

Abstract
The mechanisms that coordinate the regulation of autophagy with developmental signaling during multicellular organism development remain largely unknown. Here, we show that impaired function of ribosomal protein RPL-43 causes an accumulation of SQST-1 aggregates in the larval intestine, which are removed upon autophagy induction. Using this model to screen for autophagy regulators, we identify 139 genes that promote autophagy activity upon inactivation. Various signaling pathways, including Sma/Mab TGF-β signaling, lin-35/Rb signaling, the XBP-1-mediated ER stress response, and the ATFS-1-mediated mitochondrial stress response, regulate the expression of autophagy genes independently of the TFEB homolog HLH-30. Our study thus provides a framework for understanding the role of signaling pathways in regulating autophagy under physiological conditions.
Subject Categories: Autophagy & Cell Death; Signal Transduction; Membrane & Intracellular Transport
Keywords: autophagy, Caenorhabditis elegans, ER stress, mitochondrial stress, Rb
Introduction
Autophagy is an evolutionarily conserved lysosome-mediated degradation process. It involves the formation of a cup-shaped membrane sac, known as the isolation membrane, which expands and seals to form an enclosed double-membrane autophagosome. In higher eukaryotes, autophagosomes mature by fusing with endosomes before fusing with lysosomes to form degradative autolysosomes 1–3. Distinct steps of autophagosome formation require different sets of genes 2,4. The Atg1/Atg13 kinase complex and the Vps34/Atg6 class III PI(3)K complex are required for the induction and nucleation of isolation membranes. Expansion and closure of the autophagosome is regulated by the Atg8-phosphatidylethanolamine (PE) and Atg12-Atg5 ubiquitin-like conjugation systems 2. Progression of autophagosomes into autolysosomes requires components of the endocytic pathways and the SNARE complex 3,5.
Autophagy is activated by a variety of stress conditions, including nutrient starvation, energy deprivation, and reactive oxygen species 6. The mammalian target of rapamycin serine/threonine kinase complex 1 (mTORC1) and the Vps34 PI(3)K complex are major nodes for integrating various signaling pathways with autophagy regulation 7–9. Transcriptional regulation of autophagy genes confers another layer of regulation. The master positive regulator TFEB and the master repressor ZKSCAN3 transcriptionally regulate a network of genes involved in autophagosome and lysosome biogenesis 10,11. The forkhead transcription factor FoxO also activates the transcription of multiple autophagy genes and promotes autophagy activity 6. Although numerous factors have been uncovered that regulate autophagy activity, the mechanisms that integrate developmental signals into the autophagic machinery during multicellular organism development are poorly understood.
Here, we established Caenorhabditis elegans as a genetic model to study autophagy regulation and identified 139 genes which, when inactivated, enhance autophagy activity. We showed that autophagy activity is activated by various stress-induced conditions and is tightly controlled by various developmental signaling pathways.
Results and Discussion
Loss of rpl-43 activity results in accumulation of SQST-1 aggregates in the larval intestine
The C. elegans p62 homolog SQST-1 is removed by autophagy during development. In wild-type animals, SQST-1::GFP is weakly and diffusely expressed in the cytoplasm from embryonic to adult stages (Fig 1A and B; Supplementary Fig S1A and B) 12. In autophagy mutants, numerous SQST-1::GFP aggregates accumulate in almost all cells during embryogenesis and in multiple larval tissues, including hypodermis, intestine, and neurons (Supplementary Fig S1C and E–L). Intestinal SQST-1 aggregates, which are spherical and dispersed in the cytoplasm, accumulate throughout larval stages in autophagy mutants (Supplementary Fig S1E–L). bp399 was isolated in a genetic screen for mutants with defective degradation of SQST-1::GFP (Fig 1C–E). Unlike autophagy mutants, bp399 mutants showed the accumulation of SQST-1 aggregates strictly in the intestine in a distinct temporal pattern. SQST-1::GFP aggregates were absent in bp399 embryos, but started to form in L1 larvae and increased in number and size throughout larval development (Fig 1C and D; Supplementary Fig S1D and M–T). SQST-1::GFP aggregates in bp399 were heterogeneous in size and some were much larger than those in autophagy mutants (Supplementary Fig S1E–T). Endogenous SQST-1 also accumulated in the intestine in bp399 mutants (Supplementary Fig S1U–X). SQST-1::GFP aggregates in bp399 mutants were co-stained by anti-ubiquitin, suggesting the accumulation of ubiquitinated proteins (Supplementary Fig S1Y–B2).
Figure 1. Genome-wide RNAi screen for gene inactivations that suppress the accumulation of SQST-1::GFP aggregates in rpl-43 mutants.

A–D SQST-1::GFP is very weakly expressed and diffusely localized in the cytoplasm in wild-type larval intestine, but is expressed at much higher levels and accumulates into numerous aggregates in the intestine in bp399 mutant larvae. (A) and (C) represent DIC images of the animals in (B) and (D), respectively.
E Compared to wild-type, SQST-1::GFP levels are much higher in bp399 and bp412 mutant larvae as shown by immunoblotting assay. SQST-1::GFP levels in bp399 mutants are greatly reduced after 12 h of starvation. 200 young adult animals for each genotype were collected for analysis.
F Mapping and cloning of bp399. LG II: linkage group II.
G Protein sequence of RPL-43. The mutated residue in bp399 is highlighted in red.
H, I SQST-1::GFP aggregates in rpl-43(bp399) mutants are separable from LAAT-1::Cherry-labeled lysosomes under normal conditions, but are delivered to lysosomes upon starvation. Some aggregates are enclosed by lysosomes (inserts). Scale bar: 20 μm.
J–Q RNAi inactivation of let-363, rpn-2, cogc-1, or hgrs-1 suppresses the accumulation of SQST-1::GFP aggregates in rpl-43(bp399) mutants. (J), (L), (N), and (P): DIC images of the animals in (K), (M), (O), and (Q). Scale bar: 20 μm.
R SQST-1::GFP levels in rpl-43(bp399) mutants are greatly reduced by inactivation of rpt-3 and rpn-2. 200 young adult animals for each genotype were collected for analysis.
S Functional classification of the identified genes. The number of genes in each functional class is shown in parentheses.
A transgene containing the single gene rpl-43 rescued the SQST-1::GFP accumulation phenotype in bp399 mutants (Fig 1F). The glycine at position 15 of RPL-43 was mutated to arginine in bp399 mutants (Fig 1G). rpl-43 encodes the 60S ribosomal protein L37. RNAi inactivation of other ribosomal subunits, ribosomal maturation factors, and translation initiation factors resulted in the same phenotype as rpl-43(bp399) (Supplementary Table S1; Supplementary Fig S1C2–F2). This suggests that impaired protein synthesis causes the accumulation of SQST-1 aggregates in the larval intestine.
Autophagic degradation of other autophagy substrates was unaffected in rpl-43 mutants (Supplementary Fig S2A–P) 13. Levels of the C. elegans Atg8 homolog LGG-1, which has been widely used to measure autophagy activity, were unaltered in rpl-43 mutants and no intestinal LGG-1 puncta were detected (Supplementary Fig S2Q–T). Thus, rpl-43 is not an essential component of the autophagy pathway.
SQST-1 aggregates in rpl-43 mutants are degraded by elevated autophagy activity
Starvation or inactivation of Tor signaling resulted in the degradation of SQST-1 aggregates in rpl-43 mutants, but not in autophagy mutants (Fig 1E, J, and K; Supplementary Fig S3A–N). Intestinal SQST-1 aggregates persisted in rpl-43; atg-3 mutants after starvation or let-363/mTOR inactivation (Supplementary Fig S3O and P). We then followed the dynamic distribution of SQST-1::GFP aggregates and lysosomes. SQST-1::GFP aggregates and LAAT-1::cherry-labeled lysosomes were separable in rpl-43 mutants (Fig 1H). Upon starvation, SQST-1::GFP aggregates in rpl-43 mutants fused and became encircled by lysosomes (Fig 1I). Thus, the SQST-1 aggregates in rpl-43 mutants are degraded upon autophagy induction.
Genome-wide RNAi screen for gene inactivation elevating autophagy activity
We screened a C. elegans RNAi feeding library targeting 16,749 genes (~87% of C. elegans ORFs) to identify RNAi clones that significantly suppressed the SQST-1::GFP aggregate accumulation phenotype in rpl-43 mutants (Fig 1L–R). The RNAi clones that reproducibly suppressed rpl-43 were subjected to several additional screens. First, a GFP reporter driven by the sqst-1 promoter was screened to exclude genes that transcriptionally repress sqst-1 (Supplementary Fig S3Q–T). Second, to exclude RNAi inactivations that cause disappearance of SQST-1 aggregates by mechanisms independent of autophagy induction, we screened for RNAi clones that did not affect the accumulation of intestinal SQST-1 aggregates in atg-9 mutants. Third, we identified gene inactivations that promoted the degradation of another autophagy substrate W07G4.5::GFP in the intestine (Supplementary Fig S3U–X) 14. After these screens, 139 genes were identified as suppressors of rpl-43 (Table 1; Supplementary Table S2).
Table 1.
List of some RNAi inactivations that lead to suppression of the accumulation of SQST-1::GFP aggregates in the intestine in rpl-43 mutants
| Process | Sequence name | Gene name | Brief description of gene product |
|---|---|---|---|
| Signaling | F54C8.5 | rheb-1 | Orthologous to the mammalian Rheb and Rheb1 GTPases |
| Signaling | R13F6.9 | sma-3 | A Smad protein |
| Signaling | R12B2.1 | sma-4 | A Smad protein |
| Signaling | F02A9.6 | glp-1 | N-glycosylated transmembrane protein |
| Signaling | T20F10.1 | wts-1 | Warts/lats-like serine threonine kinase |
| Signaling | F42G8.8 | Serine/threonine-protein phosphatase PP1 | |
| Signaling | C36B1.8 | gls-1 | Transducer of the stress-activated PKC1-MPK1 signaling pathway |
| Signaling | Y54E10BL.6 | mek-2 | Mitogen-activated protein kinase kinase |
| Protein turnover | F23F12.6 | rpt-3 | An AAA ATPase subunit of the 19S regulatory particle of the 26S proteasome |
| Protein turnover | F19B6.2 | ufd-1 | Protein involved in the recognition of polyubiquitinated proteins |
| Protein turnover | F59E12.5 | npl-4.2 | Ubiquitin-binding protein involved in protein degradation |
| Protein turnover | C06A1.1 | cdc-48.1 | An AAA ATPase homologous to yeast Cdc48 and mammalian p97/VCP |
| Transcription | F47D12.4 | hmg-1.2 | HMG box-containing protein |
| Transcription | F31E3.1 | ceh-20 | Homeobox family member |
| Transcription | C01B7.1 | ztf-12 | Zinc finger transcription factor family |
| Transcription | R06C7.7 | lin-61 | Polycomb group protein SCM/L(3)MBT |
| Transcription | C32F10.2 | lin-35 | The Caenorhabditis elegans retinoblastoma protein (Rb) ortholog |
| Transcription | JC8.6 | lin-54 | Metallothionein-like protein |
| Transcription | F52C12.5 | elt-6 | GATA-4/5/6 transcription factor |
| Transcription | F44C4.2 | nhr-37 | Nuclear hormone receptor |
| Transcription | D2021.1 | utx-1 | General transcriptional co-repressor |
| Transcription | C33D3.1 | elt-2 | GATA-type transcription factor |
| Protein trafficking | C07G1.5 | hgrs-1 | Membrane trafficking and cell signaling protein HRS |
| Protein trafficking | ZK652.2 | tomm-7 | Translocase of outer mitochondrial membrane complex |
| Protein trafficking | E04A4.5 | Mitochondrial import inner membrane translocase | |
| Protein trafficking | F35H10.4 | vha-5 | Subunit a of the vacuolar H + -ATPase V0 sector |
| Protein trafficking | CD4.4 | vps-37 | A member of the endosomal sorting ESCRT-I complex |
| Metabolism | F54D8.3 | alh-1 | Aldehyde dehydrogenase |
| Metabolism | C28H8.11 | Tryptophan 2,3-dioxygenase | |
| Metabolism | F37E3.1 | ncbp-1 | Nuclear cap-binding protein complex |
| Metabolism | K04E7.2 | pep-2 | A low-affinity/high-capacity oligopeptide transporter |
Based on annotated biological function, the 139 genes were enriched for signal transduction, protein turnover, intracellular trafficking, and cellular metabolism processes (Fig 1S). According to gene ontology, 111 of the genes have clear human homologs.
We identified genes encoding proteins involved in the ubiquitin-proteasome degradation system (UPS), including components of the proteasome and the Cdc48/Ufd1/Npl4 complex involved in ER-associated degradation (ERAD), and also genes encoding transcription factors, metabolic enzymes, and transporters (Table 1; Fig 1L, M, and R). Genes encoding factors involved in intracellular trafficking included the central and lobe A subunits of the conserved oligomeric Golgi (COG) complex, clathrin adaptor protein complex AP-2, and components of the endosomal sorting ESCRT complex (Fig 1N–Q). Our screen identified genes involved in developmental signaling pathways, including TGF-β signaling (sma-3, sma-4), Hippo signaling (wts-1), PKC1-MPK1 signaling (gls-1), Notch signaling (glp-1), and the ERK MAPK pathway (mek-2).
Genome-wide functional interaction maps have been created for C. elegans based on gene expression, physical and genetic interactions, and functional annotation 15,16. Among the 139 genes, 72 genes (nodes) formed at least one interaction (edge) with other genes, and a total of 103 edges were identified (Supplementary Fig S3Y). As a control, we randomly selected four groups of 139 genes from the same library, which showed 3, 0, 4, and 5 interactions, respectively.
Sma TGF-β signaling regulates autophagy
The Sma/Mab and Dauer TGF-β signaling pathways in C. elegans, consisting of distinct components, regulate different developmental processes (Supplementary Fig S4A) 17. In addition to sma-3 and sma-4 (Smads) identified in our screen (Fig 2A–D), knockdown of other components in the Sma TGF-β pathway, including dbl-1 (ligand), sma-6 (type I receptor), sma-2 (Smad), and sma-9 (cofactor), also suppressed SQST-1 aggregates in rpl-43 mutants (Supplementary Fig S4B). Inactivation of Sma/Mab TGF-β signaling suppresses the rpl-43 mutant phenotype independent of its role in regulating body size (Supplementary Fig S4B–D). Loss of function of components in the Dauer TGF-β signaling pathway, including daf-7 (ligand), daf-1 (type I receptor), daf-8 (Smad), and daf-3 (Smad), did not suppress SQST-1 aggregates in rpl-43 mutants (Supplementary Fig S4B, E and F).
Figure 2. Loss of function of a variety of signaling pathways suppresses the rpl-43 phenotype.

A, B SQST-1::GFP aggregates are absent in the rpl-43(bp399); sma-3(wk20) mutant intestine.
C Percentage of the indicated animals with different levels of SQST-1::GFP aggregates. S: many SQST-1::GFP aggregates in all intestinal cells, as in rpl-43(bp399) mutants; M: fewer SQST-1::GFP aggregates in some but not all intestinal cells; N: no or very few SQST-1::GFP aggregates in intestinal cells. For each genotype, > 30 animals were examined.
D SQST-1::GFP levels in rpl-43(bp399) mutants are dramatically decreased in a Western blot when sma-3, lin-35, or daf-2 is simultaneously depleted. Loss of function of daf-16 partially restores SQST-1 levels in rpl-43(bp399); daf-2(RNAi) mutants.
E Endogenous LGG-1 is elevated in a Western blot in dbl-1(wk70), sma-2(e502), and sma-3(wk20) mutants.
F mRNA levels of bec-1, epg-8, lgg-1, and atg-7 are upregulated in dbl-1(wk70), sma-2(e502), and sma-3(wk20) mutants. **P < 0.01. 500 young adult animals were collected for analysis. Error bars indicate s.d. from three experiments.
G, H No SQST-1::GFP aggregates are observed in the lin-35(n745); rpl-43(bp399) mutant intestine.
I, J Numerous SQST-1::GFP aggregates are formed in rpl-43(bp399); lin-15A(n767) mutants.
K Endogenous LGG-1 levels are increased in lin-9(n112), lin-15B(n744), and lin-35(n745) mutants.
L mRNA levels of bec-1, epg-8, lgg-1, and atg-7 are upregulated in lin-9(n112), lin-15B(n744), and lin-35(n745) mutants. **P < 0.01. 500 young adult animals were collected for analysis. Error bars indicate s.d. from three experiments.
M, N rpl-43(bp399); daf-2(RNAi) mutant larvae contain no intestinal SQST-1::GFP aggregates.
O, P SQST-1::GFP aggregates are partially restored in daf-16(mu86); rpl-43(bp399); daf-2(RNAi) animals.
Data information: 200 young adult animals for each genotype were collected for analysis in (D), (E), and (K). Scale bars: 20 μm (A, B, G–J, M–P). (A), (G), (I), (M), and (O): DIC images of the animals in (B), (H), (J), (N), and (P), respectively.
Expression of endogenous LGG-1 and GFP::LGG-1 was greatly increased and a large number of GFP::LGG-1 puncta formed in dbl-1 and sma-3 mutants compared to wild-type animals (Fig 2E; Supplementary Fig S4G and H). mRNA levels of lgg-1 and other autophagy genes, including bec-1, epg-8, and atg-7, were increased in dbl-1, sma-2, and sma-3 mutants (Fig 2F), indicating that Sma TGF-β signaling regulates autophagy at least partially through transcriptional control of autophagy genes.
The LIN-35/Rb pathway regulates autophagy
Our screen identified several class B synthetic multivulva (SynMuvB) genes, including lin-35 (the Rb homolog), lin-54, and lin-61 (Fig 2C, D, G, and H; Supplementary Fig S5A–D). Endogenous SQST-1 aggregates were absent in rpl-43; lin-35 mutants (Supplementary Fig S5E and F). SynMuvB genes function redundantly with class A or class C SynMuv genes to antagonize specification of the vulva cell lineage 18. RNAi knockdown of other SynMuvB genes, including lin-15B and lin-36, components of a Rb-containing DRM complex (dpl-1, lin-9, lin-37, lin-53), a SynMuvB heterochromatin complex (hpl-2, lin-13), and a SUMO-recruited Mec complex (mep-1 and let-418), all suppressed the accumulation of SQST-1::GFP aggregates in rpl-43 mutants (Supplementary Fig S5G). However, RNAi inactivation of class A (lin-15A, lin-8, and lin-56) and class C (trr-1 and mys-1) SynMuv genes had no effect (Fig 2I and J; Supplementary Fig S5G). Knockdown of genes essential for SynMuvB function in vulval development, RNAi, and soma-germ fate determination did not cause reappearance of SQST-1 aggregates in lin-35; rpl-43 mutants (Supplementary Fig S5H–M). Thus, SynMuvB genes regulate autophagy activity independent of their role in other processes.
The number of GFP::LGG-1 puncta and levels of endogenous LGG-1 were dramatically elevated in lin-35, lin-15B, and lin-9 mutants (Fig 2K; Supplementary Fig S5N and O). Levels of bec-1, epg-8, lgg-1, and atg-7 mRNA were increased in lin-35, lin-15B, lin-9, and mep-1 mutants (Fig 2L and data not shown). Thus, the lin-35/Rb pathway transcriptionally regulates autophagy genes.
daf-2 insulin/IGF signaling regulates autophagy activity
Reduction of daf-2 signaling elevates autophagy activity 19. Accumulation of SQST-1 aggregates in rpl-43 mutants was suppressed by the loss of function of daf-2 and pdk-1 (Fig 2C, D, M, and N; Supplementary Fig S6A and B). Reduced daf-2 activity results in nuclear translocation of the forkhead transcription factor DAF-16, which further activates the expression of downstream target genes. SQST-1 aggregates were partially restored in daf-16; rpl-43; daf-2 mutants (Fig 2C, D, O, and P), indicating that daf-2 regulates autophagy activity partially through DAF-16. daf-16 is not required for suppression of rpl-43 by inactivation of let-363, sma-3, and lin-35 (Supplementary Fig S6C–H).
daf-2 mutants did not show elevated LGG-1 levels by immunoblotting (Supplementary Fig S6I). mRNA levels of lgg-1, but not of other autophagy genes, were slightly increased in daf-2 mutants (Supplementary Fig S6J). Upregulation of lgg-1 was suppressed by the loss of daf-16 activity (Supplementary Fig S6J). DAF-16 negatively regulates daf-15 transcription 20. Knockdown of mTOR or raptor did not increase mRNA levels of autophagy genes (Supplementary Fig S6K). These results suggest that daf-2 may regulate autophagy by converging on DAF-15, as in the regulation of dauer formation and fat accumulation 20.
ER stress activates autophagy activity
Accumulation of unfolded/misfolded proteins in the endoplasmic reticulum (ER) triggers the ER unfolded protein response (UPR) to ameliorate the stress 21. bp399 did not cause evident ER stress, as shown by very low expression of the ER stress marker Phsp-4::GFP (Fig 3A and B; Supplementary Fig S7A and B). Treatment of rpl-43 mutants with tunicamycin and DTT induced ER stress and strongly suppressed the accumulation of SQST-1 aggregates (Fig 3C; Supplementary Fig S7E-H). Phsp-4::GFP expression was also greatly increased by RNAi inactivation of 24 genes identified in our screen, including components of the proteasome and the ERAD complex (Supplementary Table S3; Fig 3D and E; Supplementary Fig S7I and J). Among the 24 ER stress-inducing genes, 10 are functionally connected and share 19 interactions (edges) in total (Fig 3L).
Figure 3. XBP-1-mediated ER stress suppresses the rpl-43 phenotype.

A, B Phsp-4::GFP is very weakly expressed in wild-type animals.
C Percentage of indicated animals with different levels of SQST-1::GFP aggregates. S: strong. M: medium. N: none. >30 animals were examined for each genotype.
D, E Phsp-4::GFP expression is much higher in rpt-3(RNAi) animals.
F, G Upregulation of Phsp-4::GFP expression in rpt-3(RNAi) animals is suppressed by the xbp-1(zc12) mutation.
H, I SQST-1::GFP aggregates are absent in rpl-43(bp399); rpt-3(RNAi) animals.
J, K Loss of function of xbp-1 restores SQST-1::GFP aggregates in rpl-43(bp399); rpt-3(RNAi) animals.
L Functional interaction of ER stress-induced genes.
M The increase in bec-1, epg-8, lgg-1, and atg-7 mRNA levels in npl-4.1, rpt-3, and pep-2 RNAi animals is dependent on xbp-1. *P < 0.05, **P < 0.01. 500 young adult animals were collected for analysis. Error bars indicate s.d. from three experiments.
Data information: (A), (D), (F), (H), and (J): DIC images of the animals in (B), (E), (G), (I), and (K), respectively. Scale bars: 100 μm (A, B, D–G); 20 μm (H–K).
In C. elegans, IRE-1-XBP-1, the PERK kinase homolog PEK-1, and activating transcription factor-6 (ATF-6) sense ER stress and activate the UPR. To determine whether UPR activation plays a causative role in autophagy induction, we examined the role of xbp-1, pek-1, and atf-6 in suppression of the rpl-43 phenotype by the 24 identified genes. Loss of activity of ire-1, xbp-1, pek-1, or atf-6 had no evident effect on the expression of Phsp-4::GFP or the number of SQST-1 aggregates in rpl-43 mutants (Supplementary Fig S7C, D, M, and N). Simultaneous inactivation of xbp-1 blocked upregulation of Phsp-4::GFP in ER stress-induced mutant animals (Fig 3F and G; Supplementary Fig S7K and L). In rpl-43; xbp-1 double mutants, SQST-1 aggregates persisted upon DTT treatment or knockdown of identified ER stress-induced genes except unc-89 (Fig 3H–K; Supplementary Fig S7O-R). SQST-1::GFP aggregates were restored in unc-89; rpl-43 mutants by atf-6 inactivation (Supplementary Fig S7S–V). Inactivation of pek-1 or atf-6 did not affect other identified ER stress-induced genes. Thus, XBP-1 mediates the induction of autophagy by ER stress. SQST-1 aggregates in rpl-43; xbp-1 mutants were still suppressed by the inactivation of TOR signaling, daf-2, sma-3, and lin-35 (Supplementary Fig S7W–Z), indicating that impaired ER stress did not block autophagy induction by other signaling pathways.
We determined the expression of autophagy genes in ER stress-induced mutants. GFP::LGG-1 forms a large number of puncta in the intestine after knockdown of ufd-1, npl-4.2, and pep-2 (Supplementary Fig S7A2 and B2). Levels of bec-1, epg-8, lgg-1, and atg-7 mRNA were increased in npl-4.1-, rpt-3-, and pep-2-knockdown animals (Fig 3M). Simultaneously depleting xbp-1 abolished the upregulation of autophagy genes in these same mutants (Fig 3M).
Mitochondrial stress upregulates autophagy activity
Perturbation of the protein-folding environment in the mitochondrial matrix activates the mitochondrial unfolded protein response (UPRmt) to re-establish protein homeostasis 22. Inactivation of spg-7 (not included in the RNAi feeding library), encoding C. elegans paraplegin, induced mitochondrial stress and suppressed SQST-1::GFP aggregates in rpl-43 mutants (Supplementary Fig S8A, B, J and K). RNAi inactivation of 6 genes identified in our screen, namely egl-46, gfi-1, vha-15, tomm-7, tomm-22, and E04A4.5, increased the expression of the mitochondrial stress marker Phsp-60::GFP (Fig 4A and B; Supplementary Fig S8C and D).
Figure 4. ATFS-1-mediated mitochondrial stress suppresses the rpl-43 phenotype.
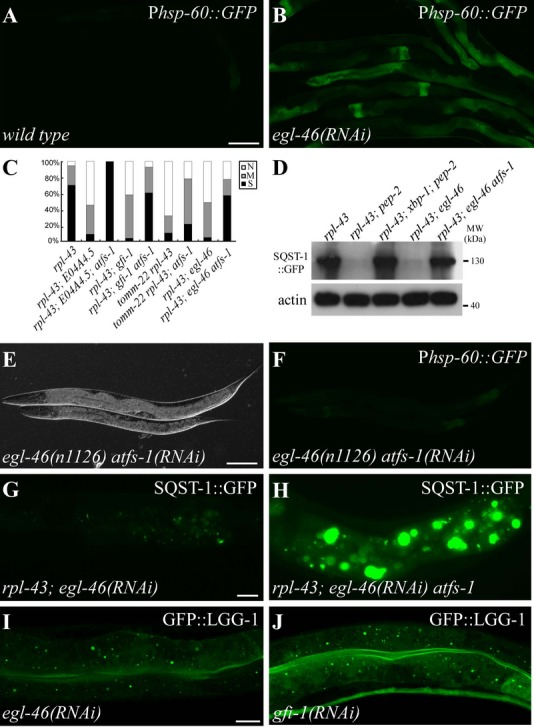
A Phsp-60::GFP is weakly expressed in wild-type animals.
B egl-46(RNAi) causes upregulation of Phsp-60::GFP.
C Percentage of indicated mutant animals with different levels of SQST-1::GFP aggregates. S: strong. M: medium. N: none. >30 animals were examined in each group.
D Levels of SQST-1::GFP in various genetic mutants. 200 young adult animals for each genotype were collected for analysis.
E, F Loss of function of atfs-1 suppresses the upregulation of Phsp-60::GFP in egl-46(n1126) mutants. (E): DIC image of (F).
G, H The dramatic decrease in the number of SQST-1::GFP aggregates in the intestine in rpl-43(bp399); egl-46(RNAi) mutants (G) is restored by simultaneous loss of function of atfs-1(gk3094) (H).
I, J GFP::LGG-1 forms numerous puncta in the intestine in egl-46(RNAi) (I), gfi-1(RNAi) (J) animals.
Data information: Scale bars: 100 μm (A, B, E, F); 20 μm (G, H); 10 μm (I, J).
The bZip transcription factor ATFS-1 is essential for UPRmt activation in C. elegans 22. atfs-1(RNAi) suppressed the elevated expression of Phsp-60::GFP in spg-7 and 6 other identified mutants and also rescued the suppression effect of SQST-1 aggregates in rpl-43 mutants (Fig 4C-H; Supplementary Fig S8E-P). atfs-1 is not required for the suppression effect caused by the loss of activity of sma-3, lin-35, and daf-2 (Supplementary Fig S8Q–V). Inactivation of daf-16 and xbp-1 had no effect on mitochondrial stress-induced autophagy (Supplementary Fig S8W–Z). LGG-1 levels and LGG-1::GFP puncta accumulated in egl-46 and gfi-1 mutants (Fig 4I and J; Supplementary Fig S8A2). Transcription of bec-1, epg-8, lgg-1, and atg-7 was upregulated in egl-46, gfi-1, and E04A4.5 mutants and reduced by simultaneous loss of atfs-1 activity (Supplementary Fig S8B2). Thus, mitochondrial stress activates autophagy through ATFS-1-mediated upregulation of autophagy genes.
The TGF-β, lin-35/Rb, ER stress, and mitochondrial stress pathways activate autophagy activity independent of HLH-30
The C. elegans TFEB homolog HLH-30 activates the expression of autophagy genes during starvation 23,24. Suppression of the rpl-43 mutant phenotype by ER stress, by mitochondrial stress, or by the loss of activity of lin-35 and sma-3 was not affected by simultaneous depletion of hlh-30 (Supplementary Fig S9A-H). The transcription factor MXL-3 acts antagonistically to HLH-30 to repress the expression of lysosomal lipases 23. Inactivation of mxl-3 did not affect the accumulation of SQST-1::GFP aggregates in rpl-43 mutants (Supplementary Fig S9I and J), indicating that autophagy activity was not elevated in the mxl-3 mutant intestine. Thus, TGF-β, lin-35/Rb, ER stress, and mitochondrial stress signaling function in parallel to HLH-30 to control autophagy gene expression.
Here, we established the degradation of SQST-1 aggregates in the rpl-43 mutant intestine as a genetic model system to investigate autophagy regulation. We demonstrated that autophagy activity is elevated by various stresses. ER stress and mitochondrial stress activate autophagy activity by transcriptional upregulation of autophagy genes in C. elegans. Components of the endocytic pathway, including the AP2 complex, the COG complex, and the ESCRT complex, were identified in our screen probably because their partial loss of function may not impair their normal function in the autophagy pathway 3, but impose a stress on the intestine which in turn activates autophagy.
In addition to these stress-induced gene inactivations, we found that the loss of activity of several developmental signaling pathways, including TGF-β, lin-35/Rb, glp-1, daf-2, mTOR, and LIN-45/MEK-2/MPK-1 MAPK signaling, promotes autophagy activity. TGF-β lin-35/Rb and glp-1 signaling specify distinct biological processes by transcriptionally regulating distinct target genes and also transcriptionally promote autophagy activity 24. Therefore, various developmental signaling factors integrate with the autophagy machinery during animal development.
Materials and Methods
See Supplementary Materials and Methods for strains used and additional protocols.
Identification, mapping, and cloning of rpl-43
bp399 was isolated in a genetic screen for mutants with ectopic accumulation of SQST-1::GFP aggregates. bp399 was mapped between the polymorphic markers pkp2118 (II: +21.2) and pkp2113 (II: +22.9). Fosmids in this region were used for transformation rescue. Accumulation of SQST-1::GFP aggregates in rpl-43(bp399) animals was rescued by a transgene expressing the single gene y48b6a.2 (including an approximately 1.5-kb promoter region, the entire ORF, and the approximately 1-kb 3′ UTR).
Preparation and induction of RNAi bacterial clones
The RNAi feeding library was purchased from Geneservice. The library contains bacterial clones expressing dsRNA designed to individually inactivate 16,749 genes (targeting about 87% of the predicted genes) 25. RNAi bacterial clones were grown on LB agar plates supplemented with 100 mg/ml ampicillin and 30 mg/ml tetracycline and then inoculated into LB medium containing 50 mg/ml ampicillin and cultured for 6 h at 37°C. 300 μl of each bacterial culture was dispensed onto 10 cm NGM agar plates containing 5 mM IPTG. dsRNA transcription was induced overnight at 25°C. Synchronized L1 sqst-1::gfp; rpl-43 animals were plated onto RNAi feeding plates with about 15 worms per plate and were grown at 20°C. The F1 progeny or arrested larvae or sterile adults were examined for expression of the sqst-1::gfp reporter.
Immunofluorescence staining
For worm immunofluorescence staining, animals were permeabilized by freeze-cracking and then fixed, blocked, and incubated with diluted antibody at room temperature for 2–4 h. The animals were then washed three times and incubated with rhodamine-conjugated or FITC-conjugated secondary antibody. Fluorescence was examined by a fluorescence microscope (Zeiss Axioplan 2 image) or a confocal microscope (Zeiss LSM 710 Meta plus Zeiss Axiovert zoom).
Statistical analysis
Data are shown as mean ± SD. Unpaired t-tests were performed for statistical analysis.
Acknowledgments
We are grateful to Dr. Isabel Hanson for editing work. This work was supported by the National Basic Research Program of China (2013CB910100, 2011CB910100) and also a grant from the NSFC (31225018) to H.Z. The research of Hong Zhang was supported in part by an International Early Career Scientist Grant from the Howard Hughes Medical Institute.
Author contributions
BG, XXH, PPZ, and HZ designed the experiments. BG, XXH, QQL, PPZ, LXQ, XBZ, JH, and BF performed the experiments. WRH and JHH performed bioinformatic analyses. BG, XXH, and HZ wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://embor.embopress.org
References
- Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–467. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759–774. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- Lu Q, Wu F, Zhang H. Aggrephagy: lessons from C. elegans. Biochemical J. 2013;452:381–390. doi: 10.1042/BJ20121721. [DOI] [PubMed] [Google Scholar]
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256–1269. doi: 10.1016/j.cell.2012.11.001. [DOI] [PubMed] [Google Scholar]
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res. 2014;24:42–57. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski MM, Hoffman G, Ng A, Zhou W, Py BF, Hsu E, Liu X, Eisenberg J, Liu J, Blenis J, et al. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell. 2010;18:1041–1052. doi: 10.1016/j.devcel.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan S, Goodwin JG, Chauhan S, Manyam G, Wang J, Kamat AM, Boyd DD. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol Cell. 2013;50:16–28. doi: 10.1016/j.molcel.2013.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Li ZP, Hu WQ, Ren HY, Tian E, Zhao Y, Lu Q, Huang XX, Yang PG, Li X, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010;141:1042–1055. doi: 10.1016/j.cell.2010.04.034. [DOI] [PubMed] [Google Scholar]
- Zhang YX, Yan LB, Zhou Z, Yang PG, Tian E, Zhang K, Zhao Y, Li ZP, Song B, Han JH, et al. SEPA-1 mediates the specific recognition and degradation of P granule components by autophagy in C. elegans. Cell. 2009;136:308–321. doi: 10.1016/j.cell.2008.12.022. [DOI] [PubMed] [Google Scholar]
- Lin L, Yang PG, Huang XX, Zhang H, Lu Q, Zhang H. The scaffold protein EPG-7 links cargo-receptor complexes with the autophagic assembly machinery. J Cell Biol. 2013;201:113–129. doi: 10.1083/jcb.201209098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Sternberg PW. Genome-wide prediction of C. elegans genetic interactions. Science. 2006;311:1481–1484. doi: 10.1126/science.1123287. [DOI] [PubMed] [Google Scholar]
- Lee I, Lehner B, Crombie C, Wong W, Fraser AG, Marcotte EM. A single gene network accurately predicts phenotypic effects of gene perturbation in Caenorhabditis elegans. Nat Genet. 2008;40:181–188. doi: 10.1038/ng.2007.70. [DOI] [PubMed] [Google Scholar]
- Savage-Dunn C. TGF-beta signaling. WormBook. 2005:1–12. doi: 10.1895/wormbook.1.22.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Shi Z, Cui M, Han M, Ruvkun G. Repression of germline RNAi pathways in somatic cells by retinoblastoma pathway chromatin complexes. PLoS Genet. 2012;8:e1002542. doi: 10.1371/journal.pgen.1002542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meléndez A, Tallóczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- Cao SS, Kaufman RJ. Unfolded protein response. Curr Biol. 2012;22:R622–R626. doi: 10.1016/j.cub.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta. 2013;1833:410–416. doi: 10.1016/j.bbamcr.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke EJ, Ruvkun G. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat Cell Biol. 2013;15:668–676. doi: 10.1038/ncb2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre LR, De Magalhaes Filho CD, McQuary PR, Chu CC, Visvikis O, Chang JT, Gelino S, Ong B, Davis AE, Irazoqui JE, et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun. 2013;4:2267. doi: 10.1038/ncomms3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.