

Abstract
Recently identified IDH mutations lead to the production of 2-hydroxyglutarate (2HG), an onco-metabolite aberrantly elevated in selected cancers. We developed a facile and inexpensive fluorimetric microplate assay for quantitation of 2HG and performed an unbiased small molecule screen in live cells to identify compounds capable of perturbing 2HG production. Zaprinast, a PDE5 inhibitor, was identified as an efficacious modulator of 2HG production and confirmed to lower 2HG levels in vivo. The mechanism of action was not due to cGMP stabilization, but rather, profiling of metabolites upstream of mutant IDH1 pointed to targeted inhibition of the enzyme glutaminase (GLS). Zaprinast treatment reversed histone hypermethylation and soft agar growth of IDH1 mutant cells, and treatment of glutamine-addicted pancreatic cancer cells reduced growth and sensitized cells to oxidative damage. Thus, Zaprinast is efficacious against glutamine metabolism and further establishes the therapeutic linkages between GLS and 2HG-mediated oncogenesis.
Introduction
Altered glutamine metabolism can maintain oncogenic transformation and support rapid growth in some cancer cells (1). Oncogenic MYC regulates glutamine metabolism by increasing both the uptake of glutamine and its catabolism through microRNA-driven regulation of glutaminase (GLS). As such, MYC-transformed cells are dependent on glutamine for growth (2, 3). Additionally, RAS-driven reprogramming of cellular metabolism shunts glutamine toward NADPH-generating reactions to maintain oxidative balance (4). Glutamine, through glutamate, is also a precursor for cellular α-ketoglutarate (αKG), which can undergo further metabolism through the Krebs cycle or, of particular interest to our study, can be further metabolized to 2-hydroxyglutarate (2HG) by mutant isocitrate dehydrogenase (IDH) (5).
Heterozygous somatic mutations in IDH enzymes are present in over 80% of grade II and III gliomas as well as secondary glioblastomas (6, 7). Mutations have also been detected in acute myeloid leukemia, chondrosarcomas, and cholangiocarcinoma among others (8–10). Nearly all identified mutations are in arginine residues 100 and 132 of IDH1 or residues 140 and 172 of IDH2, all of which are located in the active sites of the enzymes (11). Instead of inhibiting the enzymatic activity of IDH, these mutations alter the catalytic activity such that the normal product, αKG, is metabolized to R-2HG in a reaction that consumes NADPH. While endogenous levels of 2HG are normally low, gliomas harboring mutant IDH1 or IDH2 accumulate millimolar quantities of R-2HG (5).
Structural similarities between 2HG and αKG suggested that 2HG could modulate the function of αKG-dependent dioxygenases to promote transformation and alter differentiation. Indeed, 2HG was found to inhibit the TET family of methylcytosine dioxygenases as well as several members of the JmjC family of histone demethylases (12, 13). In addition, R-2HG stimulates EglN, driving the degradation of hypoxia inducible factor (HIF) (14). Modulating the activity of these various dioxygenases drives DNA and histone hypermethylation, blocks differentiation, and promotes transformation (15–17). Importantly, recent findings indicate that some of these events are reversible. Withdrawal of cell permeable 2HG or treatment with a small molecule inhibitor targeting IDH2 R140Q restores differentiation of leukemia cells, while inhibition of IDH1 R132H in transformed cells reduces histone methylation and soft-agar growth (16, 18, 19). Thus, reducing 2HG could provide therapeutic benefit in patients with malignancies harboring gain-of-function IDH mutations.
With this in mind, we developed a fluorimetric microplate assay for measuring 2HG with the intent to screen for compounds capable of perturbing 2HG production in live cells. We reasoned that this approach could identify novel drug interactions that ultimately lead to diminished mutant IDH1 activity, and perhaps perturb other related vital pathways such as glutamine metabolism, with the potential to repurpose drugs originally developed for alternative disorders. After validation and optimization of assay conditions, we performed an unbiased screen of a small molecule library using HT1080 cells, which harbor an endogenous IDH1 R132C mutation, and identified Zaprinast, a phosphodiesterase 5 (PDE5) inhibitor, as a candidate modulator of the 2HG metabolic pathway. Rather than being cGMP-mediated, the mechanism of action of Zaprinast centered on inhibition of GLS upstream of mutant IDH1.
Results
Novel Coupled Fluorescence Assay for Quantitation of 2HG
Conventional methods to measure 2HG in cell culture rely on time consuming and costly chromatography platforms that are not amenable to high throughput screening experiments. Instead, we developed an enzymatically-coupled fluorescence assay to directly quantify the total levels of 2HG in cultured cells. Our strategy involved a primary enzymatic reaction that is specific to 2HG and produces NADH, which is coupled to a second reaction that consumes NADH with mitochondrial diaphorase. The E coli K-12 enzyme 3-phosphoglycerate dehydrogenase (PHGDH) is known to have dehydrogenase activity against human R-2HG and uses NAD+ as a co-factor (23). Published reaction conditions for the 2HG dehydrogenase activity of E coli PHGDH provided a basis for its use in our fluorescence assay. The NADH produced by E coli PHGDH is subsequently oxidized by the diaphorase enzyme to reduce resazurin to resorufin, a highly fluorescent red-shifted molecule with an emission peak at 587 nm, thereby minimizing auto-fluorescence (Figure 1a). In the process, NADH is recycled back to NAD+ and resorufin is produced in stoichiometric proportion to the amount of 2HG present in the sample (24). The assay is rapid, inexpensive, quantitative and non-destructive if measuring secreted metabolites, making it ideal for large scale screening applications.
Figure 1.

Microplate assay detects changes in 2HG levels. a) Assay schematic showing the coupling of NADH production by PHGDH to resorufin production by mitochondrial diaphorase. Resorufin accumulates in proportion to 2HG metabolite turnover. b) Accumulation of resorufin with increasing 2HG dissolved in buffer produces a pink solution. c) Quantification of assay fluorescence of 2HG dissolved in complete culture media showing linearity up to 100 µM. Fluorescence is plotted as Arbitrary Units (A.U.). (d–f) Western blot showing overexpression of exogenous IDH1 in NHA cells (d) and HEK293T cells (f) and quantification of 2HG by (d) GC-MS and (e,f) the fluorimetric assay. g) Western blot of shRNA-mediated knockdown of IDH1 in HT1080 cells with endogenous mutant IDH1as well as corresponding fluorescent assay signal. 2HG GC-MS values were obtained from cell extracts and were normalized to internal standard and total protein, while the 2HG fluorescent assay was performed on conditioned cell culture media and normalized to cell viability as quantified by Alamar Blue. (n = 3, **P < .05, two-tailed Student’s t-test).
After optimization of assay conditions, we detected a change in fluorescent signal in direct proportion to added 2HG concentration (Figures 1b,c). Exogenous 2HG was detected when dissolved in culture media in a concentration-dependent manner, indicating that secreted 2HG could not only be detected, but be quantified (Figure 1c). However, signal background was measurably higher in the fluorescent assay when compared to mass spectrometry. By assaying a 20 µl aliquot of culture media, we determined that assay activity was linear to 100 µM and saturated at 2HG concentrations of 1 mM or higher; the lower limit of detection was approximately 4 µM (Figure S1). We then determined the specificity of the assay for 2HG by testing the effect of metabolites within the IDH metabolic pathway, including glutamine, glutamate, αKG, and isocitrate and found that they do not interfere with assay function (Figure S2). Next, to determine if the assay could detect physiological changes in 2HG production by cultured cells, we stably transduced immortalized human astrocytes with vector, wild type IDH1, or R132H IDH1. Accumulation of 2HG in astrocytes expressing R132H IDH1 was first confirmed by conventional GC-MS (Figure 1d). Then, by the fluorimetric assay, media from astrocytes expressing mutant IDH1 showed significantly higher signal when compared to cells expressing either vector or wild type IDH1 (Figure 1e). This was also verified in HEK293T cells (Figure 1f). In addition, to determine if the fluorimetric assay could detect a reduction in 2HG levels, we performed shRNA knockdown of IDH1 in the human fibrosarcoma cell line HT-1080, which harbors an endogenous heterozygous gain-of-function R132C IDH1 mutation. Using two different hairpin sequences, we assayed 2HG levels in the culture media and showed a significant reduction in signal in proportion to IDH1 protein knockdown (Figure 1g).
High-Throughput Screen for Modulators of 2HG Metabolism
Using the assay to quantify 2HG in media, we screened a library of 480 bioactive compounds targeting a wide variety of cellular processes in IDH1 mutant HT-1080 cells to identify targets capable of affecting the 2HG metabolic pathway. Screen results are plotted in Figure 2a as 2HG fluorescence versus viability. A global linear trend was observed between 2HG and viability indicating that most 2HG reduction was driven by compound toxicity. However, screen hits in the top left quadrant reduced 2HG but had little effect on viability over the time course of the assay (Figure 2a). Among the top candidate compounds were Zaprinast, a phosphodiesterase type 5 (PDE5) inhibitor, HBDDE, a PKC inhibitor, and Dantrolene, a calcium release inhibitor (Figure 2a). Zaprinast (5-(2-propoxyphenyl)-1H-[1,2,3]triazolo[4,5-d]pyrimidin-7(4H)-one) was the most potent compound identified in the screen and showed a concentration-dependent reduction in extracellular 2HG (Figure 2b). To confirm that the effects of Zaprinast were not an artifact caused by an inhibition of the assay itself (i.e., E. coli PHGDH or diaphorase activity), secondary validation using mass spectrometry in extracts of HT1080 and NHA cells ectopically expressing R132H IDH1 and treated with Zaprinast was performed. As anticipated, production of 2HG was modulated in a concentration-dependent manner (Figure 2c,d).
Figure 2.
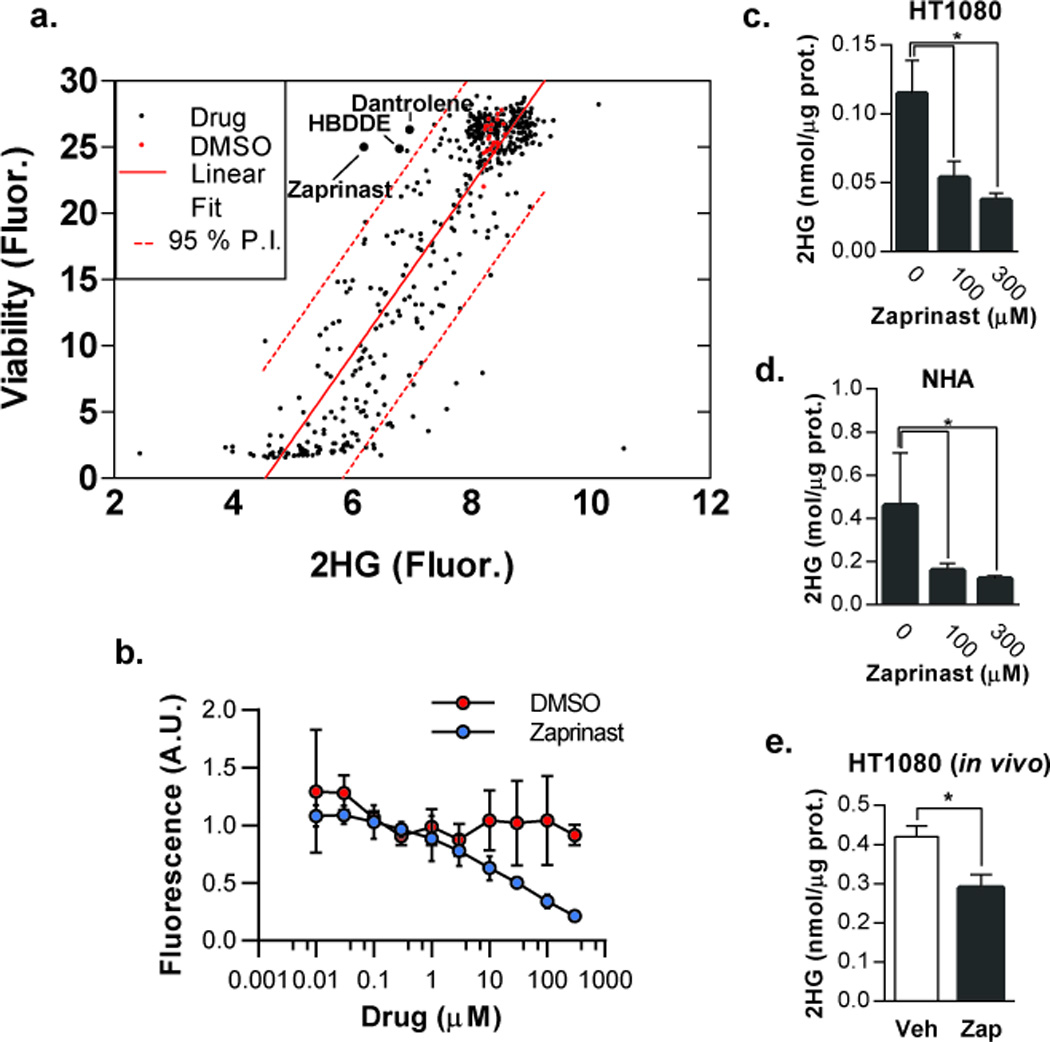
Zaprinast lowers 2HG levels in IDH1 mutant cells in culture and tumors in vivo. a) HT1080 high-throughput screen results plotted as 2HG vs viability. Data points represent the average of screens performed in triplicate. Linear fit and 95% predictive interval (P.I.) show a correlation between 2HG and viability. Compounds in the top left quadrant outside the 95% P.I. produced a reduction in 2HG that was not predicted to be a consequence of general drug toxicity. b) HT1080 cells were treated with the indicated concentrations of Zaprinast for 48 hours and secreted 2HG was measured in the media using the fluorescent assay (n=3; data are plotted as fold-viability over fold-untreated). (c–d) HT1080 cells (c) and NHA cells (d) ectopically expressing IDH1 R132H treated with Zaprinast for 48 hours were analyzed for intracellular 2HG by GC-MS (n=3). e) GC-MS quantification of 2HG in HT1080 tumor xenografts treated with vehicle or Zaprinast (n=6 for vehicle and n=7 for drug; error bars represent S.E.M; *P < .05).
To determine if Zaprinast could lower 2HG concentrations in vivo, we generated flank tumor xenografts of HT1080 cells in nu/nu mice. Because the pharmacokinetics of Zaprinast for this application have not been characterized, we performed direct intratumoral injection to assure delivery of the compound. Tumors treated with Zaprinast showed a modest but significant reduction of 2HG levels relative to vehicle as quantified by GC-MS (Figure 2e).
PDE5 hydrolyzes cGMP and is the target of several clinically-approved inhibitors that function by elevating intracellular cGMP (25). However, we observed no change in levels of 2HG when treating HT1080 cells with Sildenafil and Tadalafil, two clinically-approved PDE5 inhibitors (Figure 3a). We then asked if either cAMP or cGMP was sufficient to reduce 2HG levels in HT1080 cells. Treatment with the cell-permeable analogues 8-bromo-cAMP and 8-bromo-cGMP did not cause a reduction in 2HG (Figure 3b). To confirm that the lack of 2HG reduction upon treatment with cell-permeable cGMP was not due to structural differences between the analogue and its endogenous counterparts, we sought to elevate endogenous cGMP by expressing constitutively active soluble guanylyl-cyclase (sGC) (26). Co-expression of the wild type α1 subunit and the constitutively active β1 C105H mutant that comprise the sGC heterodimer resulted in a significant increase in basal cGMP levels as measured by ELISA (Figure 3c,d). 2HG levels, however, were unaffected by elevation of intracellular cGMP (Figure 3e), indicating that the effects of Zaprinast on 2HG production were likely not a consequence of altered cGMP levels.
Figure 3.
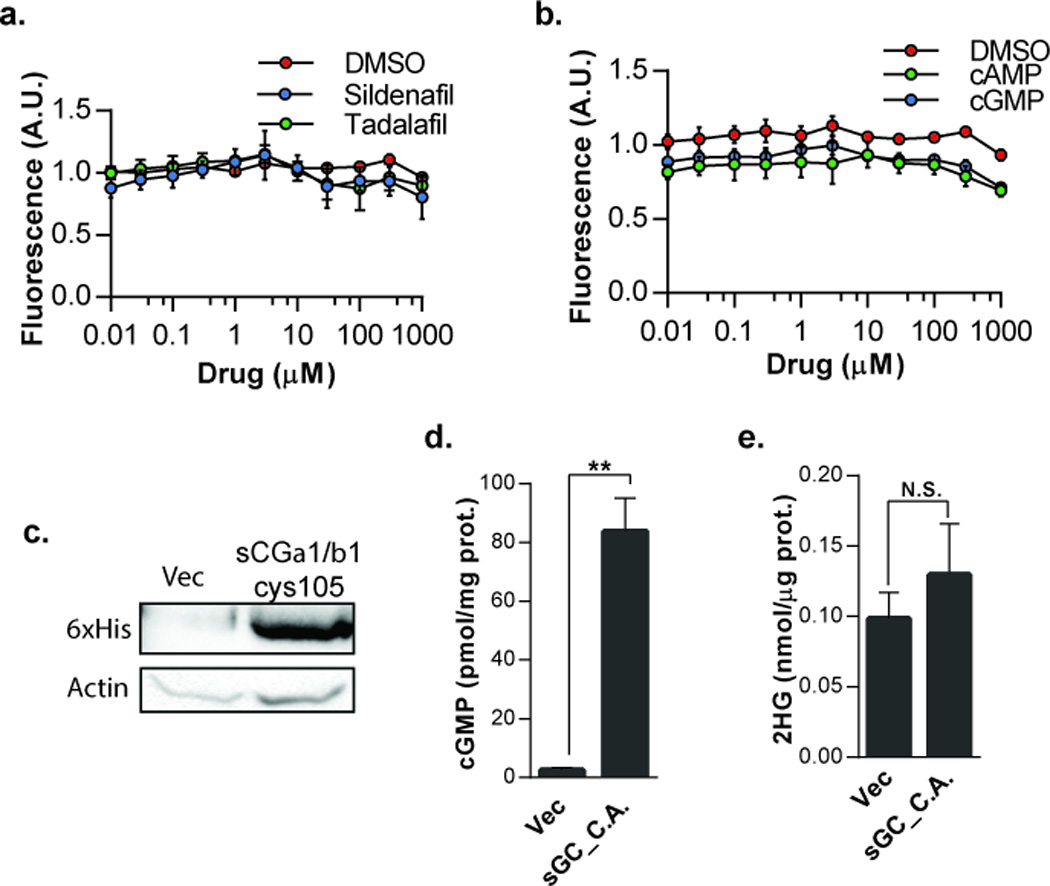
Effects of Zaprinast on 2HG are not cGMP mediated. HT1080 cells treated with a) PDE5 inhibitors Sildenafil or Tadalafil, or (b) 8-bromo-cAMP or 8-bromo-cGMP, were analyzed for secreted 2HG (n=3; data are plotted as fold-viability over fold-untreated). c) Western blot of 6×His tag in HT1080 cells expressing sGCα1/βcys105. d) cGMP levels as measured by ELISA in HT1080 cells expressing vector or sGCα1/βcys105 plasmids (n=3; **P < .05). e) GC-MS of 2HG in extracts of HT1080 cells expressing vector or sGCα1/βcys105 plamids (n=6, error bars represent S.E.M.). C.A., constitutively active.
Mechanism of Action of Zaprinast
The reported IC50 for Zaprinast against PDE5 is approximately 0.15 µM. However, approximately 100 – 300 µM were required to observe a significant reduction in 2HG. This suggested, along with the finding that the Zaprinast-mediated effects appeared to be cGMP-independent and not mimicked by other PDE5 inhibitors, that Zaprinast blocks 2HG production through an off-target effect. To define the mechanism of action of Zaprinast, we examined changes in metabolites upstream of 2HG. The predominant source of 2HG is cellular glutamine. Glutaminase (GLS) metabolizes glutamine to glutamate, which is then converted to alpha-ketoglutarate by glutamate dehydrogenase, followed by mutant IDH1 metabolism of alpha-ketoglutarate to 2HG (Figure 4a) (5). We reasoned that by measuring upstream metabolites, we could identify a candidate target enzyme whose activity was either directly or indirectly inhibited by Zaprinast. Using mass spectrometry, we measured cellular levels of glutamine, glutamate, alpha-ketoglutarate, and 2-hydroxyglutarate in vehicle and Zaprinast treated HT1080 cells. Unexpectedly, in addition to 2HG, we found that levels of alpha-ketoglutarate and glutamate were also reduced in HT1080 cells treated with Zaprinast, but intracellular levels of glutamine were unaffected. We repeated this in NHA immortalized astrocytes expressing R132H IDH1 and detected the same pattern of metabolite level changes (Figure 4b). These data suggested that Zaprinast directly or indirectly inhibited the activity of GLS, causing a reduction in the level of downstream metabolites. Treatment of HT1080 cells with BPTES, a known GLS inhibitor, produced a similar pattern of metabolite modulation (Figure 4c). Furthermore, in cells treated with Zaprinast, adding back a cell permeable form of alpha-ketoglutarate restored 2HG back to baseline levels (Figure 4d), consistent with traversing a block at the level of GLS. To determine if Zaprinast modulates 2HG by directly inhibiting GLS, we purified human GLS and utilized an in vitro assay to measure its enzymatic activity. The enzymatic activity of GLS was directly inhibited by Zaprinast with an IC50 of ~200 µM, which corresponded to the concentrations at which a significant reduction in 2HG was observed in cells (Figure 4e). GLS kinetic analysis determined that Zaprinast acted as a noncompetitive inhibitor (Ki = 220 µM) (Figure 4f,g). In a control experiment, cGMP had no direct effect on GLS enzymatic activity (Figure S3).
Figure 4.
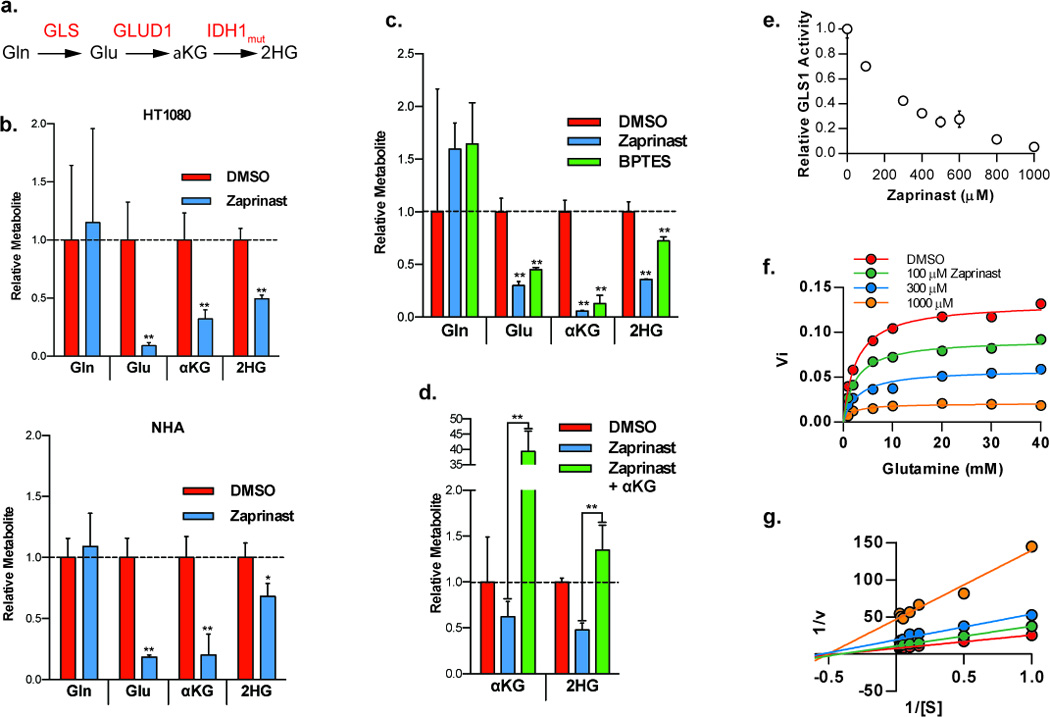
Zaprinast inhibits GLS. a) Metabolic pathway of glutamine metabolism to 2HG. Abbreviations are as follows: Gln, glutamine; GLS, glutaminase; Glu, gluamate; GLUD1, glutamate dehydrogenase; αKG, α-ketoglutarate; IDH1, isocitrate dehydrogenase; 2HG, 2-hydroxyglutarate. b) GC-MS quantification of 2HG and upstream metabolites in HT1080 and NHA cells expressing IDH1 R132H treated for 48 hours with Zaprinast. c) GC-MS quantification of 2HG and upstream metabolites in HT1080 cells treated for 48 hours with 300 µM Zaprinast or 10 µM BPTES. d) GC-MS quantification of 2HG and αKG in HT1080 cells treated with Zaprinast alone or with 5 mM cell permeable dimethyl 2-oxoglutarate. Values in panels b–d are normalized to vehicle treatment. e) Enzyme activity of purified, full length, human GLS exposed to Zaprinast. f) Kinetic activity of human GLS exposed to varying concentrations of Zaprinast and glutamine. g) Double-reciprocal Lineweaver-Burk plot of data from panel f showing noncompetitive inhibition of GLS by Zaprinast with respect to glutamine. n = 3 or 4, error bars represent S.D.; *P< .05, **P < .0l, two-tailed Student’s t-test.
Zaprinast-Mediated Modulation of the 2HG Phenotype
Having shown that Zaprinast reduces 2HG levels by blocking flux through the pathway at the level of GLS, we next explored if Zaprinast treatment could reverse the effects of 2HG on cells. 2HG is structurally similar to αKG, differing only in the oxidation state of the C2 carbon (11). Because of this similarity, 2HG has been shown to act as either an inhibitor or activator of various αKG-dependent enzymes. The JmjC family of histone demethylases have been shown to be inhibited by 2HG and expression of mutant IDH1 was shown to cause an elevation in methylated histone lysine residues and lead to a block in cellular differentiation (12, 15). As expected, expression of R132H IDH1 in human astrocytes caused an increase in histone methylation marks (H3K9Me2, H3K9Me3, H3K27Me3, and H3K79Me2), and treatment with Zaprinast caused a marked reduction toward baseline methylation levels (Figure 5a,b). By contrast, Zaprinast treatment of IDH1 WT expressing astrocytes did not produced such a marked reduction in methylation levels of either H3K9Me2 or H3K9Me3 (Figure S4). In addition, mutant IDH1 was recently shown to promote soft agar colony formation of immortalized human astrocytes (14). As expected, IDH1 R132H expression increased colony formation of human astrocytes when compared to vector or IDH1 wild type. Importantly, Zaprinast treatment reduced colony formation in IDH1 R132H astrocytes down to the level of vector and wild type cells (Figure 5c,d).
Figure 5.
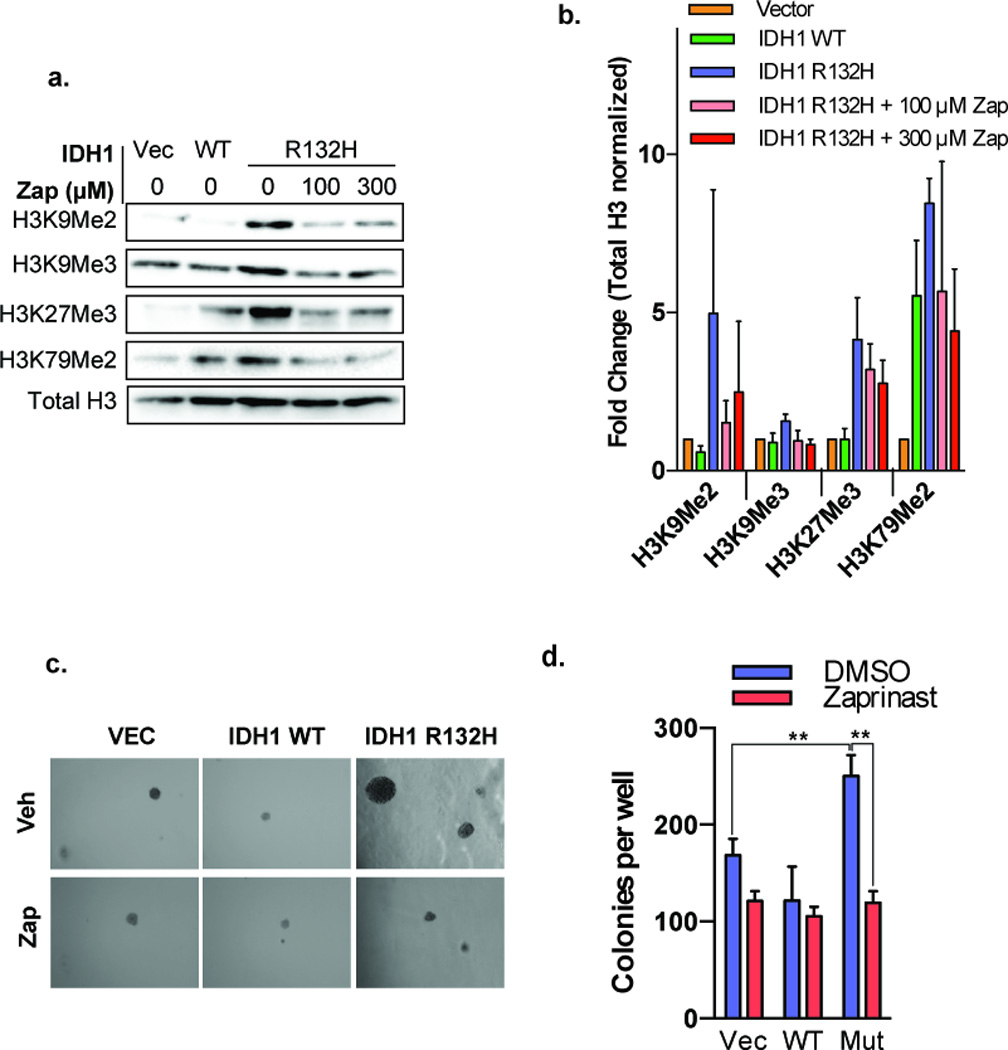
Zaprinast reverses 2HG-associated phenotypes. a) Immortalized NHA cells were transduced with vector, wild type IDH1, or IDH1 R132H plasmids and treated with vehicle or Zaprinast at the indicated concentrations. Histone extraction was then performed. A representative Western blot is shown to assess the levels of indicated histone lysine residues. b) Quantification of histone lysine methylation levels from Western blots performed in three independent experiments. Data were normalized to total H3 and vector control. c) Soft agar colony assay of NHA cells expressing vector, wild type IDH1, or IDH1 R132H plasmids and treated with vehicle or Zaprinast. d) Quantification of triplicate colony assay experiments. n = 3, error bars represent S.D.; **P < .05, two-tailed Student’s t-test.
Zaprinast Inhibits Glutamine-Dependent Tumor Cell Proliferation
Because of the requirement for glutamine in 2HG production, we hypothesized that disruption of glutamine metabolism with Zaprinast could abrogate the growth of glutamine-addicted cells with normal IDH activity. In cells undergoing the Warburg effect, whereby glucose is shunted toward lactate production in the presence of oxygen, glutamine often functions in anaplerotic reactions to replenish citric acid cycle intermediates (27). A recent report by Son et al. showed that HRAS-driven pancreatic ductal adenocarcinoma (PDAC) cells use glutamine in a different manner. PDAC cells are dependent on glutamine metabolism to maintain redox balance by shuttling glutamine carbon through the transaminase GOT1 and ultimately malic enzyme (ME) (4). Using two PDAC cell lines, Panc1 and MiaPaca2, which are glutamine-addicted, but show minimally-detectable levels of 2HG (data not shown), we sought to determine if Zaprinast could perturb redox balance in a manner similar to glutamine deprivation. First, we confirmed that Panc1 and MiaPaca2 cells were dependent on extracellular glutamine for their growth, saturating at glutamine concentrations of 2 mM (Figure 6a). Furthermore, treatment with Zaprinast led to significant reductions in cellular pools of glutamate and αKG and an increase in glutamine as measured by mass spectrometry, indicating that Zaprinast blocks GLS activity in live PDAC cells (Figure 6b). Interestingly, glutamine levels are increased more dramatically in PDAC cells treated with Zaprinast when compared to NHA or HT1080 cells treated with Zaprinast (Figure 4b). This is likely a consequence of RAS-driven metabolic reprogramming in PDAC cells wherein glutamine metabolic fluxes are high to maintain oxidative balance and promote transformation. Growth in the presence of 100 µM and 300 µM Zaprinast was significantly reduced (Figure 6c). To confirm that growth inhibition in the presence of Zaprinast was due to GLS inhibition, glutamate supplementation of the media was performed, producing a restoration of growth (Figure 6d). Consistent with previous findings that glutamine metabolism maintains oxidative homeostasis in PDAC, Zaprinast treatment of PDAC cells caused an increase in reactive oxygen species (ROS) in a concentration-dependent manner (Figure 6e,f). This resulted in increased susceptibility to oxidative damage as cells pretreated with Zaprinast were significantly more susceptible to hydrogen peroxide stress than cells treated with vehicle (Figure 6g). Moreover, adding back glutamate to Zaprinast-treated cells nearly completely abolished the increased sensitivity to oxidative damage (Figure 6g), again consistent with blockade at the level of GLS. These findings suggest that Zaprinast could increase the sensitivity of PDAC to oxidative stress, as might be therapeutically induced by radiation or chemotherapy.
Figure 6.
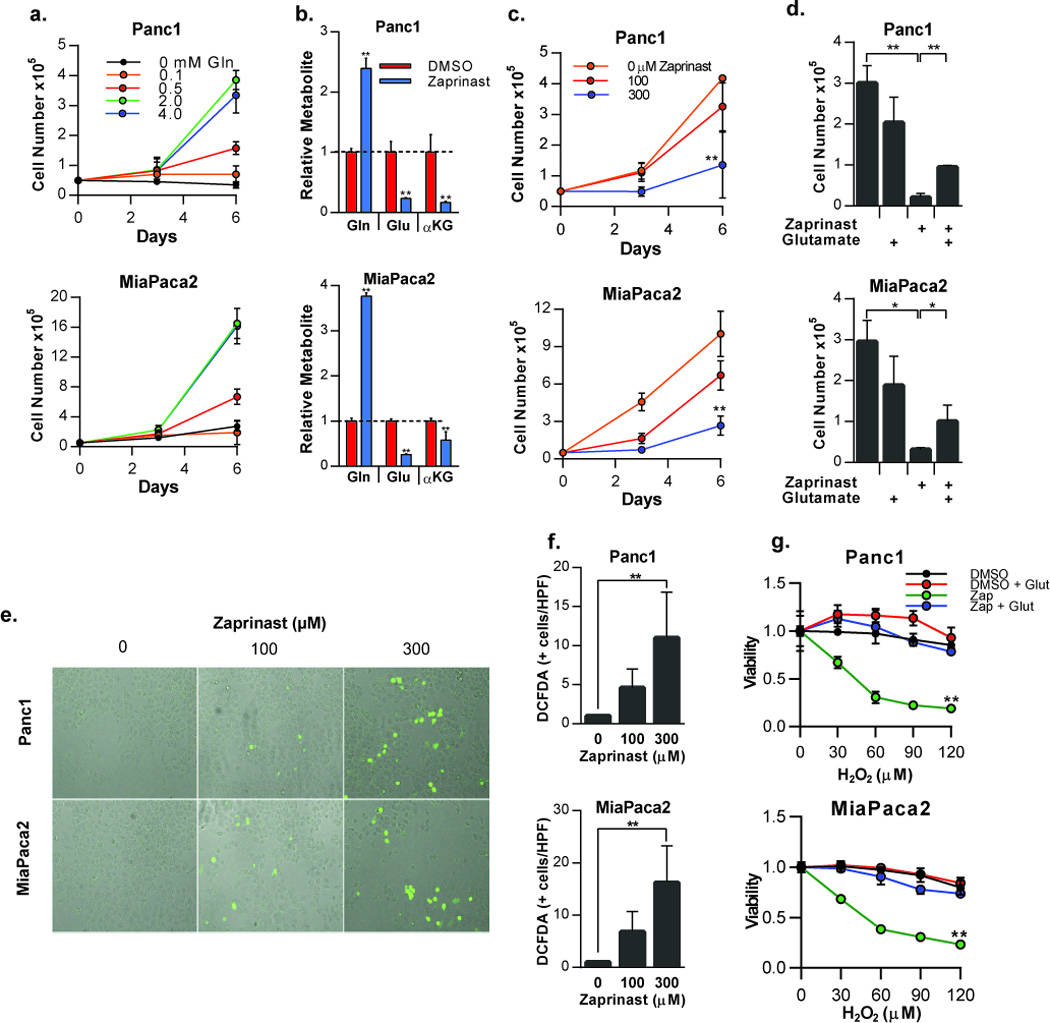
Zaprinast inhibits growth of PDAC cells and sensitizes them to oxidative stress. a) Growth of PDAC cells Panc1 and MiaPaca2 in glutamine-free media or in the presence of increasing concentrations of glutamine. b) Relative levels of glutamine, glutamate, and αKG in PDAC cells treated with 300 µM Zaprinast or vehicle control for 48 hours (normalized to cellular protein). c) Growth of PDAC cells treated with Zaprinast at the indicated concentrations. d) Growth of PDAC cells grown in complete culture media containing 2 mM glutamine. Cells were treated with vehicle or Zaprinast alone or in the presence of 5 mM glutamate. e) Microscopic images of DCFDA-positive PDAC cells following treatment with the indicated concentrations of Zaprinast. f) Quantification of DCFDA-positive PDAC cells. g) PDAC cells treated with vehicle or Zaprinast in the absence or presence of 5 mM glutamate were exposed to increasing concentrations of H2O2. Cell viability was assessed with Alamar Blue and values were normalized to vehicle-treated cells. n = 3, error bars represent S.D.; *P <0.05,**P <0.0l ; two-tailed Student’s t-test used in panels b, d, and f, while two-way ANOVA was applied to panels c, and g.
Discussion
Accumulation of 2HG in tumors harboring IDH1 and IDH2 mutations modulates the activity of several αKG-dependent dioxygenases and leads to histone and DNA hypermethylation, blocked differentiation, and cellular transformation (11). Herein we have developed a fluorimetric assay capable of detecting changes in 2HG levels and allows for rapid, high-throughput quantification when compared to mass spectrometry. Our objective was to identify alternative cellular targets or mechanisms for reducing total cellular 2HG levels. An assay using (d)-2-hydroxyglutarate dehydrogenase as the driver enzyme was recently published by Balss et al. to quantify 2HG in patient serum and tumor tissues with mutant IDH (28). Compared to E. coli PHGDH, whose primary substrate is 3-phosphoglycerate (3PG), (d)-2-hydroxyglutarate dehydrogenase is thermodynamically more specific for 2HG and could provide an alternative driver enzyme for future experiments. Nonetheless, glutaminase inhibition with either BPTES or Zaprinast did not cause an elevation in cellular 3PG, thus reducing the likelihood of interference within the context of our PHGDH-based assay signals (Figure S5). The development and optimization of our fluorimetric assay was focused on high-throughput robotic screening and lead to the successful screening of a library of 480 bioactive compounds against live cells overproducing 2HG. We identified Zaprinast, a PDE5 inhibitor, as a modulator of 2HG production. The ease of the fluorimetric assay in the high-throughput setting, coupled with its quantitative capacity and low cost, is encouraging from a drug discovery perspective. While we screened a relatively modest library of 480 compounds, interrogation of much larger compound libraries as well as siRNA libraries are also feasible by this method and provide important strategies to identify targets that reduce 2HG for therapeutic purposes and define mechanisms of regulation.
Among the other compounds identified were a calcium channel modulator and a PKC inhibitor. We chose to further follow up on Zaprinast because the drug showed the most dramatic reduction in 2HG levels and several other PDE5 inhibitors have undergone clinical approval. Additional follow up will be required to characterize the mechanism and efficacy of other promising screen hits. Specifically, several PKC inhibitors have undergone clinical trial development for use in cancer, heart failure, coronary artery disease, and diabetic retinopathy (29). Further characterization of their effects on 2HG and metabolism may allow for drug repurposing or generation of more potent analogs. We determined that the effects of Zaprinast are likely not mediated by cGMP, as neither the cell-permeable cGMP analogue, 8-bromo-cGMP, nor expression of constitutively active sGC reduced 2HG levels. Instead, we identified a previously unkown off-target effect of Zaprinast against GLS, the first enzyme of glutaminolysis that ultimately supplies αKG for mutant IDH reactions.
Losman et al. recently showed that withdrawal of cell-permeable 2HG following transformation induced by treatment of cell-permeable 2HG restores dependence on growth factors and differentiation of leukemia cells (16). Similarly, treatment with small molecule inhibitors specifically targeting IDH1 R132H and IDH2 R140Q showed that many of the effects of mutant IDH, including histone hypermethylation, colony formation, and differentiation, were indeed reversible (18, 19). Here we show that indirectly blocking 2HG production by inhibiting GLS (and thus flux through mutant IDH1), is capable of reversing histone hypermethylation and soft agar growth in human astrocytes, indicating that indirectly blocking mutant IDH1 activity by inhibiting GLS may serve as an alternative therapeutic strategy in appropriately reprogrammed cells. Additional studies are needed to fully validate the therapeutic potential of GLS inhibition in IDH mutant cancer, including dissecting relevant reprogrammed metabolic fluxes, further characterization of selective GLS inhibitors, and use of improved cell permeable analogues of R-2HG. This is of particular interest with regard to mutant IDH1-induced histone hypermethylation since inhibition of GLS simultaneously reduced both cellular 2HG and αKG. In the face of opposing metabolite actions (2HG inhibits αKG-dependent dioxygenases such as the JmjC histone demethylases, while αKG activates), histone hypermethylation states were nonetheless reversed overall by Zaprinast treatment, suggesting that the activity of histone demethylases could be more sensitive to reductions in 2HG than αKG. How the ratio of 2HG to αKG determines the activity of these enzymes or regulation of histone methylation through other routes requires further study. Zaprinast is among the first small molecule inhibitor not directly targeting mutant IDH to show this effect and broadens the targets in the pathway amenable to modulation. Selective targeting of GLS may be important for subsets of patients with IDH1 or IDH2 mutations not affected by small molecule inhibitors developed against target-specific IDH mutations or patients displaying resistance to IDH1-targeted therapies.
Interestingly, Seltzer et al. showed that GLS inhibition by BPTES preferentially limits growth of D54 glioblastoma cells expressing IDH1 R132H when compared to wild type IDH1 (30). However, 2HG levels were unaffected by BPTES. This may be attributed to differences in metabolic wiring that allows some cells to overcome GLS inhibition. Indeed, Cheng et al. showed that some cells utilize pyruvate carboxylase for anapleurosis under conditions of glutamine deprivation or silencing of GLS (31). Thus, it is possible that differences in expression of pyruvate carboxylase may dictate responsiveness to pharmacological inhibition of GLS in IDH1 mutant tumors.
GLS expression has been shown to correlate with tumor growth and tumor grade independent of IDH mutations (32). c-Myc-driven expression of GLS promotes glutaminolysis and glutamine dependence, and furthermore, inhibition of GLS was shown to cause a marked reduction in growth of several glutamine-dependent cell lines and xenograft models (2, 3, 33). Son et al. showed that PDAC cells rely on glutamine metabolism for maintenance of redox state (4). We examined the effect of Zaprinast on Panc1 and MiaPaca2 cells and showed that Zaprinast inhibits cell growth, caused an increase in ROS levels, and sensitized cells to oxidative damage by hydrogen peroxide, all in a manner which can be rescued by extracellular glutamate. Thus, our data indicate that Zaprinast mimics the effects of glutamine deprivation in PDAC cells, and point to a targeted small molecule strategy amenable to translation.
Thus, our work and that of others highlight the potential utility of pharmacologically targeting GLS in cancer and further stress the need for potent and safe inhibitors (33, 34). We have identified a new chemical structure capable of directly inhibiting the activity of purified GLS noncompetitively, which typically implies allosteric binding of compound to target enzyme. Importantly, treatment of multiple independent cell lines with Zaprinast altered cellular metabolite levels in a manner most consistent with GLS inhibition as the mechanism of action. In addition, in Zaprinast-treated cells, addition of cell permeable glutamate rescued growth and abolished the heightened sensitivity to oxidative damage, providing further evidence for a GLS-targeted mechanism in live cells. However, given the higher IC50 of Zaprinast for GLS relative to PDE5, doses needed to block GLS in vivo may not be achievable clinically without also producing vascular side-effects. While pharmacokinetic modulation and dosing regimens may assist in clinical translation, it is more likely that Zaprinast can serve as a convenient tool compound with improved drug-like molecular properties compared to other candidate GLS inhibitors, such as BPTES. Zaprinast may be useful for preclinical mechanistic studies of the linkages between GLS and 2HG-mediated oncogenesis as well as a new scaffold from which alternate analogs with increased potency and selectively against GLS can be generated. Future structure-activity relationship studies are needed to tease out the binding site and active pharmacophores required for rational drug design and the development of more potent and selective GLS inhibitors.
Materials and Methods
Plasmid construction
IDH1 cDNA clone (BC012846.1) was purchased from ATCC in the pCMV-Sport6 backbone. Site-directed mutagenesis was carried out to introduce a g395a mutation (R132H) and sequence verified. The ORF of both wild type and R132H IDH1 were then subcloned into pcDNA 3.1 and pLVX-IRES-Hyg vectors using standard molecular biology techniques. Histidine-6× tagged E coli 3-phosphoglycerate dehydrogenase (PHGDH) plasmid was a kind gift from Dr. Gregory Grant (Washington University School of Medicine). sGCα and sGCβcys105 plasmids were a kind gift from Dr. Emil Martin (University of Texas Health Sciences Center, Houston). Human GLS cDNA (BC126537) was purchased from Thermo-Scientific and the ORF was subcloned into pSV281 containing an N-terminal 6× His tag using forward and reverse restriction sites, BamH1 and HindIII, respectively. pLKO.1-puro shRNA constructs were provided by the Washington University Genome Institute and used for RNA interference against IDH1. Sequences for the shRNAs were as follows: 1) 5’-CCTTTGTATCTGAGCACCAAA-3’ and 2) 5’-GCTGCTTGCATTAAAGGTTTA-3’.
Cell culture, transfection, and generation of stable cell lines
HT1080, HEK293T, Panc1, and MIA PaCa-2 cells were obtained from the American Type Culture Collection (ATCC). Normal human astrocytes (NHA) immortalized with E6/E7/hTERT were a kind gift from Dr. Russell O. Pieper (University of California, San Francisco). All cells tested negative for mycoplasma infection. HT1080 cells were authenticated by genotyping for IDH1 and were confirmed to harbor a heterozygous IDH1 R132C mutation by Sanger sequencing of genomic DNA products. The remaining cell lines were not further authenticated. HEK293T, NHA, PANC-1, and MIA Paca-2 cells were maintained with complete DMEM containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). HT1080 cells were maintained in αMEM containing 10% FBS and 1% P/S. Glutamine-free DMEM (Life Technologies) was supplemented with 10% FBS and 1% P/S and spiked with varying amounts of L-glutamine (Cellgro) for glutamine growth curves. Zaprinast (Sigma-Aldrich) and H2O2 were added to the appropriate fresh media as indicated.
For production of lentivirus, 1×106 HEK293T cells were co-transfected with pCMV-VSVS-G, pCMVΔR8.2, and either pLKO.1-puro for RNA interference experiments or pLVX-IRES-Hyg constructs for stable over-expression using Fugene 6 (Promega) for 48 hours after which viral supernatants were collected and transferred to cells of interest. Cells transduced with pLKO.1-puro were then selected in batch with 1 µg/mL puromycin for 72 hours, while cells transduced with pLVX-IRES-Hyg were selected in batch with 100 µg/mL hygromycin for 1 week.
2HG fluorimetric assay
N-terminal 6×-His tagged PHGDH was expressed and purified from chemically competent BL-21 E.Coli using Ni-NTA agarose (Qiagen) (20). Purified protein was then dialyzed in 40 mM KPO4 buffer at 4°C overnight. Conditioned culture media (DMEM or αMEM) containing 2HG was neutralized with 60 mM HCL for 10 minutes followed by 60 mM Tris base. A 20 µL aliquot of neutralized conditioned media was then mixed with 90 µL of optimized assay reaction mix consisting of 5 µg purified PHGDH enzyme, 12.5 µM resazurin (Sigma-Aldrich), 0.125 u/ml diaphorase (Sigma-Aldrich), 1 mM NAD (Sigma-Aldrich), 40 mM Tris-HCL pH 8.8, and incubated for 90 minutes in a 96-well plate. Resorufin fluorescence was then quantified using a FLUOstar OPTIMA fluorescence plate reader (BMG Labtech) with excitation and emission peaks of 544 nm and 590 nm, respectively. Measurements were background subtracted (neutralized fresh media) and normalized either to total protein or viability as measured by Alamar Blue (21).
High-throughput screening
Screening was carried out using the Beckman Coulter (Fullerton, CA) Core Robotics system, including an FX liquid handler, controlled by the Sagian graphical method development tool (SAMI scheduling software). The Institute of Chemistry and Cell Biology (ICCB) Known Bioactives Library (Enzo Lifescience, BML-2840-0100) was diluted 1:200 in αMEM and 50 µL were added to pre-plated HT1080 cells. After 48 hours of incubation, media was removed and 2HG in the media was measured using the fluorimetric assay. Fresh 2HG fluorimetric assay reaction mix was prepared immediately before assaying each screen plate to limit background modulation. Cell viability was determined using Alamar Blue as described previously (21).
To identify screen hits and account for differences in compound toxicity, 2HG fluorescence was first median centered around DMSO treatment controls within each screen plate. 2HG fluorescence was then plotted against viability such that a correlation between the two measurements could be visualized. A linear fit and 95% predictive interval (P.I.) were then determined and plotted using GraphPad Prism (GraphPad Software).
Animal studies
All animal experiments were approved by the Institutional Animal Care and Use Committee at Washington University in St. Louis School of Medicine. To generate tumor xenografts, 8-week-old nu/nu mice (Taconic) were injected subcutaneously in the flank with 3 × 106 HT1080 cells in 100 µL αMEM. Tumors were allowed to grow for 11 days before treatments were started. Intratumoral injection was performed on days 11, 13, and 15 with 20 µL of solution containing vehicle (DMSO) or Zaprinast (600 µM) (Sigma-Aldrich). Tumor volume was calculated as l × w × h of tumor dimensions obtained by caliper measurements. On day 15, tumors were extracted and snap frozen for follow up GC-MS analysis.
Histone extraction and Western blot analysis
For extraction of total cellular protein, cells were lysed in RIPA buffer (150 mM NaCl, 50 mM Tris, 5 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, pH 7.4), supplemented with protease inhibitor cocktail (Roche), sodium orthovanadate (1 mM), and PMSF (1 mM). Whole-cell lysates were normalized for protein content by BCA assay (Pierce). Histone extraction was performed using an EpiQuick Total Histone Extraction Kit (Epigentek) and extracted protein was quantified using a Bradford assay.
Proteins were resolved by SDS-PAGE, transferred to a PVDF membrane and probed with the following antibodies: IDH1 (Origene, TA500610), 6×His (Abcam, ab1187), H3K9Me2 (Cell Signaling, 4658p), H3K9Me3 (Abcam, ab8898), H3K27Me3 (Millipore, 07-449), H3K79Me2 (Cell Signaling, 9757p), total H3 (Cell Signaling, 4499p), and Actin. Secondary anti-mouse and anti-rabbit horseradish peroxidase-conjugated IgG antibodies were used for detection.
GC-MS analysis
Metabolite extraction from cultured cells was performed as described previously by Figueroa et al. (13). Briefly, cells were rinsed in ice cold PBS and rapidly quenched with 80% methanol spiked with 3-hydroxy-1,5-pentanedioic-2,2,3,4,4-d5 acid (CDN Isotopes, Quebec Canada) as an internal standard. Extracts were then incubated at −80°C for 20 minutes, sonicated on ice, centrifuged at 14,000 × g for 20 minutes at 4°C to clear precipitate proteins, and supernatants transferred to vials for drying under N2. Derivatizing reagent (MSTFA (N-methyl-N-(trimethylsilyl) trifluoroacetamide): pyridine:acetonitrile 1:1:2) was added to the vials, which were then heated at 70°C for 15 minutes.
Derivatized samples were analyzed on an Agilent 7890A gas chromatograph interfaced to an Agilent 5975C mass spectrometer. The GC column used for the study was a HP-5MS (30 m, 0.25mm i.d., 0.25µm film coating). A linear temperature gradient was used. The initial temperature of 80°C was held for 2 minute and increased to 300°C at 10°C/minute. The temperature was held at 300°C for 2 minutes. The samples were run by electron ionization (EI) and the source temperature, electron energy and emission current were 200°C, 70 eV and 300 µA, respectively. The injector and transfer line temperatures were 250°C. 3-hydroxy-1,5-pentanedioic-2,2,3,4,4-d5 acid was used as the internal standard in SIM mode for quantitation of 2-hydroxyglutaric acid. Quantitation was carried out by monitoring the ions at m/z 347 (glutamine and ±KG), 348 (glutamate), 349 (2-hydroxyglutaric acid), and 354 (3-hydroxy-1,5-pentanedioic-2,2,3,4,4-d5).
Glutaminase assay
N-terminal 6×-His tagged GLS was expressed and purified from chemically competent BL-21 E. Coli using Ni-NTA agarose beads (Qiagen). Purified protein was then dialyzed in a 50 mM Tris-phosphate buffer containing 1 mM dithiothreitol at 4°C overnight. A previously described two-step assay was used to measure glutaminase activity (22). Briefly, 30 µL of purified glutaminase (660 µg/ml) were incubated with various concentrations of Zaprinast at 37°C for 15 minutes. Zaprinast was dissolved in DMSO and 3 µL was added to each glutaminase aliquot to maintain a constant concentration of DMSO across samples. Initial reaction mix (30 µl) containing 20 mM glutamine, 0.15 M KPO4, 0.2 mM EDTA, and 50 mM Tris-acetate (pH 8.6) was then added and samples were incubated at 37°C for 10 minutes after which the reaction was rapidly quenched with 6 µL of 3 N HCL. To generate blank samples, glutaminase and Zaprinast solutions were inactivated with 3 N HCL before the addition of initial reaction mix. For the second step of the assay, 20 µL of completed and quenched initial reaction were transferred to a new plate and 200 µL of secondary reaction mix containing 0.4 mg bovine liver glutamate dehydrogenase (Sigma-Aldrich), 0.09M Tris-acetate (pH 9.4), 0.2 M hydrazine, 0.25 mM ADP, and 2 mM NAD was added. Samples were incubated for 40 minutes at room temperature and absorbance of NADH (340 nm) was measured using a microplate spectrophotometer.
Quantification of intracellular cGMP
HT1080 cells were rinsed with ice cold PBS, lysed with 0.1 mM HCL, and centrifuged at 1,000 × g for 10 minutes. Supernatant was then assayed for cGMP using a cGMP Enzyme Immunoassay kit (Cayman Chemical, 581021) according to the manufacturer protocol.
ROS quantification
Reactive oxygen species levels in PDAC cells were quantified using 2,7-dichlorofluorescein diacetate (DCFDA). Following 48 hours of drug treatment in a 96-well plate, cells were washed with PBS, stained for 30 minutes at 37°C with 20 µM DCFDA, and imaged using an InCell Analyzer 1000. A 10× objective was used to collect 16 fluorescent and bright field images per well with a 10% overlap to allow image stitching. Stained cells were then quantified using the GE InCell Investigator software package with Developer Toolbox.
Soft agar assay
A bottom layer of 0.6% soft agar with complete DMEM and 10% FBS was set in 6-well plates and allowed to solidify. Approximately 8,000 cells were then suspended in 0.4% soft agar and plated on top. Colonies were allowed to develop over 3–4 weeks and stained with crystal violet.
Statistical analysis
Statistical significance was evaluated using Students t-test or two way ANOVA as indicated. Error bars represent S.D. unless otherwise indicated.
Supplementary Material
Statement of Significant.
Gain-of-function IDH mutations are common events in glioma, AML, and other cancer types, which lead to the accumulation of the onco-metabolite 2HG. We show that the drug Zaprinast is capable of reducing cellular 2HG levels by inhibiting the upstream enzyme GLS, thus identifying a new strategy to target 2HG production in selected IDH mutant cancers.
Acknowledgements
We thank Julie Prior for technical assistance with animal experiments. This work was supported by National Institutes of Health NCI Molecular Imaging Center Grant P50 CA94056. The HTC obtains support from the Siteman Cancer Center, supported in part by a NCI Cancer Center Support Grant (P30 CA91842). The Washington University Biomedical Mass Spectrometry core obtains support from the following sources: P41 RR000954, P60 DK020579, and P30 DK056341.
Footnotes
The authors have no conflicts of interest to disclose.
References
- 1.DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224:334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 10.Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17:72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 20.Grant GA, Xu XL, Hu Z. The relationship between effector binding and inhibition of activity in D-3-phosphoglycerate dehydrogenase. Protein Sci. 1999;8:2501–2505. doi: 10.1110/ps.8.11.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naik S, Dothager RS, Marasa J, Lewis CL, Piwnica-Worms D. Vascular Endothelial Growth Factor Receptor-1 Is Synthetic Lethal to Aberrant {beta}-Catenin Activation in Colon Cancer. Clin Cancer Res. 2009;15:7529–7537. doi: 10.1158/1078-0432.CCR-09-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kenny J, Bao Y, Hamm B, Taylor L, Toth A, Wagers B, et al. Bacterial expression, purification, and characterization of rat kidney-type mitochondrial glutaminase. Protein expression and purification. 2003;31:140–148. doi: 10.1016/s1046-5928(03)00161-x. [DOI] [PubMed] [Google Scholar]
- 23.Zhao G, Winkler ME. A novel alpha-ketoglutarate reductase activity of the serA-encoded 3-phosphoglycerate dehydrogenase of Escherichia coli K-12 and its possible implications for human 2-hydroxyglutaric aciduria. J Bacteriol. 1996;178:232–239. doi: 10.1128/jb.178.1.232-239.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu A, Romero R, Petty HR. An enzymatic fluorimetric assay for glucose-6-phosphate: application in an in vitro Warburg-like effect. Anal Biochem. 2009;388:97–101. doi: 10.1016/j.ab.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sandner P, Hutter J, Tinel H, Ziegelbauer K, Bischoff E. PDE5 inhibitors beyond erectile dysfunction. International journal of impotence research. 2007;19:533–543. doi: 10.1038/sj.ijir.3901577. [DOI] [PubMed] [Google Scholar]
- 26.Martin E, Sharina I, Kots A, Murad F. A constitutively activated mutant of human soluble guanylyl cyclase (sGC): implication for the mechanism of sGC activation. Proc Natl Acad Sci U S A. 2003;100:9208–9213. doi: 10.1073/pnas.1633590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balss J, Pusch S, Beck AC, Herold-Mende C, Kramer A, Thiede C, et al. Enzymatic assay for quantitative analysis of (D)-2-hydroxyglutarate. Acta Neuropathol. 2012;124:883–891. doi: 10.1007/s00401-012-1060-y. [DOI] [PubMed] [Google Scholar]
- 29.Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discov. 2012;11:937–957. doi: 10.1038/nrd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70:8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cassago A, Ferreira AP, Ferreira IM, Fornezari C, Gomes ER, Greene KS, et al. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci U S A. 2012;109:1092–1097. doi: 10.1073/pnas.1112495109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18:207–219. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shukla K, Ferraris DV, Thomas AG, Stathis M, Duvall B, Delahanty G, et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem. 2012;55:10551–10563. doi: 10.1021/jm301191p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.