

Abstract
Proteins function mainly through interactions, especially with DNA and other proteins. While some large-scale interaction networks are now available for a number of model organisms, their experimental generation remains difficult. Consequently, interolog mapping—the transfer of interaction annotation from one organism to another using comparative genomics—is of significant value. Here we quantitatively assess the degree to which interologs can be reliably transferred between species as a function of the sequence similarity of the corresponding interacting proteins. Using interaction information from Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster, and Helicobacter pylori, we find that protein–protein interactions can be transferred when a pair of proteins has a joint sequence identity >80% or a joint E-value <10–70. (These “joint” quantities are the geometric means of the identities or E-values for the two pairs of interacting proteins.) We generalize our interolog analysis to protein–DNA binding, finding such interactions are conserved at specific thresholds between 30% and 60% sequence identity depending on the protein family. Furthermore, we introduce the concept of a “regulog”—a conserved regulatory relationship between proteins across different species. We map interologs and regulogs from yeast to a number of genomes with limited experimental annotation (e.g., Arabidopsis thaliana) and make these available through an online database at http://interolog.gersteinlab.org. Specifically, we are able to transfer ∼90,000 potential protein–protein interactions to the worm. We test a number of these in two-hybrid experiments and are able to verify 45 overlaps, which we show to be statistically significant.
The ultimate goal of functional genomics is to determine the functions of all gene products in newly sequenced genomes. Unfortunately, although there is a deluge of sequence data available, only a small fraction has been functionally characterized (Andrade and Sander 1997). Nevertheless, for some genomes belonging to experimentally tractable model organisms, such as Saccharomyces cerevisiae, Caenorhabditis elegans, and Helicobacter pylori, scientists have elucidated the functions of many of their gene products. Given the quantity of sequence and structural data available, a major method for assigning functions is to transfer the existing annotation of a known gene to the newly sequenced gene product. This is based on the concept that sequence and structural similarities between gene products suggest functional similarities (Bork et al. 1994, 1998; Fraser et al. 1995, 1998; Wilson et al. 2000; Hegyi and Gerstein 2001).
The transfer of structural annotations is well characterized. It has been shown that structural similarity (measured as the Root Means Square [RMS] of matching Cα backbone atoms) between two proteins decreases exponentially with increased sequence divergence (measured as percent identity; Chothia and Lesk 1986, 1987). Thus, the reliability of a homology-based structural annotation depends on the level of sequence similarity between homologous proteins.
Several groups have recently examined the dependency of functional similarity on sequence and structural similarity (Bork et al. 1994, 1998; Marcotte et al. 1999). The best matching sequences in a database search are often used as the basis for initial annotations (Fraser et al. 1995, 1998). However, further work has provided the potential for more robust annotation transfer, including analyzing patterns of protein family occurrence in different phylogenetic groups (Pellegrini et al. 1999) and associating key sequence motifs with particular functions (Bairoch et al. 1996; Attwood et al. 1997). Other work has also shown that, in general, protein function is conserved for sequence identities down to 40% for single-domain proteins that share the same structural fold; however, for multidomain proteins, the pattern of functional conservation is more complex: Proteins are most likely to share functions if they contain similar domain combinations (Brenner 1999; Wilson et al. 2000; Hegyi and Gerstein 2001).
It is difficult to evaluate the relationship between sequence homology and function, because no clear measure of functional similarity exists between any two proteins, and the definition of “function” itself is often vague (Bork et al. 1998; Wilson et al. 2000; Lan et al. 2002, 2003). Previous studies, based on hierarchical classification systems, such as ENZYME (Webb 1992), MIPS (Mewes et al. 2000), and GO (Ashburner et al. 2000), determine functional similarity by comparing both proteins' respective levels in the hierarchy. This is a rough definition underlying the difficulties inherent in the earlier work. However, an important aspect of protein function is the physical interactions of proteins with other molecules, in particular, with other proteins or with DNA. No previous work has addressed this issue. With recent genome-wide studies on protein–protein and protein–DNA interactions (Ito et al. 2000; Uetz et al. 2000; Iyer et al. 2001; Gavin et al. 2002; Ho et al. 2002; Horak et al. 2002; Lee et al. 2002), it is now possible to examine the degree to which protein–protein and protein–DNA interactions are transferred between different organisms as a function of the underlying sequence similarities of the interacting proteins.
To this end, Walhout et al. (2000) introduced the concept of “interologs”: orthologous pairs of interacting proteins in different organisms. In this study, we extend and assess this concept in detail. We present a large-scale quantitative assessment on conservation of protein–protein and protein–DNA interactions between proteins and organisms. Compared with the previous survey, our investigation has greater statistical weight and precision. In our calculations, we use almost all available genome-wide interaction data sets from four model organisms (14,911 interactions total). Moreover, we generalize the interolog concept and propose that there are at least two kinds of interologs: protein–protein interologs and protein–DNA interologs. Based on the latter idea, we also introduce a new concept, the “regulog.” Furthermore, we calibrate the ability of interologs to reliably map interactions across different organisms. Combining our interolog and regulog mapping with available large-scale interaction data for yeast, we construct genome-wide interaction maps and regulatory networks for several organisms.
METHODS
Definitions and Formalism for Protein–Protein Interologs
Homologs and Orthologs
Homologs are proteins with significant sequence similarity. Operationally, this can be defined as having an E-value ≤10–10 from BLASTP (Altschul et al. 1990). This is a similar cutoff to that used previously (Matthews et al. 2001).
Orthologs are proteins in different species that evolved from a common ancestor “by speciation” (Tatusov et al. 1997). Orthologous proteins in different organisms usually have the same functions. Operationally, the ortholog of a protein is usually defined as its best-matching homolog in another organism. Here we define orthologs as:
Candidates with a significant BLASTP E-value (≤10–10).
Having ≥80% residues in both sequences included in the BLASTP alignment.
Having one candidate as the best-matching homolog of the other candidate in the corresponding organism.
Conditions 1, 2, and 3 must be true reciprocally.
It is obvious that this operational definition of ortholog by sequence homology is not perfect. Actually, orthologs are not always determined as the best-matching homologs (Tatusov et al. 1997).
Interologs
Based on Walhout et al. (2000), if interacting proteins A and B in one organism have interacting orthologs A′ and B′ in another species, the pair of interactions A–B and A′–B′ are called interologs (see Fig. 1A).
Figure 1.




Schematic illustration of protein–protein interologs and the mapping methods. (A) Original interolog mapping. Theoretically, A-A′ and B-B′ should be orthologs between the two organisms. Operationally, only best-matching homologs are required. (B) Generalized interolog mapping. Proteins A1′, A2′, A3′, and A4′ in the target organism are all homologs of protein A in the source organism. These proteins form the A′ family. Likewise, protein B's homologs (B1′, B2′, B3′) form the B′ family in the target organism. If we know that protein A interacts with B, we can predict that the A′ family and the B′ family are interacting families. All possible pairs between these two families are considered as the generalized interologs (shown as black, dashed lines with arrows). (C) Comparison with the gold standards. After the interactions in the source organism are mapped onto the target organism, the predictions (i.e., generalized interologs) are compared with the gold standard positives and negatives. True positives are the predictions that overlap with the gold standard positives. False positives are those that overlap with the gold standard negatives. (D) Schematic illustration of protein–DNA interologs and regulogs. In the source organism, TF A binds to its binding site (SA) and regulates the downstream gene B. To perform the regulog mapping, TF A′ in the target organism needs to be the ortholog of A. Proteins B and B′ should also be orthologs. The DNA sequence upstream of gene B′ needs to contain the same motif (SA′) as SA. However, practically TF A and A′ only need to share ≥30% identity. The interaction between TF A′ and SA′ is the protein–DNA interolog of that between A and SA. The regulatory relationships between A → B and A′ → B′ are regulogs.
Joint Sequence Similarity
A goal of this work is to measure the transferability of interactions based on sequence similarity. In the case of protein–protein interactions, sequence similarities to homologs of both interacting partners are important. We therefore use joint sequence similarity (J) between protein pairs. There are many potential ways to define joint sequence similarity, but our results show that different definitions of J do not matter much. Here, we use two major definitions of J.
Joint Sequence Identity (JI) as the Geometric Mean of Individual Percent Identities
Percent identity is routinely used to measure the sequence similarity between proteins. Therefore, joint similarity is first defined as the geometric mean of individual percent identities:
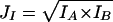 |
Given that protein A is known to bind to protein B, IA represents the individual sequence identity of protein A and its homolog. Likewise, IB is the individual sequence identity of protein B and its corresponding homolog. We calculate individual sequence identities based on the sequence alignment using the Smith-Waterman algorithm in FASTA (Pearson and Lipman 1988).
Joint E-Value (JE) as the Geometric Mean of Individual E-Values
Measuring homology by percent identity has certain disadvantages (Wilson et al. 2000). For instance, the length of the matching sequences is not considered. Naturally, the shorter the sequence is, the higher the chance of randomly finding similar sequences. Furthermore, it has become more common to use statistical scoring schemes, especially E-values in BLAST, to measure the statistical significance of the homology in order to determine the orthologs across organisms (Tatusov et al. 1997; Brenner et al. 1998). Therefore, we also calculate the joint similarity as a joint E-value, that is, the geometric mean of the individual E-values:
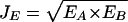 |
where EA represents the BLASTP E-value of protein A and its homolog, and EB is the individual BLASTP E-value of protein B and its homolog.
Joint Similarity as the Minimal Individual Similarity
Calculating the joint similarity using the geometric mean of the individual similarities places equal weight on each of the two similarities. However, the joint similarity could also be defined as the smaller of the two individual similarities:
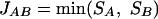 |
where SA and SB represent the individual similarities, respectively, of protein A and its homolog and of protein B and its homolog. In this manner, JAB measures the minimal similarity level necessary for the reliable transfer of interaction information between protein pairs. Individual similarities can also be determined as percent identities by FASTA or E-values by BLASTP.
Source and Target Organisms
In the “source organism,” there is a set of known interactions. The “target organism” is a fully sequenced organism onto which the known interactions in the source organism are mapped (as described below) based on sequence similarities (see Fig. 1C).
Interolog Mapping
“Interolog mapping” is a process that maps interactions in the source organism onto the target organism to find possible interactions (i.e., interologs) in that organism (see Fig. 1A). To assess the performance of mapping methods, one can use known interacting and noninteracting protein pairs (positives and negatives) in the target organism as benchmarks.
Original Interolog Mapping Method: Best-Match Mapping
Previously, Matthews et al. (2001) proposed a best-match mapping method to transfer yeast interactions onto the worm proteome. Simply put, their method selects all best-matching homologs between two organisms (E-value < 10–10). In worm, all pairs of best-matching homologs of interacting yeast proteins are considered as potential interologs. Using two-hybrid systems, they tested 216 worm protein pairs and 72 yeast protein pairs. Their results showed that only 16% to 32% of interologs predicted experimentally determined interactions correctly.
A New Method: Reciprocal Best-Match Mapping
A more stringent derivative of this original method would be to use only the reciprocal best matches in mapping interologs between organisms (Li et al. 2004). In this paper, we present results from both approaches.
Generalized Interolog Mapping
Both interolog mapping methods, using only the best matches, suffer from low coverage of the total interactome and low prediction accuracy. This is discussed further in the next section. To address the problem of low coverage, we introduce a new “generalized interolog mapping” method using all possible homologs of interacting proteins. For any given protein in one organism, all of its homologs in another organism are considered as a homolog family (or simply family). Two families of two interacting proteins are called interacting families, that is, at least one member of one family interacts with a member of the other family. All possible protein pairs between the two interacting families are called generalized interologs (see Fig. 1B). This method has the advantage of sidestepping some of the ambiguities in defining orthologs.
Gold Standard Target Data Sets
Set of Gold Standard Positives P
To assess the performance of interolog mapping, we need a group of known interactions as positives in the target organism. This set is called the gold standard positives and is denoted by P. The total number of elements in this set is |P|.
As the most extensive and reliable interaction data sets exist for S. cerevisiae, we use it first as the target organism. In S. cerevisiae, the MIPS complex catalog, which contains 8250 unique interacting protein pairs, has previously been used as a standard reference for known interactions (Mewes et al. 2000; Edwards et al. 2002; von Mering et al. 2002; Jansen et al. 2003). Therefore, we consider the MIPS interactions as gold standard positives in the next section. To compile a reference data set with the lowest false-positive rate, we consider two proteins as interaction partners if and only if they are in the same complex of the highest level in the catalog. At the end of the paper, we reverse this situation and use S. cerevisiae as the source organism and map its reliable interaction information (from the complex catalog) onto other eukaryotes (such as Arabidopsis thaliana) to build an interolog database.
It should also be noted that proteins in the same complex do not necessarily interact with each other directly. Here, we use the term “interaction” to signify “complex association,” that is, two protein subunits may belong to the same quaternary complex but not physically interact. Therefore, the number of complex associations of a protein may be larger than the number of its pairwise physical associations.
To probe the direct physical interactions more closely, we constructed a refined, smaller data set comprising 1867 interactions between 1391 proteins. In parallel to our “gold standard” nomenclature, we call this the platinum standard data set. Briefly, the data set contains physical interactions from complex protein structures in the Protein Data Bank (Westbrook et al. 2003), verified interactions from small-scale experiments (Mewes et al. 2000; Xenarios et al. 2002; Bader et al. 2003), and protein pairs from small MIPS catalog complexes (≤4 subunits). The data set and a detailed explanation of its construction are available from our Web site. The platinum standard data set is of equally high quality as the gold standard set, but differs as it describes physical pairwise interactions between proteins rather than complex associations. As shown below, the two data sets yield very similar results, indicating a good correspondence between physical interactions and complex associations. However, because better statistics are obtained from a larger data set, we perform the bulk of the analysis in this paper using the gold standard interactions.
Set of Gold Standard Negatives N
We also need a set of negatives (i.e., noninteracting proteins) in the target organism to assess our method. This set is called gold standard negatives and is denoted by N.
Previously, Jansen et al. (2003) considered pairs of proteins in different subcellular compartments as good estimates for noninteracting pairs (Kumar et al. 2002). In total, there are 2,708,746 such protein pairs.
However, sometimes not all interolog features could be defined for each of the pairs in the gold standard. In this case, we use alternate sets P′ and N′, subsets of P and N with defined features.
Source Data Sets
To assess the interolog mapping method, we need source organisms with known interaction data. In this paper, C. elegans, D. melanogaster, and H. pylori are used as source organisms. We then map the interactions in these organisms onto the S. cerevisiae genome. These are the only three organisms, besides S. cerevisiae, for which large-scale interaction data sets are available.
C. elegans Interaction Data Set
For C. elegans, there are 410 interactions from two-hybrid experiments (Walhout et al. 2000; Davy et al. 2001; Boulton et al. 2002).
D. melanogaster Interaction Data Set
For D. melanogaster, there are 4786 interaction pairs from two-hybrid experiments (Giot et al. 2003).
H. pylori Interaction Data Set
For H. pylori, there are 1465 interaction pairs from two-hybrid experiments (Rain et al. 2001).
Assessment Parameters
As shown in Figure 1C, based on interactions in the source organisms, all generalized interologs with joint similarities larger than a certain cutoff (J) are considered possible interactions in the target organism. We then assess these predictions (thin red solid lines) against gold standard positives (thick, black, solid lines) and negatives (dashed lines) in the target organism. The assessment parameters are as follows.
G(J)
The set of generalized interologs in the target organism at a certain joint similarity level (J) is denoted by G(J).
T(J)
The set of the true positives in G(J) is denoted by T(J), that is, T(J) = G(J) ∩ P. We define the number of true positives at a given J as TP = |T(J)|.
F(J)
The set of false positives in G(J) is denoted by F(J), that is, F(J) = G(J) ∩ N. We define the number of false positives at a given J as FP = |F(J)|.
V(J)
We denote V(J) as the percentage of verified predictions among generalized interologs at a certain joint similarity level J, which is calculated as:
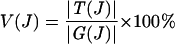 |
We also call V a level of verification (or loosely, an accuracy). Please note that V calculated here may be a lower bound estimate because the MIPS complex catalog is not complete.
L(J)
We denote L(J) as the likelihood ratio for a generalized interolog, with a certain joint similarity (J), to be a true prediction. L(J) can be calculated by a Bayesian approach. This is a straightforward extension of the formalism described previously (Jansen et al. 2003). If we know the number of positives (Np) among the total number of protein pairs (Nt), the probability of finding an interacting pair in the genome, P(pos), can be defined as Np/Nt. Therefore, the “prior” odds of finding a positive are:
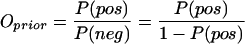 |
In contrast, the “posterior” odds are the odds of finding a positive given that, in another organism, its generalized interolog with a joint similarity J is a known interaction:
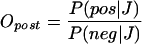 |
The likelihood ratio L defined as
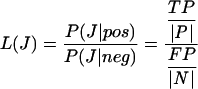 |
relates prior and posterior odds according to Bayes' rule:
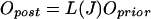 |
As Oprior is fixed for a given organism, Opost is proportional to L(J), that is, the higher the likelihood ratio, the more likely the prediction is true. In a naive Bayesian network where there are no correlations between features, this procedure can be iterated. Specifically, Opost can be multiplied again by another L for a different feature. In doing so, one could combine many different features within a uniform framework of likelihood ratios. In particular, it would allow us to combine our likelihood ratios from interologs with the other features in Jansen et al. (2003).
Definitions and Formalism for Protein–DNA Interologs and Regulogs
Protein–DNA Interologs and Mapping
If transcription factor (TF) A with binding site SA has, in another species, an ortholog A′ with binding site SA′ of identical DNA sequence, A′-SA′ is a protein–DNA interolog of A-SA (see Fig. 1D).
We can extend protein–protein interolog mapping to protein–DNA interolog mapping. In this process, we transfer the DNA-binding information of a given TF A to its ortholog A′ as a function of the sequence similarity between A and A′.
Regulogs
TFs bind to DNA to regulate the expression of downstream genes. Therefore, there is a regulatory relationship between a given TF and its target. Suppose that TF A and its target B in one organism have orthologs A′ and B′, respectively, in another organism. Furthermore, suppose that in the second organism, A′ is also a TF regulating B′, then we call A′ → B′ a regulog of A → B.
Source Data Sets
For practical calculations, we used TF families as described previously (Luscombe and Thornton 2002). Target-binding sequences of individual factors were obtained from the TRANSFAC database (Wingender et al. 2001). All known protein–DNA interactions are considered as positives. We do not have negative data sets for protein–DNA interologs and regulogs.
Assessment Parameters
The parameters involved in assessing the conservation of protein–DNA interologs are analogous to those for protein–protein interologs. They are given as follows:
G(I)
The set of predicted protein–DNA interologs with the sequence identities between TFs larger than a certain cutoff (I) is denoted by G(I).
T(I)
The set of the transcription factor pairs that share the same DNA-binding sites in G(I) is denoted by T(I).
V(I)
We denote V(I) as the percentage of verified predictions among the predicted protein–DNA interologs at a certain sequence identity level, I. This is calculated as:
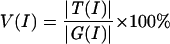 |
We calculate Vs both for TFs within each family separately and for all TFs together (see Fig. 1D). Due to the relatively small amount of TF-binding data, we aggregate all of our predictions. This procedure is described in the Supplemental material.
RESULTS AND DISCUSSION
Assessment of Interologs on Current Interaction Data Sets
Conservation of Generalized Interologs
Relationships Between V and J
To measure the conservation of interactions between homologous protein pairs, we assessed the chance (V) that two proteins interact with each other as a function of their joint sequence identities (JI) with other known interacting pairs. First, we mapped only worm interactions onto the yeast genome. As there are not many data points, we grouped all the generalized interologs into three bins based on their joint identities: low, medium and high. Figure 2A shows a clear monotonic relationship between V and JI. This confirms that the higher the joint identity, the more likely the predicted interolog is true.
Figure 2.

Conservation of protein–protein interactions between homologous protein pairs. (A,B) Relationships between V and JI. (C,D) Relationships between V and JE.(E,F) Relationships between L and JE. (A,C,E) Calculated based on the results from worm-yeast mapping. (B,D,F) The weighted average obtained when the interactions in all four organisms (i.e., S. cerevisiae, C. elegans, D. melanogaster, and H. pylori) were mapped onto yeast. (A) Low: JI ≤ 10%; Medium: 20% ≤ JI ≤ 30%; High: JI ≥ 40%. (C,D) Low: 10–40 ≤ JE ≤ 10–10; Medium: 10–100 ≤ JE ≤ 10–50; High: JE ≤ 10–110. Error bars represent 95% CI calculated by a resampling algorithm (see Supplemental material).
To get better statistics, we mapped interactions in S. cerevisiae, C. elegans, D. melanogaster, and H. pylori onto the S. cerevisiae genome, assessing them against our gold standards described above. (In this case, S. cerevisiae functions as both a source and a target organism.) In Figure 2B, the relationship between V and JI is the weighted average (based on the total number of true positives in each data set) of the relationships in all four mapping processes. The plot exhibits a sigmoidal relationship with a sharp decrease around 80% JI. This indicates that all protein pairs having JI ≥ 80% with a known interacting pair will interact with each other, whereas few pairs interact at JI < 40%. These results confirm that pairs of proteins with sufficient sequence similarity tend to share the annotation of protein–protein interactions.
Furthermore, we performed a similar analysis using joint E-values (JE). Figure 2C shows the same monotonic relationship as that in Figure 2A, when we mapped worm interactions onto yeast genome. In Figure 2D, the weighted average curve also has a sigmoidal characteristic. Overall, more than half of the protein pairs with JE ≤ 10–70 indeed bind to each other. Therefore, JE of 10–70 could be used as a good threshold to reliably transfer the annotation of interactions.
Relationships Between L and J
The above approach (i.e., assessing the transferability of a property between organisms by calculating the fraction sharing the property with certain similarity) has been generally used for similar purposes (Wilson et al. 2000; Hegyi and Gerstein 2001). Here, we apply a Bayesian network approach to further evaluate the transferability of interactions. Likelihood ratios (L) are more directly related to probabilities and are, therefore, more quantitative and precise in describing the transferability of the interactions.
As we did for V above, we calculated the relationships between L and JE for two mappings: worm-to-yeast and a weighted average of all four organisms to yeast (Fig. 2E and 2F, respectively). Both figures exhibit positive relationships between L and JE, suggesting that the better the joint E-values, the higher the likelihood ratios. This further confirms the relationships found in Figure 2, A–D, and the validity of using joint similarities.
Conservatively, the total number of interactions in yeast genome is ∼30,000 (Kumar and Snyder 2002). Given that there are ∼18 million yeast protein pairs in total, the prior odds (Oprior) would be roughly 1/600. Therefore, only protein pairs with L > 600 would have a >50% chance of interaction. As shown in Figure 2F, protein pairs with JE ≤ 10–50 have L > 600. The JE threshold (10–70), determined previously, easily satisfies this criterion. If we were to use L to perform the mapping methods, cross-validation could be applied in choosing the optimal L cutoff as described previously (Jansen et al. 2003).
We examine the correspondence between direct, physical interactions and complex associations, by repeating the calculations for Figure 2, B, D, and F, using the platinum standard data set. The results show similar trends to the gold standard data set (Supplemental Fig. 1), indicating the high correspondence between the two data sets. Due to its smaller size, the statistics for the platinum standard data set are not as good as for the gold standard. Owing to the similarity of results, and better statistics, we therefore use the MIPS complex catalog as the main reference data set in this paper.
Results of J as the Minimal Sequence Similarity Remain the Same
As discussed above, we could also use the minimal individual similarity instead of the geometric mean to calculate J. We repeated all calculations in Figure 2 using this new definition of J. The results show that the new definition has little effect (Supplemental Fig. 2). Therefore, for the remaining discussion, J is defined as the geometric mean of the individual E-values (i.e., JE).
Comparison of Different Interolog Mapping Methods
To compare different mapping methods, C. elegans was used as the source organism, and its interactions were mapped onto S. cerevisiae genome by three different mapping methods as discussed above. We compared the predicted interologs produced by the different methods above against the gold standard positives and negatives. The results are as follows:
Best-Match Mapping Method
From 410 interacting pairs in worm, we found 84 corresponding interolog candidates in yeast. Only 25 of these pairs overlapped with gold standard positives, corresponding to V ≈ 30% (i.e., loosely 30% accuracy). This agrees with previous results (Matthews et al. 2001).
Reciprocal Best-Match Mapping Method
In total, we determined 33 interolog candidates based on the 410 worm interactions, among which 18 pairs (54%) were true positives.
Generalized Interolog Mapping Method
Based on the 410 interacting pairs, we found 92 pairs of interacting families in yeast, 91 of which contain at least one true interaction. In total, we predicted 9317 interactions (i.e., generalized interologs), among which 162 pairs (2%) are true positives. In Figure 3, it is evident that the fraction of true positives clearly increases as JE decreases. When only the top 5% pairs with the best JE values are selected, V increases to 31% (35 true positives out of 112 predictions), resulting in even better accuracy than that of the best-match mapping method (30%).
Figure 3.

Distribution of the number of generalized interologs as a function of joint E-value (JE). The dashed line represents the number of all predictions above a given JE, that is, G(J). The solid line represents the number of true positives above a given JE, that is, TP.
Previously, four large-scale experimental interaction data sets in yeast have been combined into a “PIE” (i.e., Probabilistic Interactome Experimental), in which each interaction is associated with a particular L (Jansen et al. 2003). To assess the performance of our method in relation to known standards, we compared our results against the PIE. We show our comparison as a TP/|P′| versus TP/FP graph, a close analog of the conventional ROC curve. As shown in Figure 4, the coverage and accuracy of interolog mapping are roughly comparable to those of the large-scale experiments.
Figure 4.

Comparison of generalized interolog mapping with PIE. In this figure, the plot (TP/|P′| versus TP/FP) is analogous to an ROC plot (TP/P vs. FP/N). Based on this curve, the performance of our method is comparable to that of the large-scale experimental data sets.
Examples of Protein–Protein Interologs
The Ste5-MAPK complex is a key six-subunit complex in the yeast mating-pheromone response pathway (Posas et al. 1998). The interaction partners of worm MAPK (F43C1.2a) were determined experimentally (see Supplemental Table 1). In total, there are 26 known partners for F43C1.2a, none of which is involved in this MAPK signal transduction pathway. However, using the generalized interolog mapping method, we successfully predicted five of the six subunits in yeast based on only one MAP kinase in worm. This illustrates the power and utility of our method (see Supplemental material).
Assessment of Protein–DNA Interologs and Regulogs
Conservation of Protein–DNA Interologs
As shown in Figure 5, the relationship between V and I is sigmoidal, with a sharp decrease in target site conservation between 30% and 60% sequence identity. This indicates that all TFs within a certain range of identities invariably share the same target sequence. The specific threshold for the identities is highly family-dependent, ranging from 30% to 60%. The hormone receptor and LacI repressor families have a higher threshold of ∼60%, whereas the other families diverge at lower thresholds of 30%. The C2H2-zinc finger family is an exception, and sequence recognition is barely conserved even for close homologs (threshold identity 80%). The main reason for this is that the binding domains of C2H2-zinc fingers are often very short (∼30–90 amino acids in length) and, therefore, only a few mutations are required to alter its specificity.
Figure 5.

Conservation of protein–DNA interactions between homologous TFs. The conservation is measured as the relationships between V and I. The legend appears as an inset on the graph. The red, bold curve was calculated for all TFs in the source data sets (see Supplementary material). Error bars represent 95% CI calculated by the resampling algorithm.
The fact that TF families have different thresholds reflects the regulatory diversity of different families. Families with high thresholds contain factors that regulate many different processes, whereas those with low thresholds regulate only a few different processes (Luscombe and Thornton 2002).
We further assessed the general transferability of protein–DNA binding properties between homologous protein sequences by calculating the relationship between V and I for all TFs. As shown in Figure 5, ∼60% of homologous TFs share the same binding sites at 30% sequence identity; at 50% sequence identity, 80% of TFs share the same binding sites. Therefore, if two proteins have ≥30% sequence identity, they can be predicted to share the same binding sites. The confidence level of the prediction is shown as a function of sequence identity in Figure 5.
Protein–DNA Interolog (Regulog) Mapping Method
When a protein–DNA interaction is transferred across species, the regulatory relationship between the TF and its target is also implicitly transferred. Based on our calculations, at least three conditions are necessary for regulogs to be transferred (see Fig. 1D):
TF A and its homolog A′ must have ≥30% sequence identity. (Note that formally A and A′ should be orthologs. However, practically this is defined here by this sequence similarity criterion.)
Target gene B and its homolog B′ must be orthologs.
The DNA sequence upstream of B′ must contain the same binding site as that of B.
Unfortunately, we only have large-scale transcriptional regulatory networks in S. cerevisiae for eukaryotes and in Escherichia coli for prokaryotes. Because the transcription machinery differs radically between eukaryotes and prokaryotes, the performance of our regulog mapping method cannot currently be assessed on a large scale. However, we would like to discuss one specific example of regulogs between S. cerevisiae and D. melanogaster to illustrate the process of regulog mapping and its underlying logic.
In S. cerevisiae, Cyc1 is a mitochondrial protein with electron-transport function. The Hap2–Hap3 heteromeric TF complex binds to the UAS2 activation sequence (GTTGG) upstream of CYC1 and then activates transcription of this gene (Olesen et al. 1987; Hahn and Guarente 1988). Using the above-mentioned three conditions, we define potential regulogs in D. melanogaster:
CG10447 (a TF) and CG17618 (function unknown) are fly homologs of yeast proteins Hap2 and Hap3 with 30% and 40% sequence identities, respectively.
CG17903 (CD4) is a fly ortholog of Cyc1. It shows electron-transport activities and is located in the mitochondria (Limbach and Wu 1985).
The same UAS2 activation sequence (GTTGG) is also found in the promoter regions of CG17903 at the appropriate position (∼ –200 bp).
Based on the above, we predict that CG10447 and CG17618 may also regulate the expression of CG17903. This regulatory relationship is the fly regulog of its counterpart involving the yeast proteins Hap2–Hap3, and CYC1. Elucidating this allows us to predict the function of an unannotated fly protein, CG17618. Furthermore, the interactions between the two fly TFs and the UAS2 DNA sequence are the fly protein–DNA interologs of those between Hap2, Hap3, and the UAS2 sequence. More interestingly, because Hap2 and Hap3 interact with each other, their fly homologs CG10447 and CG17618 may also interact. This fly interaction is a potential protein–protein interolog of that between Hap2 and Hap3.
Database of Interologs and Regulogs
Finally, having proven the feasibility of the generalized interolog mapping method, we applied this method on the MIPS complex data set in yeast to predict protein–protein interactions in several other important eukaryotic organisms, including C. elegans, Candida albicans, D. melanogaster, and A. thaliana. In each organism, the top 1% of predicted generalized interologs with the best JEs are considered as highly reliable interologs. Simple statistics relating to the interolog database are shown in Table 1.
Table 1.
Statistics of the Interolog/Regulog Database
| Organisms | Total protein—protein interactions | JE cutoff for highly reliable interologs | Total TFs | Total targets | Total connectionsa |
|---|---|---|---|---|---|
| S. cerevisiae | 8250 | N/A | 148 | 3380 | 6765 |
| C. albicans | 20,470 | 10-105 | 66 | 1085 | 2349 |
| C. elegans | 91,224 | 10-75 | 36 | 601 | 1625 |
| D. melanogaster | 101,920 | 10-90 | 33 | 621 | 2936 |
| A. thaliana | 201,754 | 10-90 | 19 | 165 | 328 |
A connection is a TF—target pair.
To assess the accuracy of our database, we compared our predicted worm interactions with those from independent and on-going large-scale worm two-hybrid experiments. A total of 3730 interaction pairs were generated. Because only one splicing form was used for each gene in these experiments, we removed all alternative splicing forms and our prediction of yeast-to-worm interologs decreased from 91,224 (in Table 1) to 55,223 pairs. Among these, 45 pairs were confirmed experimentally. We use a hypergeometric model (see Supplemental material) to evaluate the significance of this overlap. The calculated P-value is smaller than 10–10. The P-value is the probability of finding a certain overlap between two independent data sets by chance within the whole worm interactome. Therefore, the experimental results support and validate our predictions.
More interestingly, the experimentally determined interaction pairs can be further divided into different groups involved in different pathways, for example, the 26S proteasome (Davy et al. 2001), DNA-damage repair (DDR; Boulton et al. 2002), and vulval development (Walhout et al. 2000). The overlaps between these groups and our predictions vary considerably, as shown in Figure 6. For groups known to be well conserved in eukaryotes, such as the proteasome and DDR (Larsen and Finley 1997; Davy et al. 2001), the overlaps are much better than those that are not. The nonsignificant P-value for the group “others” is also attributable to the fact that the baits in this group are specially selected to ensure they have no yeast homologs. Thus, Figure 6 further confirms the biological relevance of our database.
Figure 6.

Percentage of the overlaps between the predictions and different groups. (All) All experimentally determined interaction pairs; (Proteasome) interaction pairs involved in the 26S proteasome; (DDR) interaction pairs involved in DNA-damage repair; (Vulval-dev) interaction pairs involved in vulval development; (Others) interaction pairs involved in germ line, meiosis, metazoan, mitotic machinery, dauer formation, Chromosome III, chromatin remodeling, pharynx, and immunity. The P-values measuring the statistical significance of the overlaps between different groups and the predictions are given on top of each bar, which are calculated using the hypergeometric models (see Supplementary material).
We also applied our regulog mapping method to yeast transcriptional regulation data sets (Wingender et al. 2001; Horak et al. 2002; Lee et al. 2002). The results suggest potential regulatory networks in other eukaryotic organisms. Owing to variable TF-binding sites and insufficient information on binding sequences, we transferred the yeast regulatory networks using only the first two conditions, that is, sequence homology for both TFs and targets. In general, distant organisms share smaller sets of TFs and targets. Using D. melanogaster as an example, our regulog method determined 33 TFs, 621 targets, and 2936 regulatory connections (see Table 1). If the requirement of having the same binding sites is included, we were only able to determine 29 connections between 13 TFs and 5 target genes.
The results of the interolog and regulog mapping are recorded in an interolog/regulog database at http://genecensus.org/interactions/interolog/ (see Fig. 7). To find possible physical or regulatory interaction partners of one's favorite protein, the user simply inputs the names of the organism and the protein. For the protein–protein interolog database, all predicted interaction partners will be shown and ranked by JE. Our database also links each protein to an external Web resource such as SGD (Christie et al. 2004), WormBase (Harris et al. 2004), or FlyBase (The FlyBase Consortium 2002). For the regulog database, all predicted TFs and their targets are ranked by sequence homologies between query TFs and their yeast homologs. The layout of the Web page is similar to that of the interolog database.
Figure 7.

Screenshot of the interolog/regulog database.
Conclusion
In this study, we comprehensively assessed the transferability of protein–protein and protein–DNA interactions by analyzing the relationships between sequence similarity and interaction conservation. A total of 14,911 interactions in four organisms are included in our investigation. In general, the conservation of both interaction types shows a sigmoidal relationship with sequence similarity. For these four organisms, protein–protein interactions are well conserved between protein pairs with at least 80% JI or 10–70 JE. For protein–DNA interactions, the specific threshold of sequence identity is highly family-dependent. In general, 60% of TFs with 30% or more sequence identity share the same target sites.
Previously, Walhout et al. (2000) proposed an interolog concept to transfer protein–protein interactions across species. Here, we develop this concept into a concrete interaction prediction approach, the generalized interolog mapping method. This is readily expandable to any newly completed genomes. Using generalized interolog mapping method, we construct several genome-wide protein–protein interaction maps.
We further introduce a new regulog concept to map regulatory relationships between TFs and their targets across organisms. We apply the regulog mapping to produce genome-wide regulatory networks for several eukaryotic organisms. The results of the newly produced interaction maps and regulatory networks are stored in an interolog/regulog database.
Future Directions
There are several directions to extend this work. With respect to the conservation of protein–protein interactions, there are many more sequenced genomes without known genome-wide interaction networks. We will apply our method to these genomes to gain insight into their protein–protein interactions, and eventually to shed light on their functions. However, our analysis is still hampered by not having sufficient interaction data for other organisms. Once such large-scale interaction data sets are available, we can repeat our calculations taking into consideration the new information, which will give results with better statistical precision. For the regulog mapping method, we are unable to evaluate its performance at this time. When genome-wide regulatory net works are created in other organisms, we will evaluate the feasibility and accuracy of the regulog mapping method in a similar fashion to that of the protein–protein interolog mapping method.
Acknowledgments
The authors thank the referees for insightful comments that helped improve the manuscript. M.G. acknowledges support from the NIH grant 5P50GM062413.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1774904.
Footnotes
[Supplemental material is available online at www.genome.org. The interologs and regulogs mapped from yeast to other genomes are available online at http://interolog.gersteinlab.org.]
References
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Andrade, M.A. and Sander, C. 1997. Bioinformatics: From genome data to biological knowledge. Curr. Opin. Biotechnol. 8: 675–683. [DOI] [PubMed] [Google Scholar]
- Ashburner, M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M., Davis, A.P., Dolinski, K., Dwight, S.S., Eppig, J.T., et al. 2000. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood, T.K., Beck, M.E., Bleasby, A.J., Degtyarenko, K., Michie, A.D., and Parry-Smith, D.J. 1997. Novel developments with the PRINTS protein fingerprint database. Nucleic Acids Res. 25: 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader, G.D., Betel, D., and Hogue, C.W. 2003. BIND: The Biomolecular Interaction Network Database. Nucleic Acids Res. 31: 248–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch, A., Bucher, P., and Hofmann, K. 1996. The PROSITE database, its status in 1995. Nucleic Acids Res. 24: 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork, P., Ouzounis, C., and Sander, C. 1994. From genome sequences to protein function. Curr. Opin. Struct. Biol. 4: 393–403. [Google Scholar]
- Bork, P., Dandekar, T., Diaz-Lazcoz, Y., Eisenhaber, F., Huynen, M., and Yuan, Y. 1998. Predicting function: From genes to genomes and back. J. Mol. Biol. 283: 707–725. [DOI] [PubMed] [Google Scholar]
- Boulton, S.J., Gartner, A., Reboul, J., Vaglio, P., Dyson, N., Hill, D.E., and Vidal, M. 2002. Combined functional genomic maps of the C. elegans DNA damage response. Science 295: 127–131. [DOI] [PubMed] [Google Scholar]
- Brenner, S.E. 1999. Errors in genome annotation. Trends Genet. 15: 132–133. [DOI] [PubMed] [Google Scholar]
- Brenner, S.E., Chothia, C., and Hubbard, T.J. 1998. Assessing sequence comparison methods with reliable structurally identified distant evolutionary relationships. Proc. Natl. Acad. Sci. 95: 6073–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia, C. and Lesk, A.M. 1986. The relation between the divergence of sequence and structure in proteins. EMBO J. 5: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia, C. and Lesk, A.M. 1987. The evolution of protein structures. Cold Spring Harb. Symp. Quant. Biol. 52: 399–405. [DOI] [PubMed] [Google Scholar]
- Christie, K.R., Weng, S., Balakrishnan, R., Costanzo, M.C., Dolinski, K., Dwight, S.S., Engel, S.R., Feierbach, B., Fisk, D.G., Hirschman, J.E., et al. 2004. Saccharomyces Genome Database (SGD) provides tools to identify and analyze sequences from Saccharomyces cerevisiae and related sequences from other organisms. Nucleic Acids Res. 32: D311–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davy, A., Bello, P., Thierry-Mieg, N., Vaglio, P., Hitti, J., Doucette-Stamm, L., Thierry-Mieg, D., Reboul, J., Boulton, S., Walhout, A.J., et al. 2001. A protein–protein interaction map of the Caenorhabditis elegans 26S proteasome. EMBO Rep. 2: 821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, A.M., Kus, B., Jansen, R., Greenbaum, D., Greenblatt, J., and Gerstein, M. 2002. Bridging structural biology and genomics: Assessing protein interaction data with known complexes. Trends Genet. 18: 529–536. [DOI] [PubMed] [Google Scholar]
- The FlyBase Consortium. 2002. The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res. 30: 106–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, C.M., Gocayne, J.D., White, O., Adams, M.D., Clayton, R.A., Fleischmann, R.D., Bult, C.J., Kerlavage, A.R., Sutton, G., and Kelley, J.M. 1995. The minimal gene complement of Mycoplasma genitalium. Science 270: 397–403. [DOI] [PubMed] [Google Scholar]
- Fraser, C.M., Norris, S.J., Weinstock, G.M., White, O., Sutton, G.G., Dodson, R., Gwinn, M., Hickey, E.K., Clayton, R., Ketchum, K.A., et al. 1998. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science 281: 375–388. [DOI] [PubMed] [Google Scholar]
- Gavin, A.C., Bosche, M., Krause, R., Grandi, P., Marzioch, M., Bauer, A., Schultz, J., Rick, J.M., Michon, A.M., Cruciat, C.M., et al. 2002. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: 141–147. [DOI] [PubMed] [Google Scholar]
- Giot, L., Bader, J.S., Brouwer, C., Chaudhuri, A., Kuang, B., Li, Y., Hao, Y.L., Ooi, C.E., Godwin, B., Vitols, E., et al. 2003. A protein interaction map of Drosophila melanogaster. Science 302: 1727–1736. [DOI] [PubMed] [Google Scholar]
- Hahn, S. and Guarente, L. 1988. Yeast HAP2 and HAP3: Transcriptional activators in a heteromeric complex. Science 240: 317–321. [DOI] [PubMed] [Google Scholar]
- Harris, T.W., Chen, N., Cunningham, F., Tello-Ruiz, M., Antoshechkin, I., Bastiani, C., Bieri, T., Blasiar, D., Bradnam, K., Chan, J., et al. 2004. WormBase: A multi-species resource for nematode biology and genomics. Nucleic Acids Res. 32: D411–D417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi, H. and Gerstein, M. 2001. Annotation transfer for genomics: Measuring functional divergence in multi-domain proteins. Genome Res. 11: 1632–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, Y., Gruhler, A., Heilbut, A., Bader, G.D., Moore, L., Adams, S.L., Millar, A., Taylor, P., Bennett, K., Boutilier, K., et al. 2002. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415: 180–183. [DOI] [PubMed] [Google Scholar]
- Horak, C.E., Luscombe, N.M., Qian, J., Bertone, P., Piccirrillo, S., Gerstein, M., and Snyder, M. 2002. Complex transcriptional circuitry at the G1/S transition in Saccharomyces cerevisiae. Genes & Dev. 16: 3017–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, T., Tashiro, K., Muta, S., Ozawa, R., Chiba, T., Nishizawa, M., Yamamoto, K., Kuhara, S., and Sakaki, Y. 2000. Toward a protein–protein interaction map of the budding yeast: A comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc. Natl. Acad. Sci. 97: 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, V.R., Horak, C.E., Scafe, C.S., Botstein, D., Snyder, M., and Brown, P.O. 2001. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature 409: 533–538. [DOI] [PubMed] [Google Scholar]
- Jansen, R., Yu, H., Greenbaum, D., Kluger, Y., Krogan, N.J., Chung, S., Emili, A., Snyder, M., Greenblatt, J.F., and Gerstein, M. 2003. A Bayesian networks approach for predicting protein–protein interactions from genomic data. Science 302: 449–453. [DOI] [PubMed] [Google Scholar]
- Kumar, A. and Snyder, M. 2002. Protein complexes take the bait. Nature 415: 123–124. [DOI] [PubMed] [Google Scholar]
- Kumar, A., Agarwal, S., Heyman, J.A., Matson, S., Heidtman, M., Piccirillo, S., Umansky, L., Drawid, A., Jansen, R., Liu, Y., et al. 2002. Subcellular localization of the yeast proteome. Genes & Dev. 16: 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan, N., Jansen, R., and Gerstein, M. 2002. Toward a systematic definition of protein function that scales to the genome level: Defining function in terms of interactions. Proc. IEEE 90: 1848–1858. [Google Scholar]
- Lan, N., Montelione, G.T., and Gerstein, M. 2003. Ontologies for proteomics: Towards a systematic definition of structure and function that scales to the genome level. Curr. Opin. Chem. Biol. 7: 44–54. [DOI] [PubMed] [Google Scholar]
- Larsen, C.N. and Finley, D. 1997. Protein translocation channels in the proteasome and other proteases. Cell 91: 431–434. [DOI] [PubMed] [Google Scholar]
- Lee, T.I., Rinaldi, N.J., Robert, F., Odom, D.T., Bar-Joseph, Z., Gerber, G.K., Hannett, N.M., Harbison, C.T., Thompson, C.M., Simon, I., et al. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298: 799–804. [DOI] [PubMed] [Google Scholar]
- Li, S., Armstrong, C.M., Bertin, N., Ge, H., Milstein, S., Boxem, M., Vidalain, P.O., Han, J.D., Chesneau, A., Hao, T., et al. 2004. A map of the interactome network of the metazoan C. elegans. Science 303: 540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbach, K.J. and Wu, R. 1985. Characterization of two Drosophila melanogaster cytochrome c genes and their transcripts. Nucleic Acids Res. 13: 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe, N.M. and Thornton, J.M. 2002. Protein–DNA interactions: Amino acid conservation and the effects of mutations on binding specificity. J. Mol. Biol. 320: 991–1009. [DOI] [PubMed] [Google Scholar]
- Marcotte, E.M., Pellegrini, M., Thompson, M.J., Yeates, T.O., and Eisenberg, D. 1999. A combined algorithm for genome-wide prediction of protein function. Nature 402: 83–86. [DOI] [PubMed] [Google Scholar]
- Matthews, L.R., Vaglio, P., Reboul, J., Ge, H., Davis, B.P., Garrels, J., Vincent, S., and Vidal, M. 2001. Identification of potential interaction networks using sequence-based searches for conserved protein–protein interactions or “interologs.” Genome Res. 11: 2120–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewes, H.W., Frishman, D., Gruber, C., Geier, B., Haase, D., Kaps, A., Lemcke, K., Mannhaupt, G., Pfeiffer, F., Schuller, C., et al. 2000. MIPS: A database for genomes and protein sequences. Nucleic Acids Res. 28: 37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen, J., Hahn, S., and Guarente, L. 1987. Yeast HAP2 and HAP3 activators both bind to the CYC1 upstream activation site, UAS2, in an interdependent manner. Cell 51: 953–961. [DOI] [PubMed] [Google Scholar]
- Pearson, W.R. and Lipman, D.J. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. 85: 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini, M., Marcotte, E.M., Thompson, M.J., Eisenberg, D., and Yeates, T.O. 1999. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc. Natl. Acad. Sci. 96: 4285–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posas, F., Takekawa, M., and Saito, H. 1998. Signal transduction by MAP kinase cascades in budding yeast. Curr. Opin. Microbiol. 1: 175–182. [DOI] [PubMed] [Google Scholar]
- Rain, J.C., Selig, L., De Reuse, H., Battaglia, V., Reverdy, C., Simon, S., Lenzen, G., Petel, F., Wojcik, J., Schachter, V., et al. 2001. The protein–protein interaction map of Helicobacter pylori. Nature 409: 211–215. [DOI] [PubMed] [Google Scholar]
- Tatusov, R.L., Koonin, E.V., and Lipman, D.J. 1997. A genomic perspective on protein families. Science 278: 631–637. [DOI] [PubMed] [Google Scholar]
- Uetz, P., Giot, L., Cagney, G., Mansfield, T.A., Judson, R.S., Knight, J.R., Lockshon, D., Narayan, V., Srinivasan, M., Pochart, P., et al. 2000. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 403: 623–627. [DOI] [PubMed] [Google Scholar]
- von Mering, C., Krause, R., Snel, B., Cornell, M., Oliver, S.G., Fields, S., and Bork, P. 2002. Comparative assessment of large-scale data sets of protein–protein interactions. Nature 417: 399–403. [DOI] [PubMed] [Google Scholar]
- Walhout, A.J., Sordella, R., Lu, X., Hartley, J.L., Temple, G.F., Brasch, M.A., Thierry-Mieg, N., and Vidal, M. 2000. Protein interaction mapping in C. elegans using proteins involved in vulval development. Science 287: 116–122. [DOI] [PubMed] [Google Scholar]
- Webb, E.C. 1992. Enzyme Nomenclature 1992, Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology. Academic Press, New York.
- Westbrook, J., Feng, Z., Chen, L., Yang, H., and Berman, H.M. 2003. The Protein Data Bank and structural genomics. Nucleic Acids Res. 31: 489–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, C.A., Kreychman, J., and Gerstein, M. 2000. Assessing annotation transfer for genomics: Quantifying the relations between protein sequence, structure and function through traditional and probabilistic scores. J. Mol. Biol. 297: 233–249. [DOI] [PubMed] [Google Scholar]
- Wingender, E., Chen, X., Fricke, E., Geffers, R., Hehl, R., Liebich, I., Krull, M., Matys, V., Michael, H., Ohnhauser, R., et al. 2001. The TRANSFAC system on gene expression regulation. Nucleic Acids Res. 29: 281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xenarios, I., Salwinski, L., Duan, X.J., Higney, P., Kim, S.M., and Eisenberg, D. 2002. DIP, the Database of Interacting Proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 30: 303–305. [DOI] [PMC free article] [PubMed] [Google Scholar]