxd12 gene showed expected expression, with many reads from both of the two exons, as well as reads that spanned the single intron (Fig. 4C). The positions of the Hoxd11 and Hoxd12 genes are shown at the bottom of Fig. 4C, as represented in the UCSC genome browser. Surprisingly, cell 29 shows the expression of two novel exons that splice to the standard second exon of Hoxd11. The resulting 1927-base cDNA has limited coding potential. The longest open reading frame encodes a possible 133 amino acid protein, with no conserved domains. In another reading frame, however, this transcript encodes a short 37 amino acid peptide that includes the homeodomain DNA-binding recognition helix, with potentially interesting regulatory ramifications.
Cell 4 shows a novel intergenic splicing pattern. The Hoxd12 first exon and part of the second exon are spliced to the second exon of Hoxd11. The resulting 1341-base cDNA is particularly interesting as it provides an in frame connection between most of the coding sequence of Hoxd12 and the homeodomain-coding region of Hoxd11. Homeobox swap splicing for these two genes was the predominant transcription/processing pattern in this one cell, but it was not seen in any of the other cells.
Expression of differentiation-related genes
The RNA-seq data also validated the microarray results showing that the E11.5 MM cells expressed genes associated with differentiated cell types. We have previously defined the gene expression profiles of podocytes as a function of development, thereby identifying genes with particularly elevated expression in these unique cells (Brunskill et al., 2011a). We examined the single cell resolution expression of a number of podocyte related genes in the E11.5 kidney.
For some podocyte genes, including Nphs1, Nphs2 and Tdrd5, none of the E11.5 cells showed expression. At E11.5, these genes appear to be firmly repressed. For another group of podocyte genes, however, we observed seeming stochastic expression in a small number of cells. Synpo and Gpsm3 were each expressed in two cells, while Clic5, Rhpn1 and Sgip1 were each expressed in separate single cells. Similar to the array results, the expression of these genes appeared randomly distributed among the cells, with no single cell showing a strong podocyte signature.
Mafb was strongly expressed in eight cells, but for seven of these cells the detected RNA-seq reads were again restricted to the 3′ UTR, similar to what we observed for Foxd1 and Six2. The Mafb transcripts, therefore, appeared to often be partially degraded and noncoding. It is important to note that the RNA-seq data was filtered on post alignment read metrics to remove any reads that aligned to more than one place in the genome. The reads assigned to the 3′ UTRs of genes were therefore not the result of the presence of repetitive elements, with actual transcription occurring elsewhere in the genome.
E12.5 single cell RNA-seq
To further examine the progression of nephrogenesis, we carried out RNA-seq analysis of 86 cells of the E12.5 total kidney. To better define the process of CM induction, we compared gene expression profiles of uninduced (Six2 plus Cited1) and induced (Six2 only) cells, identifying 450 genes with differential expression [fold change (FC)>2] (supplementary material Table S2). Increasing the stringency to require FC>5 reduced the gene list to 129 (Fig. 5). It is interesting to note that at E12.5 the general pattern of gene activation/repression is distinct from that observed at E11.5. At E11.5 the predominant feature was the association of induction with gene repression. By contrast, at E12.5 there were more genes activated than repressed in the induced CM. This suggest that the first stage of induction is primarily driven by gene repression, whereas the second stage includes a predominant element of gene activation, as the CM cells begin to form pretubular aggregate/renal vesicles and undergo nephrogenesis.
Fig. 5.
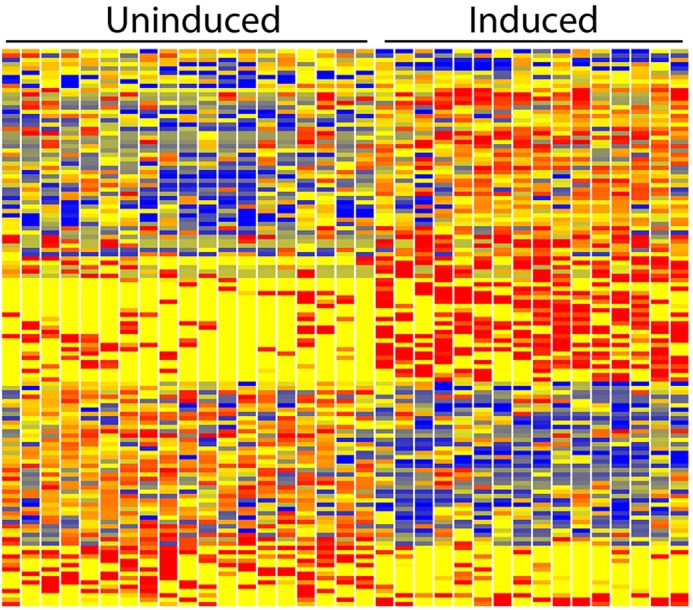
Heatmap of differential gene expression of E12.5 uninduced and induced CM single cells. Of interest, at this stage the induction process shows a combination of both strong gene activation and repression. This is in contrast with E11.5, where the predominant feature was gene repression. Red, blue and yellow indicate high, low and intermediate expression levels.
Gene ontology analysis of the 183 genes with higher expression (FC>2) in the uninduced CM revealed 21 genes encoding DNA-binding proteins (Smad4, Ddit3, Kdm6a, Safb, Tcf7L1, Meis3, Meox1, Foxc1, Prdm5, Zfhx2, Pole3, Mybbp1a, Etv6, Smg6, Mta1, Kmt2A, Calcoco1, Cited1, Mterfd3, Klf11, Zfhx2) and nine genes implicated in some manner in the Wnt signaling pathway (Ddit3, Ndel1, Kdm6a, Tcf7l1, Tmem237, Csnk1g1, Cited1, Emd, Calcoco1).
The ten genes with highest FC elevated expression in the uninduced CM compared with induced were Cited1 (75 FC), Ctsz (17 FC), Osr1 (17 FC), Coq9 (12 FC), Gprasp1 (10 FC), Arf2 (9 FC) Mllt11 (9 FC), Rmi1 (8 FC), Cpxm1 (7 FC) and Ccm2 (7 FC). Cited1 is a commonly used marker of the uninduced CM, Ctsz encodes the lysosomal proteinase cathepsin Z and Osr1 is a key regulatory transcription factor of kidney development (Xu et al., 2014). It is interesting that the strongly upregulated Ctsz, Gprasp1, Sort1, Cyth2 and Arf2 are all involved in protein vesicular trafficking and degradation. It is clear that as CM induction takes place there is a considerable shift in the functions of these pathways.
The ten genes with the highest fold change elevated expression in induced CM were Tmem100 (14 FC), G6pc3 (11 FC), Peo1 (11 FC), Ablim1 (11 FC), Tagln2 (10 FC), Zfp27 (9.6 FC), Hddc2 (9 FC), Nit1 (9 FC), Golph3l (9 FC) and Nsdhl (9 FC). The GUDMAP database confirms elevated expression of Tmem100 in induced CM (GUDMAP:14173).
Of interest, only three of the single cells showed significant Wnt4 expression. One of these (cell 43) was grouped as induced, without Cited1 expression, whereas two (cells 56 and 79) surprisingly did show Cited1 transcription and were therefore grouped as uninduced. This was confirmed by mating Wnt4-Cre mice to Rosa26 reporter mice, with some of the resulting labeled cells localized to regions of uninduced CM (supplementary material Fig. S2). A GO analysis of the genes showing higher expression in induced CM identified RNA modification activity as a key upregulated molecular function, including the RNA methylases Alkbh8, Rnmt, Tfb2m and Trmt5, and the RNA editing protein Adar.
E12.5 single cell CM versus stroma
We compared the E12.5 RNA-seq profiles of the 25 CM cells (Six2 positive) to the 12 stromal cells (Foxd1 positive), identifying 573 differentially expressed genes (FC>5) (supplementary material Table S3). A few of the interesting genes with strongly elevated (>10 FC) stromal expression included annexin A2 (Anxa2), secreted frizzled related protein 1 (Sfrp1), sphingosine-1-phosphate receptor 3 (S1pr3), snail2 (Snai2), Pdgfra and Wnt5a. Genes with elevated CM expression included Crym, Six2 and Eya1, each with FC>100, as well as Fgf10, Shroom3, Pdgfc, Itga8 and Meox2 (FC>10).
Single cell analysis of the renal vesicle
The renal vesicle is the first epithelial derivative of the CM progenitor cells, and contributes all of the epithelial components of the nephron (Boyle et al., 2008; Kobayashi et al., 2008a). How does this hollow ball of cells become a nephron? What are the gene expression programs driving its polarization and development?
Previous work using in situ hybridizations has shown that the RV already displays proximal/distal patterning at the gene expression level. It has been reported that Pou3f3 (Nakai et al., 2003), Dll1 (Kobayashi et al., 2008b), Sox9 (Reginensi et al., 2011), Dkk1, Papss2, Greb1, Pcsk9, Lhx1 and Bmp2 (Georgas et al., 2009) are expressed specifically in the distal RV, whereas Cdh6 (Cho et al., 1998), Wt1 (Georgas et al., 2008) and Tmem100 (Georgas et al., 2009) are expressed in the proximal RV. In addition, Ccnd1, Jag1, Ccdc86 and Wnt4 were reported to be expressed throughout the RV, but with higher expression in distal than proximal regions (Georgas et al., 2009).
In order to construct a more global definition of RV polarity, we again used single-cell RNA-seq gene expression profiling. P4 RV cells were isolated as previously described (Brunskill and Potter, 2012b). We carried out RNA-seq gene expression profiling of 58 single cells. Clustering produced 12 distal, 16 proximal and 29 equatorial cells.
The resulting single-cell RNA-seq data provides, to our knowledge, the first global view of the gene expression basis of early RV patterning. We identified 463 genes with proximal/distal polarized expression (FC>5) (supplementary material Table S4). Increasing the stringency to FC>10 gave 133 genes (Fig. 6). The RV cells expressed constellations of genes that were quite consistent with previous in situ hybridization results. Tmem100 (173 FC) and Wt1 (96 FC) showed elevated expression in the proximal RV cells. Genes with strongly elevated expression in the distal cells included Dkk1 (49 FC), Papss2 (220 FC), Dll1 (88 FC), Greb1 (28 FC), Sox9 (131 FC), Pou3f3 (Brn1) (31 FC), Lhx1 (5 FC) and Jag1 (2.4 FC), again consistent with previous in situ hybridization studies. The single cell RNA-seq data and previous in situ hybridization studies were therefore very strongly cross validating.
Fig. 6.
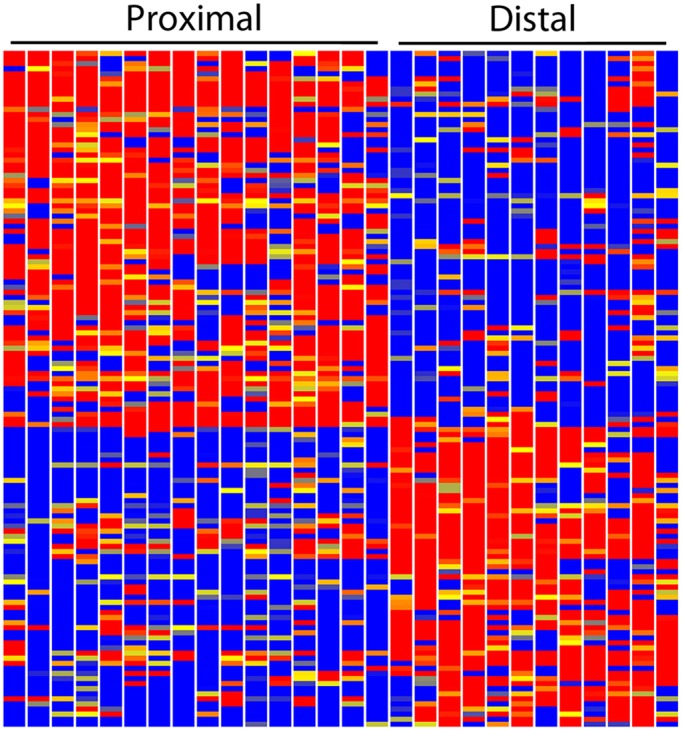
Heatmap showing polarized gene expression of the renal vesicle. The 16 single-cell replicates of the proximal RV and the 12 single-cell replicates of the distal RV show remarkably consistent differences in the expression levels of a large number of genes. Red, blue and yellow indicate high, low and intermediate expression levels.
It is also important to note that the gene expression profiles of the different single cells serve to cross validate each other. For example, we defined the RNA-seq gene expression profiles of 16 individual proximal RV cells, representing 16 biological replicates. The observed profound commonalities of the resulting 16 gene expression profiles provide key cross-confirmation.
There were, however, a few discrepancies between the RNA-seq and previous in situ hybridization datasets. Cdh6 (Cho et al., 1998), Bmp2, Wnt4, Ccdc86, Pcsk9 and Ccnd1 (Georgas et al., 2009) showed no significant proximal/distal RV polarity in the RNA-seq data. The possible causes of the differences include failure of the RNA-seq amplification method used to detect expression of a small percentage of genes (e.g. no expression of Pcsk9 was seen), the noisy nature of single cell profiling and/or possible problems with the in situ hybridization experiments.
The 463 genes with >5 FC polarized expression in the RV can be ranked according to FC. The 17 annotated genes with the greatest elevated expression in the distal RV (>20 FC) were Papss2, Sox9, Dll1, Pcdh7, Prnp, Dkk1, Tpd52, Etv5, Pou3f3, Greb1, Rbm47, Adamts9, Fam171a1, Sim1 Frmd4a, Etl4 and Prom1. Prnp encodes prion protein, and has been implicated in neuronal development and synaptic plasticity. In situ hybridization confirms that it is expressed strongly in a restricted manner in the developing kidney, consistent with the predicted distal RV (Gudmap:9112, 9113) (Eurexpress). Another distal gene of particular interest was Lrp4. Mutation of Lrp4 causes Cenanzi-Lenz syndrome, with limb abnormalities as well as renal hypoplasia or agenesis (Li et al., 2010). It is thought to work through the negative regulation of Wnt signaling, as an antagonist of LRP6.
Gene ontology analysis of the distal RV genes identified several biological processes of interest, including locomotion, response to external stimulus, chemotaxis, neurogenesis, ephrin signaling, regulation of cell proliferation and regulation of branching. Twenty genes involved in receptor binding were expressed in distal RV cells, including Bmp7, Dkk1, Dll1, Efna5, Epha4, Epha7, Itga1, Itga6, Sema4d, Sema6a and Socs2.
As demonstrated by the elegant studies of Lassar (Davis et al., 1987) and Yamanaka (Takahashi et al., 2007), it is possible to use a restricted set of transcription factors to reprogram cells to differentiate down different pathways, or to de-differentiate. The transcription factor code of the distal RV includes Bach2, Hoxa10, Hoxb6, Hoxb7, Mecom, Myc, Pou3f3 (Brn1), Sim1, Sox9, Txhz2, Uncx, Vopp1 and Yeats4.
The 20 genes with strongest upregulation in the proximal, compared with distal, RV, were Tmem100 (173 FC), H19 (115 FC), Wt1 (96 FC), Pmp22 (65 FC), Sincaip (47 FC), Sulf1 (42 FC), Nid1 (38 FC), Mafb (37 FC), Plat (36 FC), Unc5c (34 FC), Prr5l (29 FC), Car12 (29 FC), Efnb2 (25 FC), Napa (24 FC), Tshz1 (24 FC), Acaa2 (23 FC), Msra (23 FC), Dab1 (23 FC), Rasl11a (22 FC) and Tspan15 (22 FC). As observed for earlier developmental time points, the RNA-seq data suggested that for some genes the observed transcripts were largely noncoding. For Mafb, we confirmed this by immunostaining, with no Mafb protein detected in the proximal RV cells (supplementary material Fig. S3).
The transcription factor code for the proximal RV includes Eya2, Foxc2, Hey2, Jun, Tshz1, Wt1 and Zeb2. In addition, Meis1, which is generally thought to be a marker of stromal and mesangial cells, showed robust transcription in 5/16 proximal RV cells. Gene ontology analysis produced top biological processes for proximal enriched genes, including neurogenesis (interestingly, similar to distal RV), blood vessel morphogenesis, kidney development, regulation of locomotion and epithelium development.
Is the RV pre-patterned?
Analysis of the of RV single cell expression patterns of segment specific markers could support a homunculus model, with the RV representing a pre-patterned nephron. Perhaps the individual RV cells have already chosen their developmental directions. Alternatively, the RV could be a blank slate, partially polarized but awaiting appropriate external growth factor cues to drive regional differentiation.
In favor of a pre-patterned model, most of the proximal RV cells already showed a strong podocyte gene expression signature. Wt1, a marker of podocytes in the adult kidney, was expressed in all proximal RV cells. We observed Mafb transcripts in 11 proximal cells, nine equatorial cells and no distal cells. Other podocyte-associated genes, including Sncaip, Sulf1 and Plat 1 (Brunskill et al., 2011a), showed similar expression patterns, with elevated expression in many proximal RV cells, some equatorial and very few distal cells. Seventeen cells expressed at least four of the five podocyte-associated genes, Mafb, Wt1, Sulf1, Sncaip and Plat (Fig. 7, yellow diamonds). This did not, however, extend to all podocyte-marker genes. For example Nphs2, Podxl, Pdpn and Synpo, were not detectably expressed in any of the RV cells. These genes apparently are only activated as a function of further podocyte differentiation.
Fig. 7.
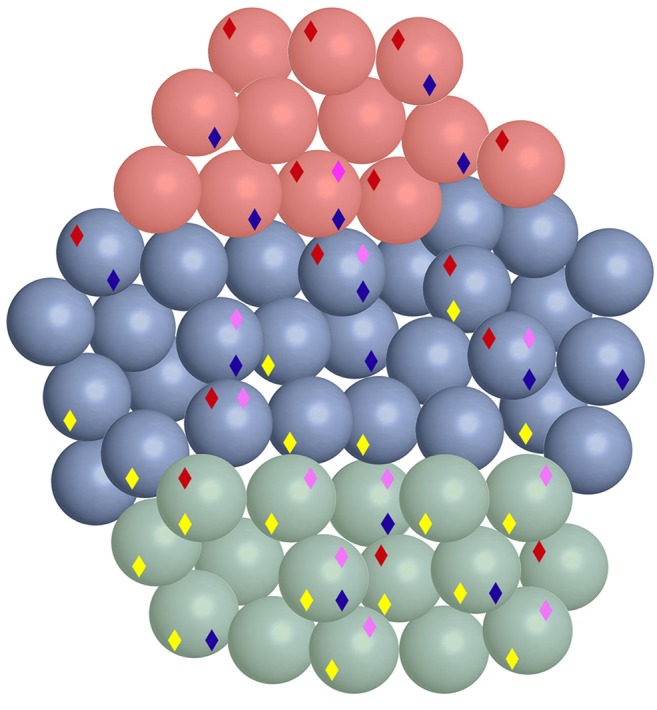
Multilineage priming of renal vesicle cells. The 58 RV single cells are color coded: proximal (green), distal (red) and equatorial (blue). Diamonds indicate gene expression patterns reflecting distinct differentiated cell types: yellow, podocytes; purple, proximal tubules; blue, parietal epithelial cell; red, distal tubule. Of interest, many cells show dual character, indicating multilineage priming.
It was surprising that such a large percentage of RV cells appeared developmentally primed to make the podocyte, which represents only a single cell type of the glomerulus of the adult nephron. To pursue this further, we examined the expression patterns of additional genes associated with other nephron cell types. We used GUDMAP data (gudmap.org) (Brunskill et al., 2008) to identify 19 genes expressed in the RV that were also strongly associated with the proximal tubules (Clcn5, Akr1c18, Dhcr7, Dll1, Erdr1, Gcfc1, Grm4, Higd1c, Irx3, Lama1, Mir17hg, Pcsk9, Pdss1, Slc11a2, Slc30a6, Slc37a4, Tagap, Timm9, Tmem144 and Zfand1). We then examined the gene expression profiles of each of the individual 58 RV cells with regard to these markers. Eleven single cells expressed at least six of these genes and therefore appeared lineage primed to make proximal tubule (Fig. 7, purple diamonds).
This analysis showed that many cells expressed combinations of markers, suggesting simultaneous lineage priming in more than one direction. Cells 22 and 31, for example, both expressed many markers of both proximal tubules and podocytes. This general result was further confirmed by examining expression patterns for other cell type-specific markers. For example, Cdh6 is expressed in parietal epithelial cells (Cho et al., 1998). Fifteen of the 58 total RV cells showed Cdh6 expression (Fig. 7, blue diamonds). Another cadherin, Cdh16, is a marker of the distal tubule (Shen et al., 2005). Thirteen of the 58 RV cells expressed Cdh16 (Fig. 7, red diamonds). The striking observation was the large number of cells expressing genes associated with multiple differentiation lineages. The results strongly suggest that the progenitor cells of the RV can simultaneously prime in multiple developmental directions.
The single-cell view of the RV shown in Fig. 7 is simplified. For example, in order to be marked with the yellow podocyte diamond a cell must express four out of the five (Sulf1, Sncaip, Plat, Mafb, Wt1) podocyte-associated genes examined. Additional cells express a smaller fraction of these genes and therefore have a more modest podocyte character. Use of a les stringent definition of lineage character would create a more-complex view, with many more cells showing gene expression signatures of additional cell types. Indeed when each cell is examined for expression levels of each individual marker, it is apparent that essentially all cells express multiple genes associated with distinct differentiation lineages (supplementary material Fig. S4).
DISCUSSION
We have created a single cell resolution atlas of gene expression during early kidney development. Global gene expression profiles were defined for 235 single cells from E11.5 and E12.5 total kidneys as well as from P4 renal vesicles. The results provide a marker-free unbiased view of the gene expression patterns driving nephrogenesis. The data can be analyzed using a rich variety of methods. All of the transcription factors, growth factors and receptors expressed in each compartment can be determined and compared. In addition, this single cell resolution resource defines the complete repertoire of transcription/RNA-processing patterns present, revealing surprising heterogeneities that could not be found by studies examining ensemble averages of populations of cells.
Multilineage priming
The single cell RNA-seq data can be used to distinguish two alternative models of development. In the first model, progenitor cells would start as a blank slate, not expressing differentiation marker genes associated with any lineage. Then as the cells were induced to move in a particular developmental direction, they would gradually transcribe increasing numbers of genes associated with that specific linage. The single cell results, however, strongly support a different model, with progenitor cells expressing marker genes associated with multiple distinct lineages, thereby showing multilineage priming. Developmental progression would then require a combination of the activation of additional genes specific to the chosen differentiated state, and more complete repression of genes involved in alternative pathways.
This model is consistent with previous studies. A single cell analysis study examined pluripotent epiblast and primitive endoderm formation within the inner cell mass of the mouse blastocyst (Ohnishi et al., 2014). At E3.25, there was stochastic expression of markers of both lineages in most cells, but by E3.5 there was a combination of gene repression and activation that began to separate the two cell types, and by E4.5 there were two distinct populations of pluripotent epiblast and primitive endoderm cells, with each showing highly restricted expression of lineage specific genes.
The hematopoietic system provides another example of multilineage priming. Single cell RT-PCR was used to show that hematopoietic stem cells ‘prime several different lineage affiliated programs of gene activity prior to unilineage commitment and differentiation’ (Hu et al., 1997). The results showed that hematopoietic lineage specification is preceded by a phase of ‘promiscuous’ multilineage gene expression. Additional single cell studies have further refined our understanding of this process, showing that as subsequent populations of progenitors are formed there is a combined repression of genes associated with some lineages and activation of genes associated with others that then reflect their more restricted developmental potential (Akashi et al., 2000; Mansson et al., 2007; Miyamoto et al., 2002).
Also consistent with our findings, it has been shown that during development important regulator genes often show bivalent active (H3K4me3) and repressive (H3K27me3) histone modifications, marking genes that are poised for expression (Azuara et al., 2006; Mikkelsen et al., 2007). Studies of early mouse embryos found bivalent histone modifications associated with developmental regulator genes that were generally repressed (Rugg-Gunn et al., 2010). These observations are in agreement with our nephron progenitor results, showing, for example, that only a small percentage of E11.5 MM cells show strong expression of the podocyte marker gene Mafb. In a population pool analysis of these cells, Mafb would look strongly repressed, but in a single cell study it is found that a few cells escape repression and show strong Mafb transcription.
Noncoding transcripts
The single cell RNA-seq analysis of early nephrogenesis described in this report defines several distinct types of noncoding RNAs. LincRNAs are found enriched in the nucleus, show very little evidence of coding potential and are under evolutionary selective constraint, although weaker than for protein-coding genes (Derrien et al., 2012). In ES cells, about 60% of LincRNAs appear to originate from divergent transcription of promoters of protein-coding genes (Sigova et al., 2013). We describe one such example, the Six2 gene, that shows concordant expression of an opposite strand LincRNA arising from divergent transcription of a single promoter region (Fig. 4). Each individual cell showed a strongly preferred transcription/RNA-processing for the Six2 opposite strand transcript. In a study of pools of cells this would not have been determined.
We were surprised to observe that for a number of protein-coding genes almost all of the detected RNA-seq reads were restricted to the 3′ noncoding UTR. This did not appear to be the result of general RNA degradation, as many other genes from the same cell, including the housekeeping Gapdh, showed reads extending relatively uniformly across the exons, including the 5′ ends. It therefore likely reflects selective RNA degradation. These results remind us that the presence of RNA does not always equate with protein. For some genes, there are abundant partially degraded transcripts that cannot encode protein.
An earlier study examined gene expression patterns of single cells of the embryonic and adult pancreas (Chiang and Melton, 2003). Although the E10.5 pancreatic epithelium is morphologically uniform, it was possible to define gene expression heterogeneities that divided the cells into six groups and to propose a developmental progression of gene expression. Of interest, some cells showed unexpected combinations of expressed genes. These could not all be confirmed by immunostaining, however, suggesting that there ‘may be translational controls’. Our results suggest that noncoding/partially degraded transcripts could provide an explanation for their discordant RNA versus protein observations. It is also interesting to note that many enhancers are transcribed (De Santa et al., 2010; Kim et al., 2010), with evidence suggesting the functional importance of enhancer transcripts in gene regulation (Li et al., 2013).
RNA transcription/processing patterns
The single cell RNA-seq data provides a valuable resource for the study of alternative patterns of RNA processing. We observed surprising single-cell specificity in RNA-processing patterns, with the Hox genes providing dramatic illustration. We focused on Hoxd11 and Hoxd12, which showed alternative exon use as well as astonishing in-frame intergenic splicing that swapped the homeodomain of Hoxd11 into the Hoxd12 protein. These two genes, however, represent only two examples. Others include the almost exclusive use of an alternative exon for Hoxa10, splicing of Mir196b transcript into a cryptic splice acceptor of Hoxa10 and the presence of very abundant polyadenylated unspliced intergenic transcripts from the Hox clusters in RV cells. And these are only examples from the Hox clusters. The single cell RNA-seq data clearly provides a rich resource for the analysis of gene transcription and RNA processing.
MATERIALS AND METHODS
Single cell suspensions
We generated E11.5 and E12.5 total mouse kidney single cell suspensions by first mincing with a razor blade followed by digestion with 0.25% trypsin-EDTA at 37°C for 5 min, with vigorous trituration. Trypsin was stopped with 1% FBS/PBS. Cells were passed through 100 μm, 40 μm and 20 μm mesh filters, pelleted at 500 g for 5 min, resuspended in 1 ml Red Blood Cell Lysis buffer (Sigma) and pelleted again. This was repeated until no red cells were visible in the pellet. Cells were then resuspended in 1% FBS/PBS, and the concentration adjusted to 350,000/ml. P4 RV cells were prepared using FACS as previously described (Brunskill and Potter, 2012b).
Single cell gene expression profiling
The manual/microarray single cell gene expression profiling of E11.5 kidney was carried out as previously described (Brunskill et al., 2011b). Microfluidics/RNA-seq analysis of E11.5 and E12.5 kidney, as well as P4 RV, was carried out using the Fluidigm C1 system coupled with the Clontech UltraLow SMARTer amplification chemistry, and Illumina/Nextera tagmentation-barcoding, as per Fluidigm recommended protocols. Sequencing with the Illumina HiSeq2500 was carried out with single-end, 100 base reads, and an average per cell read depth of 2.6 million. The data are available at GEO under accession numbers GSE59127, GSE59129, GSE59130.
Mice and antibodies
Mice used were Wnt4GFPcre (knock-in); Wnt4tm3(EGFP/cre)Amc, Gt(ROSA)26Sortm1Sor and Foxd1-Cre (gifts from Andrew P. McMahon); Jax: B6;129S4-Foxd1tm1(GFP/cre)Amc/J, Rosa26-lacZ, Jax: B6N.129S4(B6)-Gt(ROSA)26Sortm1Sor/CjDswJ and Foxd1 GCE (knock-in Foxd1 CRE GFP ERT2; MGI:4437895); and Foxd1-GFP/CRE (transgenic; MGI:3848354). The tamoxifen concentration used was 3 mg per 40 g body weight.
Primary antibodies used were against Jag1 (DSHB, TS1.15H; 1:20), pan-cytokeratin (Sigma, C2562; 1:200), β-galactosidase (MPbio, #559761; 1:15,000), Six2 (Proteintech, 11562-1-AP; 1:500), GFP (Aves, GFP-1020; 1:500), MafB (Sigma, HPA005653; 1:500), annexin A2 (Cell Signaling, 8235S; 1:100). Secondary antibodies used for supplementary material Fig. S2 were A555 anti-rabbit (Invitrogen, A31572), A647 anti-mouse IgG1 (Invitrogen, A21126), A488 anti-rat (Invitrogen, A21208); for Anxa2: Invitrogen Alexa Fluor 488 anti-rabbit; for MafB: Jackson Laboratories Alexa Fluor 488 anti-chicken.
Data analysis
The pipeline used for the generation of RNA-seq Bam files has been previously described (Brunskill and Potter, 2012b). We primarily used GeneSpring 12.6.1 for data analysis. We filtered to remove duplicate reads, as well as reads that aligned to more than one place in the genome. We then removed genes with only low expression by requiring a minimum of three nRPKM in at least one sample. Cells were in some cases grouped according to the expression of predefined marker genes. Differential expression was determined by replicate analysis, unpaired t-test, P≤0.05, without correction, and with subsequent screening based on fold change. Gene ontology analysis was typically carried out in duplicate, with both GeneSpring and ToppGene (http://toppgene.cchmc.org/enrichment.jsp), with similar results.
Supplementary Material
Acknowledgements
We thank Hung-Chi Liang and Shawn Smith for carrying out the Fluidigm-based single-cell RNA-seq data generation.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
E.W.B. carried out the isolation of tissues, preparation of single cells, manual SCAMP amplifications for microarray analysis, analysis of Six2/Foxd1 double-positive cells, and helped in the analysis of data and writing of the paper. J.-S.P. and E.C. performed immunostains and helped in the writing of the paper. F.C. was responsible for the germline Foxd1-Cre/floxed stop lacZ reporter experiments showing Foxd1 expression in the nephron epithelial lineage. B.M. performed immunostains. S.S.P. was primarily responsible for data analysis and writing of the paper.
Funding
This work was supported by the National Institutes of Health (NIH) [RC4 DK090891]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.110601/-/DC1
References
- Akashi K., Traver D., Miyamoto T., Weissman I. L. (2000). A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404, 193-197 10.1038/35004599 [DOI] [PubMed] [Google Scholar]
- Azuara V., Perry P., Sauer S., Spivakov M., Jørgensen H. F., John R. M., Gouti M., Casanova M., Warnes G., Merkenschlager M., et al. (2006). Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 8, 532-538 10.1038/ncb1403 [DOI] [PubMed] [Google Scholar]
- Boyle S., Misfeldt A., Chandler K. J., Deal K. K., Southard-Smith E. M., Mortlock D. P., Baldwin H. S., de Caestecker M. (2008). Fate mapping using Cited1-CreERT2 mice demonstrates that the cap mesenchyme contains self-renewing progenitor cells and gives rise exclusively to nephronic epithelia. Dev. Biol. 313, 234-245 10.1016/j.ydbio.2007.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill E. W., Potter S. S. (2010). Gene expression programs of mouse endothelial cells in kidney development and disease. PLoS ONE 5, e12034 10.1371/journal.pone.0012034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill E. W., Potter S. S. (2012a). Changes in the gene expression programs of renal mesangial cells during diabetic nephropathy. BMC Nephrol. 13, 70 10.1186/1471-2369-13-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill E. W., Potter S. S. (2012b). RNA-Seq defines novel genes, RNA processing patterns and enhancer maps for the early stages of nephrogenesis: Hox supergenes. Dev. Biol. 368, 4-17 10.1016/j.ydbio.2012.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill E. W., Aronow B. J., Georgas K., Rumballe B., Valerius M. T., Aronow J., Kaimal V., Jegga A. G., Grimmond S., McMahon A.P., et al. (2008). Atlas of gene expression in the developing kidney at microanatomic resolution. Dev. Cell 15, 781-791 10.1016/j.devcel.2008.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill E. W., Georgas K., Rumballe B., Little M. H., Potter S. S. (2011a). Defining the molecular character of the developing and adult kidney podocyte. PLoS ONE 6, e24640 10.1371/journal.pone.0024640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill E. W., Sequeira-Lopez M. L. S., Pentz E. S., Lin E., Yu J., Aronow B. J., Potter S. S., Gomez R. A. (2011b). Genes that confer the identity of the renin cell. J. Am. Soc. Nephrol. 22, 2213-2225 10.1681/ASN.2011040401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang M.-K., Melton D. A. (2003). Single-cell transcript analysis of pancreas development. Dev. Cell 4, 383-393 10.1016/S1534-5807(03)00035-2 [DOI] [PubMed] [Google Scholar]
- Cho E. A., Patterson L. T., Brookhiser W. T., Mah S., Kintner C., Dressler G. R. (1998). Differential expression and function of cadherin-6 during renal epithelium development. Development 125, 803-812. [DOI] [PubMed] [Google Scholar]
- Costantini F., Kopan R. (2010). Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev. Cell 18, 698-712 10.1016/j.devcel.2010.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R. L., Weintraub H., Lassar A. B. (1987). Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51, 987-1000 10.1016/0092-8674(87)90585-X [DOI] [PubMed] [Google Scholar]
- De Santa F., Barozzi I., Mietton F., Ghisletti S., Polletti S., Tusi B. K., Muller H., Ragoussis J., Wei C.-L., Natoli G. (2010). A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 8, e1000384 10.1371/journal.pbio.1000384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D. G., et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775-1789 10.1101/gr.132159.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgas K., Rumballe B., Wilkinson L., Chiu H. S., Lesieur E., Gilbert T., Little M. H. (2008). Use of dual section mRNA in situ hybridisation/immunohistochemistry to clarify gene expression patterns during the early stages of nephron development in the embryo and in the mature nephron of the adult mouse kidney. Histochem. Cell Biol. 130, 927-942 10.1007/s00418-008-0454-3 [DOI] [PubMed] [Google Scholar]
- Georgas K., Rumballe B., Valerius M. T., Chiu H. S., Thiagarajan R. D., Lesieur E., Aronow B. J., Brunskill E. W., Combes A. N., Tang D., et al. (2009). Analysis of early nephron patterning reveals a role for distal RV proliferation in fusion to the ureteric tip via a cap mesenchyme-derived connecting segment. Dev. Biol. 332, 273-286 10.1016/j.ydbio.2009.05.578 [DOI] [PubMed] [Google Scholar]
- Harding S. D., Armit C., Armstrong J., Brennan J., Cheng Y., Haggarty B., Houghton D., Lloyd-MacGilp S., Pi X., Roochun Y., et al. (2011). The GUDMAP database--an online resource for genitourinary research. Development 138, 2845-2853 10.1242/dev.063594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M., Krause D., Greaves M., Sharkis S., Dexter M., Heyworth C., Enver T. (1997). Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev. 11, 774-785 10.1101/gad.11.6.774 [DOI] [PubMed] [Google Scholar]
- Humphreys B. D., Lin S.-L., Kobayashi A., Hudson T. E., Nowlin B. T., Bonventre J. V., Valerius M. T., McMahon A. P., Duffield J. S. (2010). Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 176, 85-97 10.2353/ajpath.2010.090517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.-K., Hemberg M., Gray J. M., Costa A. M., Bear D. M., Wu J., Harmin D. A., Laptewicz M., Barbara-Haley K., Kuersten S., et al. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182-187 10.1038/nature09033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A., Valerius M. T., Mugford J. W., Carroll T. J., Self M., Oliver G., McMahon A. P. (2008a). Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3, 169-181 10.1016/j.stem.2008.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Terada Y., Kuwana H., Tanaka H., Okado T., Kuwahara M., Tohda S., Sakano S., Sasaki S. (2008b). Expression and function of the Delta-1/Notch-2/Hes-1 pathway during experimental acute kidney injury. Kidney Int. 73, 1240-1250 10.1038/ki.2008.74 [DOI] [PubMed] [Google Scholar]
- Li Y., Pawlik B., Elcioglu N., Aglan M., Kayserili H., Yigit G., Percin F., Goodman F., Nürnberg G., Cenani A., et al. (2010). LRP4 mutations alter Wnt/beta-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am. J. Hum. Genet. 86, 696-706 10.1016/j.ajhg.2010.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Notani D., Ma Q., Tanasa B., Nunez E., Chen A. Y., Merkurjev D., Zhang J., Ohgi K., Song X., et al. (2013). Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498, 516-520 10.1038/nature12210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Månsson R., Hultquist A., Luc S., Yang L., Anderson K., Kharazi S., Al-Hashmi S., Liuba K., Thorén L., Adolfsson J., et al. (2007). Molecular evidence for hierarchical transcriptional lineage priming in fetal and adult stem cells and multipotent progenitors. Immunity 26, 407-419 10.1016/j.immuni.2007.02.013 [DOI] [PubMed] [Google Scholar]
- McMahon A. P., Aronow B. J., Davidson D. R., Davies J. A., Gaido K. W., Grimmond S., Lessard J. L., Little M. H., Potter S. S., Wilder E. L., et al. (2008). GUDMAP: the genitourinary developmental molecular anatomy project. J. Am. Soc. Nephrol. 19, 667-671 10.1681/ASN.2007101078 [DOI] [PubMed] [Google Scholar]
- Mikkelsen T. S., Ku M., Jaffe D. B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.-K., Koche R. P., et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553-560 10.1038/nature06008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T., Iwasaki H., Reizis B., Ye M., Graf T., Weissman I. L., Akashi K. (2002). Myeloid or lymphoid promiscuity as a critical step in hematopoietic lineage commitment. Dev. Cell 3, 137-147 10.1016/S1534-5807(02)00201-0 [DOI] [PubMed] [Google Scholar]
- Mugford J. W., Yu J., Kobayashi A., McMahon A. P. (2009). High-resolution gene expression analysis of the developing mouse kidney defines novel cellular compartments within the nephron progenitor population. Dev. Biol. 333, 312-323 10.1016/j.ydbio.2009.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai S., Sugitani Y., Sato H., Ito S., Miura Y., Ogawa M., Nishi M., Jishage K.-i., Minowa O., Noda T. (2003). Crucial roles of Brn1 in distal tubule formation and function in mouse kidney. Development 130, 4751-4759 10.1242/dev.00666 [DOI] [PubMed] [Google Scholar]
- Ohnishi Y., Huber W., Tsumura A., Kang M., Xenopoulos P., Kurimoto K., Oleś A. K., Araúzo-Bravo M. J., Saitou M., Hadjantonakis A.-K., et al. (2014). Cell-to-cell expression variability followed by signal reinforcement progressively segregates early mouse lineages. Nat. Cell Biol. 16, 27-37 10.1038/ncb2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginensi A., Clarkson M., Neirijnck Y., Lu B., Ohyama T., Groves A. K., Sock E., Wegner M., Costantini F., Chaboissier M.-C., et al. (2011). SOX9 controls epithelial branching by activating RET effector genes during kidney development. Hum. Mol. Genet. 20, 1143-1153 10.1093/hmg/ddq558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugg-Gunn P. J., Cox B. J., Ralston A., Rossant J. (2010). Distinct histone modifications in stem cell lines and tissue lineages from the early mouse embryo. Proc. Natl. Acad. Sci. USA 107, 10783-10790 10.1073/pnas.0914507107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S. S., Krishna B., Chirala R., Amato R. J., Truong L. D. (2005). Kidney-specific cadherin, a specific marker for the distal portion of the nephron and related renal neoplasms. Mod. Pathol. 18, 933-940 10.1038/modpathol.3800373 [DOI] [PubMed] [Google Scholar]
- Sigova A. A., Mullen A. C., Molinie B., Gupta S., Orlando D. A., Guenther M. G., Almada A. E., Lin C., Sharp P. A., Giallourakis C. C., et al. (2013). Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc. Natl. Acad. Sci. USA 110, 2876-2881 10.1073/pnas.1221904110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861-872 10.1016/j.cell.2007.11.019 [DOI] [PubMed] [Google Scholar]
- Xu J., Liu H., Park J.-S., Lan Y., Jiang R. (2014). Osr1 acts downstream of and interacts synergistically with Six2 to maintain nephron progenitor cells during kidney organogenesis. Development 141, 1442-1452 10.1242/dev.103283 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.