

Abstract
Recent technological advances in accurate mass spectrometry and data analysis have revolutionized metabolomics experimentation. Activity-based and global metabolomic profiling methods allow simultaneous and rapid screening of hundreds of metabolites from a variety of chemical classes, making them useful tools for the discovery of novel enzymatic activities and metabolic pathways. By using the metabolome of the relevant organism or close species, these methods capitalize on biological relevance, avoiding the assignment of artificial and non-physiological functions. This review discusses state-of-the-art metabolomic approaches and highlights recent examples of their use for enzyme annotation, discovery of new metabolic pathways, and gene assignment of orphan metabolic activities across diverse biological sources.
Keywords: enzyme annotation, mass spectrometry, metabolomics, pathway discovery
Introduction
The functional annotation of the wealth of genetic information generated during the genomic era is currently considered one of the grand challenges in molecular biology. Although bioinformatics and modelling have contributed considerably to the functional assignment of proteins, significant portions of sequenced genomes (between 30 and 40% of genes by a recent estimate 1) remain unannotated or are ascribed a putative function. Accurate genome annotation is essential for developing a comprehensive and detailed understanding of cellular physiology and is therefore a primary concern in almost every avenue of biological research.
In silico sequence homology-based methods have been the driving force behind most genome annotation endeavours to date 2. Despite their strengths in automation and sample throughput, such techniques are unable to identify the functions of novel gene sequences that have little to no homology with pre-existing database entries or may lead to the misannotation of gene products that share very high homology but catalyse fundamentally different reactions. Gene misannotations in particular are a prevalent consequence of automated in silico methods, and the propagation of such misannotations is a serious and growing threat to the accuracy and reliability of genome and protein databases 3–5. Automated genome annotation remains the only viable option to efficiently process genetic sequences at their current rate of influx, although a more comprehensive and experimentally determined understanding of the relationship between primary sequence and function is clearly required in order to improve annotation accuracy.
A significant proportion of unannotated or misannotated genes encode enzymes, the catalytic activity of which is usually fundamental to their physiological function. Techniques that can directly exploit or monitor the native activity of a candidate enzyme are therefore powerful tools in accurate functional assignment of unannotated gene products: several approaches that have historically been used to annotate enzyme function, as well as newly developed techniques, are described in Fig 1 and Table 1. However, many activity-based assays—for example, those performed in vitro with purified enzyme preparations—require at least a prior basic knowledge of the type of reaction catalysed by or substrate specificity of the candidate enzyme and therefore lack broad applicability. Furthermore, testing individual putative substrates on a case-by-case basis is time-consuming and expensive and relies on the native substrate being commercially available. Alternatively, screens performed in situ in the host organism are less biased as they evaluate enzyme activity within a physiologically relevant milieu. Forward and reverse genetic screens, for example, are fundamental tools in uncovering the function of unknown gene products, but ultimately rely on the emergence of observable phenotypic traits for successful gene assignment, which does not occur for many mutants 6,7. An activity-based proteomics approach (activity-based proteomic profiling; ABPP) has been recently used to identify class-specific enzymes within a complex mixture (including in situ in cells) in an unbiased manner using covalent active-site-directed probes 8. Its potential as an enzyme function discovery tool has been illustrated by the successful assignment of mechanistic class and function to previously unannotated enzymes that lack sequence homology with canonical members of their enzyme class 9,10. Despite its dependence on unique synthetic chemistry tools and limited scope in native substrate identification, this technique demonstrates the usefulness of physiologically relevant sequence- and phenotype-independent tools in modern functional genomics.
Figure 1. Approaches used to uncover the function of orphan enzymes.
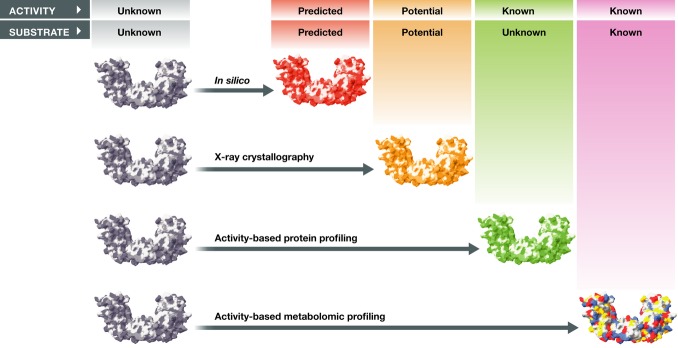
Table 1.
Strategies for enzyme function discovery
| Techniques available | Enzyme/genetic requirements | Purposes | Advantages | Inconveniences | Key technologies |
|---|---|---|---|---|---|
| In vitro activity-based metabolomic profiling | Purified, homogeneous enzyme | Track enzyme-induced changes in a complex metabolite extract | High throughput (hundreds to thousands of metabolites can be screened). | Enzymes have to be purified to homogeneity. | Protein purification |
| Physiologically relevant library of substrates and co-factors. | Host organism or related species has to be cultured. | LC/GC/CE-MS | |||
| No a priori knowledge of the types and number of substrates and products involved. | Recombinant expression might lead to loss of native partner or post-translational modifications required for activity. | NMR | |||
| No a priori knowledge of the type of chemistry catalysed. | Substrates might not be present at quantifiable levels in molecular extract. | Libraries of spectral data | |||
| Direct identification of potential substrates and products. | |||||
| Ex vivo metabolomic profiling – genetically modified/chemically treated organism | None or verified genetic knockout/over-expression strain of organism of interest | Identify one enzymatic reaction or pathway that is disturbed upon deletion/alteration of levels of a particular enzyme | High throughput (hundreds to thousands of metabolites can be screened). | Host organism or related species has to be cultured and genetically tractable. | Genetic manipulation LC/GC/CE-MS |
| No knowledge of the types and number of substrates and products involved required. | Candidate substrates and products might constitute secondary effect changes. | NMR | |||
| No knowledge of the type of chemistry catalysed required. | Levels of substrates/products might be tightly controlled and not change. | Libraries of spectral data | |||
| No enzyme purification required | Chemical with a clear phenotype must be available. | ||||
| Preservation of native enzyme partners and post-translational modifications. | |||||
| Activity-based protein profiling | None | Track activity of a specific class of enzymes towards a probe | High throughput (several dozen enzymes can be screened). | Highly selective and specific probe needs to be synthesized. | Chemical probe |
| Identifies active enzymes. | Identification of physiological substrates needs to be carried out subsequently. | Gel electrophoresis | |||
| Highly specific for the chemistry and enzyme class to which the probe has been developed. | Host organism or related species has to be cultured. | Imaging | |||
| No enzyme purification or genetic modification required | Active enzyme of interest needs to be identified. | Protein identification | |||
| Preservation of native enzyme partners and post-translational modifications. | |||||
| Computational enzymology | High-resolution structure | Identification of putative substrates, products and intermediates based on structural determinants | High throughput in silico approach can be applied to any enzyme type. | Relies on strength of ligand docking software and accuracy of crystal structure. | Docking |
| No a priori knowledge of substrate specificity or type of chemistry catalysed required. | Identified compounds might not exist in the host organism. | Virtual libraries | |||
| Computation | |||||
| X-ray crystallography | Purified, homogeneous enzyme | Identify co-purified small molecules associated with purified enzyme | Tightly bound ligands can directly lead to the identity of substrates/products/intermediates. | Enzymes have to co-purify with a tightly bound metabolite. | Protein purification |
| High-resolution structure | Enzymes have to be crystallized and the structure has to be solved at sufficiently high resolution. | Crystallization | |||
| Bound ligand structure has to be determined. | Structure determination | ||||
| Bound ligand might not be present in the host organism or be related to the native function. |
NMR, nuclear magnetic resonance; GC/LC/CE, gas or liquid chromatography or capillary electrophoresis.
Metabolites constitute the substrates and products of enzymatic reactions, and the study of the total metabolite pool of a given organism or cell type is known as metabolomics. As metabolites represent the final outcome of gene expression and activity, the metabolome can be perceived as the ultimate readout of the biochemical and physiological state of a cell, that is, a direct link between mechanistic biochemistry and cellular phenotype 11. The concept of metabolomic profiling for assessing cellular or bodily function is not new 12; however, the analytical and computational technologies have only recently become sufficiently powerful and widely accessible to allow routine and unbiased investigations of cellular metabolite pools. With the latest improvements in mass spectrometry (MS), for example, it is now possible to sample hundreds to thousands of unique ion peaks, assign putative (or verified) molecular formula to these peaks, and even extrapolate native intracellular concentrations, all from a small (even single cell analysis 13) quantity of starting material 14,15 (Sidebar A). Due to these recent innovations, metabolomics is rapidly becoming a routine discipline in diverse areas of biological research, including disease biomarker and drug target discovery, drug pharmacodynamics, and metabolic engineering 16–21. The ability to impartially monitor metabolic transformations from a variety of biological sources has also allowed metabolomics to thrive within the context of enzyme function discovery, typically in combination with recombinant genetic and protein expression tools. The focus of this review is the application of activity-based metabolomic strategies in three principal areas of functional genomics: the discovery of novel metabolic functions and pathways, the functional assignment of unannotated genes, and the assignment of gene sequences to orphan metabolic activities.
Sidebar A: Mass spectrometry for metabolomics.
The diversity and quantity of metabolites that comprise a metabolome varies depending on the organism, and can range from several hundred (bacteria) to several thousand (mammals, plants) unique low-molecular-weight (< 1000 Da) chemical entities 82,83. The relative scarcity of metabolomic investigations, relative to other ‘omics strategies’, is a consequence of the difficulties associated with the unbiased analysis of the wide physicochemical heterogeneity apparent across these compounds. Although procedures for analysing specific chemical classes have been employed for some time (‘targeted’ metabolomics 84,85), the ability to assess a range of classes simultaneously and impartially (‘untargeted’) has only become possible with recent technological progress in certain analytical platforms; in particular nuclear magnetic resonance (NMR) and mass spectrometry (MS). NMR offers advantages through milder sample processing and a more robust quantitative output, but MS-based methods have dominated the field due to their superior mass resolution, sensitivity (atto- to zemptomolar 86), and mass accuracy (sub millidalton 87). Furthermore, MS can be combined with sample separation techniques such as gas or liquid chromatography or capillary electrophoresis (GC/LC/CE) to enhance the detection of individual species. Another major benefit of MS-based platforms is the increasing number and quality of publicly available databases dedicated to mass spectral profiles of authentic standards, enhancing confidence in matching experimental data peaks to molecular structures 88–91. The intricacies of MS function, sample preparation and data analysis are beyond the scope of this review, and have been excellently covered elsewhere 11,88,92.
Discovery of New Metabolic Functions and Pathways
A significant impediment to large-scale assignation of enzymatic function is our incomplete understanding of primary and secondary metabolism, even among well-studied organisms such as Escherichia coli. Consequently, the discovery of novel metabolic pathways is an ongoing effort 22–25. The ability of metabolomics, particularly in combination with stable-isotope probing (Sidebar B), to follow the metabolic fate of target compounds and their flux through specific pathways underlies its utility in the identification of new biochemical reactions occurring within cells. In this context, new pathways and metabolites can be identified without the need for targeted genetic modification or recombinant protein studies, simplifying the workflow and allowing greater flexibility in the conditions and test organisms used. Several approaches to metabolite and pathway discovery using activity-based metabolomics methodologies are outlined below.
Sidebar B: Stable isotope labelling.
Stable isotope labelling or SIL (mainly with 13C and 15N) is an essential component of most studies utilizing discovery metabolomic techniques. Two types of information can be uniquely gathered from well-designed SIL experiments: (a) metabolic fate, and (b) metabolic pace.
Metabolic fate defines the relationship between the reaction identified and its exact position in the metabolic network to which it belongs. This is especially relevant when substrates and/or products of the candidate enzyme overlap multiple metabolic pathways, obscuring the true physiological function of the enzyme. For example, if enzyme X converts succinic semialdehyde (SSA) into succinate, there are at least two known potential metabolic pathways in which this enzyme could serve (Fig 2A). The first is the γ–aminobutyric acid (GABA) shunt 93 which is involved in glutamate and GABA metabolism and is of particular importance in neurotransmitter regulation in the mammalian central nervous system. The second involves detoxification of SSA generated by the α–ketoglutarate dehydrogenase complex under non-optimal Krebs cycle conditions, which can be caused by mutations or environmental stimuli such as oxidative stress 94,95. Therefore, by measuring the extent of succinate labelling following selective supplementation of growth media with either labelled glutamate or a labelled glycolytic intermediate (e.g. dextrose), one can unambiguously define the specific pathway in which enzyme X is involved (Fig 2A and B), and by consequence gain a more detailed indication of the enzyme’s physiological role (e.g. neurotransmitter metabolism versus mitochondrial defect).
Metabolic pace refers to the flux (rate) of biochemical reactions and pathways in living cells (Fig 2C). Time-course experiments with SIL can therefore provide direct evidence for variations in the flux of specific biochemical pathways that have been perturbed by chemical or genetic means. Metabolic rate studies of newly discovered/annotated enzymes are particularly informative when the concentration of a determined metabolite (pool size) does not change considerably upon gene deletion or drug treatment, but the flux through the pathway is affected 96–98.
Figure 2. Labelling experiments can probe pathway(s) and the metabolic context of newly identified enzymatic activities.
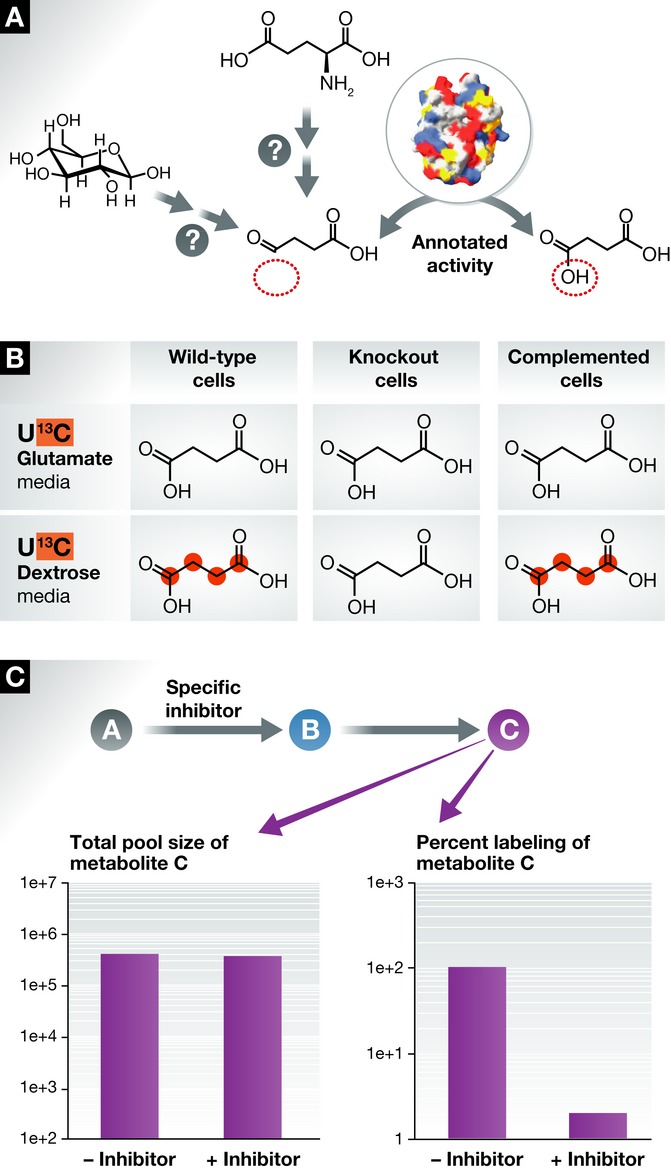
(A) An enzymatic reaction is identified by activity-based metabolic profiling or another method. The structure is of Mycobacterium tuberculosis CitE 99. (B) Possible outcome of a labelling experiment designed to probe the origin of the carbon backbone of succinate. In this case, as glutamate labelling does not generate labelled succinate, a classic GABA shunt starting from glutamate is ruled out. Labelling with dextrose would therefore indicate a mitochondrial role for this newly identified enzyme. (C) Labelling experiment designed to confirm that the enzymatic activity identified belongs to the pathway described. The specific inhibition of synthesis of compound B leads to no change in the pool size of metabolite C. However, it drastically diminishes its labelling, supporting the relationship between A and C.
Our current understanding of central metabolism has been influenced by studies performed in a small number of model organisms under a limited set of test conditions, suggesting that conditionally induced pathways are poorly represented in metabolic charts. Varying growth conditions, or applying abiotic or biotic stimuli to cultures of interest, are therefore useful tactics for the elucidation of cryptic pathways. Farag et al recently used a metabolomics approach to delineate several novel isoflavonoid and phenylpropanoid pathways in the leguminous plant species Medicago truncatula after stimulation with methyl jasmonate and yeast elicitor, two agents known to influence plant secondary metabolite synthesis 26. The use of stable-isotope tracers in combination with metabolomic profiling allowed the authors to define the precise biosynthetic origin, metabolic pathway, and molecular formula of each identified compound, as well as the complex regulatory patterns governing the expression of each pathway. Many plant secondary metabolites have important pharmacological and biotherapeutic properties, and it is therefore crucial to understand their biosynthetic routes and regulation. Metabolomics-based strategies should have a central role in achieving this goal.
Similarly, new pathways and functions are likely to be found in non-model organisms, particularly those inhabiting unique physical environments—such as thermophiles and halophiles. Importantly, non-canonical metabolic processes that occur in these organisms, or those able to operate under non-mesophilic conditions, are often highly sought after as biotechnological or industrial tools. Stable-isotope probing combined with metabolomics and fluxomics was recently used to uncover an unusual isoleucine biosynthesis pathway in Geobacter metallireducens 27 and the formation of a non-canonical (R-) stereoisomer of citrate in the Krebs cycle of Desulfovibrio vulgaris 28, two environmentally important microorganisms involved in global element cycling pathways. In addition, the long predicted ethylmalonyl-CoA pathway for growth on reduced single carbon compounds was finally demonstrated to exist through the combined application of metabolomics and stable-isotope flux analysis in the methylotrophic bacterium Methylobacterium extorquens 29. Understanding how microorganisms are able to convert single carbon compounds into complex biomolecules is of major interest in biotechnology, particularly for biofuel production, and these studies exemplify the potential of metabolomics to uncover hitherto unknown metabolic pathways.
Alternatively, molecular genetics can be used in combination with metabolomics as an effective hypothesis-generating tool to discover new metabolic functions: the disruption of a pathway of interest might lead to the upregulation of alternative pathways to compensate for the lost function(s). For example, despite the essentiality of central carbon metabolism in all living systems, studies have shown that just 4 of the more than 70 enzymes involved in E. coli glycolysis and the tricarboxylic acid (TCA) cycle are essential for growth under standard laboratory conditions 30. Such genetic redundancy indicates the presence of alternative compensatory pathways or isoenzymes that operate in the event of primary pathway disruption 31. Nakahigashi et al recently analysed the biochemical mechanisms of pathway compensation in E. coli strains deficient in primary enzymes of central carbon metabolism 32. One unexpected observation was the lack of a growth defect in a transaldolase-deficient mutant when xylose was supplied as sole carbon source, as transaldolase provides the link between the pentose phosphate pathway and glycolysis. Metabolomic profiling revealed the appearance of a molecular species equivalent to sedoheptulose-1,7-bisphosphate only in the mutant strain, which was further confirmed through genetic screening and metabolic flux analysis and found to be a novel side reaction of 6-phosphofructokinase I (pfkA). The combination of genetic tools and metabolomics has also been used to identify a previously unrecognized uridine monophosphate (UMP) degradation pathway in E. coli strains that are deficient in a negative feedback regulator of pyrimidine homeostasis 33. In this case, metabolomic profiling led to the identification of not only a novel enzymatic activity (UMP phosphatase), but also a unique ‘directed overflow’ regulatory mechanism involved in pyrimidine catabolism. These examples illustrate the power of metabolomics approaches to delineate both the enzymatic activities and regulatory features responsible for the correct function of metabolic pathways.
Co-Expression Analysis: Combining Metabolomics with Transcriptomics
The understanding of novel metabolic activities and pathways is incomplete without the identification of the genes responsible. As already mentioned, untargeted investigations of new biochemical pathways can rarely associate metabolic reactions with the enzymes that catalyse them without subsequent genetic intervention or recombinant protein studies. One method that can overcome this is co-expression analysis. This transcriptomics-based method allows the identification of genes involved in a defined metabolic pathway based on their co-expression with genes of known function, through the assumption that genes involved in the same biochemical pathway are co-regulated 34. For example, by comparing transcriptome data sets of a test organism cultured under various conditions, sets of genes that are commonly co-expressed are predicted to operate in a single metabolic pathway. As a stand-alone technique, co-expression analysis has been an instrumental tool in functional genomics, allowing high-throughput genome-wide and inter-species predictions of protein function 35,36. However, protein functional assignment is usually restricted to the overall pathway level, and specific enzymatic functions can rarely be extrapolated from the co-expression data alone. The recent combination of co-expression analysis with metabolomics-based platforms has increased the potential of this approach. This allows the use of changes in metabolomic profiles that correlate with changes in transcriptomic profiles to predict putative associations between genes and defined metabolic functions. For example, this approach has been successfully applied in the assignment of sulfotransferase and glucosyltransferase functions to genes involved in glucosinolate and flavonoid biosynthesis in Arabidopsis, respectively 37,38. Importantly, the predicted enzyme functions were validated through subsequent genetic and recombinant enzyme-based assays. Although effective in both pathway discovery and gene assignment, a significant pitfall of this approach is the reliance on a direct and positive correlative relationship between gene expression levels and metabolic activity. As post-transcriptional modifications and regulation of enzyme function have substantial effects on most, if not all, metabolic pathways, many of the causal effects between transcript and metabolite levels may be lost in the resulting data sets.
Enzyme Annotation
Activity-based metabolomic profiling approaches
In vitro studies with purified recombinant protein often provide the most definitive proof of a bona fide activity for a specific enzyme, as consumption of substrate and generation of product can be monitored directly in an isolated system, free of contaminating species. The disadvantages of this approach are that putative substrates need to be tested individually (or in small batches) and are often not commercially available, or, if so, prohibitively expensive. A possible solution is to engage the metabolome of the parent organism as a substrate library substitute. The metabolome represents a chemically diverse and rich mixture of physiologically relevant compounds, including those that are unknown or not commercially available, and is easily and cheaply obtained in large quantities (depending on the culturability of the parent organism). After the incubation of a recombinant enzyme of interest with a cellular metabolome extract (including any required co-factors or co-substrates), mass changes can be monitored by mass spectrometry to identify putative native substrates, which decrease in abundance, and products, which increase in abundance (Fig 3). Furthermore, reactions are run in near-native conditions (in the absence of metabolite or protein tagging or labelling) and genetic modification of the host organism is not required, simplifying the overall workflow and improving accessibility to non-specialist laboratories and non-model organisms. The identification of enzyme-induced spectral changes among the complex metabolomic profile obtained by MS can be eased by the use of dedicated MS data analysis software, such as XCMS 39. This general approach was first implemented by Saito et al, who used capillary electrophoresis (CE)-MS on mixtures of recombinant enzyme and E. coli small molecular extract to identify the phosphatase and phosphotransferase activities of two E. coli uncharacterized enzymes, YbhA and YbiV 40. Similar workflows have subsequently provided mechanistic insights for unannotated or misannotated enzymes from a variety of functional classes and biological origins 41–44. Several cases of particular interest are outlined below.
Figure 3. Activity-based metabolomic profiling of recombinant enzymes.

(A) In vitro experimental set-up. (B) Common computational analysis steps.
The human cytochrome P450 monooxygenase (hP450) family of enzymes comprise more than 50 members and have significant roles in the normal physiological metabolism of a variety of lipids, sterols, vitamins and xenobiotics 45. Their diversity, however, has complicated the functional assignment for each member, and currently more than a quarter of annotated hP450s have unknown functions 46. Guengerich et al have extensively characterized the substrate specificity of hP450 enzymes, with successful application of metabolome-based in vitro assays in many cases 47–49. One approach they pioneered is the use of stable-isotope-labelled co-substrates in metabolome-based in vitro reactions to facilitate the positive identification of substrates and products after analysis by MS 50,51. In this case, the reaction containing cellular extract and enzyme is allowed to proceed in the presence of a 50:50 mixture of 18O and 16O-labelled oxygen gas, causing the resulting enzymatic product to emit a 1:1 ratio of native (M) and isotopically labelled (M + 2) m/zs on a mass spectrum plot. Although this approach is applicable to enzymes that have other common co-substrates, such as ATP, NAD(P)H, or SAM (as long as isotopically labelled versions are available), the prerequisite of knowing the identity of the co-substrate limits its application to putative enzymes of known functional class.
Genetic techniques are powerful tools in functional genomics, allowing target gene disruption and subsequent assessment of the resulting phenotype or metabolic status to infer important information on enzymatic function. However, genetic methods are not always applicable, such as when the candidate gene is essential for growth or the host organism is not genetically tractable. In these cases, in vitro metabolomic methods are particularly useful for characterizing enzyme function. For example, although genetic tools are relatively well developed for the human pathogen Mycobacterium tuberculosis, the slow growth rate (~20 h doubling time) of this organism makes gene knockout protocols cumbersome. Consequently, we have used in vitro metabolomic methods to identify the functions of two uncharacterized M. tuberculosis gene products. One is a 2-hydroxy-3-oxoadipate synthase that was previously annotated as an oxoglutarate decarboxylase component of the Krebs cycle oxoglutarate dehydrogenase complex 43 and the other a haloacid dehalogenase superfamily member with glycerol-3-phosphate phosphatase activity involved in the recycling of cell-wall lipid polar heads 44.
Metabolomic profiling approaches
Metabolic enzymes do not function in isolation. Indeed, the core of biochemical network regulation relies upon a multiplicity of complex and dynamic interactions between the various enzymatic players and metabolites involved 52. Both catalytic competency and substrate specificity can be significantly modified via post-translational modifications or allosteric interactions with other biomolecules, according to metabolic requirements 53,54. As such, in vitro technologies are generally insufficient, in isolation, to attain an accurate and detailed picture of the physiological function of a candidate gene. Instead, in or ex vivo approaches are becoming increasingly popular as primary or complementary screens in enzyme function or pathway discovery, with metabolomics at the forefront of these advances. As already mentioned, ex vivo metabolomics has long been a major tool in the delineation of cellular metabolic circuitry and is still a key experimental platform in pathway and metabolite discovery 25. However, the lack of integration between metabolomic and genetic data in such workflows largely precludes the ability to unequivocally match gene products with individual chemical transformations. Instead, the application of ex vivo metabolomics to conditional knock-down, stable genetic mutants, over-expression strains or to chemical knock-downs of a candidate gene product allows a more precise and specific assessment of the function of a gene in cellular metabolism (Fig 4). This methodology was initially used to analyse gene deletion strains of Sacchromyces cerevisiae; whole-cell metabolomes from strains deficient in genes of unknown function were globally compared—by principal components analysis—to those deficient in genes of known function that spanned a variety of metabolic pathways 55. Data sets that clustered similarly provided the necessary evidence to assign a gene product to a specific metabolic pathway, without defining a precise enzymatic function. Improvements in metabolomic data resolution and processing power subsequently allowed Saghatelian et al 56 to precisely identify several natural substrates of the mammalian enzyme fatty acid amide hydrolase (FAAH) by comparing the brain metabolomes of wild-type and FAAH null mutant mouse strains, thus providing the first successful integration of ex vivo metabolomics and gene function discovery. The succeeding decade has witnessed the application of similar workflows to the discovery of natural substrates and catalytic functions for a variety of metabolic enzymes from a range of cell types/organisms 57–62.
Figure 4. Global profiling of cellular metabolome.

(A) Activity-based ex vivo metabolomic profiling of genetically or chemically modified cells. (B) Common computational analysis steps.
Ex vivo metabolomic profiling has seen considerable success in the assessment of enzymes involved in secondary metabolite biosynthesis, in particular non-ribosomal peptide (NRPS) and polyketide synthase (PKS) pathways, the products of which are of substantial biomedical interest due to their pharmacological properties and roles in virulence. In these cases, bioinformatics can reliably predict gene clusters likely to code for NRPS or PKS operons, and subsequent metabolomics of individual gene mutants provides the necessary means of both identifying the final metabolite and assigning each of the chemical steps involved in its biosynthesis to individual gene products 63–65. The strength of using an untargeted metabolomics approach in these situations is evident when considering the myriad of diverse chemical transformations involved in secondary metabolite biosynthesis. Few alternative technologies would have the capacity to simultaneously monitor such a distinct array of enzymatic reactions using a single analytical platform. For example, Schroeder et al recently used genetics and metabolomics to identify the catalytic roles of each of the eight gene products responsible for the production of a new siderophore and virulence factor, hexahydroastechrome, in the filamentous fungus Aspergillus fumigatus 66. By systematically profiling the metabolomes of individual gene deletion strains, the authors were able to assign P450-like hydroxylation, O-methyl transfer, prenylation, and FAD-dependent carbon–carbon double-bond formation activities to each gene product in the pathway.
In addition to the canonical approach described above, several groups have demonstrated that gene functional assessment by ex vivo metabolomics need not require genetic disruption of the gene of interest. This is particularly useful in cases where gene function is essential for cell survival, the host organism does not have readily available genetic tools, or where genetic inactivation of the candidate gene does not cause significant metabolomic changes due to inherent genetic robustness of the test organism 67. For example, Chiang et al recently used a mechanism-based inhibitor to inactivate and interrogate the physiological function of an integral membrane hydrolase (KIAA1363) that had been shown to be upregulated in certain aggressive cancers 68. The metabolomic impact of the inhibitor on target cells permitted the identification of 2-acetyl monoalkylglycerol as the physiological substrate for this enzyme. Although effective in this case, the reliance on synthetic chemistry resources and/or known inhibitors of the candidate enzyme will likely limit the broad applicability of this approach. Furthermore, generic inhibitors that target specific functional enzyme classes are likely to have pleiotropic effects on cellular metabolism, complicating subsequent data analysis. Transient over-expression of candidate enzymes in test cell lines in situ and subsequent metabolomics has been used by Cravatt et al to investigate the function of a series of unannotated human serine hydrolase family proteins 69. Depletion of substrate and overabundance of product in resulting mass spectrum traces allowed the identification of phosphatidylcholine phospholipase activity for one of the test enzymes, ABHD3. As the authors stated, however, care must be taken when interpreting results obtained through this approach, due to possible metabolic artefacts arising from the artificial over-expression system, or the absence of a physiological substrate due to the use of a heterologous host.
Enzyme Discovery: Assigning Genes to Known Metabolic Activities
The experimental workflows and examples discussed above implemented a ‘reverse genetics’ approach, whereby the coding gene of interest is initially chosen by bioinformatic means and the cognate function subsequently sought with activity-based metabolomics. Although this is a logical process for assigning function to unannotated genes, the reverse process, that is, assigning gene sequences to predetermined orphan metabolic activities or pathways, is largely excluded.
Orphan metabolic activities are an alternative and growing problem within functional genomics, a consequence of many decades of pre-genomics enzymology research, as well as the recent and rapid rise in untargeted metabolomics data accumulation. For example, recent studies found that 30–40% of all enzymatic activities currently assigned an enzyme commission (EC) number are not associated with a defined genetic locus 70,71. Although the methodology is not as well developed as for gene annotation, activity-based metabolomics is beginning to show promise as a valuable platform for de-orphaning metabolic activities, as illustrated by two recent studies. Both of these relied on a ‘forward genetics’ approach, whereby random mutagenesis of an organism’s genome was followed by metabolomics to identify specific mutant strains lacking the activity of interest. The main disadvantage of this process is the requirement to screen thousands to tens of thousands of genetic mutants in order to achieve full coverage of the test organism’s genome. Typical MS-based metabolomic strategies are not suited to high-throughput analysis (typical sample processing time >30 min 72,73). Messerli et al overcame this problem by initially selecting for mutants with a known phenotype that was predicted to correlate with the metabolic pathway of interest, in their case impaired starch metabolism in Arabidopsis thaliana. Subsequent metabolomics was then used to compare, by principal components analysis, mutants of known starch metabolizing genes with the experimentally derived mutants and subsequently to discriminate between strains with a bona fide deficiency in starch metabolism and those containing mutations in alternative pathways that coincidentally had pleiotropic effects on starch metabolism 74. They were thus able to identify genetic loci not previously associated with starch metabolism. Although specific metabolic activities were not linked to these genes in this case, a more detailed analysis of the metabolomics data would likely pinpoint precise metabolic deficiencies for each tested strain, as previously discussed.
Sidebar C: In need of answers.
How can we best prioritize large-scale metabolomic-based functional genomics efforts?
Which organisms have a large number of orphan enzymes and are therefore good models for the discovery of new enzymatic functions and new metabolic pathways?
Which are the most complementary methods to confirm metabolomic results?
Which enzyme classes present the larger potential for new catalytic functions?
Defined phenotypic traits are not always associated with the presence or absence of metabolic activities of interest, nor are the global metabolic pathways to which they belong commonly known. Baran et al recently used a technique termed ‘metabolic footprinting’ to substantially enhance the throughput of metabolomic analysis of a library of randomly mutagenized bacterial strains of interest, for the identification of genes involved in the metabolism of test compounds 75. Transposon mutagenesis libraries of E. coli and Shewanella oneidensis (more than 8,000 mutants) were grown in media containing compounds of interest, and a shortened LC-MS protocol (2 min per sample) was then used to analyse the supernatant from spent cultures. This streamlined methodology was possible due to the substantially reduced quantity and diversity of metabolites present in the supernatant relative to cell lysates. Strains that had reduced metabolism (higher remaining levels) of the test compound of interest were analysed further to confirm the role of the target gene in the predicted metabolic pathway(s). This approach allowed the authors to assign ergothioneine histidase activity to the S. oneidensis gene SO3057.
Future Direction: Imaging Mass Spectrometry
An intrinsic disadvantage of in vitro and ex vivo metabolomic experiments is the loss of information regarding the subcellular and cellular localization of the metabolites/pathways under scrutiny. For example, compartmentalized metabolite pools (such as mitochondrial versus cytoplasmic acetyl-CoA) and/or cellular heterogeneity among groups of cells in a tissue cannot be differentiated in a typical metabolomic experiment. In practical terms, the changes in abundance of putative substrates and products are diminished due to dilution of non-reacting pools of other cellular or tissue compartments. Imaging mass spectrometry (IMS) is a powerful technique that can complement traditional metabolomic experiments and provide information about spatial distribution, in addition to mass and abundance data (reviewed in 76–78). IMS techniques allow spatial mapping of metals, lipids, polar metabolites, peptides, drugs and even proteins in fixed samples, without a priori extraction and loss of cellular architecture. Two types of IMS methods are the most used, matrix-assisted laser desorption ionization (MALDI) imaging and secondary ion mass spectroscopy (SIMS) imaging. MALDI imaging currently has a spatial resolution of 5–50 μm, allowing analysis of single cells within tissue samples, for example. A lateral resolution under 50 nm can be achieved using SIMS, and up to five different ions can be detected simultaneously, making SIMS-based ‘ion microscopy’ a unique technique to follow the fate and organization of metabolic pathways inside and outside cells. However, with the available technology, only mono- or diatomic ions can be detected, in contrast to entire molecular ion detection obtained with MALDI imaging. Direct monitoring of new enzymatic activities with subcellular resolution is a goal that is yet to be achieved, but holds the promise to improve our understanding of cellular physiology and enzyme function at their correct compartment within cells.
Conclusions
Enzymes control metabolism and are responsible for the formation of many of the most elaborate and interesting chemical structures in the natural world. Tools that enable rapid and unbiased functional assignment of unannotated enzymes are therefore valuable commodities in almost every field of natural science research. Metabolomics has recently emerged as a leading technology in functional genomics, where in combination with modern genetic tools and recombinant protein techniques, it has enabled a unique perspective on the identification of the mechanistic properties of test enzymes either in vitro or ex vivo. The future of metabolomics as a tool in enzyme functional assessment is inextricably linked to the future of MS as an analytical platform. As the technology improves, its reliability and accuracy as a functional genomics tool will follow suit. Similarly, methodological improvements in how metabolomics experiments are executed are constantly being sought, including increases in the breadth of metabolic diversity detected with a single analytical method 79, increases and automation in sample throughput 80, and increases in analytical sensitivity towards single-cell levels 81. Enzyme function discovery will also likely benefit from a more holistic systems approach, in which genomics, transcriptomics and proteomics will complement and validate the hypotheses and conclusions regarding protein function derived from metabolomics data sets 67. Overall, the capacity of metabolomics-based approaches to not only explore unforeseen and diverse aspects of enzyme-catalysed chemical transformations, but also associate them to global metabolic pathways and physiological functions, will likely ensure its prominence as a key technology in functional genomics strategies in the foreseeable future.
Acknowledgments
We apologize to our colleagues whose studies we were not able to cover in this review due to space limitations and focus on recent work. We would like to thank Dr Cesira De-Chiara and Dr Thomas Lerner for critical reading of the manuscript. Research in the Carvalho Laboratory related to the subject of this review is supported by the Medical Research Council (MC_UP_A253_1111).
Glossary
- 13C, 15N, 18O
stable heavy isotope of specified element
- ATP
adenosine triphosphate
- CE
capillary electrophoresis
- CoA
co-enzyme A
- EC
enzyme commission
- FAAH
fatty-acid amide hydrolase
- FAD
flavin adenine dinucleotide
- GABA
gamma-amino butyric acid
- GC
gas-chromatography
- HAD
haloacid dehalogenase
- hP450
human cytochrome P450
- IMS
imaging mass spectrometry
- LC
liquid-chromatography
- MALDI
matrix-assisted laser desorption ionization
- MS
mass spectrometry
- NAD(P)H
nicotinamide adenine dinucleotide (phosphate)
- NMR
nuclear magnetic resonance
- NRPS
non-ribosomal peptide synthase
- PKS
polyketide synthase
- SAM
S-adenosyl methionine
- SIL
stable isotope labelling
- SIMS
secondary ion mass spectrometry
- SSA
succinic semi-aldehyde
- TCA
tricarboxylic acid
- TIC
total ion current
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Galperin MY, Koonin EV. ‘Conserved hypothetical’ proteins: prioritization of targets for experimental study. Nucleic Acids Res. 2004;32:5452–5463. doi: 10.1093/nar/gkh885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Redfern O, Orengo C. Predicting protein function from sequence and structure. Nat Rev Mol Cell Biol. 2007;8:995–1005. doi: 10.1038/nrm2281. [DOI] [PubMed] [Google Scholar]
- Jones CE, Brown AL, Baumann U. Estimating the annotation error rate of curated GO database sequence annotations. BMC Bioinformatics. 2007;8:170. doi: 10.1186/1471-2105-8-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol. 2009;5:e1000605. doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilks WR, Audit B, de Angelis D, Tsoka S, Ouzounis CA. Percolation of annotation errors through hierarchically structured protein sequence databases. Math Biosci. 2005;193:223–234. doi: 10.1016/j.mbs.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Thatcher JW, Shaw JM, Dickinson WJ. Marginal fitness contributions of nonessential genes in yeast. Proc Natl Acad Sci USA. 1998;95:253–257. doi: 10.1073/pnas.95.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barglow KT, Cravatt BF. Activity-based protein profiling for the functional annotation of enzymes. Nat Methods. 2007;4:822–827. doi: 10.1038/nmeth1092. [DOI] [PubMed] [Google Scholar]
- Borodovsky A, Ovaa H, Kolli N, Gan-Erdene T, Wilkinson KD, Ploegh HL, Kessler BM. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem Biol. 2002;9:1149–1159. doi: 10.1016/s1074-5521(02)00248-x. [DOI] [PubMed] [Google Scholar]
- Hekmat O, Kim Y-W, Williams SJ, He S, Withers SG. Active-site peptide ‘fingerprinting’ of glycosidases in complex mixtures by mass spectrometry. Discovery of a novel retaining beta-1,4-glycanase in Cellulomonas fimi. J Biol Chem. 2005;280:35126–35135. doi: 10.1074/jbc.M508434200. [DOI] [PubMed] [Google Scholar]
- Patti GJ, Yanes O, Siuzdak G. Innovation: metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol. 2012;13:263–269. doi: 10.1038/nrm3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L, Robinson AB, Teranishi R, Cary P. Quantitative analysis of urine vapor and breath by gas-liquid partition chromatography. Proc Natl Acad Sci USA. 1971;68:2374–2376. doi: 10.1073/pnas.68.10.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemes P, Rubakhin SS, Aerts JT, Sweedler JV. Qualitative and quantitative metabolomic investigation of single neurons by capillary electrophoresis electrospray ionization mass spectrometry. Nat Protoc. 2013;8:783–799. doi: 10.1038/nprot.2013.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautenhahn R, Cho K, Uritboonthai W, Zhu Z, Patti GJ, Siuzdak G. An accelerated workflow for untargeted metabolomics using the METLIN database. Nat Biotechnol. 2012;30:826–828. doi: 10.1038/nbt.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Gruber T, Barry CE, 3rd, Boshoff HI, Rhee KY. Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science. 2013;339:88–91. doi: 10.1126/science.1228980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser GA, de Carvalho LPS. Metabolomics reveal d-alanine:d-alanine ligase as the target of d-cycloserine in Mycobacterium tuberculosis. ACS Med Chem Lett. 2013;4:1233–1237. doi: 10.1021/ml400349n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, Laxman B, Mehra R, Lonigro RJ, Li Y, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457:910–914. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Rabinowitz JD, Purdy JG, Vastag L, Shenk T, Koyuncu E. Metabolomics in drug target discovery. Cold Spring Harb Symp Quant Biol. 2011;76:235–246. doi: 10.1101/sqb.2011.76.010694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro MS, Carvalho M, Bastos ML, Guedes de Pinho P. Metabolomics analysis for biomarker discovery: advances and challenges. Curr Med Chem. 2013;20:257–271. doi: 10.2174/092986713804806621. [DOI] [PubMed] [Google Scholar]
- Vanholme R, Storme V, Vanholme B, Sundin L, Christensen JH, Goeminne G, Halpin C, Rohde A, Morreel K, Boerjan W. A systems biology view of responses to lignin biosynthesis perturbations in Arabidopsis. Plant Cell. 2012;24:3506–3529. doi: 10.1105/tpc.112.102574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KD, Gyaneshwar P, Markenscoff Papadimitriou E, Fong R, Kim K-S, Parales R, Zhou Z, Inwood W, Kustu S. A previously undescribed pathway for pyrimidine catabolism. Proc Natl Acad Sci USA. 2006;103:5114–5119. doi: 10.1073/pnas.0600521103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qualley AV, Widhalm JR, Adebesin F, Kish CM, Dudareva N. Completion of the core β-oxidative pathway of benzoic acid biosynthesis in plants. Proc Natl Acad Sci USA. 2012;109:16383–16388. doi: 10.1073/pnas.1211001109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Xiao H, Bonanno JB, Kalyanaraman C, Brown S, Tang X, Al-Obaidi NF, Patskovsky Y, Babbitt PC, Jacobson MP, et al. Structure-guided discovery of the metabolite carboxy-SAM that modulates tRNA function. Nature. 2013;498:123–126. doi: 10.1038/nature12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G-F, Sadhukhan S, Tochtrop GP, Brunengraber H. Metabolomics, pathway regulation, and pathway discovery. J Biol Chem. 2011;286:23631–23635. doi: 10.1074/jbc.R110.171405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farag MA, Huhman DV, Dixon RA, Sumner LW. Metabolomics reveals novel pathways and differential mechanistic and elicitor-specific responses in phenylpropanoid and isoflavonoid biosynthesis in Medicago truncatula cell cultures. Plant Physiol. 2008;146:387–402. doi: 10.1104/pp.107.108431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YJ, Chakraborty R, Martín HG, Chu J, Hazen TC, Keasling JD. Flux analysis of central metabolic pathways in Geobacter metallireducens during reduction of soluble Fe(III)-nitrilotriacetic acid. Appl Environ Microbiol. 2007;73:3859–3864. doi: 10.1128/AEM.02986-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Pingitore F, Mukhopadhyay A, Phan R, Hazen TC, Keasling JD. Pathway confirmation and flux analysis of central metabolic pathways in Desulfovibrio vulgaris Hildenborough using gas chromatography-mass spectrometry and Fourier transform-ion cyclotron resonance mass spectrometry. J Bacteriol. 2007;189:940–949. doi: 10.1128/JB.00948-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyraud R, Kiefer P, Christen P, Massou S, Portais J-C, Vorholt JA. Demonstration of the ethylmalonyl-CoA pathway by using 13C metabolomics. Proc Natl Acad Sci USA. 2009;106:4846–4851. doi: 10.1073/pnas.0810932106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2(2006):0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong SS, Nanchen A, Palsson BO, Sauer U. Latent pathway activation and increased pathway capacity enable Escherichia coli adaptation to loss of key metabolic enzymes. J Biol Chem. 2006;281:8024–8033. doi: 10.1074/jbc.M510016200. [DOI] [PubMed] [Google Scholar]
- Nakahigashi K, Toya Y, Ishii N, Soga T, Hasegawa M, Watanabe H, Takai Y, Honma M, Mori H, Tomita M. Systematic phenome analysis of Escherichia coli multiple-knockout mutants reveals hidden reactions in central carbon metabolism. Mol Syst Biol. 2009;5:306. doi: 10.1038/msb.2009.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaves ML, Young BD, Hosios AM, Xu Y-F, Rabinowitz JD. Pyrimidine homeostasis is accomplished by directed overflow metabolism. Nature. 2013;500:237–241. doi: 10.1038/nature12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Hirai MY, Yonekura-Sakakibara K. Decoding genes with coexpression networks and metabolomics - ‘majority report by precogs’. Trends Plant Sci. 2008;13:36–43. doi: 10.1016/j.tplants.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Wu LF, Hughes TR, Davierwala AP, Robinson MD, Stoughton R, Altschuler SJ. Large-scale prediction of Saccharomyces cerevisiae gene function using overlapping transcriptional clusters. Nat Genet. 2002;31:255–265. doi: 10.1038/ng906. [DOI] [PubMed] [Google Scholar]
- Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- Tohge T, Nishiyama Y, Hirai MY, Yano M, Nakajima J, Awazuhara M, Inoue E, Takahashi H, Goodenowe DB, Kitayama M, et al. Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J. 2005;42:218–235. doi: 10.1111/j.1365-313X.2005.02371.x. [DOI] [PubMed] [Google Scholar]
- Hirai MY, Klein M, Fujikawa Y, Yano M, Goodenowe DB, Yamazaki Y, Kanaya S, Nakamura Y, Kitayama M, Suzuki H, et al. Elucidation of gene-to-gene and metabolite-to-gene networks in Arabidopsis by integration of metabolomics and transcriptomics. J Biol Chem. 2005;280:25590–25595. doi: 10.1074/jbc.M502332200. [DOI] [PubMed] [Google Scholar]
- Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. XCMS Online: a web-based platform to process untargeted metabolomic data. Anal Chem. 2012;84:5035–5039. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito N, Robert M, Kitamura S, Baran R, Soga T, Mori H, Nishioka T, Tomita M. Metabolomics approach for enzyme discovery. J Proteome Res. 2006;5:1979–1987. doi: 10.1021/pr0600576. [DOI] [PubMed] [Google Scholar]
- Saito N, Robert M, Kochi H, Matsuo G, Kakazu Y, Soga T, Tomita M. Metabolite profiling reveals YihU as a novel hydroxybutyrate dehydrogenase for alternative succinic semialdehyde metabolism in Escherichia coli. J Biol Chem. 2009;284:16442–16451. doi: 10.1074/jbc.M109.002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscombe DK, Usera AR, O’Connor SE. Homolog of tocopherol C methyltransferases catalyzes N methylation in anticancer alkaloid biosynthesis. Proc Natl Acad Sci USA. 2010;107:18793–18798. doi: 10.1073/pnas.1009003107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Carvalho LPS, Zhao H, Dickinson CE, Arango NM, Lima CD, Fischer SM, Ouerfelli O, Nathan C, Rhee KY. Activity-based metabolomic profiling of enzymatic function: identification of Rv1248c as a mycobacterial 2-hydroxy-3-oxoadipate synthase. Chem Biol. 2010;17:323–332. doi: 10.1016/j.chembiol.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrouy-Maumus G, Biswas T, Hunt DM, Kelly G, Tsodikov OV, de Carvalho LPS. Discovery of a glycerol 3-phosphate phosphatase reveals glycerophospholipid polar head recycling in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2013;110:11320–11325. doi: 10.1073/pnas.1221597110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- Guengerich FP, Wu Z-L, Bartleson CJ. Function of human cytochrome P450s: characterization of the orphans. Biochem Biophys Res Commun. 2005;338:465–469. doi: 10.1016/j.bbrc.2005.08.079. [DOI] [PubMed] [Google Scholar]
- Cheng Q, Lamb DC, Kelly SL, Lei L, Guengerich FP. Cyclization of a cellular dipentaenone by Streptomyces coelicolor cytochrome P450 154A1 without oxidation/reduction. J Am Chem Soc. 2010;132:15173–15175. doi: 10.1021/ja107801v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Guengerich FP. Metabolomic analysis and identification of a role for the orphan human cytochrome P450 2W1 in selective oxidation of lysophospholipids. J Lipid Res. 2012;53:1610–1617. doi: 10.1194/jlr.M027185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich FP, Tang Z, Cheng Q, Salamanca-Pinzón SG. Approaches to deorphanization of human and microbial cytochrome P450 enzymes. Biochim Biophys Acta. 2011;1814:139–145. doi: 10.1016/j.bbapap.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Martin MV, Guengerich FP. Elucidation of functions of human cytochrome P450 enzymes: identification of endogenous substrates in tissue extracts using metabolomic and isotopic labeling approaches. Anal Chem. 2009;81:3071–3078. doi: 10.1021/ac900021a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Salamanca-Pinzón SG, Wu Z-L, Xiao Y, Guengerich FP. Human cytochrome P450 4F11: heterologous expression in bacteria, purification, and characterization of catalytic function. Arch Biochem Biophys. 2010;494:86–93. doi: 10.1016/j.abb.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy I, Michnick SW. Visualization of biochemical networks in living cells. Proc Natl Acad Sci USA. 2001;98:7678–7683. doi: 10.1073/pnas.131216098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobe B, Kemp BE. Active site-directed protein regulation. Nature. 1999;402:373–376. doi: 10.1038/46478. [DOI] [PubMed] [Google Scholar]
- Moellering RE, Cravatt BF. Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science. 2013;341:549–553. doi: 10.1126/science.1238327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raamsdonk LM, Teusink B, Broadhurst D, Zhang N, Hayes A, Walsh MC, Berden JA, Brindle KM, Kell DB, Rowland JJ, et al. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat Biotechnol. 2001;19:45–50. doi: 10.1038/83496. [DOI] [PubMed] [Google Scholar]
- Saghatelian A, Trauger SA, Want EJ, Hawkins EG, Siuzdak G, Cravatt BF. Assignment of endogenous substrates to enzymes by global metabolite profiling. Biochemistry (Mosc) 2004;43:14332–14339. doi: 10.1021/bi0480335. [DOI] [PubMed] [Google Scholar]
- Ooga T, Ohashi Y, Kuramitsu S, Koyama Y, Tomita M, Soga T, Masui R. Degradation of ppGpp by nudix pyrophosphatase modulates the transition of growth phase in the bacterium Thermus thermophilus. J Biol Chem. 2009;284:15549–15556. doi: 10.1074/jbc.M900582200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Reuss SH, Bose N, Srinivasan J, Yim JJ, Judkins JC, Sternberg PW, Schroeder FC. Comparative metabolomics reveals biogenesis of ascarosides, a modular library of small-molecule signals in C. elegans. J Am Chem Soc. 2012;134:1817–1824. doi: 10.1021/ja210202y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quanbeck SM, Brachova L, Campbell AA, Guan X, Perera A, He K, Rhee SY, Bais P, Dickerson JA, Dixon P, et al. Metabolomics as a hypothesis-generating functional genomics tool for the annotation of Arabidopsis thaliana genes of ‘unknown function’. Front Plant Sci. 2012;3:15. doi: 10.3389/fpls.2012.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagore DM, Nolte WM, Neveu JM, Rangel R, Guzman-Rojas L, Pasqualini R, Arap W, Lane WS, Saghatelian A. Peptidase substrates via global peptide profiling. Nat Chem Biol. 2009;5:23–25. doi: 10.1038/nchembio.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Saint PaulV, Zhang W, Kanawati B, Geist B, Faus-Kessler T, Schmitt-Kopplin P, Schäffner AR. The Arabidopsis glucosyltransferase UGT76B1 conjugates isoleucic acid and modulates plant defense and senescence. Plant Cell. 2011;23:4124–4145. doi: 10.1105/tpc.111.088443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura-Sakakibara K, Tohge T, Matsuda F, Nakabayashi R, Takayama H, Niida R, Watanabe-Takahashi A, Inoue E, Saito K. Comprehensive flavonol profiling and transcriptome coexpression analysis leading to decoding gene-metabolite correlations in Arabidopsis. Plant Cell. 2008;20:2160–2176. doi: 10.1105/tpc.108.058040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Barona-Gomez F, Corre C, Xiang L, Udwary DW, Austin MB, Noel JP, Moore BS, Challis GL. Type III polyketide synthase beta-ketoacyl-ACP starter unit and ethylmalonyl-CoA extender unit selectivity discovered by Streptomyces coelicolor genome mining. J Am Chem Soc. 2006;128:14754–14755. doi: 10.1021/ja065247w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo S, Sydor PK, Corre C, Alhamadsheh MM, Stanley AE, Haynes SW, Song L, Reynolds KA, Challis GL. Elucidation of the Streptomyces coelicolor pathway to 2-undecylpyrrole, a key intermediate in undecylprodiginine and streptorubin B biosynthesis. Chem Biol. 2008;15:137–148. doi: 10.1016/j.chembiol.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Forseth RR, Amaike S, Schwenk D, Affeldt KJ, Hoffmeister D, Schroeder FC, Keller NP. Homologous NRPS-like gene clusters mediate redundant small-molecule biosynthesis in Aspergillus flavus. Angew Chem Int Ed Engl. 2013;52:1590–1594. doi: 10.1002/anie.201207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W-B, Baccile JA, Bok JW, Chen Y, Keller NP, Schroeder FC. A nonribosomal peptide synthetase-derived iron(III) complex from the pathogenic fungus Aspergillus fumigatus. J Am Chem Soc. 2013;135:2064–2067. doi: 10.1021/ja311145n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii N, Nakahigashi K, Baba T, Robert M, Soga T, Kanai A, Hirasawa T, Naba M, Hirai K, Hoque A, et al. Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science. 2007;316:593–597. doi: 10.1126/science.1132067. [DOI] [PubMed] [Google Scholar]
- Chiang KP, Niessen S, Saghatelian A, Cravatt BF. An enzyme that regulates ether lipid signaling pathways in cancer annotated by multidimensional profiling. Chem Biol. 2006;13:1041–1050. doi: 10.1016/j.chembiol.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Long JZ, Cisar JS, Milliken D, Niessen S, Wang C, Trauger SA, Siuzdak G, Cravatt BF. Metabolomics annotates ABHD3 as a physiologic regulator of medium-chain phospholipids. Nat Chem Biol. 2011;7:763–765. doi: 10.1038/nchembio.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Vitkup D. Distribution of orphan metabolic activities. Trends Biotechnol. 2007;25:343–348. doi: 10.1016/j.tibtech.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Pouliot Y, Karp PD. A survey of orphan enzyme activities. BMC Bioinformatics. 2007;8:244. doi: 10.1186/1471-2105-8-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajad SU, Lu W, Kimball EH, Yuan J, Peterson C, Rabinowitz JD. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J Chromatogr A. 2006;1125:76–88. doi: 10.1016/j.chroma.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Pesek JJ, Matyska MT, Fischer SM, Sana TR. Analysis of hydrophilic metabolites by high-performance liquid chromatography-mass spectrometry using a silica hydride-based stationary phase. J Chromatogr A. 2008;1204:48–55. doi: 10.1016/j.chroma.2008.07.077. [DOI] [PubMed] [Google Scholar]
- Messerli G, Partovi Nia V, Trevisan M, Kolbe A, Schauer N, Geigenberger P, Chen J, Davison AC, Fernie AR, Zeeman SC. Rapid classification of phenotypic mutants of Arabidopsis via metabolite fingerprinting. Plant Physiol. 2007;143:1484–1492. doi: 10.1104/pp.106.090795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran R, Bowen BP, Price MN, Arkin AP, Deutschbauer AM, Northen TR. Metabolic footprinting of mutant libraries to map metabolite utilization to genotype. ACS Chem Biol. 2013;8:189–199. doi: 10.1021/cb300477w. [DOI] [PubMed] [Google Scholar]
- Musat N, Foster R, Vagner T, Adam B, Kuypers MMM. Detecting metabolic activities in single cells, with emphasis on nanoSIMS. FEMS Microbiol Rev. 2012;36:486–511. doi: 10.1111/j.1574-6976.2011.00303.x. [DOI] [PubMed] [Google Scholar]
- Lanni EJ, Rubakhin SS, Sweedler JV. Mass spectrometry imaging and profiling of single cells. J Proteomics. 2012;75:5036–5051. doi: 10.1016/j.jprot.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watrous JD, Dorrestein PC. Imaging mass spectrometry in microbiology. Nat Rev Microbiol. 2011;9:683–694. doi: 10.1038/nrmicro2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanisevic J, Zhu Z-J, Plate L, Tautenhahn R, Chen S, O’Brien PJ, Johnson CH, Marletta MA, Patti GJ, Siuzdak G. Toward ‘omic scale metabolite profiling: a dual separation-mass spectrometry approach for coverage of lipid and central carbon metabolism. Anal Chem. 2013;85:6876–6884. doi: 10.1021/ac401140h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald JC, Heux S, Zamboni N. High-throughput quantitative metabolomics: workflow for cultivation, quenching, and analysis of yeast in a multiwell format. Anal Chem. 2009;81:3623–3629. doi: 10.1021/ac900002u. [DOI] [PubMed] [Google Scholar]
- Zenobi R. Single-cell metabolomics: analytical and biological perspectives. Science. 2013;342:1243259. doi: 10.1126/science.1243259. [DOI] [PubMed] [Google Scholar]
- Van der Werf MJ, Overkamp KM, Muilwijk B, Coulier L, Hankemeier T. Microbial metabolomics: toward a platform with full metabolome coverage. Anal Biochem. 2007;370:17–25. doi: 10.1016/j.ab.2007.07.022. [DOI] [PubMed] [Google Scholar]
- DellaPenna D, Last RL. Genome-enabled approaches shed new light on plant metabolism. Science. 2008;320:479–481. doi: 10.1126/science.1153715. [DOI] [PubMed] [Google Scholar]
- Bochner BR, Ames BN. Complete analysis of cellular nucleotides by two-dimensional thin layer chromatography. J Biol Chem. 1982;257:9759–9769. [PubMed] [Google Scholar]
- Tempest DW, Meers JL, Brown CM. Influence of environment on the content and composition of microbial free amino acid pools. J Gen Microbiol. 1970;64:171–185. doi: 10.1099/00221287-64-2-171. [DOI] [PubMed] [Google Scholar]
- Kandiah M, Urban PL. Advances in ultrasensitive mass spectrometry of organic molecules. Chem Soc Rev. 2013;42:5299–5322. doi: 10.1039/c3cs35389c. [DOI] [PubMed] [Google Scholar]
- Cortés-Francisco N, Flores C, Moyano E, Caixach J. Accurate mass measurements and ultrahigh-resolution: evaluation of different mass spectrometers for daily routine analysis of small molecules in negative electrospray ionization mode. Anal Bioanal Chem. 2011;400:3595–3606. doi: 10.1007/s00216-011-5046-8. [DOI] [PubMed] [Google Scholar]
- Zhu Z-J, Schultz AW, Wang J, Johnson CH, Yannone SM, Patti GJ, Siuzdak G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat Protoc. 2013;8:451–460. doi: 10.1038/nprot.2013.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo AC, Jewison T, Wilson M, Liu Y, Knox C, Djoumbou Y, Lo P, Mandal R, Krishnamurthy R, Wishart DS. ECMDB: the E. coli metabolome database. Nucleic Acids Res. 2013;41:D625–D630. doi: 10.1093/nar/gks992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewison T, Knox C, Neveu V, Djoumbou Y, Guo AC, Lee J, Liu P, Mandal R, Krishnamurthy R, Sinelnikov I, et al. YMDB: the yeast metabolome database. Nucleic Acids Res. 2012;40:D815–D820. doi: 10.1093/nar/gkr916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, et al. HMDB 3.0–the human metabolome database in 2013. Nucleic Acids Res. 2013;41:D801–D807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayavekhin N, Saghatelian A. Untargeted metabolomics. Curr Protoc Mol Biol. 2010;30 doi: 10.1002/0471142727.mb3001s90. Chapter: Unit 30.1.1–24. [DOI] [PubMed] [Google Scholar]
- Yogeeswari P, Sriram D, Vaigundaragavendran J. The GABA shunt: an attractive and potential therapeutic target in the treatment of epileptic disorders. Curr Drug Metab. 2005;6:127–139. doi: 10.2174/1389200053586073. [DOI] [PubMed] [Google Scholar]
- Rustin P, Bourgeron T, Parfait B, Chretien D, Munnich A, Rötig A. Inborn errors of the Krebs cycle: a group of unusual mitochondrial diseases in human. Biochim Biophys Acta. 1997;1361:185–197. doi: 10.1016/s0925-4439(97)00035-5. [DOI] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci. 2005;360:2335–2345. doi: 10.1098/rstb.2005.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G, Krömer JO. Fluxomics - connecting ‘omics analysis and phenotypes. Environ Microbiol. 2013;15:1901–1916. doi: 10.1111/1462-2920.12064. [DOI] [PubMed] [Google Scholar]
- Winder CL, Dunn WB, Goodacre R. TARDIS-based microbial metabolomics: time and relative differences in systems. Trends Microbiol. 2011;19:315–322. doi: 10.1016/j.tim.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Klein S, Heinzle E. Isotope labeling experiments in metabolomics and fluxomics. Wiley Interdiscip Rev Syst Biol Med. 2012;4:261–272. doi: 10.1002/wsbm.1167. [DOI] [PubMed] [Google Scholar]
- Goulding CW, Bowers PM, Segelke B, Lekin T, Kim C-Y, Terwilliger TC, Eisenberg D. The structure and computational analysis of Mycobacterium tuberculosis protein CitE suggest a novel enzymatic function. J Mol Biol. 2007;365:275–283. doi: 10.1016/j.jmb.2006.09.086. [DOI] [PubMed] [Google Scholar]