

Abstract
Importance
Expanded hexanucleotide repeats in C9ORF72 are a common genetic cause of frontotemporal dementia and amyotrophic lateral sclerosis. Repeat expansions have also been detected infrequently in other disorders, including Alzheimer’s disease, dementia with Lewy bodies and Parkinsonian disorders.
Objective
To assess the incidence of the expanded C9ORF72 repeat in cases of depressive pseudodementia.
Design
An immunohistochemical screen of autopsied brains collected between 1998 and 2013.
Setting
Brain bank at Mayo Clinic Florida, a large tertiary care research institution.
Participants
Thirty one neuropathologically normal individuals (no atrophy, neuronal loss, or gliosis beyond what would be expected for age) with an antemortem clinical history or diagnosis of depression and/or dementia.
Main Outcome Measures
Presence of the hexanucleotide repeat was established using immunohistochemistry with a highly disease-specific antibody (C9RANT), and was further validated in carriers using repeat-primed polymerase chain reaction and Southern blotting.
Results
Of the 31 cases studied, 2 (6.45%) individuals harbored the C9ORF72 repeat expansion. Both patients were men with refractory depression. One patient experienced drug-induced Parkinsonism and sudden-onset dementia, while the other patient had a more insidious disease course suspected to be Alzheimer’s disease. Clinical and neuropathologic features are described.
Conclusions and Relevance
This report expands the range of clinicopathologic presentations of C9ORF72 expanded hexanucleotide repeat to include psychiatric disorders such as depressive pseudodementia.
INTRODUCTION
An expanded GGGGCC hexanucleotide repeat in a gene on chromosome 9, C9ORF72, is a common genetic cause of frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS), the general class of disorders referred to as c9FTD/ALS.1, 2 Pathogenic C9ORF72 expansions have been reported infrequently in clinically diagnosed3 and rarely in autopsy-confirmed Alzheimer’s disease,4 as well as clinically probable dementia with Lewy bodies,5 Parkinson’s disease,6 and corticobasal or ataxia syndromes.6, 7 Psychiatric presentations have also been reported, including psychosis and depression.8 Moreover, depression may be more frequent in c9ALS than in sporadic ALS.9 In addition to system-specific neuronal loss and gliosis, the hallmark neuropathologic feature of c9FTD/ALS is presence of neuronal inclusions immunopositive for ubiquitin and ubiquitin-binding proteins,10 some of which also contain dipeptide repeat polymers11 possibly generated by repeat-associated non-ATG translation. Dipeptide repeat polymers can be detected with a c9FTD/ALS disease-specific antibody, C9RANT.12 The clinicopathologic spectrum of C9ORF72-related disease and discussion of disease mechanisms have recently been reviewed by Cruts and colleagues.13
Here, we present clinical and pathologic findings of two patients with depression clinically thought to have dementia, where the differential diagnosis included depressive pseudodementia, a controversial term for cognitive deficits thought to be due to a treatable, often psychiatric condition.14 The prognosis of pseudodementia is contentious, with some studies reporting conversion to irreversible organic dementia and other studies reporting no conversion, but rather a disease course resembling the primary underlying psychiatric disorder.15, 16 In the present study, we describe patients in whom there is no overt evidence of neurodegeneration, based upon macroscopic and microscopic studies with routine histologic and fluorescence microscopy. We screened 31 cases meeting these criteria and found two, both with a family history of neurodegenerative disease, which had dipeptide repeat pathology characteristic of c9FTD/ALS, but lacked significant brain atrophy or neuronal loss in neuronal populations usually vulnerable to c9FTD/ALS. This study illustrates the C9ORF72 repeat expansion should be considered in patients with depressive pseudodementia, especially if they have a family history of neurodegenerative disease.
METHODS
Case selection for C9RANT screening
We screened a consecutive series of 31 cases from the brain bank for neurodegenerative disorders at Mayo Clinic in Jacksonville for evidence of C9ANT immunoreactive inclusions using immunohistochemistry of cerebellar sections with a previously characterized antibody (C9RANT (Rb5823, 1:5000).4 Demographic features of the cases is summarized in Table 1. The average age of the cohort was 77, ranging from 50 to 102. The average brain weight (determined by doubling the weight of the fixed hemibrain) was 1,277 g for men and 1,079 g for women. Cases were included in the study if routine neuropathologic evaluation assessing macroscopic atrophy and microscopic evidence of neuronal loss and gliosis was negative. They also had to have an antemortem diagnosis of dementia, depression or both. Three cases had a clinical antemortem history or diagnosis of dementia (mild cognitive impairment, Alzheimer’s disease, FTD, vascular dementia, or dementia with Lewy bodies), six cases had a clinical history or diagnosis of a depression, and twenty cases had a clinical history or diagnosis of both dementia and depression. They had to have minimal or no Alzheimer type pathology as assessed with thioflavin-S fluorescence microscopy and no α-synuclein pathology, or at most sparse Lewy bodies consistent with incidental Lewy body disease, on α-synuclein immunohistochemistry. They also could not have significant cerebrovascular disease that would be consistent with vascular ischemic dementia.
Table 1.
Pseudodementia clinicopathological study cohort
| Case # | C9 repeat | Age at Death | Sex | Brain weight | Dep | Dem |
|---|---|---|---|---|---|---|
| 1 | Y | 66 | M | 1380 | + | + |
| 2 | Y | 71 | M | 1360 | + | + |
| 3 | N | 64 | F | 1320 | + | + |
| 4 | N | 74 | F | 1060 | + | + |
| 5 | N | 75 | F | 960 | + | + |
| 6 | N | 79 | F | UNK | + | + |
| 7 | N | 92 | F | 1060 | + | + |
| 8 | N | 102 | F | 960 | + | + |
| 9 | N | 50 | M | 1320 | + | + |
| 10 | N | 63 | M | 1440 | + | + |
| 11 | N | 66 | M | 1500 | + | + |
| 12 | N | 69 | M | 1060 | + | + |
| 13 | N | 78 | M | 1340 | + | + |
| 14 | N | 78 | M | 1400 | + | + |
| 15 | N | 79 | M | 1260 | + | + |
| 16 | N | 80 | M | 1200 | + | + |
| 17 | N | 82 | M | 1200 | + | + |
| 18 | N | 85 | M | 1260 | + | + |
| 19 | N | 87 | M | 1160 | + | + |
| 20 | N | 88 | M | 1180 | + | + |
| 21 | N | 89 | M | 1340 | + | + |
| 22 | N | 94 | M | 1310 | + | + |
| 23 | N | 66 | F | 1140 | + | |
| 24 | N | 63 | M | 1400 | + | |
| 25 | N | 64 | M | 1320 | + | |
| 26 | N | 81 | M | 1320 | + | |
| 27 | N | 82 | M | 1160 | + | |
| 28 | N | 85 | M | 1270 | + | |
| 29 | N | 55 | F | 1050 | + | |
| 30 | N | 83 | M | 1350 | + | |
| 31 | N | 85 | M | 1020 | + |
Dep = depression, Dem = dementia, F = female, M = male, N = no repeat, UNK = unknown, Y = C9ORF72 repeat expansion, + = positive clinical history/diagnosis
Brains were obtained from patients from whom autopsies were performed after informed consent by the legal next-of-kin. Clinical information was obtained by review of available from medical records supplied to the brain bank, which operates under protocols approved by the Mayo Clinic IRB, in accordance with HIPAA guidelines after authorization by legal next-of-kin.
Neuropathologic characterization of C9RANT positive cases
Two cases were found to have C9RANT immunoreactive inclusions. In addition to routine studies, for these cases sections of cortex, hippocampus, amygdala, basal forebrain, thalamus, medulla, pons, and cerebellum were studied with immunohistochemistry for phosphotau (CP13, 1:1000), p62 (p62-lck ligand, 1:250), and TDP-43 (pS409/410, 1:5000) as previously described.4
Genetic methods
Frozen cerebellar tissue was processed for repeat-primed polymerase chain reaction (PCR) and Southern blot as previously described.1
RESULTS
We screened cerebellar sections of 31 brains for C9RANT pathology if they had no obvious structural basis of dementia, a history of depression or both. As expected, most brains lacking macroscopic and microscopic evidence of neurodegeneration were negative for C9RANT inclusions, extending the results of previous studies in which C9RANT failed to detect pathology in over 97 neurologically normal cases. Two of the 31 cases had neuronal cytoplasmic inclusions (NCI) in the cerebellum with C9RANT immunohistochemistry.
Neuropathology of cases with C9RANT pathology
As expected given the inclusions criteria, neither of the cases with C9RANT pathology had macroscopic evidence of cortical atrophy and no foci of softening or discoloration (Fig 1A, B). The calculated brain weights (1380 g and 1360 g) were within normal limits. Blood vessels at the base of the brains had no atherosclerosis. In Patient 1, the frontal and temporal horns of the lateral ventricle were not enlarged (Fig 1C) and in Patient 2, there is only mild widening (Fig. 1D). In both individuals, the cortical gray mantle had normal thickness and distribution. The hippocampal formation, amygdala, basal ganglia, thalamus, midbrain, pons, and medulla were unremarkable. The substantia nigra and locus ceruleus had visible pigmentation (Fig 1E, F).
Figure 1.
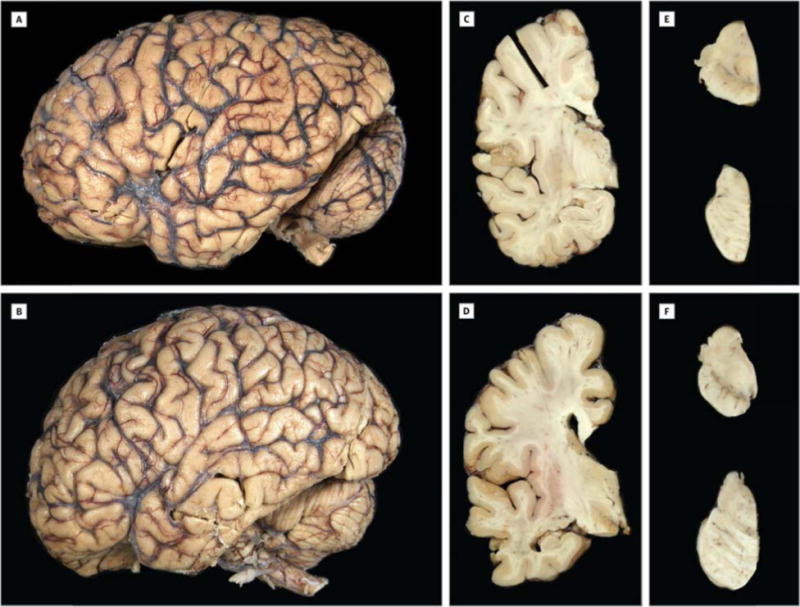
Macroscopic features of the C9ORF72-associated pseudodementia patients. (A, B) Fixed left hemibrain of the pseudodementia patients both show no cortical atrophy. Coronal sections show a thick cortical ribbon, corpus callosum and a normal appearing thalamus and hippocampus with no ventricular enlargement in Patient 1 (C) and minimal enlargement in Patient 2 (D). (E, F) There is visible pigment in the substantia nigra (midbrain; upper) and locus ceruleus (pons; lower) of both individuals.
Microscopically, all regions examined showed normal neuronal populations and no gliosis. Specifically absent was evidence of motor neuron disease in cortex and brainstem nuclei. There was also no cortical spongiosis and no hippocampal or amygdala neuronal loss. Neither case had hippocampal sclerosis. Thioflavin S fluorescent microscopy did not reveal any senile plaques or amyloid angiopathy, and only isolated neurofibrillary tangles in the neocortex (Patient 1), hippocampus (Patients 1 and 2), entorhinal cortex (Patient 1), and basal forebrain (Patients 1 and 2). In Patient 1, no inclusions were detected with α-synuclein immunohistochemistry. Immunohistochemistry with α-synuclein revealed a few Lewy neurites in the substantia nigra, raphe and periaqueductal gray, mesopontine tegmentum, and medullary tegmentum in Patient 2. Isolated Lewy bodies were detected in the basal forebrain, hypothalamus, and amygdala, but not in the basal ganglia. No cortical Lewy bodies were detected in the cerebral cortex. Given the absence of neuronal loss in areas with α-synuclein pathology, the findings were interpreted as incidental Lewy bodies. Tau immunohistochemistry revealed only mild age-related medial temporal pretangles and neuropil threads. A few neurons in the well-populated hippocampal dentate fascia had neuronal cytoplasmic inclusions (NCI) with TDP-43 immunohistochemistry. Sparse NCI were also present in entorhinal cortex, occipitotemporal cortex, and amygdala of Patient 1. There was no TDP-43 pathology in basal ganglia or in any of the vulnerable brainstem nuclei. In Patient 2, isolated TDP-43-immunoreactive NCI and dystrophic neurites were found in the frontal and temporal cortices with rare NCI found in the basal ganglia and substantia nigra. The density of TDP-43 pathology was not much greater than detected in a subset of neurologically normal individuals.17
Neuronal inclusions immunoreactive with C9RANT and p62 were more numerous than with TDP-43 immunohistochemistry in both brains. There were C9RANT- and p62-positive NCI in the dentate fascia and the pyramidal layer of the hippocampus (Fig 2B). C9RANT and p62 inclusions were detected in neocortex, amygdala, basal ganglia, and thalamus, with the highest burden in the cerebellum (Fig 2B). On the other hand, there was no neuronal loss or gliosis in the cerebellar cortex or deep nuclei. C9RANT and p62 pathology was more numerous in Patient 2 compared to Patient 1. Additional neuropathologic documentation is included in eFigures 1 and 2.
Figure 2.
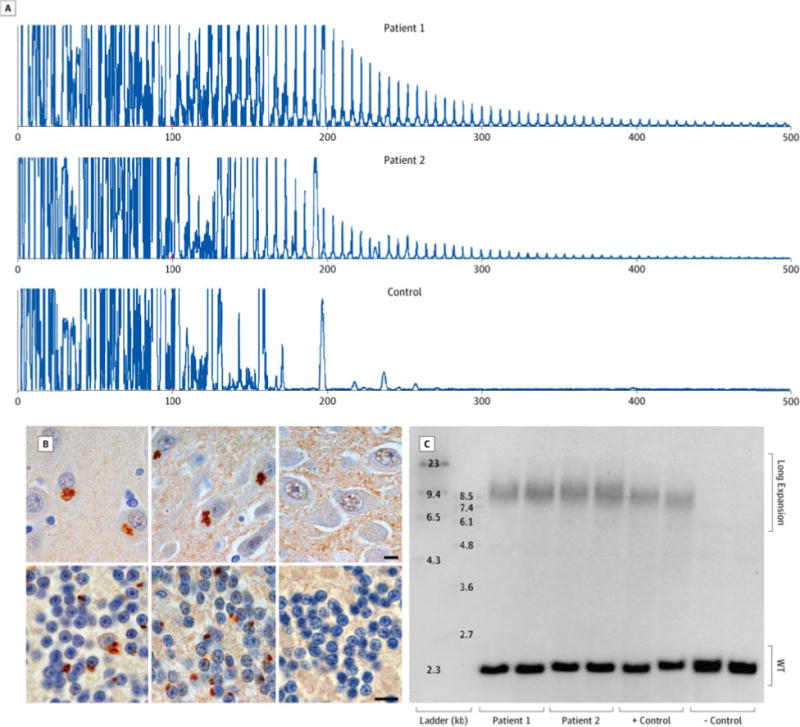
Pseudodementia patients meets diagnostic criteria for C9ORF72 hexanucleotide repeat expansion. (A) Repeat-primed PCR shows stutter amplification of the GGGGCC repeat in the pseudodementia cases (Patient 1, upper; Patient 2, middle), but not in non-carrier negative controls (lower). (B) NCI immunoreactive for C9RANT antibodies are seen in primary neurons of the hippocampus (upper panels) and granule cells of the cerebellum (lower panels) of the pseudodementia cases (Patient 1, left panels; Patient 2, middle panels) but not in other non-carrier pseudodementia controls (right panels)(10 μm bars). (C) Southern blotting of cerebellar tissue from the pseudodementia cases (in duplicate; Patient 1 in lanes 3 & 4; Patient 2 in lanes 5 & 6), two positive frontotemporal dementia controls (lanes 7 & 8), and two negative non-carrier controls (lanes 9 & 10) reveals a wild-type non-expanded allele at 2.5 kilobases (kb) for the cases and all controls as well as an expanded allele at approximately 8.5 kb (1,000–1,400 repeat units) for the cases and positive controls.
Genetics
Molecular genetic studies with repeat-primed PCR and Southern blot analysis for both patients demonstrated the characteristic C9ORF72 saw-tooth, stutter amplification-repeat pattern on PCR (Fig 2A) and high molecular weight bands on Southern blot at approximately 8.5 kilobases, which corresponds to 1,000–1,400 hexanucleotide repeat units. This expansion size is comparable to positive c9FTD controls (Fig 2C).
Clinical features of two individuals with C9RANT immunoreactive inclusions
Patient 1
This66-year-old, right-handed man presented first for neurologic evaluation at the age of 57 with a one-year history of toe numbness. He was a retired junior high school teacher. He did not smoke, and he drank minimal coffee and alcohol. He had a medical history of hypertension and hypercholesterolemia. Family history was notable for Parkinsonism and depression in his deceased father. He also had a 5-year history of depression with stable appetite and weight. Physical and neurologic examinations were normal, except for mild distal sensory loss, suggestive of an early polyneuropathy; however, a nerve conduction study was within normal limits. Toe sensation and numbness gradually improved. Six months later, he had worsening depression. His escitalopram dosage was increased, and bupropion was added. An MRI revealed findings suggestive of an old left basal ganglia lacunar infarct and small vessel ischemic changes. Carotid sonography revealed minimal plaque formation.
At age 61, he began electroconvulsive therapy with his family reporting benefits in his mood. CT and MRI at the time both showed no significant changes. Three years later, he had worsening and treatment-refractory depression, with lethargy, social withdrawal, and subjective memory complaints. He was treated with aripiprazole, lamotrigine, and nortriptyline for his depression. He developed drug-induced parkinsonism with hypomimia, intermittent resting leg tremor, decreased right arm spontaneous movements, bradykinesia and minimal increased tone with facilitation in his arms. Rapid repetitive movements and upper extremity reflexes were normal. Lower extremity reflexes were absent. He had a normal gait, and he was able to tandem walk without difficulty. The Romberg test was negative. On neuropsychological testing, he scored 30/30 on the Mini-Mental State Examination (MMSE) and a 93/100 on the Modified MMSE, with errors on timed naming, similarities and delayed recall. Trail Making Tests A and B were completed in 30 and 55 seconds, respectively, both without errors.
At age 65, he became lost while wandering at night and was found the next day in a catatonic state. He was unable to speak intelligently. Many of his medications were discontinued, and he was placed on risperidone, venlafaxine, fluvoxamine, and benztropine., He was not speaking, and substituting words when he did. He had a slightly shuffling gait, paucity of movement, masked facies, and he was withdrawn. CT and MRI revealed very mild cerebral atrophy and white matter changes. The patient died at the age of 66 with a final clinical diagnosis of possible dementia versus “all psychiatric manifestations” (pseudodementia inferred) and drug-induced Parkinsonism.
Patient 2
This 71-year-old right-handed man who first presented for neurologic evaluation at 66 years of age with depression and complaints of mild memory loss over the past year. He was a retired, college-educated banker who was working as a real estate sales associate. He was a pack-a-day smoker from the ages 19 to 59 and a frequent drinker who quit at the age of 70. He had a medical history of coronary artery disease with a bypass graft surgery at age 58. Family history was positive for an unspecified dementia in his deceased mother. He reported difficulties with short-term memory, rare word-finding difficulties, and social withdrawal. These issues did not impact sense of direction or work-related memory and cognition. He attributed these changes to a series of financial issues which have caused stress and depression. Physical and neurological exams were normal, and he scored a 28/30 on the MMSE. He was placed on Lexapro (10 mg daily) for his depression. Subsequent MRI revealed an essentially normal brain with very slight symmetric atrophy of the cerebral cortex.
Throughout the following year, he had some lingering difficulties with depression and a decrease in activity and motivation, but reported improved mood. Lexapro dosage was increased to 20 mg daily. Aricept was prescribed, but the patient did not tolerate it and it was discontinued after 3 months. Patient 2 was subsequently prescribed topical Exelon (4.6 mg/day) which was tolerated. At age 67, he underwent neuropsychological evaluation in which he scored within acceptable limits in domains of executive function, had borderline deficits in attention and concentration, visuospatial skills, and motor processing speed, and more severe impairments in learning and memory. He scored 19/30 on the Montreal Cognitive Assessment (MOCA; below normal cutoff), 7/10 on the Clock Drawing Test, 11/30 on the Geriatric Depression Scale, suggestive of mild depression, and 9/20 on the Geriatric Anxiety Inventory, indicating the absence of an anxiety disorder. Patient 2 scored a best of 5 on the 10 word list and 0 on delayed (0% recall). A diagnosis of probable Alzheimer’s disease was made at age 68.
The disease course was slowly progressive for 1.5 years with no changes in activities of daily living, but declining insight of cognitive deficits. Exelon became intolerable and he was prescribed Namenda. MOCA was 14/30 in at age 70. Later that year, his dementia began to worsen with delirium, increased anxiety, behavioral decompensation, and inappropriate outbursts. CT scan showed mild atrophy and white matter changes. Eight months later, delirium had subsided and dementia began to stabilize. Patient 2 died 5 month later at the age of 71 with a final clinical diagnosis of moderate Alzheimer’s disease.
DISCUSSION
These patients had unique clinicopathological features for C9ORF72 repeat expansion carriers, most notably brains that ostensibly had no evidence of neurodegeneration, except for disease-specific neuronal inclusions with C9RANT immunohistochemistry and sparse NCI with TDP-43 immunohistochemistry. TDP-43 immunoreactive lesions, usually dystrophic neurites, are uncommon in normal individuals and most often in advanced age.17 In these patients most of the abnormal TDP-43 was in NCI, not dystrophic neurites and both men were much younger than subjects with incidental TDP-43 pathology. Nevertheless, TDP-43 pathology, including NCI, has been previously reported in the neocortex of individuals with psychiatric diseases,18 and given the mild burden of pathology in comparison to TDP-43 seen in frontotemporal lobar degeneration, it is reasonable that the TDP-43 seen in these two cases is within the limits of normal aging and psychosis. On the other hand, they had abundant C9RANT immunoreactive inclusions that are specific to c9FTD/ALS and have not been detected in any other disorder or in normal controls. While Patient 1 had Parkinsonism, the lack of substantia nigra or basal ganglia pathology is consistent with drug-induced Parkinsonism. Patient 2 did not present with Parkinsonism, but had sparse Lewy bodies and neurites in vulnerable brainstem nuclei, basal forebrain, and amygdala. The extent of this pathology and the lack of clinical phenotype suggest that this pathology is merely coincidental and did not contribute to neuronal loss and, likely, the disease process. Despite neuroimaging findings of mild atrophy and white matter changes, no cerebrovascular pathology was observed during the neuropathological evaluation.
Notably, C9RANT immunoreactive inclusions were not found in the cerebella of 3 neuropathologically normal cases with dementia, 6 neuropathologically normal cases with depression, or 20 other cases with neuropathologic depressive pseudodementia in this series and in 97 cases from a previous series of neurologically normal controls. The C9ORF72 expansion was found in 2 of the 22 pseudodementia cases (9.1%) in this series. Further screening of additional cases is needed to further establish the frequency of the C9ORF72 repeat expansion in pseudodementia.
Psychiatric clinical features have been reported in c9FTD/ALS5, 19 and it is possible that C9ORF72 repeat expansion-related dysfunction is directly or indirectly related to depression in these patients. The most severe C9RANT pathology was in the cerebellum. The clinical significance of cerebellar pathology in c9FTD/ALS is unknown, but increasingly the cerebellum is thought to have roles in cognitive and affective functions. A study of resting state functional connectivity in major depression suggests that the cerebellum should be considered as a possible node in the distributed disease-related brain network in depression.20 As previously noted, one study has reported a higher frequency of depression in c9ALS kindreds compared to sporadic ALS kindreds.9 It should be noted, however, that due to the high prevalence of depression, this phenotype may be unrelated to C9ORF72-associated pathology.
It is possible that these patients may have had prodromal or subclinical c9FTD/ALS and coincidental depressive pseudodementia. Alternatively, it is possible that the repeat expansion in these patients represents incomplete penetrance, which is relatively common in c9FTD/ALS and may explain some of the so-called sporadic cases of c9FTD/ALS. As in other neurodegenerative diseases, the present case raises questions about the significance of neuronal inclusions visible with ubiquitin, ubiquitin-binding proteins, and even C9RANT, immunohistochemistry with respect to certain clinical manifestations of the C9ORF72 hexanucleotide repeat expansion. While the C9RANT pathology remains disease-specific, until the neurotoxic species is defined in c9FTD/ALS, this question will remain open. Importantly, these patients broaden the range of clinicopathologic presentations of c9FTD/ALS and emphasized the need to consider C9ORF72 in patients with a family history of psychiatric disorders.
Supplementary Material
Acknowledgments
We are grateful to all patients, family members, and caregivers who agreed to brain donation, without which these studies would have been impossible. We also acknowledge expert technical assistance of Linda Rousseau, Virginia Phillips, and Monica Castanedes-Casey for histology and Beth Marten for brain banking. This study was funded by Mayo Clinic Foundation, National Institutes of Health [R01 AG026251 (LP), R01 NS063964-01 (LP), R01 NS077402 (LP), ES20395-01 (LP), R01 NS080882 (RR), P50-AG16574 (DWD & RR), P50-NS72187 (DWD & RR), P01-AG03949], and the ALS Therapy Alliance (RR).
Funding Contributions: All of the aforementioned funding was used in the collection of data, and did not contribute to the design and conduct of the study, management, analysis, and interpretation of the data, preparation, review, or approval of the manuscript, or decision to submit the manuscript for publication.
Dr. Dickson had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Conflict of Interest Disclosure: RR has a pending patent on the identification of the C9ORF72 repeat expansion. LP has a patent for the C9RANT antibodies.
Authors’ Contributions: KFB and DWD were responsible for the study design. KFB, MvB, and MCB were involved in data collection and analysis. KFB, RR, LP, and DWD were involved in interpretation of the data. KFB wrote the report with all other authors assisting in drafting and revising the manuscript for intellectual content.
References
- 1.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron. 2011 Oct 20;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renton AE, Majounie E, Waite A, et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron. 2011 Oct 20;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Majounie E, Abramzon Y, Renton AE, et al. Repeat Expansion in C9ORF72 in Alzheimer’s Disease. New England Journal of Medicine. 2012 Jan 19;366(3):283–284. doi: 10.1056/NEJMc1113592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bieniek KF, Murray ME, Rutherford NJ, et al. Tau pathology in frontotemporal lobar degeneration with C9ORF72 hexanucleotide repeat expansion. Acta Neuropathologica. 2013 Feb;125(2):289–302. doi: 10.1007/s00401-012-1048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snowden JS, Rollinson S, Lafon C, et al. Psychosis, C9ORF72 and dementia with Lewy bodies. J Neurol Neurosurg Psychiatry. 2012 Oct;83(10):1031–1032. doi: 10.1136/jnnp-2012-303032. [DOI] [PubMed] [Google Scholar]
- 6.Lesage S, Le Ber I, Condroyer C, et al. C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain. 2013 Feb;136(Pt 2):385–391. doi: 10.1093/brain/aws357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindquist SG, Duno M, Batbayli M, et al. Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clinical Genetics. 2013 Mar;83(3):279–283. doi: 10.1111/j.1399-0004.2012.01903.x. [DOI] [PubMed] [Google Scholar]
- 8.Arighi A, Fumagalli GG, Jacini F, et al. Early onset behavioral variant frontotemporal dementia due to the C9ORF72 hexanucleotide repeat expansion: psychiatric clinical presentations. J Alzheimers Dis. 2012;31(2):447–452. doi: 10.3233/JAD-2012-120523. [DOI] [PubMed] [Google Scholar]
- 9.Byrne S, Heverin M, Elamin M, et al. Aggregation of neurologic and neuropsychiatric disease in amyotrophic lateral sclerosis kindreds: A Population-Based Case-Control Cohort Study of Familial and Sporadic Amyotrophic Lateral Sclerosis. Annals of Neurology. 2013 Jul 9; doi: 10.1002/ana.23969. [DOI] [PubMed] [Google Scholar]
- 10.Al-Sarraj S, King A, Troakes C, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathologica. 2011 Dec;122(6):691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- 11.Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013 Mar 15;339(6125):1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 12.Ash PEA, Bieniek KF, Gendron TF, et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to c9FTD/ALS. Neuron. 2013 Feb 20;77(4):639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cruts M, Gijselinck I, Van Langenhove T, van der Zee J, Van Broeckhoven C. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends in Neurosciences. 2013 Aug;36(8):450–459. doi: 10.1016/j.tins.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 14.Caine ED. Pseudodementia. Current concepts and future directions. Arch Gen Psychiatry. 1981 Dec;38(12):1359–1364. doi: 10.1001/archpsyc.1981.01780370061008. [DOI] [PubMed] [Google Scholar]
- 15.Saez-Fonseca JA, Lee L, Walker Z. Long-term outcome of depressive pseudodementia in the elderly. Journal of Affective Disorders. 2007 Aug;101(1–3):123–129. doi: 10.1016/j.jad.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Sachdev PS, Smith JS, Angus-Lepan H, Rodriguez P. Pseudodementia twelve years on. J Neurol Neurosurg Psychiatry. 1990 Mar;53(3):254–259. doi: 10.1136/jnnp.53.3.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnold SJ, Dugger BN, Beach TG. TDP-43 deposition in prospectively followed, cognitively normal elderly individuals: correlation with argyrophilic grains but not other concomitant pathologies. Acta Neuropathologica. 2013 Jul;126(1):51–57. doi: 10.1007/s00401-013-1110-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geser F, Robinson JL, Malunda JA, et al. Pathological 43-kDa Transactivation Response DNA-Binding Protein in Older Adults With and Without Severe Mental Illness. Archives of Neurology. 2010 Oct;67(10):1238–1250. doi: 10.1001/archneurol.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takada LT, Sha SJ. Neuropsychiatric features of C9orf72-associated behavioral variant frontotemporal dementia and frontotemporal dementia with motor neuron disease. Alzheimers Res Ther. 2012 Oct 3;4(5):38. doi: 10.1186/alzrt141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma Q, Zeng LL, Shen H, Liu L, Hu D. Altered cerebellar-cerebral resting-state functional connectivity reliably identifies major depressive disorder. Brain Res. 2013 Feb 7;1495:86–94. doi: 10.1016/j.brainres.2012.12.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.