

Abstract
We present an EST sequence assembler that specializes in reconstruction of pristine mRNA transcripts, while at the same time detecting and classifying single nucleotide polymorphisms (SNPs) occuring in different variations thereof. The assembler uses iterative multipass strategies centered on high-confidence regions within sequences and has a fallback strategy for using low-confidence regions when needed. It features special functions to assemble high numbers of highly similar sequences without prior masking, an automatic editor that edits and analyzes alignments by inspecting the underlying traces, and detection and classification of sequence properties like SNPs with a high specificity and a sensitivity down to one mutation per sequence. In addition, it includes possibilities to use incorrectly preprocessed sequences, routines to make use of additional sequencing information such as base-error probabilities, template insert sizes, strain information, etc., and functions to detect and resolve possible misassemblies. The assembler is routinely used for such various tasks as mutation detection in different cell types, similarity analysis of transcripts between organisms, and pristine assembly of sequences from various sources for oligo design in clinical microarray experiments.
On the way to understand the function of all genes of an organism, it is now clear that the genome sequence alone may be not enough, especially if the organism shows a high degree of complexity. Analysis of the genome must be supported by efforts on understanding its transcription—the transcriptome—occurring in cells. Citing Camargo et al. (2001), the “most definitive approach to the elucidation of transcripts remains their direct sequencing.” This corresponds with earlier findings of Bonfield et al. (1998), who concluded that “direct sequencing is required to define the precise location and nature of any [mutation] change”, as this method ensures the highest reliability and quality regarding the definition of single nucleotide polymorphisms (SNPs).
Several approaches have been proposed to assemble ESTs and detect SNPs in the resulting alignments, among these are TRACE-DIFF by Bonfield et al. (1998), polyphred by Nickerson et al. (2000), pta and AGENT by Paracel, Inc. (Paracel 2002b), the TGICL system developed by Pertea et al. (2003), and autoSNP by Barker et al. (2003). The most significant shortcoming common to all of these methods is the fact that they determine potential SNP positions from assemblies that align all available sequences together, regardless of whether they contain differing SNP positions or originate from different sources such as, for example, organisms, strains, cell types, etc. Unfortunately, the intrinsic properties of alignment algorithms can, and do lead to misassemblies, especially when the sequences involved are highly similar. This, in turn, leads to wrongly assembled transcripts, and these can cause false or nonexistent proteins to be predicted as is shown in Figure 1. As a side effect, nonexistent SNP positions are also generated.
Figure 1.

Example of a misassembled transcript when SNPs are disregarded. Assembly of three input sequences are shown at left; the resulting transcripts of this assembly are shown at right. The three sequences s1, s1*, and s2 contain different homologous parts, represented by the different shades of gray, and exactly one SNP position. A normal assembly algorithm will assemble first s1, then s2 (because of the long overlapping alignment in the white part), and then might try to align s1*, but fail because of the large mismatch. The SNP position with G in sequence s1 and A in s2 is treated as typical noise in the alignment algorithms and ignored. The resulting transcript sequences are therefore wrong, as they do not represent the sequences found in vivo: t1 is a mix of two transcripts and does not code a true protein.
To address these problems, the method we have devised and implemented, the miraEST assembler, consists of an iterative multiple-pass system that focuses on observed data. The key processing steps first assemble the sequences gained by EST clone analysis into pristine transcripts by performing SNP detection during the assembly and then classifying the SNPs into types. Therefore, information about potential SNP sites detected de novo within the ongoing assembly process is used, as well as possibly supplied additional information for each sequence-like known SNPs, known motifs, or strain (or cell or organism) types. The importance of detecting SNPs in an assembly process can be seen by comparing Figure 1 with Figure 2, in which we show how ignored SNP positions can lead to wrongly assembled transcripts in one example and how honored SNP information leads to a correct transcript assembly in the second example.
Figure 2.

The same example as in Figure 1, but in this example, the assembly algorithm honors SNP positions that were detected during earlier iterations of the assembly process. The alignment between s1 and s2 will, therefore, not be made, as there is a mismatch at the SNP position, even with the long overlap between both sequences. Instead, the assembler will align s1 and s1*, as they do not contain mismatches at SNP positions. The result is a correct representation of the transcriptome.
A key element of miraEST is an integrated automatic assembly editor that uses trace files (if provided) to correct alignment mismatches produced by base-calling errors in the sequences. This feature greatly improves the quality of assemblies and facilitates automatic differentiation of SNPs from base-calling problems. In contrast to the TGICL system and autoSNP, which both use only redundancy information, and pta that uses only quality and template information, the miraEST assembly and SNP detection algorithms use a combination of base-calling error probabilities, trace file analysis, template information, and redundancy analysis. The result is a number of—sometimes partial—real mRNA transcripts, which were present in the clone libraries and consecutively sequenced.
It is important to note that miraEST is an assembler and not a clustering tool, that is, it is used routinely to reconstruct the pristine mRNA transcript sequences gathered in EST sequencing projects, which can be a reliable basis for subsequent analysis steps like clustering or exon analysis. This means that even genes that contain only one transcripted SNP on different alleles are first treated as different transcripts. However, the optional last step of the assembly process can be configured as a simple clusterer that can assemble transcripts containing the same exon sequence, but only differ in SNP positions, into one consensus sequence. Such SNPs can then be analyzed, classified, and reliably assigned to their corresponding mRNA transcriptome sequence.
RESULTS
In three very different projects we present our approach to achieving an accurate assembly and subsequent SNP scanning of transcript sequences with the miraEST assembler. The non-normalized libraries contain ESTs sequenced from the plant Vitis vinifera Linnaeus (Plantae: Spermatophyta: Rosopsida/Dicotyledoneae), and two animal taxa, the sponge Suberites domuncula Olivi (Metazoa: Porifera: Demospongiae), and the vertebrate Canis lupus familiaris Linnaeus (Metazoa: Chordata: Vertebrata).
Although these three multicellular organisms are eukaryotes, they are only distantly related. In general, plants split off first from the common ancestor, ∼1000 million years ago (Mya). Later, the Metazoa evolved, (700 Mya) with Porifera as the oldest still extant phylum, and finally the Chordata appeared (500 Mya; for review, see Kumar and Rzhetsky 1996; Müller 2001). Until recently, the Porifera were an enigmatic taxon (see Müller 2001). Only the analyses of the molecular sequences from sponges, both cDNA and genomic ones, gave strong evidence that all metazoan phyla originated from one ancestor. Therefore, ESTs from this taxon were included in this study in order to obtain a first estimation about the abundance of particular genes in such a collection.
The assembled ESTs from the S. domuncula (sponge) should allow a further elucidation of the evolutionary novelties that emerged during the transition from the fungi to the Metazoa. Likewise, the data gathered here from the V. vinifera (grapevine) and the mammal C. lupus familiaris (dog) should provide an understanding of the change of gene pool in organisms under domestication. Whereas the dog and sponge project had only ESTs sequenced from one strain (respectively, cell type), the grapevine project had ESTs that were collected from a multitude of cell types, ranging from root cells to berry cells. Table 1 shows an overview of these projects together with some of the more interesting statistics of the assembly.
Table 1.
Summary of Results From EST Assembly of Sponge, Dog, and Grapevine Sequences
| Sponge | Dog | Grapevine | |
|---|---|---|---|
| Input sequences | 9747 | 10,863 | 32,776 |
| Strains/cell types | 1 | 1 | 10 |
| Step 1: transcript SNP separation assembly | |||
| Total transcripts | 4401 | 5921 | 12,380 |
| thereof singlets | 3151 | 4204 | 7904 |
| thereof contigs | 1250 | 1717 | 4476 |
| Max cov/occurred | 145/1 | 106/1 | 812/1 |
| Min cov/occurred | 2/637 | 2/885 | 2/2143 |
| Total transcript len. | 3,342,596 | 3,941,124 | 7,082,719 |
| Step 3: transcript SNP classification assembly | |||
| Total unified transcr. | 4077 | 5901 | 8547 |
| thereof singlets | 3780 | 5811 | 6131 |
| thereof contigs | 297 | 90 | 2416 |
| thereof with SNPs | 285 | 81 | 2103 |
| Total transcript len. | 3,120,847 | 3,897,635 | 4,872,333 |
| Transcript SNP types | |||
| Intra strain/cell | 2158 | 461 | 959 |
| Inter strain/cell | — | — | 1505 |
| Intra and Inter s./c. | — | — | 7221 |
| Total SNP sites | 4653 | 927 | 9685 |
Step 1: result sequences are transcripts separated by SNPs, but not by strain. The number of contigs, the classification numbers on maximum and minimum coverage (and the times they occurred) within the contigs as well as the number of singlets, give a rough idea about the asymmetrical distributions of EST reads in the different contigs.
Step 3: `assembly of pristine mRNA transcripts' to analyze SNP sites and types. The transcripts' sequences gained there can be seen as a consensus of the (hopefully) pristine transcripts gained in the previous steps of the assembly. Classification of SNPs (see also the subsection of the same name in the Methods section) is also performed in this step: Intra means that SNPs occur only with a strain or cell type, SNPs of type Inter occur only when comparing different strains or cell types, and the Intra and Inter SNP type is a combination of the first two types. Intermediary results from step 2 are not shown, as sponge and dog do not use this step, and the grapevine results are too extensive.
Depending on the projects, sequences we used were obtained by capillary electrophoresis on ABI 3100 or ABI 3700 machines, with each project having specific sequencing vectors. For this study, all project sequences were preprocessed and cleaned using standard computational methods; we used TraceTuner 2.0.1 (Paracel, Inc.) for extracting the bases. Data sets were cleaned by using PFP as described in Paracel (2002a), masking of known sequencing vectors, filtering against contaminant vectors present in the UniVec core database, filtering of possible Escherichia coli, and other bacterial contamination and masking of poly(A)/poly(T) tails in sequences. Repeats and known standard motifs were not masked, as these are integral parts of the data and contain valuable information. Sequences that were shorter than 80 bases were removed from the projects. The remaining sequences used in the three projects total 53,386 sequences with 54,303,071 bases.
For each project, the miraEST assembler's integrated standard parameter set was used. This set is configured as a three-pass assembly:
Classification of the sequences by SNP type using all sequences from all strains/cell types, etc. The motivation for performing a first pass that separates only by SNP and not also directly by strain/cell type is the simple observation in which the assembler still can find useful SNP on rarely expressed genes when looking at the entirety of the available data within alignments. Interesting sequence features found in this first pass are valuable for the two subsequent passes in which the algorithms will benefit from them.
Additional step if strain information is available, separation of the sequences by strain (resp. cell type) and SNP. This results in clean mRNA transcript sequences that represent the actual state of the transcriptome of a strain/cell type as it is present in the clone library. Although the results of this step are interesting on their own, their major importance is the fact that they are used as pristine input for the following third pass.
Production of a combined SNP-strain assembly. If strain information was available, this step uses results from step 2, or else from step 1. The result of this assembly has the exact SNP positions and types tagged in the mRNA transcript sequences that form an alignment of the resulting consensus.
Each pass had a standard set of options activated to enhance the preprocessed reads by trimming for quality, unifying areas of masked bases at read-ends, clipping sequencing vector relicts, and tagging remaining poly(A)/poly(T) stretches in sequences (see Data preprocessing in the Methods section for more detail). Trace data was used in the assembly to edit base-calling errors in sequences and assess bases and possible SNP sites when available. Table 2 shows computer requirements in conjunction with project complexity aspects.
Table 2.
Runtime and Memory Consumption of the Study Projects Using an Intel 2.4 GHz Xeon P4/HT PC With 512 K L2 Cache and 2 G RDRAM
| Sponge | Dog | Grapevine | |
|---|---|---|---|
| Peak memory usage | 250 M | 280 M | 1.7 G |
| Runtime in minutes | |||
| Step 1 | 27 | 14 | 735 |
| Step 2 | 20 | 10 | 101 |
| Step 3 | 3 | 4 | 35 |
| Total | 137 | 69 | 871 |
| Number of contig reassemblies | |||
| Step 1 | 577 | 250 | 3827 |
| Step 2 | 51 | 18 | 1927 |
| Step 3 | 0 | 0 | 0 |
| Total reassemblies | 628 | 268 | 5754 |
Comparison of the sponge and dog project, which have roughly the same number of sequences showing a clear relationship between the runtime and the number of detected contig reassemblies (which were triggered by newly detected SNP sites).
The reduced runtime from step 1 to step 2 is due to potentially problematic regions with SNP sites that were detected in the first step. These SNPs give additional information to the second step, which then prevents misassemblies that involve those sites. Hence, the lower number of reassemblies reduced runtime.
In general, step 3 has less transcript sequences to assemble than step 1 and step 2, also leading to reduced runtimes.
Comparing the projects led to some interesting insights both on the behavior of miraEST and on the data itself. Although the sponge and the dog projects have about the same numbers of sequences used as input (9747 vs. 10,863), the assembly runtimes of the sponge project took about twice as long to complete than the dog project. When analyzing log files and intermediary results from both projects, we found that there are two main causes for this behavior:
The more assembled transcript contigs contain SNPs, the more the assembler will have to break those up and reassemble them in steps 1 and 2, leading to higher assembly times.
The more similar sequences from one or several gene families are present, the higher is the probability for an increased number of iterations needed to get the transcripts assembled cleanly.
Both of these factors can be seen as predominant indicators for the complexity of a project. The sequences of the sponge project contain 285 mRNA transcript contigs (7.0% of the transcripts) with SNPs. These total 2158 SNP sites, which is ∼7.5 SNPs per mRNA transcript that contains SNPs. The sequences of the dog project, however, lead to only 81 mRNA transcript contigs (1.4% of the transcripts) with SNPs. These total only 461 SNP sites, which is ∼5.7 SNPs per mRNA transcript that contains SNPs. The sequenced sponge EST sequences, therefore, not only contain more transcripts with polymorphisms than the dog sequences, they generally also contain more SNPs per transcript.
Comparing the grapevine project with the two other projects also yielded some interesting discoveries. First, the contig with the maximum coverage that occurred in step 1 contained 812 reads compared with 145 for the sponge and 106 for the dog. The grapevine data also contained several additional high-coverage contigs, which meant that this project contained a number of genes or gene families that were, in absolute numbers, more expressed, and thus sequenced, than in the dog and sponge project. The second interesting discovery was the decrease in total transcripts from step 1 to step 3; the sponge project had a 7.4% reduction (from 4401 clean transcripts to 4077 unified transcript consensi), and the dog only 0.3% (from 5921 to 5901), but the grapevine project had a 31% reduction (from 12,380 down to 8547) in the number of transcripts. This meant that many gene transcripts of the grapevine project differed only in a few SNP bases and were assembled together in step 3, forming transcript consensi that allowed the classification of SNPs whether they occur within a cell type, between different cell types, or both. On the other hand, the 9685 SNPs found were dispersed over 2103 transcripts, which is ∼4.6 SNPs per transcript containing SNPs and, therefore, less than the sponge or even the dog project.
The exact reason for these high transcript redundancy numbers in this project is currently under investigation, but preliminary results indicated that a significant number of almost identical common basic housekeeping genes are expressed and were sequenced in different cell types, and that several of them contain SNPs. For example, we found a transcript family in 9 of 10 cell types formed by 147 Metallothionein transcripts with no less than 98 positively identified SNP sites over a length of 650 bases. The SNPs are in the coding region and the 3′ UTR, with many of the SNPs leading to a mutation in the amino acid sequence of the protein.
DISCUSSION
We have developed miraEST concurrently to the mira assembler for genome sequences presented in Chevreux et al. (1999, 2000), which enables us to use basic algorithms for both branches of the assembly system. This has also allowed us to concentrate on developing and improving those algorithms that are specifically needed to tackle the slightly different assembly problems of genome and EST sequences once the basic facilities were in place.
We discovered very early in the development process that using high-quality sequence data first in the assembly process was a very viable way to proceed, as it substantially reduced computing time. This permitted us to reinvest this saved time into other algorithms that increased the actual quality of the final results – resolving detected misassembly conflicts, analysis and detection of previously unknown relevant sequence features (e.g., SNPs), and detection and elimination of conflicts caused by misassemblies. The ever-increasing computing power permitted the design of exact iterative algorithms instead of relying on makeshift algorithms when assembly problems occurred. That is, we clearly chose not to trade off quality for speed when the loss in quality was deemed to be substantial. Furthermore, we think that integrating an automated trace editor into the assembly process was the correct choice, as results showed that spurious base-calling errors are reliably detected and removed in an alignment, and the assembler can also use the trace analysis routines to perform in-depth and multilevel analysis on problematic regions in alignments.
To our knowledge no other assembly system, be it for genomic or transcript data, contains a comparable mix of algorithms that enables the assembler to dependably detect by itself and use the information about special base positions that differentiate between repetitive stretches within sequences, as is the case for SNP bases in EST assemblies or repetitive elements in genome assemblies. We would like to reiterate our stance regarding the importance of discovering such base positions during the assembly; they allow our assembler to perform a reliable separation of almost identical sequences, which may ultimately differ only in one single position within two single sequences, into their true original transcriptome. This is significantly more sensitive and specific than other methods such as the one presented by Tammi et al. (2002), which needs at least two differences in reads to distinguish them from sequencing errors.
Additionally, corrections performed by the integrated automatic editor resolve errors in alignments produced by base-calling problems. This makes SNP detection much less vulnerable to sequence-specific electrophoresis glitches and base-calling errors, as is the case for, for example, the AG-problem known with the ABI 373 and 377 machines, where a G preceeded by an A is often unincisive or only weakly pronounced.
In contrast to other transcript assemblers or SNP detection programs, like pta, the TGICL system, polyphred, or autoSNP, the approach we devised uses strict separation of sequences by SNP bases according to their respective mRNA transcript. Ultimately, this is the only way to ensure that the transcripts' sequences produced as a result correspond to the real transcriptome sequence. Our method permits us to use these results directly for the design of further investigative studies with high-quality and precision requirements such as, for example, the design of oligo probes for specific SNP detection in clinical microarray hybridization screening experiments.
The possibility to export the assembled projects together with the analysis of SNP sites to a variety of standard formats, for example, gap4-directed assembly or phrap.ace, opens the door to visual inspection of the results, as well as integrating the tool into more complex and semiautomated-to-automated laboratory workflows.
Our goal was to show that the combination of those methods and algorithms leads to a system that accomplishes the given task of reconstructing a transcriptome from sequenced ESTs in such a way that the detection, analysis, and classification of SNPs prevents grave misassemblies that occur in other systems. However, use of miraEST assembler on a daily basis in production environments shows that some algorithms still need a form of fine tuning.
In the future, our primary focus will shift to enable parallel execution in portions of the algorithms, in order to take advantage of multiple processor architectures. Until now, the program uses only one processor on a given machine, and this clearly represents a bottleneck when several hundreds of thousands or even millions of ESTs are to be assembled. Fortunately, most of the methods presented can be parallelized using a divide-and-conquer strategy, so that distributing the work load across different threads, processes, and even machines is one of the targets we are currently pursuing. Another point we are looking into is that usage of the C++ standard template library (STL) currently leads to unexpected high memory consumption in some parts of the algorithms. We traced this back to memory-pooling strategies of the STL. First experiments with a combination of adapted algorithms together with better behavior prediction (data not shown) lead to a significant reduction of these side-effects.
METHODS
Assembly Strategies
The extensively studied reconstruction of the unknown, correct contiguous nucleic acid sequence by inferring it through the help of a number of fragments is called the assembly problem. The devil is in the details; however, errors in base sequences gained by electrophoresis, combined with the sometimes exacerbating fact that mRNA tends to contain highly repetitive stretches with only very few bases differing across different locations, impedes the assembly process in an awesome way and leads to the necessity of using fault-tolerant and alternatives-seeking algorithms that can cope with the sometimes extreme high coverage data that results from using non-normalized EST clone libraries.
Referring to Dear et al. (1998), a “sequence assembly is essentially a set of contigs, each contig being a multiple alignment of reads.” In the case of EST assembly, each of these contigs8 is a mRNA transcript of a few hundred to thousands base-pairs long. Each contig can, depending of the clone library type and the frequency with which this transcript was sequenced, consists of two to a few hundreds or even thousands of fragments.
A number of different strategies have been proposed to tackle the assembly problem, ranging from simple greedy pairwise alignments, sometimes using additional information (Peltola et al. 1984), using a whole set of refinements (Huang 1996; Paracel 2002b), performing coverage analysis (Kececioglu and Myers 1992)—to weak AI methods like genetic algorithms (Parsons et al. 1993; Notredame and Higgins 1996; Zhang and Wong 1997). Most of these deal with genomic sequence assembly, but the underlying problem is similar enough to be applied to EST sequence assembly.
A common characteristic of all existing assemblers is that they rely on the quality values with which the bases have been attributed by a base caller. Within this process, an error probability is computed by the base caller to express the confidence with which the called base is thought to be the true base. Although methods for storing alternative base calls and other data have been suggested (Allex et al. 1996; Walther et al. 2001), this approach produces results that are simple to handle and apparently “good enough,” so that it has imposed itself as standard over the years. The positive aspect is the possibility for assemblers to decide in favor of the best, most probable bases when a discrepancy occurs. The negative aspects of currently used base callers is their inability to write confidence values for optional, uncalled bases at the same place.
The miraEST assembler is a purpose-built modification for EST assembly and SNP detection that is concurrently developed with our mira genome assembler that we presented in Chevreux et al. (1999, 2000). The modification combines and substantially extends the strengths of approaches mentioned above and copies assembly analysis and SNP detection strategies done by human experts.
We used four criteria when designing the assembler:
Insuring the quality aspect of the final result. By making cautious use of the available data, the assembler will start with high-confidence regions (HCR) in the nucleic acid sequence to ensure a firm base and good basic building blocks. Low confidence regions (LCR) of the sequences can be automatically used later on if needed.
Using additional information like quality values, known SNP sites, repeat stretches, template insert sizes, etc., as checking mechanisms to confirm the basic alignments.
Implementing discovery and usage within the assembly process of previously unknown facts such as, for example, new SNP sites, by analyzing the available information within the whole assembly context and not only on a sequence-by-sequence basis.
Resolving misassembly conflicts, which can always happen, by using the information about the misassemblies and the sequences that caused it, to prevent the same errors in future iterations.
Another important approach is that we combined the assembler with capabilities of an automatic editor. Both the assembler and the automatic editor are separate programs and can run separately, but we view the task of assembly, assembly validation, and finishing to be closely related enough for both parts to include routines from each other (see also Chevreux et al. 1999; Pfisterer and Wetter 1999). In this symbiosis, the signal-analysis-aided assembler acquires two substantial advantages, compared with a sequential-base-caller-and-assembler strategy:
The assembler gains the ability to perform signal analysis on partially assembled data. Analyzing trace data at precise points with a given hypothesis in mind9 helps to discern possible base-caller errors from errors due to misassemblies or errors due to SNPs, where simple base-error probabilities alone could not help.
During the assembly process, reads in temporarily finished contigs can be automatically edited to increase their quality if the traces of the alignment support the hypotheses of an error that occurred in the base-calling step at that position. The edited reads, in turn, can be used to increase assembly quality in the ongoing assembly process.
Alignment Optimality
Different authors have proposed different sets of acceptance criteria for the optimality of an alignment (see Chan et al. 1992). Traditionally, “the objective of this (assembly) problem has been to produce the shortest string that contains all of the fragments as substrings, but in case of repetitive target sequences, this objective produces answers that are overcompressed” (Myers 1995). In the case of EST clone library assembly, however, and especially for non-normalized libraries, certain genes get expressed more often than others (e.g., cytochromes), resulting in more mRNA clones, and thus, sequence fragments of these sequences. This means that the overcompression criterion cannot be used to track misassembled transcripts.
As a result of this, we conceived the strategy of the least number of unexplainable errors not supported by signal values present in an alignment resp. assembly to be optimal. That is, EST sequences get assembled together as long as the bases and their signals support the assembly without clashes. We worked out a multiphased concept to have our assembler perform the task of sequence alignments (see Fig. 3). In the following sections, we describe each phase of the assembly process and its interaction with other phases, giving a short overview on the algorithms and their expected results, without going into algorithmic details, as this would go beyond the scope of this article
Figure 3.

The multipass and iterative nature of the assembler becomes clear as in this schematic diagram of the phases of a miraEST assembly. Previously unknown information (like possible SNP sites) can be discovered and taken into account throughout all of the assembly stages. Solid arrows show imperative pathways, dashed arrows denote optional pathways that may or may be not taken, depending on assembly parameter values and the actual data.
Data Preprocessing
Strictly speaking, data preprocessing does not belong to the actual assembler, as almost every laboratory has its own means of defining good quality within reads and already uses existing programs to perform this task.10 But as this preprocessing step directly influences the quality of the results obtained during the assembly, defining the scope of the expected data is desirable. Moreover, it can explain strategies implemented to eventually handle incorrectly preprocessed data.
The most important part in the sequenced fragments (apart from the target sequence itself) is the sequencing vector data, which will invariably be found at the start of each read, and sometimes, for short inserts, at the end. These parts of any cloned sequence must be marked or removed from the assembly. In analogy to the terms used in the GAP4 package, we will refer to marked or removed parts as hidden data (Staden et al. 1997), other terms frequently used are masked out or clipped data.
Errors occurring during the base-calling step or simply quality problems with a clone can lead to more or less spurious errors occurring in the gained sequences. These, in turn, sometimes interfere with the ability of preprocessing programs to correctly recognize and clip the offending sequence parts. Therefore, the miraEST assembler incorporates a number of routines across all steps of the assembly that saves sequences that were incorrectly preprocessed. Although we give a brief algorithmical overview of implemented methods within the scope of this section, please refer to the documentation available on the project homepage (http://www.chevreux.org/projects_mira.html) for a full description of all available options. The routines that we implemented and that can be used by the assembler are:
Standard quality clipping routines: Clipping is done with a modified sliding-window approach known from literature as in Staden et al. (1997) and Chou and Holmes (2001), where a window of a defined length l is slided across the sequence until the average of the quality values attains a threshold t. Usual values for this procedure are l = 30 and t = 20 when using quality values in phred style. An additional backtracking step is implemented to search for the optimal cutoff point within the window once the stop criterion has been reached, discarding bases with quality values below the threshold. This is performed from both sides of the sequences.
Pooling masked areas at sequence tails: Parts of sequences that were masked (X'ed out) by other preprocessing programs sometimes contain small areas between 1 and 30 nucleotides of nonmasked characters within the masked area due to, for example, low-quality data or the usage of slightly differing sequencing vectors. If requested, the assembler will unify the masked areas into more homogeneous masking when the nonmasked sections do not exceed a given length.
Clipping of sequencing vector relicts (while differentiating them from possible splice variants): This is done by generating hit/miss histograms of all sequence alignments. Whereas the good-quality middle parts will have a high hit/miss ratio within a sequence histogram, vector leftovers at the ends will have a reversed ratio. The beginning/end of such vector fractions is marked by a relatively sharp change, a cliff, which can easily be detected. Unfortunately, different splice variants of eukaryotic genes present the same effects within histograms, so that hit/miss ratio changes are searched for only within a given window at the start and end of the good sequence parts (usually between 1 and 20 bases) to only catch such vector relicts present there.
Uncovering and tagging of poly(A) and poly(T) bases at sequence ends: Unlike other specialized transcript assemblers like pta (Paracel 2002b), our algorithms differentiate between different splice variants present in an assembly and must include poly(A)/poly(T) bases when aligning sequences. The assembler will recover those areas by comparing masked sequences with the original counterpart and uncover exactly the poly(A/T) stretches present at the end of the sequences by a simple base-by-base comparison algorithm. These stretches will furthermore be tagged with assembly internal meta information to help the algorithms in the splice detection task.
A HCR of bases within every read is selected through quality clipping as an anchor point for the next phase. Existing base callers (ABI, PHRED, TraceTuner, and others) detect bases and rate their quality quite accurately and keep increasing in their performance, but bases in a called sequence always remain afflicted by increasing uncertainty toward the ends of a read. This additional information, potentially worthful, can nevertheless constitute an impeding moment in the early phases of an assembly process, bringing in too much noise. It is therefore marked as LCR for cautious use in the assembly process.
The following shows the information the assembler will work with, any of which can be left out (except sequence and vector clippings), but will reduce the efficiency of the assembler: (1) the initial trace data, representing the gel electrophoresis signal; (2) the called nucleic acid sequence; (3) position-specific confidence values for the called bases of the nucleic acid sequence; (4) a stretch in each sequence marked as HCR; (5) general properties such as direction of the clone read and name of the sequencing template, etc.; (6) special sequence properties in different regions of a read (such as sequencing vector, known standard repeat sequence, and known SNP sites, etc.) that have been tagged or marked.
Read Scanning
A common start for an assembly is to compare every read with every other read (and its reversed complement) using a fast and fault-tolerant algorithm to detect potential overlaps. We developed and tested several different algorithms to perform this task, retaining a combination of two, SKIM and DNASAND.
The extremely fast SKIM algorithm develops ideas used by Grillo et al. (1996) to find equal subsequences by hashing and allowing for errors. More specifically, SKIM computes the relative distances for every oligo of 8 nucleotides by creating distance histograms where high peaks represent long common subsequences containing equal oligos at the same relative distance, which makes this algorithm have both a high sensitivity and specificity. The DNASAND algorithm is a modified Shift-AND text search algorithm introduced by Wu and Manber (1992), which extended the ideas of Baeza-Yates and Gonnet (1992). It is particularly useful for finding short overlaps at read ends that are riddled with errors caused by low-quality traces and problems in the base-calling process. See also Gusfield (1997) for more information on these.
Both fast-scanning algorithms do not only specify the overall type of global relationship of two sequences (total correspondence, containment, and overlapping; see Huang 1994) and the approximate degree of similarity, but they also return the approximate offset of the alignment. As a result of this first scan, a table containing information on potential overlaps of all of the fragments is generated. The direction of the potential overlap (forward-forward or forward-complement) and the approximate offset is also stored.
Systematic Match Inspection
In the next step, potential overlaps found during the scanning phase are examined with a banded Smith-Waterman-based algorithm for local alignment of overlaps. Our Smith-Waterman algorithm implementation, which runs in almost linear time and space, takes into account that, regarding the fact that today's base callers have a low error rate, the block-indel model (Giegerich and Wheeler 1996) does not apply to the alignment of EST-sequencing data (although the block-indel model can be a great help when detecting different splice forms of genes, but this is not the task of an assembler). This means that long stretches of mismatches or gaps in the alignment are less probable than small, punctual errors. We also assume a mismatch of a base against an N (symbol for an Ny base) to have no penalty, as in many cases the base caller rightfully set N for a real existing base (and not an erroneous extra one) that could not be further resolved.
Scoring
Two different scores can be computed using different Smith-Waterman weight matrices and some alignment score post-processing; the expected score Se and the computed score Sc of the overlap are calculated for each overlap of two sequences, with Sc always ≤ Se. A score is computed as follows: assume two aligned sequences to be s1 and s2 in an alignment A with the length ∥ A∥, so that A(s1(i)) and A(s2(i)) give the bases b1 and b2 of sequences the s1 and s2, respectively, at the position i of the alignment. Furthermore, assume a comparison matrix W to contain all comparison scores for two bases, so that W(b1, b2) gives the comparison score of the two bases b1 and b2. The score computing function now summarizes the comparison scores given in a matrix W of the bases at each position of the alignment:
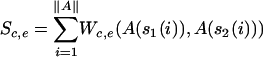 |
The difference for Sc and Se lies in the usage of different matrices; whereas the matrix for Sc uses a scoring scheme similar to (1, –1, –2) for match, mismatch, and gap, Se uses a matrix with a scheme similar to (1, 1, 1), as in a perfect alignment, all the bases of the overlap of two sequences are expected to be equal. It is now trivial to make a rough guess of the alignment quality of the overlap by calculating the score ratio Rs of the computed score compared with the expected score:
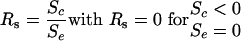 |
and therefore 0 ≤ Rs ≤ 1.
A score ratio of 0 shows that the two sequences do not form a valid alignment, whereas a ratio of 1 means a perfect alignment without gaps or base mismatches (but perhaps one or several aligns of a base against an N). Every candidate pair whose score ratio is within a configurable threshold (normally upward of from 70% to 80%) and where the length of the overlap is not too small, is accepted as true overlap; candidate pairs not matching these criteria, often due to spurious hits in the scanning phase or extremely short common motifs in the sequence, are identified and rejected from further assembly.
The sequence alignments, along with complementary data (like orientation of the aligned reads, overlap region, etc.), that passed the Smith-Waterman test are stored to facilitate and speed up the next phases. Good alternatives are also stored to enable alternative alignments to be found later on in the assembly.
Graph Building
All of the passed alignments that are above a certain alignmentscore threshold, and whose edge weight (see below) is above another threshold, form one or several weighted graphs. By setting cutoffs with the thresholds mentioned, overlap alignments that are too short or too disparate are sorted out from the weighted graphs, easing the algorithmical strain on computer memory and CPU. The graph(s) nevertheless represent almost the totality of all of the valid assembly layout possibilities of a given set of sequencing data.
The nodes of a graph are represented by the reads. An edge between two nodes indicates that these two reads are overlapping. The weight of the edges themselves are computed from a combination of the alignment score and the length of the overlap to take into account both the quality aspect and length of an alignment, a process which is called normalization.
The simplest method for a weight would be to multiply the length of the alignment with the score ratio to get the weighted length of the overlap wo = leno*Rs. The most important problem with this approach is the fact that it attributes far too much weight to the length than it does to the score ratio, which is, after all, the predominant measure for the quality in this early stage of an assembly. A simple and elegant solution to this problem is squaring the score ratio, giving it more importance in the calculation: wo = leno*Rs2. We found this simple and fast function to be as effective for the needs of an assembler than other, more complicated, and slower calculations. Among these were methods proposed by Shpaer et al. (1996) and Pearson (1995, 1998), who used different kinds of ln/ln normalization, whereas Arslan et al. (2001) devised special, but even slower, iterated Smith-Waterman computational algorithms with fractional programming.
Building Contigs
The overlaps found and verified in the previous phases must then be assembled into contigs. This is the most fundamental and intricate part of the process, especially in projects containing many common protein motifs and/or SNPs. Several basic approaches to the multiple alignment problem have been devised to tackle this problem. Although algorithms for aligning multiple sequences at once have been used with increasing success for up to ∼10–15 sequences (Stoye 1998), the amount of time needed to perform this alignment is still too unpredictable (ranging from a few seconds to several hours) to be used in regular EST sequence assembly, especially taking into account the fact that first-iteration assembly coverages with several hundreds, or even thousands of sequences are not uncommon for some non-normalized EST clone libraries.
We decided to use an iterative pairwise sequence alignment and devise new methods for searching overlap candidates and for empowering contigs to accept or reject reads presented to them during the contig-assembling process. The algorithm consists mainly of two objects that interact with each other, a pathfinder object and a contig object.
Pathfinder and Contig Interaction
Because we use an iterative approach to the multiple alignment problem—this means we always successively align the next read against an existing consensus—the result of the alignment sensitively depends on the order of pairwise alignments (Morgenstern et al. 1996). We have to make sure that we start at the position in the mRNA transcript contig where there are many reads with almost no errors. The pathfinder will thus, in the beginning, search for a node in the weighted graph having a maximum number of highly weighted edges to neighbors. The idea behind this behavior is to take the read with the longest and qualitatively best overlaps with as many other reads as possible. This ensures a good starting point, the anchor for this contig.
We first tried a simple greedy algorithm to determine the next overlap candidate to a contig, but occasionally, especially in high-coverage mRNAs, this algorithm produces substandard results. With our current algorithm, which is a width-first-depth-last n, m-recursive look-ahead algorithm with a cutoff value of normally four or five recursions, the number of misalignments could be substantially reduced. The pathfinder designates the next read to add to an existing contig by making an in-depth analysis of the weights and clone template orientations of neighboring reads. Again, non-normalized EST libraries with extremely high-coverage contigs made adapted algorithms necessary. An internal test in the development phase showed that a combination of graph pruning and a time based cutoff strategy proved to be the most successful in terms of result quality and time consumption. In the end, the pathfinder takes the edge leading to the first node that is contained in the best partial path found so far, and then presents the corresponding read (and its approximate position) to the contig object as a potential candidate for inclusion into the existing consensus.
The contig will then accept or reject the read from its alignment. The pathfinder will eventually try to add the same read to the same contig at an alternative position or, skipping it, try other reads. Once the pathfinder has no possibilities left to add unused reads to the actual contig, it will again search for a new anchor point and use this as starting point for a new EST contig. This loop continues until all of the reads have been put into contigs or, if some reads could not be assembled anywhere, form single-read contigs11 called singlets.
Contig Approval Methods: Discovering and Using Additional Information
A contig is represented by a collection of reads that have been arranged in a certain order with given offsets to form an alignment that is as optimal as possible, that is, an alignment in which the reads forming it have as few unexplained errors as possible, but still form the shortest possible alignment. To serve this purpose, a contig object has been provided with algorithms that analyze the impact of every newly added read on the existing consensus. Our assumption is now that, as the assembly started with the best overlapping reads available, the bases in the consensus will be correct at almost every position. Should the newly added read integrate well into the consensus, perhaps extending it, then the contig object will accept this read as part of the consensus. In cases representing a chance alignment, the read differs in too many places from the actual consensus, thus differing in many aspects from reads that have been introduced to the consensus before. The contig will reject such cases without further tests.
The simplest behavior of a contig could be to accept every new read that is being presented as part of the consensus without further checks. In this case, the assembler would thus rely, as layout algorithm only, on the weighted graph, the method with which the weighted edges were calculated and the algorithm that traverses this graph.
However, using additional information that is available before or even becomes available during the assembly proves useful. For example, newly discovered SNP sites may have been tagged in an earlier iteration of the assembly. It is therefore possible for the contig to apply much stricter control mechanisms in those sites of a sequence then known to be a possible SNP. The method is learned from a study by Bonfield et al. (1998), which described how to use traces and their signal values to detect mutations in ESTs.
Figure 4 shows how the miraEST assembler extracts previously unknown information from the data itself. In this example, the algorithms failed to find a valid explanation for the read discrepancies at these base positions in this contig object. The signal analysis function of the automatic editor that was called onto these disagreements also failed to resolve discrepancies by investigating probable alternatives at the fault site. A previous suggestion on incorporating electrophoresis data into the assembly process promoted the idea of capturing intensity and characteristic trace shape information and provide these as additional data to the assembly algorithm (Allex et al. 1996). We decided against such an approach, as essentially all the assembler, and with it the consensus-finding algorithm of a contig, needs to know is whether, yes or no, the signal analysis reveals enough evidence for resolving a conflict within reads by changing the bases incriminated. Signal analysis is therefore treated as a black-box decision formed by an expert system, called during the assembly when conflicts arise. Nevertheless, it provides more information than the one contained in quality values extracted from the signal of one read only (cf. with Durbin and Dear 1998; Walther et al. 2001); there is a reasonable suspicion deduced from other aligned reads on the place and the type of error where the base caller could have made a mistake in a single read.
Figure 4.

Snapshot of a contig in the sequence assembly after the first iteration (visual representation by means of the gap4 program). All sequences were assembled together. After the assembly, miraEST searched for unresolved mismatches with good signal qualities, tagging entire columns as dangerous potential SNP sites for the next iteration. miraEST tagged strong SNP sites bright red, weak sites in blue; bases differing from the consensus are shown in green by the gap4 program. Some bases were not tagged, although they cover a possible SNP site; these bases generally have trace signals of bad quality that the assembler deemed to be too dangerous to be taken as differentiation criterion. miraEST will dismantle that contig and reassemble the sequences immediately, this time using the information gained about the potential SNP sites in the previous assembly to correctly discern between different mRNA transcripts having different SNP variants. The black rectangle amidst the sequences depicts the three trace signal extracts that have been exemplarily shown below; the smaller black boxes within the rectangle depict the discrepancy bases that have also been surrounded by black boxes in the traces. All sequences have indisputable trace curves and quality values (shown as a blue bar above the traces). One can clearly see that there will be at least three different mRNA transcripts to be built, on the basis of the double-base mutation in the middle of the box, one reading CC, the next CT, and the last TC.
In Figure 4, there is not enough evidence found in the conflicting reads to allow one or several base changes to resolve each of the shown discrepancies. Consequently, the assembler will recognize that sequences from several different ESTs were assembled, and tag the bases involved as potential SNPs. The contig will be dismantled at once, and the reads will be reassembled in a different fashion, this time with the additional information of which bases have a possible SNP.
Another example for useful additional assembly information is the clone template insert size for projects using sequencing techniques that analyze the nucleic acid from both strands of a template. Knowing the approximate size allows the contig object to check whether a newly inserted read has the correct distance from the affiliated read possibly present. If the distance does not match approximately the template insert size, the newly inserted read will be rejected. Although this information is rarely useful in EST projects because mRNAs longer than 2000 bases are quite infrequent, it still can be used in those few cases.
Automatic Editing
As already stated, the assembler makes use of different available base-quality or base-probability values. This induces the possibility of using other methods for dealing with possible base-call errors that might be present in reads, and which introduce discrepancies or misassemblies in the assembly. This is entirely done by an incorporated version of the automatic editor.
The automatic editor has to meet four stringent stipulations to be of any use to the assembler: (1) it must not edit toward the consensus, because possible misassemblies would cause wrongly assembled reads to be wrongly edited; (2) the editor must be able to establish relatively complex error hypotheses, spanning several columns in the assembly to resolve conflicts due to reads containing only low quality data; (3) the editor must be able to check alternative hypotheses for error correction and pick the one with the highest probability; and (4) every edit operation made in a read must be supported by its trace signal to interdict complaisant edits.
Automatic editing is treated, like signal analysis, as a black-box expert system called during the assembly process to resolve conflicts present in contigs on the basis of trace evidence only. How the automatic editor proceeds is not of any interest to the assembler as long as the conditions described above are met. The exact methods and algorithms are described in T. Pfisterer, B. Chevreux, M. Platzer, T. Wetter, and S. Sushai, in prep.
Differentiating mRNA Transcripts by SNPs
The mira genome assembler we presented in Chevreux et al. (1999) used a method that searches for patterns on a symbolic level12 in an alignment to detect differences in repetitive sequences in a genome assembly and subsequently tag the bases, allowing discrimination of repeats. Exactly the same approach and algorithms can be used to detect SNPs in EST sequences, the only difference is the fact that the sequences assembled are not genomic, but gene transcripts, and therefore, the bases in question get other tag names. The algorithms were thoroughly revised and enhanced since, but still, the most important factor remains the same, the observable circumstance that, normally, errors in reads that cause a drop in the alignment quality do not accumulate at specific column positions. Sequences from repeats in genome projects or from different mRNAs in EST projects, however, may show column discrepancies between bases of different reads that have very low base-calling error probabilities, so that the discrepancies simply cannot be edited away; these are the potential SNP sites.
On the basis of this approach, the enhanced method uses a combined approach of sequence redundancy, base-error probabilities and column-discrepancy distribution in alignments to recognize SNPs. The better the trace signal values are, the less coverage redundancy and number of discrepancy columns is needed to locate a SNP. In the end, bases with good signal traces allow even a single differing base in a column to be seen as a hint for a SNP site. Once potential SNP positions have been detected in an alignment, the bases allowing discrimination of reads belonging to different mRNA transcripts are then marked as possible SNP marker bases by the assembler as shown exemplarily in Figure 4. Contigs containing such misassemblies are immediately dismantled and, during the subsequent reassembly, no discrepancy in alignments implicating these bases will be allowed, and hence, misassemblies of ESTs with different SNP bases are prevented.
The operations necessary for reassembly and realignment are unpredictable and depend heavily on the type of data to be assembled. The simplest assumption could be that the falsely integrated reads could be simply removed from the contig, but unfortunately, in quite a number of cases, misaligned reads change the whole EST contig structure. To make the best possible use of the improved sequences, the assembler therefore restarts the whole assembly process of the affected mRNA transcript from the beginning. This ensures an optimal new assembly without risking errors introduced by unpredictable or wrongly predicted reordering operations.
Read Extension
As the initial assembly used only high-quality parts of the reads, further information can be extracted from the alignments by examining the end of the reads that were previously unused. Although the signal-to-noise ratio quickly degrades in read traces toward the end, the data is not generally useless. These hidden parts of the reads can now be used by uncovering parts of the reads that align to the already existing consensus of the transcript and even for extending the actual consensus over the ends. Especially the last case may provide the extra bases needed to link different partial transcripts to one complete transcript.
This extension is computed concurrently by analyzing the overlap relationships characterized in the alignments computed in the earlier phase of the assembly. For every aligned sequence pair whose score ratio surpasses a defined threshold, the extension algorithm tries to realign longer subsequences, including parts of the previously unused LCR present at the ends of each read.
Performing the extension operation at this stage of the assembly process incorporates the inestimable surplus value that the reads previously assembled into contigs will have been cautiously edited at least once by the automatic editor in their actual high-confidence regions. Additionally, the sequences edited in each contig do not include sequences with contradicting SNPs, as these would have been recognized and fixed in an earlier step. The presumably few errors present in these parts of the read have thus been edited away where the trace signals and the alignment with other reads showed enough evidence to support the error hypothesis. Less errors present in a sequence help the alignment algorithm to build more accurate alignments, and thus, will increase the score ratio of aligned sequences even with parts of the LCR data included.
A window search is then performed across the new alignment to compute the optimal extension length of the new HCR up to the point where the called sequence gets too bad to be correctly aligned. The chances for a long extension are increased, because each read is present in many alignments, giving it many occasions to be extended.
The iterative enlargement procedure enables the assembler to redefine step-by-step the HCR of each read by comparing it with supporting sequences from aligned reads. This usage of information in collateral reads is the major advantage our assembler has over a simple base caller, which has only the trace information of one read to call bases and estimate their probability.
Merging Pristine Transcripts for SNP Classification
During an optional last assembly pass, the miraEST assembler will merge almost identical, strain and SNP separated, transcriptome sequences from the previous passes for a last alignment. Such an alignment shows SNP differences between the mRNA sequence transcripts. The transcript sequences used for this final assembly stage will be precisely classified and assembled at least by SNP types and, if the information was present, by organism/strain/cell type in the previous passes. Consequently, it is reasonable to assume that the transcript sequences used at this stage are pristine, that is, they code existing proteins.
It is important to note that this step, like the whole process performed by miraEST, is still an assembly and not a clustering step. That is, sequences composed by different exon structures, or which contain large indels, will not be assembled. The results obtained here are nevertheless important in the sense that they allow analysis and classification of the SNP types of nearly identical mRNA sequences that occur in one or several sequencing assembly projects.
We differentiate between three distinct types of SNPs when analyzing transcripts from one or several organisms, strains or cell types.
PAOS Polymorphisms that occur within a single organism or cell transcriptome are tagged as Possible intrAOrganism Snp.
PROS Polymorphisms that occur between different organisms or cells are tagged Possible inteR Organism Snp.
PIOS Polymorphisms that occur both within and between organisms (respectively, cell types) are tagged as Possible Intra- and inter Organism Snp.
According to the classification above, each SNP will be tagged either as PROS, PAOS, or PIOS, depending on which sequence has strain information that can contribute to the exact polymorphism. Sequences without strain information will also have the bases tagged, but only as PAOS, as they will be assigned to a default strain. Figure 5 illustrates the assembly of two strains. SNPs are classified into all three categories, the example figure showing two of them (PAOS and PIOS).
Figure 5.

The last (optional) step of the EST assembly consists of the input sequences being given strain information to show the effect when two different organism strains (named sponge1 and sponge2) are sequenced and analyzed. In this example, miraEST classified the SNPs into two categories: PROS (shown in light blue) for SNPs that occur only between strains/organisms (e.g., column 661) and PIOS (shown in light green) for SNPs that occur both within a strain as between different strains (e.g., column 662). Interestingly enough, most of the SNPs shown in this example will not cause a change in the amino acids of the resulting protein, with one notable exception, the SNP of sponge1_singlet4 at base position 662 causes a TAA codon to be expressed, which is a stop codon. The SNPs of the same sequence at position 686 and 707 would cause mutations in the amino acid sequence, but are, because of the TAA mutation earlier, in the3′ UTR of this particular mRNA transcript.
Acknowledgments
Intensive and helpful discussions with Dr. Jacqueline Weber and Dr. Andrea Hörster on the subtleties occurring in the transcriptome helped greatly in the writing of this paper. We thank the coworkers of the former Staden group at the MRC LMB Cambridge, and especially James Bonfield for providing support for the integration of mira and miraEST within GAP4. We thank Dr. John Perkins for a thorough review of this paper and Prof. Dr. Jörn Bullerdiek for providing data for this research.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1917404. Article published online before print in May 2004.
Footnotes
Contig as a short form of contiguous sequence, a term first coined for assembly of genomic data.
For example, could the base A at position 235 in read 1 be replaced by a G? (because the overall consensus at this position of the other reads suggests this possibility).
For example, quality clipping, sequencing vector, and cosmid vector removal can be controlled by the PREGAP4 environment provided with the GAP4 package (Bonfield et al. 1995; Bonfield and Staden 1996; Staden 1996) or the LUCY program from Chou and Holmes (2001); parts of these tasks can also be done with cross-match provided by the PHRAP package or other packages such as, for example, PFP from Paracel (Paracel 2002a).
Of course, a single read itself cannot be called a contig. However, putting it into the same data structure (a contig object) like the other, assembled reads is a convenient way to keep unassembled reads in the internal assembly database.
Based mainly on redundancy information in suspicious sequence stretches, using base-call error probabilities and signal analysis capabilities of the automatic editor very sparsely.
References
- Allex, C.F., Baldwin, S.F., Shavlik, J.W., and Blattner, F.R. 1996. Improving the quality of automatic DNA sequence assembly using fluorescent trace-data classifications. Intell. Systems Mol. Biol. 4: 3–14. [PubMed] [Google Scholar]
- Arslan, A.N., Egecioglu, O., and Pevzner, P.A. 2001. A new approach to sequence comparison: Normalized sequence alignment. Bioinformatics 17: 327–337. [DOI] [PubMed] [Google Scholar]
- Baeza-Yates, R.A. and Gonnet, G.H. 1992. A new approach to text searching. Commun. of the Assoc. for Comp. Mach. 35: 74–82. [Google Scholar]
- Barker, G., Batley, J., O'Sullivan, H., Edwards, K.J., and Edwards, D. 2003. Redundancy based detection of sequence polymorphisms in expressed sequence tag data using autoSNP. Bioinformatics 19: 421–422. [DOI] [PubMed] [Google Scholar]
- Bonfield, J.K. and Staden, R. 1996. Experiment files and their application during large-scale sequencing projects. DNA Seq. 6: 109–117. [DOI] [PubMed] [Google Scholar]
- Bonfield, J.K., Smith, K.F., and Staden, R. 1995. A new DNA sequence assembly program. Nucleic Acids Res. 23: 4992–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfield, J.K., Rada, C., and Staden, R. 1998. Automated detection of point mutations using fluorescent sequence trace subtraction. Nucleic Acids Res. 26: 3404–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo, A.A., Samaia, H.P., Dias-Neto, E., Simao, D.F., Migotto, I.A., Briones, M.R., Costa, F.F., Nagai, M.A., Verjovski-Almeida, S., Zago, M.A., et al. 2001. The contribution of 700,000 ORF sequence tags to the definition of the human transcriptome. Proc. Natl. Acad. Sci. 98: 12103–12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, S.C., Wong, A.K.C., and Chiu, D.K.Y. 1992. A survey of multiple sequence comparison methods. Bull. Mathem. Biol. 54: 563–598. [DOI] [PubMed] [Google Scholar]
- Chevreux, B., Wetter, T., and Suhai, S. 1999. Genome sequence assembly using trace signals and additional sequence information. Comput. Sci. Biol.: Proc. German Conference on Bioinformatics GCB'99 GCB, pp. 45–56.
- Chevreux, B., Pfisterer, T., and Suhai, S. 2000. Automatic assembly and editing of genomic sequences. In Genomics and proteomics—functional and computational aspects (ed. S. Suhai), Chap. 5, pp. 51–65. Kluwer Academic/Plenum Publishers, New York.
- Chou, H.-H. and Holmes, M.H. 2001. DNA sequence quality trimming and vector removal. Bioinformatics 17: 1093–1104. [DOI] [PubMed] [Google Scholar]
- Dear, S., Durbin, R., Hilloier, L., Marth, G., Thierry-Mieg, J., and Mott, R. 1998. Sequence assembly with CAFTOOLS. Genome Res. 8: 260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin, R. and Dear, S. 1998. Base qualities help sequencing software. Genome Res. 8: 161–162. [DOI] [PubMed] [Google Scholar]
- Giegerich, R. and Wheeler, D. 1996. Pairwise sequence alignment. http://www.techfak.uni-bielefeld.de/bcd/Curric/PrwAli/prwali.html.
- Grillo, G., Attimonelli, M., Luici, S., and Pesole, G. 1996. CLEANUP: A fast computer program for removing redundancies from nucleotide sequence databases. Comput. Appl. Biosci. 12: 1–8. [DOI] [PubMed] [Google Scholar]
- Gusfield, D. 1997. Algorithms on strings, trees and sequences: Computer science and computational biology. Cambridge University Press, Cambridge, London.
- Huang, X. 1994. On global sequence alignment. Comput. Appl. Biosci. 10: 227–235. [DOI] [PubMed] [Google Scholar]
- Huang, X. 1996. An improved sequence assembly program. Genomics 33: 21–31. [DOI] [PubMed] [Google Scholar]
- Kececioglu, J.D. and Myers, E.W. 1992. Combinatorial algorithms for DNA sequence assembly. Tech. Rep. TR 92–37, University of California at Davis, University of Arizona, Davis, AZ.
- Kumar, S. and Rzhetsky, A. 1996. Evolutionary relationships of eukaryotic kingdoms. J. Mol. Evol. 42: 183–193. [DOI] [PubMed] [Google Scholar]
- Morgenstern, B., Dress, A., and Werner, T. 1996. Multiple DNA and protein sequence alignment based on segment-to-segment comparison. Proc. Natl. Acad. Sci. 93: 12098–12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, W.E. 2001. How was metazoan threshold crossed: The hypothetical Urmetazoa (part A). Compar. Biochem. Physiol. 129: 433–460. [DOI] [PubMed] [Google Scholar]
- Myers, E.W. 1995. Toward simplifying and accurately formulating fragment assembly. J. Computat. Biol. 2: 275–290. [DOI] [PubMed] [Google Scholar]
- Nickerson, D.A., Taylor, S.L., and Rieder, M.J. 2000. Identifying single nucleotide polymorphisms (SNPs) in human candidate genes. In Research abstracts from the DOE human genome program Contractor-Grantee Workshop VIII. Feb. 27 to Mar. 2, 2000. Santa Fe, NM.
- Notredame, C. and Higgins, D.G. 1996. SAGA: Sequence alignment by genetic algorithm. Nucleic Acids Res. 24: 1515–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paracel 2002a. Paracel filtering package user manual. Paracel, Inc., Pasadena, CA.
- Paracel. 2002b. PTA: Paracel transcript assembler user manual. Paracel, Inc., Pasadena, CA.
- Parsons, R., Forrest, S., and Burks, C. 1993. Genetic algorithms for DNA sequence assembly. In Proc. of the 1st International Conference on Intelligent Systems for Molecular Biology (eds. L. Hunter et al.). AAAI, Bethesda, MD. [PubMed]
- Pearson, W.R. 1995. Comparison of methods for searching protein sequence databases. Protein Sci. 4: 1145–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, W.R. 1998. Empirical statistical estimates for sequence similarity searches. J. Mol. Biol. 276: 71–84. [DOI] [PubMed] [Google Scholar]
- Peltola, H., Söderlund, H., and Ukkonen, E. 1984. SEQAID: A DNA sequence assembling program based on a mathematical model. Nucleic Acids Res. 12: 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea, G., Huang, X., Liang, F., Antonescu, V., Sultana, R., Karamycheva, S., Lee, Y., White, J., Cheung, F., Parvizi, B., et al. 2003. TIGR gene indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 19: 651–652. [DOI] [PubMed] [Google Scholar]
- Pfisterer, T. and Wetter, T. 1999. Computer assisted editing of genomic sequences—why and how we evaluated a prototype, Lecture Notes in Artificial Intelligence; Subseries of Lecture Notes in Computer Science, pp. 201–209. Springer-Verlag, Berlin, Heidelberg, New York.
- Shpaer, E.G., Robinson, M., Yee, D., Candlin, J.D., Mines, R., and Hunkapiller, T. 1996. Sensitivity and selectivity in protein similarity searches: Comparison of Smith-Waterman in hardware. Genomics 38: 179–191. [DOI] [PubMed] [Google Scholar]
- Staden, R. 1996. The Staden sequence analysis package. Mol. Biotechnol. 5: 233–241. [DOI] [PubMed] [Google Scholar]
- Staden, R., Bonfield, J., and Beal, K. 1997. The new Staden package manual—3Part 1. Medical Research Council, Laboratory of Molecular Biology, http://staden.sourceforge.net/.
- Stoye, J. 1998. Multiple sequence alignment with the divide-and-conquer method. Gene/GC 211: 45–56. [DOI] [PubMed] [Google Scholar]
- Tammi, M.T., Arner, E., Britton, T., and Andersson, B. 2002. Separation of nearly identical repeats in shotgun assemblies using defined nucleotide positions, DNPs. Bioinformatics 18: 379–388. [DOI] [PubMed] [Google Scholar]
- Walther, D., Bartha, G., and Morris, M. 2001. Basecalling with LifeTrace. Genome Res. 11: 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, S. and Manber, U. 1992. Fast text searching allowing errors. Commun. ACM 35: 83–91. [Google Scholar]
- Zhang, C. and Wong, A.K. 1997. A genetic algorithm for multiple molecular sequence alignment. Comput. Appl. Biosci. 13: 565–581. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.chevreux.org/projects_mira.html; homepage of the MIRA V2 assembly system.
- http://www.dkfz.de/mbp-ased/; homepage of the MIRA V1 assembly system and EdIt automatic editor.