

Abstract
The syndrome of hereditary 1,25-dihydroxyvitamin D–resistant rickets (HVDRR) is a genetic disease of altered mineral homeostasis due to mutations in the vitamin D receptor (VDR) gene. It is frequently, but not always, accompanied by the presence of alopecia. Mouse models that recapitulate this syndrome have been prepared through genetic deletion of the Vdr gene and are characterized by the presence of rickets and alopecia. Subsequent studies have revealed that VDR expression in hair follicle keratinocytes protects against alopecia and that this activity is independent of the protein's ability to bind 1,25-dihydroxyvitamin D3 [1,25(OH)2D3]. In the present study, we introduced into VDR-null mice a human VDR (hVDR) bacterial artificial chromosome minigene containing a mutation that converts leucine to serine at amino acid 233 in the hVDR protein, which prevents 1,25(OH)2D3 binding. We then assessed whether this transgene recreated features of the HVDRR syndrome without alopecia. RT-PCR and Western blot analysis in one strain showed an appropriate level of mutant hVDR expression in all tissues examined including skin. The hVDR-L233S mutant failed to rescue the aberrant systemic and skeletal phenotype characteristic of the VDR null mouse due to the inability of the mutant receptor to activate transcription after treatment with 1,25(OH)2D3. Importantly, however, neither alopecia nor the dermal cysts characteristic of VDR-null mice were observed in the skin of these hVDR-L233S mutant mice. This study confirms that we have created a humanized mouse model of HVDRR without alopecia that will be useful in defining additional features of this syndrome and in identifying potential novel functions of the unoccupied VDR.
1,25-Dihydroxyvitamin D3 [1,25(OH)2D3] is a steroid hormone that serves to regulate mineral homeostasis in higher vertebrates through its actions on multiple tissues that include the intestine, kidney, and bone (1, 2). This hormone also has a regulatory impact on numerous additional biological systems found in skin (3, 4), immune cells (5–7), and cardiovascular tissue (8, 9) and may exert broad antiproliferative and/or prodifferentiative effects on cells relevant to the prevention or treatment of cancer (10). Each of these activities of 1,25(OH)2D3 is mediated by the vitamin D receptor (VDR), which functions at the genomic level to regulate the expression of genes that support these highly cell-specific, biological events (1, 11–13). Transcriptional regulation involves the ligand-mediated formation of a VDR/retinoid X receptor (RXR) heterodimer that binds to specific DNA sequences within the regulatory regions of target genes, thereby facilitating the gene-selective recruitment of chromatin-active coregulatory complexes that function at multiple levels to modulate target gene expression (12–15). Several alternative mechanisms involving the interaction of the VDR and perhaps RXR with other transcription factors have also been reported (16–20). Finally, recent genome-wide studies have now defined cistromes for the VDR and RXR partner in several cell types (13, 21–23). These studies have provided new insight into the molecular actions of the vitamin D hormone and its nuclear receptor, which include the location, quantitation, and epigenetic characterization of VDR DNA binding sites, and the realization that these cistromes are cell specific and also highly dynamic. The latter studies and those of others have added significantly to our understanding of the mechanisms through which 1,25(OH)2D3 regulates gene expression.
Early studies by Brooks et al (24) and Marx et al (25) defined the first cases of a human syndrome that involved the VDR, initially termed vitamin D–dependent rickets, type II and now referred to as hereditary 1,25-dihydroxyvitamin D–resistant rickets (HVDRR). This disease is characterized by hypocalcemia, hypophosphatemia, and secondary hyperparathyroidism and perhaps more definitively by the presence of high levels of circulating 1,25(OH)2D3 (26). The major biological consequence of this rare disease is early-onset rickets due to defective skeletal development and mineralization, and it also causes growth retardation and, in a subset of cases, a deficiency in hair growth and alopecia (26). Numerous biochemical investigations followed by genetic studies by Malloy and Feldman (26) and others ultimately confirmed that this syndrome was due to an extensive and independent series of mutations in the VDR gene that disrupt expression of the VDR or its ability to interact with 1,25(OH)2D3, DNA or key coregulatory protein mediators. Many of these mutations fully compromise the ability of the VDR to mediate 1,25(OH)2D3-regulated transcription (26). However, the precise structural nature of the VDR mutant itself now appears to be a primary determinant of the presence of alopecia (26). This discovery coupled with the absence of the alopecic phenotype in the syndrome of vitamin D dependency rickets type 1 (VDDR-1), a disease characterized by the presence of the receptor but the absence of 1,25(OH)2D3 due to mutations in the CYP27B1 gene responsible for its synthesis (27), led investigators to conclude that the VDR facilitated hair growth in a ligand-independent manner. This conclusion was reinforced in turn through the creation of analogous mouse models of VDRR-1 and HVDRR, the former through direct genetic mutation of the Cyp27b1 gene (28, 29) and the latter through a VDR deletion mutation that abrogated VDR expression (30–33); both genetic models generally phenocopied the human syndromes. Final evidence for this hypothesis has been provided more recently through the keratinocyte-targeted expression of cDNAs for both a wild-type (WT) human vitamin D receptor (hVDR) and a 1,25(OH)2D3-binding defective form containing mutations converting leucine 233 to serine (34–36); both rescued the alopecia and the epidermal features evident in the VDR-null mouse, although other vitamin D actions in the skin were not examined. Thus, the development of these genetic and transgenic mouse models has confirmed and extended our understanding of the syndromes of VDDR-l and HVDRR with alopecia. Despite these advances, an authentic, humanized mouse model of HVDRR without alopecia has not been created.
The role of the VDR as the primary determinant of response to 1,25(OH)2D3 in cells prompted early efforts to identify the structural features of the mouse and human chromosomal genes for the VDR (37, 38). More recently, we used unbiased chromatin immunoprecipitation–tiled microarray and chromatin immunoprecipitation linked to deep sequencing approaches, as summarized above, to more fully characterize these two VDR genes and to identify the regulatory regions responsible for controlling their transcriptional output (39, 40). Studies in the mouse revealed a large gene that contained several internal intronic and upstream distal regulatory domains (40). These elements mediated up-regulation by 1,25(OH)2D3, other lipophilic hormones, and PTH and bound the transcription factors that were integral to this regulation (39, 40). These control features were confirmed using large bacterial artificial chromosome (BAC)–derived minigenes stably integrated into host cells (40). Importantly, the human gene displayed similar organizational and functional features (39, 40). Perhaps most relevant, however, was our observation that both the mouse and human minigenes could serve as bone fide transgenes in mice in vivo and that each recapitulated endogenous, tissue-specific VDR gene expression in the mouse as well (41). Moreover, upon transfer to a VDR-null background, both transgenes fully restored the transcriptional response to 1,25(OH)2D3, were regulated appropriately, and rescued the complex systemic and organ-specific phenotype that was observed when the VDR was absent (41). The success of these studies confirmed the spatial extent of both the mouse and human VDR genes and their genetic regulatory sequences while leading simultaneously to the creation of a functional humanized VDR mouse model.
Based on the fidelity of the above approach, we introduced in the present study a mutation into the human VDR transgene (L233S relative to codon 233 in the mRNA) (35) and via transgenesis created 3 strains that expressed this ligand binding–defective VDR protein in a VDR-null mouse background. We report here that this mutant human VDR protein, which was expressed in a manner analogous to that of the wild-type human VDR gene in all the classic vitamin D target tissues examined, was unable to rescue the systemic and skeletal phenotype associated with the VDR-null mouse or to restore broad tissue-specific transcriptional responses to 1,25(OH)2D3 that underlie these biological activities. Despite this, the mutant hVDR fully prevented the development of alopecia and the formation of epidermal cysts observed in the skin of VDR-null mice. Thus, we have created a humanized mouse model of HVDDR without alopecia that should be useful both in assessing additional features of the human syndrome and in identifying hormone-independent functions of the VDR in skin and perhaps in other tissues.
Materials and Methods
Construction of VDR BAC clones
The WT hVDR BAC transgene was constructed from BAC clone RP11–89H19 and contained the entire human VDR gene including 82 kb of the 5′-intergenic region, the transcription unit, a neomycin selection/luciferase reporter cassette, and 48 kb of 3′-intergenic region; the actual size of the 5′- and the 3′-regions flanking the human VDR gene itself in the transgene are approximately 82 and 22 kb, respectively, due to NotI-mediated linearization to generate the transgene as described previously (41) and as depicted in Figure 1. The mutant hVDR BAC transgene (L233S hVDR BAC) (Figure 1) was derived from the WT hVDR BAC transgene through mutagenesis to alter the triplet codon for leucine (CTG) in exon 8 (amino acid 233 in the human VDR) to serine (TCG) using the galactokinase system as described previously (42).
Figure 1.
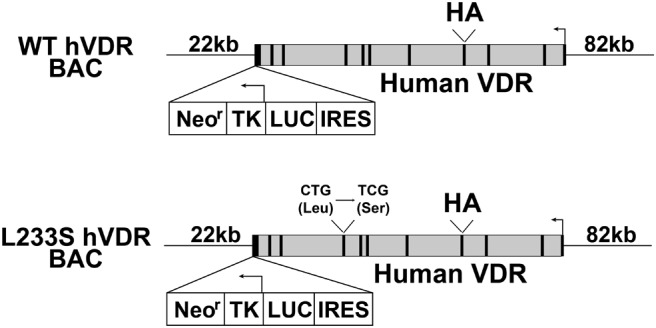
Schematic structures of the WT hVDR BAC and the L233S hVDR BAC transgenes. Human VDR BAC clones including the entire human VDR gene locus and its surrounding intergenic segments were genetically engineered by BAC recombineering techniques to contain a hemagglutinin tag (HA) at the translation start site of the human VDR gene and a cassette containing an internal ribosome entry site (IRES)–driven luciferase (LUC) reporter and a TK promoter (TK)–driven neomycin resistance gene (Neor) selection mechanism in the 3′ untranslated region of the human VDR gene, respectively. The L233S mutation was introduced through nucleotide mutagenesis (CTG to TCG) at codon 233.
Generation of BAC transgenic mice and Vdr-null mice containing the BAC transgenes
The hVDRWT transgenic mouse strain (41) and the VDR-null mouse strain (VDR−/−) created by Li et al (31) have been reported previously. Three hVDRL233S transgenic mouse strains (T805, T806, and T807) were generated as described by Lee et al (41). Transgenic strains were maintained as heterozygotes through outbreeding with C57BL/6 mice (Harlan). We used a breeding strategy as outlined previously to create crosses for both hVDRWT/VDR−/− mice and the 3 hVDRL233S/VDR−/− mouse strains (T805/VDR−/−, T806/VDR−/−, and T807/VDR−/−) (41). The genotypes of these mice were identified as reported previously (41).
Animals
WT C57BL/6 mice and C57BL/6 mice carrying either the WT hVDR BAC transgene (hVDRWT) or the mutant L233S hVDR BAC transgene (hVDRL233S) were maintained on a standard rodent chow diet (5008; Harlan Teklad). VDR-null mice and the 3 hVDRL233S/VDR−/− strains were maintained on a synthetic rescue diet containing 20% lactose, 2% calcium, and 1.25% phosphate (TD.96348; Harlan Teklad). All mice in the experimental protocols terminated at 8 to 10 weeks of age were fed standard rodent chow diet after weaning. The presence or absence of alopecia and/or histological skin defects was assessed in mice maintained on the rescue diet, which has been shown to have no effect on the alopecic phenotype. Mice were exposed to a 12-hour light-dark cycle, and all animal studies were reviewed and approved by the Research Animal Care and Use Committee of the University of Wisconsin-Madison.
RNA analysis and transcriptional response to 1,25(OH)2D3
Total RNAs were prepared and subjected to reverse transcription for VDR expression, as described previously (41). The transcriptional response to 1,25(OH)2D3 was assessed in both male and female mice after a single ip injection of either propylene glycol (vehicle) or 1,25(OH)2D3 (10 ng/g body weight in vehicle). Total RNA was isolated 6 hours after treatment and subjected to reverse transcription, as described previously (41). Gene expression was assessed by quantitative PCR using TaqMan primers (Applied Biosystems).
VDR expression, binding activity, and Western blot analysis
Tissue lysates were obtained and subjected to Western blot analysis using anti-VDR antibody (1:500 dilution, C-20, SC-1008, Santa Cruz Biotechnology) and anti-VDR antibody 9A7 (1:1000 dilution) as described previously (41) (Supplemental Table 1). Anti-β-tubulin antibody (1:1000, sc-9104; Santa Cruz Biotechnology) was used to provide a loading control. A tritiated hormone binding assay of intestinal lysates from mice expressing the hVDR-L233S mutant protein confirmed previous studies (35), indicating that this receptor was incapable of binding 1,25(OH)2D3 (data not shown).
Serum analysis
Calcium and phosphate concentrations were determined as described previously (41). The serum PTH concentration was measured in EDTA-plasma using a mouse PTH 1–84 ELISA kit (Immutopics) according to the manufacturer's instructions.
Bone mineral density (BMD)
Eight-week-old mice of both sexes were scanned and analyzed for BMD as described previously (41). Spinal BMD was obtained at L1 to L6.
Gross and histological analyses
Assessments of parathyroid gland hyperplasia, tibia length, and growth plate histology were conducted as described previously (41). The presence or absence of alopecia and/or skin defects was assessed visually and histologically at 6 and 2 months of age, respectively, in male and female mice maintained on the rescue diet. Methods for histological analysis were described previously (41).
Assessment of hair recycling
To assess hair regrowth, a dorsal patch of hair was initially shaved from individual male and female mice at 2 months of age and then depilated using wax strips under isoflurane-induced anesthesia. Images of hair regrowth in the mice were obtained 2 weeks after depilation.
Statistical analysis
All data are presented as means ± SEM. The Student unpaired t test was used to identify significant differences (P < .05).
Results
hVDRL233S mouse strains
To study the biological effects of a non-1,25(OH)2D3 binding form of the hVDR, we prepared transgenic strains of mice capable of synthesizing the hVDR-L233S mutant (T805, T806, and T807). The features of this transgene (L233S hVDR BAC), together with the parental transgene (WT hVDR BAC) used to create the hVDRWT mouse strain that was previously characterized (41) are seen in Figure 1. Three hVDRL233S strains were identified using luciferase output in the tail and then validated further for appropriate expression in bone fide vitamin D target and nontarget tissues using this marker (data not shown). Although luciferase activity varied in the three hVDRL233S strains (T805, low; T806, low to medium; and T807, high), the tissue patterns were similar. These results suggest that luciferase expression from the mutant transgene, like that observed in the hVDRWT strain (41), fully recapitulated the profile observed for expression of the endogenous mouse VDR gene in vivo. We then crossed each of these mutant strains of mice into a VDR-null background strain as described previously (41), and after expansion of these lines, examined their biological features relative to those of WT and VDR−/− mice and the hVDRWT/VDR−/− transgenic mouse strain.
Relative expression of the hVDR-L233S mutant in target tissues
We first examined hVDR-L233S mutant expression levels in the hVDRL233S/VDR−/− strains relative to those in the hVDRWT/VDR−/− strain across several bone fide target tissues. As can be seen in Figure 2A, analysis of the RNA transcript levels in the tissues of the three strains revealed an extended range of expression with the highest transcript levels observed in the T807/VDR−/− strain of mice. Although this level was generally about half or less than that seen in the hVDRWT/VDR−/− control strain, the previous observation that overall expression of hVDR-WT from the hVDRWT/VDR−/− strain was 2- to 3-fold higher than that of endogenous mouse VDR expression (41) suggested the successful preparation of at least 1 mouse strain expressing appropriate levels of VDR relative to the mouse and 2 strains with significantly lower levels of expression. To further analyze these expression profiles, we conducted a Western blot analysis of VDR protein in jejunum, colon, kidney, and skin of these 3 strains and compared hVDR expression with endogenous levels in WT mice, VDR-null mice, and the hVDRWT/VDR−/− strain. As can be seen in Figure 2B, this analysis revealed that the levels of hVDR-L233S protein expression in the 3 hVDRL233S/VDR−/− strains ranged from lower to higher levels, but that the level of expression in the T807/VDR−/− strain was similar to that observed for endogenous VDR in WT mouse and either equivalent or slightly lower than that observed in the hVDRWT/VDR−/− strain. VDR expression from VDR-null tissue was absent. The authenticity of the VDR bands shown was also confirmed using the 9A7 antibody. These results indicate that hVDR-L233S is expressed appropriately in the T807/VDR−/− strain of mice for subsequent functional studies in diverse calcium-regulating tissues and in skin and that the biological effects of low amounts of VDR expression from the T805/VDR−/− and T806/VDR−/− strains might be examined as well.
Figure 2.

Expression of hVDR-WT and hVDR-L233S in VDR-null mice by BAC transgenes. A, Basal transcriptional levels of BAC-derived hVDR in the indicated tissues from the established strains were measured by quantitative PCR. Expression levels of the VDR transcripts were normalized to Gapdh and expressed as the mean for each strain ± SEM (5–7 mice per group) normalized to the levels measured in the hVDRWT/VDR−/− mouse strain. B, Expression of the hVDR protein was examined by Western blot analysis using the indicated tissue lysates. VDR−/− and VDR+/+ mice were used as negative and positive controls, respectively, and β-tubulin was used as a loading control. Total protein from jejunum and colon (20 μg), from kidney (100 μg), and from skin (60 μg) was examined. Data shown are representative of analyses of the indicated tissues from three separate mice.
Measurement of systemic and skeletal parameters in the hVDRL233S/VDR−/− mouse strains
Given hVDR-L233S expression in the 3 mouse strains, we next explored the ability of the hVDR mutant to rescue the hypocalcemia and hypophosphatemia seen in the VDR-null mouse and the associated secondary hyperparathyroidism as well. As can be seen in Figure 3A, although these parameters were fully normalized in the hVDRWT/VDR−/− strain relative to those of WT mice, none of the hVDRL233S/VDR−/− strains were capable of restoring appropriate levels of blood calcium, phosphorus, or PTH. Interestingly, PTH values in these latter strains of mice were elevated well above those observed in the VDR−/− background strain. Importantly, as seen in Figure 3B, expression of the hVDR-L233S mutant also failed to rescue the parathyroid gland hyperplasia observed in the VDR-null mouse, whereas expression of the hVDR-WT receptor fully reversed this hyperplasia. These data suggest that the mutant hVDR-L233S was unable to facilitate the actions of 1,25(OH)2D3 to control systemic mineral metabolism. As seen in Figure 4, these deranged systemic parameters in the hVDRL233S/VDR−/− strains were also accompanied by aberrant skeletal growth, as evidenced by the reduction in tibia length (Figure 4A). Expression of the hVDR-L233S mutant also failed to normalize BMD in both males and females (Figure 4B) as measured by total body BMD and also this measurement at the femur or the spine of these mice. In fact, the presence of the mutant receptor appeared to exaggerate bone loss at all sites in both sexes. These mice were similarly unable to normalize the growth plate, as evidenced in Figure 4C by the disorganized and disaggregated structures seen histologically in sections from each of the 3 hVDRL233S/VDR−/−mouse strains. These data collectively suggest that although systemic features, tibia length, BMD, growth plate development, and parathyroid gland growth are normal in WT and hVDR-WT–expressing mice, expression of the hVDR-L233S mutant at normal levels failed to rescue and may have exaggerated a subset of these alterations in mineral homeostasis or the skeletal or parathyroid gland consequences of the altered homeostasis that were observed in the VDR-null mouse.
Figure 3.
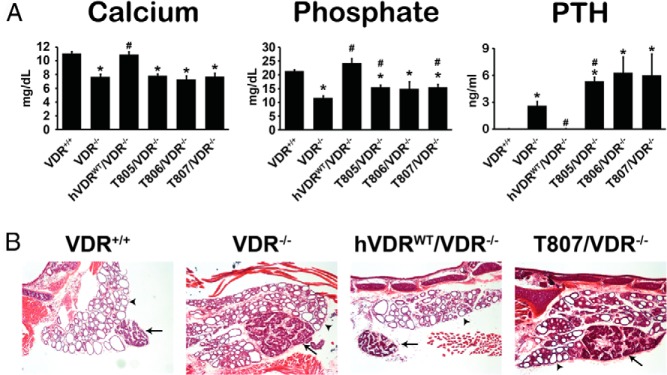
Defects in serum and parathyroid gland (PTG) from VDR-null mice expressing hVDR-L233S. A, Recovery of defects in serum was examined by measuring the levels of serum calcium, phosphate, and PTH. Each value is the average of 5 to 7 mice per strain ± SEM. *, P < .05 compared with the VDR+/+ control; #, P < .05 compared with the VDR−/− control. B, PTG sections were stained with hematoxylin and eosin. Arrows and arrowheads indicate parathyroid and thyroid tissue, respectively. Original magnification, ×40. Representative images of each strain are shown.
Figure 4.

Defects in bone from VDR-null mice expressing hVDR-L233S. A, Tibiae were collected from 10-week-old male mice of the indicated strains and images of representative tibiae from each strain are shown. B, BMDs of total body, femur, and spine of 8-week-old mice were measured. *, P < .05 compared with the VDR+/+ control; #, P < .05 compared with the VDR−/− control. C, Growth plate histology. Tibia sections were stained with hematoxylin and eosin. Original magnification, ×100. Representative images for each strain are shown.
1,25(OH)2D3-sensitive transcriptional competence in hVDR-L233S–expressing mouse strains
To examine the failure of the hVDR-L233S mutant to normalize the above phenotypic parameters at a mechanistic level, we next assessed the ability of the mutant to mediate transcriptional activation by 1,25(OH)2D3. Accordingly, we treated C57BL/6, hVDRWT/VDR−/−, and the 3 strains of hVDR-L233S–expressing mutant mice with a single ip dose of 1,25(OH)2D3, isolated RNA from the appropriate tissues, and assessed the gene response to the hormone 6 hours later. As can be seen in Figure 5A, although 1,25(OH)2D3 was capable of inducing Cyp24a1, S100g, and Trpv6 expression in distinct segments of the intestine in both WT and hVDR-WT receptor–expressing mice, no such induction of these genes was observed at the RNA level in any of the strains that expressed the mutant hVDR-L233S form of the receptor. As seen in Figure 5B, a similar evaluation of 1,25(OH)2D3 induction of Cyp24a1 and suppression of Cyp27b1 expression in the kidney and 1,25(OH)2D3 induction of Cyp24a1, Tnfsf11, Spp1, and Fgf23 in bone cells of calvaria confirmed that hVDR-L233S was transcriptionally inactive in these tissues as well. It is particularly noteworthy that Fgf23 transcript levels, although regulated by 1,25(OH)2D3 when endogenous mouse or hVDR-WT receptors were present, were exceptionally low in the absence of a transcriptionally active receptor. This would suggest that the fibroblast growth factor 23 levels in the blood, although not measured here, are likely to be low as well. We conclude that although the hVDR-L233S mutant receptor was adequately expressed in intestine, kidney, and bone, it was incapable of modulating the transcription of bone fide vitamin D target genes in the specific tissues examined. Thus, the failure to restore the gross phenotype of the VDR-null mouse is almost certainly due to the inability of hVDR-L233S to bind 1,25(OH)2D3 and therefore to activate transcription.
Figure 5.

Loss of transcriptional response of hVDR-L233S to 1,25(OH)2D3. Expression of the indicated 1,25(OH)2D3 target genes in segments of the intestinal tract (A), kidney (B), and calvaria (C) were measured by quantitative PCR. Expression levels of the target gene transcripts were normalized to Gapdh and are expressed as the mean for each strain ± SEM (5–7 mice per group) normalized to the levels measured in the vehicle-treated VDR+/+ mouse strain. *, P < .05 compared with the vehicle-treated sample of each strain. (−), vehicle-treated sample; (+), 1,25(OH)2D3-treated sample.
hVDR-L233S rescues the alopecic phenotype
Global loss of VDR expression in both humans with the syndrome of HVDRR and in mice with a genetic mutation in the VDR gene results in skin defects and alopecia (26, 30–33). Because this phenotype is not evident in either humans or mice that are unable to produce CYP27B1 (27–29), it has been concluded that normal hair cycling requires the VDR but not 1,25(OH)2D3. This concept was confirmed in subsequent experiments wherein both WT and 1,25(OH)2D3 binding-deficient hVDR-L233S cDNAs targeted selectively to keratinocytes of VDR-null mice were capable of rescuing the alopecic phenotype (34–36). Because alopecia in the VDR-null mouse was rescued in the hVDRWT/VDR−/− strain, the result suggested that we might also be able to examine the effects of the 1,25(OH)2D3-binding–deficient hVDR-L233S mutant on alopecia in the hVDRL233S/VDR−/− strains. Important advantages to this approach include the ability to target appropriately regulated VDR to skin cells using the endogenous human chromosomal gene locus and the opportunity to examine the dependence of the response on varied levels of expression of mutant hVDRs. To this end, we examined WT, VDR−/−, hVDRWT/VDR−/−, and each of the 3 hVDRL233S/VDR−/− strains of mice for evidence of alopecia. As can be seen in the photographs documented in Figure 6A, although VDR-null mice exhibited striking alopecia at 6 months of age relative to that of normal WT mice, alopecia was not evident in either the WT hVDR or the 3 mutant hVDR strains of mice. Interestingly, however, although the alopecia was fully rescued in both the T807/VDR−/− and T806/VDR−/− strains, normalization of hair growth in the T805/VDR−/− strain that expressed the lowest levels of the mutant hVDR was incomplete. Thus, hair growth appeared less full and less healthy. To explore the ability of these strains of mice to reinitiate the hair cycle, we depilated mice in the individual strains as above and assessed hair regrowth after 2 week. As can be seen in Figure 6A, although the VDR-null mouse was unable to initiate hair growth after depilation, the hair cycle was reinitiated in both the hVDR WT and the hVDR mutant-expressing strains of mice. The visual results, however, indicate that reinitiation in the T805/VDR−/− strain expressing the lowest amount of hVDR-L233S was again spotty and somewhat nonuniform. Finally, we performed a histological examination of the skin of each of these strains. As indicated in Figure 6B, although the skin of the VDR-null mouse revealed the presence of dermal cysts, virtually all of the remaining mouse strains exhibited normal epidermal morphology complete with the presence of hair follicles. These studies suggest not only that the 1,25(OH)2D3 binding–deficient hVDR-L233S mutant was fully capable of restoring the hair cycle and preventing the development of dermal cysts and alopecia in the skin but also that this inhibitory effect required very low levels of the mutant hVDR-L233S. Thus, we have created a true humanized mouse model of HVDRR without alopecia that is characterized by altered mineral homeostasis, defective skeletal mineralization, and parathyroid gland hyperplasia. These features resulted from defective regulation of transcription, yet were coincident with the absence of alopecia and the presence of normal epidermal morphology.
Figure 6.

Recovery of defects in skin of VDR-null mice by expression of hVDR-L233S. A, Top, 6-month-old mice representative of the strains indicated. Bottom, 2-month-old mice 2 weeks after depilation. Each image represents a mouse of the indicated strain. B, Histological sections obtained from the dorsal epidermis of 8-week-old mice were stained with hematoxylin and eosin. Arrows point to hair follicles and arrowheads to dermal cysts. Original magnifications, ×100. Images are representative of the strains indicated.
Discussion
We previously reported that mice carrying integrated copies of a modified BAC transgene containing the complete human VDR gene locus were capable of expressing hVDR protein at appropriate levels in all the mouse VDR-positive tissues examined (41). Importantly, this transgene when genetically crossed was also capable of rescuing the complex biological phenotype of the VDR-null mouse. In the study conducted herein, we used the existing WT hVDR BAC transgene to introduce a mutation that compromised the 1,25(OH)2D3-binding capacity of the hVDR (hVDR-L233S), inserted this transgene into the VDR-null background, and explored its properties relative to those obtained in hVDRWT transgenic mice. We discovered that this transgene was indeed expressed in a manner that was equivalent to that of the WT human transgene and that expression levels of the mutant hVDR-L233S ranged in 3 separate strains from very low to levels that were essentially equivalent to that seen for the VDR in normal mice. Continued examination of these 3 transgenic mouse strains revealed that expression of this 1,25(OH)2D3 binding–defective hVDR-L233S mutant failed to rescue the VDR-null phenotype characterized by hypocalcemia, hypophosphatemia, and hyperparathyroidism, parathyroid gland hyperplasia, shortened tibiae, and other skeletal defects linked to reduced BMD and altered growth plate morphology associated with deranged mineral homeostasis. It is of interest that the PTH levels in these hVDRL233S/VDR−/− strains of mice were elevated well beyond that seen in the VDR−/− background strain and more consistent with those observed in Cyp27b1−/− strains (Ref. 1 and Lee, S. M., and J. W. Pike, unpublished data). It is possible that although 1,25(OH)2D3-activated VDR is known to suppress elaboration of PTH from the parathyroid gland, the unliganded hVDR-L233S mutant in this instance may bind to the Pth gene and exert an opposite action to stimulate PTH synthesis and/or secretion. The inability of the hVDR-L233S mutant to rescue these specific features was probably due to its failure to mediate the 1,25(OH)2D3-induced regulation of genes such as Cyp24a1, Cyp27b1, and others in intestine, kidney, and bone. In contrast to these defects in the protein's activity, however, the hVDR-L233S mutant was fully functional in rescuing the alopecic phenotype observed in the VDR-null mouse, restoring the hair cycle, and preventing the development of dermal cysts. The ability of exceedingly low levels of hVDR-L233S protein to rescue these skin defects suggests that this particular function of the receptor is strikingly different from that as defined through transcription for the WT VDR. We conclude that the broad tissue-specific expression of the hVDR-L233S mutant in mice results in a humanized mouse model of HVDRR without alopecia.
The BAC transgene approach we used to prepare transgenic mice took advantage of our previous characterization of the mouse and human VDR genes and their regulatory boundaries as well as the discovery that the human gene was appropriately regulated in a murine background (39–41). Importantly, the human transgene, like that for the mouse, contained the natural regulatory domains that control the cell-specific expression of the VDR in mice during adult homeostasis and probably during growth and development as well (41). This feature enabled the expression of hVDR-L233S in a fashion similar to that of the endogenous mouse gene. As a result, this transgene also facilitated the study of the functional activity of the hVDR-L233S mutant in skin, revealing that despite its failure to mediate transcriptional activation in key tissues, the mutant was capable of rescuing the aberrant skin phenotype of the VDR-null mouse. We believe that these features of the human transgene augment the key observations made several years ago that the keratin-14 (K14) promoter–targeted expression of cDNAs for both the WT and L233S mutant forms of the hVDR in the keratinocyte was able to rescue the VDR-null phenotype (34–36). Whether this highly selective targeting to the keratinocyte failed to reveal a role(s) for this receptor in other cellular components of the hair cycling process is unknown as was the impact of the K14 promoter's off-target directed expression of the receptor to other non-vitamin D–sensitive tissues (36). Clearly, however, the current model should allow these issues to be addressed in more detail. Importantly, our observation that very low amounts of the hVDR-L233S mutant can also effectively rescue the skin phenotype suggests that the mechanism through which the VDR functions in a 1,25(OH)2D3-independent manner may be very different from that of its 1,25(OH)2D3-activated counterpart. Interestingly, this observation of VDR sensitivity was made earlier in the targeted keratinocyte model as well, although the mutant form of hVDR was not examined in that particular study (36). Nevertheless, the creation of this mouse model should enable future studies of this ligand-independent prototypic function of the VDR, including other studies in skin, with the potential for this type of activity in additional tissues.
The development of an authentic in vivo model for ligand-independent VDR action is important. Its value derives from the fact that although it may appear self-evident that a receptor incapable of binding hormone might not be able to regulate transcription in target tissues through currently understood mechanisms, the broad cell type–specific expression of a full-length VDR without this capability will enable the search for additional ligand-independent functions of this receptor in other tissues for which the skin may represent a prototype. None of the mouse models of VDR action available presently are capable of such a search as they do not express the VDR (30–33). There is a clear precedent for this action, because many nuclear receptors including those for the estrogens and androgens exhibit properties such that they are activated through mechanisms independent of their cognate ligands (43–47). With respect to the VDR, there are numerous hints of this possibility as well, associated directly with the receptor's ability to interact with the specific transcription factor targets of other signaling pathways both on or off DNA (18, 19); a recent report that the role of VDR in fat cell differentiation may be dissociated from the influence of 1,25(OH)2D3 [independent of 1,25(OH)2D3] may also be relevant and deserving of further exploration (48). Interestingly, a recent analysis of VDR binding sites on a genome-wide scale has revealed that although the vast majority are induced by 1,25(OH)2D3, relatively robust levels of VDR can be found in the absence of 1,25(OH)2D3 at a rather substantial number of sites on the genome (13, 21, 22, 49). The underlying mechanism(s) that permit this ligand-independent VDR DNA binding as well as its functional consequence(s) is not clear at present. Interestingly, this unique mouse model may also facilitate the search for novel VDR ligands for which intestinal lithocholic acid may be representative (50). Indeed, the possibility still exists that the VDR expressed in keratinocytes of the hair follicle may be activated through binding to a local and perhaps cell autonomously produced ligand other than 1,25(OH)2D3.
The actions of the hVDR-L233S mutant identified here in skin are both ligand-independent and exquisitely sensitive to low levels of VDR expression (actions in the T805/VDR−/− and T806/VDR−/− strains). It is important to note that not all VDR actions in the skin are ligand independent, because it has been shown that the VDR is able to induce in a 1,25(OH)2D3-sensitive manner the expression of numerous genes in the epidermis that appear to play a role in keratinocyte differentiation and barrier function (51–54). A similar likelihood exists in the ability of the VDR to control the development of tumors in the skin and perhaps elsewhere (55–57). Indeed, analogous to the phenotype of alopecia, although tumors develop in the skin in response to UV irradiation or to 7,12-dimethylbenzanthracene treatment in the absence of the VDR, genetic inactivation of Cyp27b1 and the accompanying loss of circulating 1,25(OH)2D3 in these animals does not lead to an increase in tumor formation (56, 57). Given this dual role of the VDR in both 1,25(OH)2D3-independent and -sensitive functions, the question arises as to how these two functionally distinct activities of the VDR may be coordinated. This is particularly intriguing because while actions stimulated by the ligand are gated and modulated by changing levels of 1,25(OH)2D3 in the cell, those that are independent of the ligand appear to be acutely controlled simply by the presence of low levels of VDR protein. Thus, this activity may be representative of a function that appeared during an earlier and more primitive stage of VDR evolution and certainly well before the emergence of allosteric control through ligands exemplified by 1,25(OH)2D3. We hypothesize that the actions of the unoccupied VDR are separated even further from those that involve 1,25(OH)2D3 as a result of the apparent requirement for low levels of the VDR. This separation may be particularly important in skin, where UVB irradiation to produce vitamin D from 7-dehydrocholesterol (58) and expression of the enzymes necessary for conversion of the vitamin to 25-hydroxyvitamin D3 and then to 1,25(OH)2D3 are all present, resulting potentially in significant intracellular levels of each of these forms of vitamin D (59, 60). Of course, although receptors may operate in the hair follicle in a 1,25(OH)2D3-independent manner, the effect of the ligand on the VDR relative to this action in the hair follicle is not known. The separation of 1,25(OH)2D3 bound and unbound VDR functions is indeed highlighted by the observation that the ratio of 1,25(OH)2D3-occupied to -unoccupied VDR in tissues in vivo is surprisingly low, ranging from 7% to 20% (61, 62). Thus, even in times of high circulating 1,25(OH)2D3, significant levels of unoccupied VDR are likely to be present and perhaps effectively insulated from occupancy by hormone, although alternatively they could be localized to different subcellular sites as well. Interestingly, epidermal differentiation proceeds in the absence of 1,25(OH)2D3 (4). Thus, this effect of the hormone in skin appears to represent a redundant function. On the other hand, the role of the VDR in hair cycle reinitiation appears to be critical, a dependency mirrored by the equal and absolute requirement for the presence of other proteins such as Hairless (63). We suggest that the actions of the unliganded VDR in other tissues, if such activities exist, may be subject to some of these same principles.
In conclusion, we have prepared a humanized mouse model of HVDRR that is free from alopecia and other epidermal abnormalities. This model was created by integrating into the mouse genome a DNA segment that contained the natural human VDR gene locus into which we have introduced a point mutation that abrogates the ability of the VDR to bind 1,25(OH)2D3. Importantly, although this receptor was unable to activate transcription in response to 1,25(OH)2D3 in classic target tissues or to rescue the diverse skeletal phenotype of the VDR-null mouse, it was fully functional in reestablishing the hair cycle, restoring normal cutaneous morphology, and perhaps rescuing additional selected activities in the skin. We suggest that this animal model may be useful in exploring tissues for additional biological roles of the VDR that do not require 1,25(OH)2D3, in identifying additional features of the HVDRR syndrome, and perhaps in searching for presently undiscovered ligands for the VDR.
Acknowledgments
We thank the members of the Pike Laboratory for their helpful contributions to this work. We acknowledge David Nehls, Regina Berget, and Douglas Jacobson for the extensive animal husbandry associated with this study. We also thank Ms Jean Prahl for conducting the titrated hormone binding assay that confirmed the inability of the hVDR-L233S mutant to bind to 1,25(OH)2D3.
This work was supported by the National Institute of Arthritis, Musculoskeletal and Skin Diseases (Grant AR-045173) and the National Institute of Diabetes, Digestive and Kidney Diseases (Grant DK-072281 to J.W.P.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BAC
- bacterial artificial chromosome
- BMD
- bone mineral density
- 1,25(OH)2D3
- 1,25-dihydroxyvitamin D3
- hVDR
- human vitamin D receptor
- HVDRR
- hereditary 1,25-dihydroxyvitamin D3–resistant rickets
- RXR
- retinoid X receptor
- VDDR-1
- vitamin D dependency rickets type 1
- VDR
- vitamin D receptor
- WT
- wild type.
References
- 1. Bouillon R, Carmeliet G, Verlinden L, et al. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev. 2008;29:726–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DeLuca HF. Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr. 2004;80(6 suppl):1689S–1696S. [DOI] [PubMed] [Google Scholar]
- 3. Reichrath J, Schilli M, Kerber A, Bahmer FA, Czarnetzki BM, Paus R. Hair follicle expression of 1,25-dihydroxyvitamin D3 receptors during the murine hair cycle. Br J Dermatol. 1994;131:477–482. [DOI] [PubMed] [Google Scholar]
- 4. Sakai Y, Demay MB. Evaluation of keratinocyte proliferation and differentiation in vitamin D receptor knockout mice. Endocrinology. 2000;141:2043–2049. [DOI] [PubMed] [Google Scholar]
- 5. Provvedini DM, Tsoukas CD, Deftos LJ, Manolagas SC. 1,25-Dihydroxyvitamin D3 receptors in human leukocytes. Science. 1983;221:1181–1183. [DOI] [PubMed] [Google Scholar]
- 6. Heine G, Niesner U, Chang HD, et al. 1,25-Dihydroxyvitamin D3 promotes IL-10 production in human B cells. Eur J Immunol. 2008;38:2210–2218. [DOI] [PubMed] [Google Scholar]
- 7. Chun RF, Liu PT, Modlin RL, Adams JS, Hewison M. Impact of vitamin D on immune function: lessons learned from genome-wide analysis. Front Physiol. 2014;5:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D3 is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen S, Law CS, Grigsby CL, et al. Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac hypertrophy. Circulation. 2011;124:1838–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ordonez-Moran P, Larriba MJ, Pendas-Franco N, Aguilera O, Gonzalez-Sancho JM, Munoz A. Vitamin D and cancer: an update of in vitro and in vivo data. Front Biosci. 2005;10:2723–2749. [DOI] [PubMed] [Google Scholar]
- 11. Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pike JW, Meyer MB, Bishop KA. Regulation of target gene expression by the vitamin D receptor—an update on mechanisms. Rev Endocr Metab Disord. 2012;13:45–55. [DOI] [PubMed] [Google Scholar]
- 13. Pike JW, Meyer MB. Fundamentals of vitamin D hormone-regulated gene expression [published online November 12, 2013]. J Steroid Biochem Mol Biol. doi:10.1016/j.jsbmb.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. [DOI] [PubMed] [Google Scholar]
- 15. Smith CL, O'Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25:45–71. [DOI] [PubMed] [Google Scholar]
- 16. Dhawan P, Peng X, Sutton AL, et al. Functional cooperation between CCAAT/enhancer-binding proteins and the vitamin D receptor in regulation of 25-hydroxyvitamin D3 24-hydroxylase. Mol Cell Biol. 2005;25:472–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kong J, Zhu X, Shi Y, et al. VDR attenuates acute lung injury by blocking Ang-2-Tie-2 pathway and renin-angiotensin system. Mol Endocrinol. 2013;27:2116–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Salehi-Tabar R, Nguyen-Yamamoto L, Tavera-Mendoza LE, et al. Vitamin D receptor as a master regulator of the c-MYC/MXD1 network. Proc Natl Acad Sci USA. 2012;109:18827–18832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Larriba MJ, González-Sancho JM, Bonilla F, Muñoz A. Interaction of vitamin D with membrane-based signaling pathways. Front Physiol. 2014;5:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xie Z, Chang S, Oda Y, Bikle DD. Hairless suppresses vitamin D receptor transactivation in human keratinocytes. Endocrinology. 2006;147:314–323. [DOI] [PubMed] [Google Scholar]
- 21. Meyer MB, Goetsch PD, Pike JW. Genome-wide analysis of the VDR/RXR cistrome in osteoblast cells provides new mechanistic insight into the actions of the vitamin D hormone. J Steroid Biochem Mol Biol. 2010;121:136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meyer MB, Goetsch PD, Pike JW. VDR/RXR and TCF4/β-catenin cistromes in colonic cells of colorectal tumor origin: impact on c-FOS and c-MYC gene expression. Mol Endocrinol. 2012;26:37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heikkinen S, Väisänen S, Pehkonen P, Seuter S, Benes V, Carlberg C. Nuclear hormone 1α,25-dihydroxyvitamin D3 elicits a genome-wide shift in the locations of VDR chromatin occupancy. Nucleic Acids Res. 2011;39:9181–9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brooks MH, Bell NH, Love L, et al. Vitamin-D-dependent rickets type II. Resistance of target organs to 1,25-dihydroxyvitamin D. N Engl J Med. 1978;298:996–999. [DOI] [PubMed] [Google Scholar]
- 25. Marx SJ, Spiegel AM, Brown EM, et al. A familial syndrome of decrease in sensitivity to 1,25-dihydroxyvitamin D. J Clin Endocrinol Metab. 1978;47:1303–1310. [DOI] [PubMed] [Google Scholar]
- 26. Feldman D, Malloy PJ. Mutations in the vitamin D receptor and hereditary vitamin D-resistant rickets. Bonekey Rep. 2014;3:510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feldman D, Malloy PJ, Miller WL. Genetic disorders of vitamin D synthesis and action. In: Thakker RV, Whyte MP, Eisman J, Igarashi T, eds. Genetics of Bone Biology and Skeletal Disease. San Diego, CA: Elsevier; 2013:537–552. [Google Scholar]
- 28. Dardenne O, Prud'homme J, Arabian A, Glorieux FH, St-Arnaud R. Targeted inactivation of the 25-hydroxyvitamin D3-1α-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology. 2001;142:3135–3141. [DOI] [PubMed] [Google Scholar]
- 29. Panda DK, Miao D, Tremblay ML, et al. Targeted ablation of the 25-hydroxyvitamin D 1α-hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc Natl Acad Sci USA. 2001;98:7498–7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoshizawa T, Handa Y, Uematsu Y, et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet. 1997;16:391–396. [DOI] [PubMed] [Google Scholar]
- 31. Li YC, Pirro AE, Amling M, et al. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci USA. 1997;94:9831–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Van Cromphaut SJ, Dewerchin M, Hoenderop JG, et al. Duodenal calcium absorption in vitamin D receptor-knockout mice: functional and molecular aspects. Proc Natl Acad Sci USA. 2001;98:13324–13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Erben RG, Soegiarto DW, Weber K, et al. Deletion of deoxyribonucleic acid binding domain of the vitamin D receptor abrogates genomic and nongenomic functions of vitamin D. Mol Endocrinol. 2002;16:1524–1537. [DOI] [PubMed] [Google Scholar]
- 34. Chen CH, Sakai Y, Demay MB. Targeting expression of the human vitamin D receptor to the keratinocytes of vitamin D receptor null mice prevents alopecia. Endocrinology. 2001;142:5386–5389. [DOI] [PubMed] [Google Scholar]
- 35. Skorija K, Cox M, Sisk JM, et al. Ligand-independent actions of the vitamin D receptor maintain hair follicle homeostasis. Mol Endocrinol. 2005;19:855–862. [DOI] [PubMed] [Google Scholar]
- 36. Kong J, Li XJ, Gavin D, Jiang Y, Li YC. Targeted expression of human vitamin D receptor in the skin promotes the initiation of the postnatal hair follicle cycle and rescues the alopecia in vitamin D receptor null mice. J Invest Dermatol. 2002;118:631–638. [DOI] [PubMed] [Google Scholar]
- 37. Miyamoto K, Kesterson RA, Yamamoto H, et al. Structural organization of the human vitamin D receptor chromosomal gene and its promoter. Mol Endocrinol. 1997;11:1165–1179. [DOI] [PubMed] [Google Scholar]
- 38. Jehan F, DeLuca HF. Cloning and characterization of the mouse vitamin D receptor promoter. Proc Natl Acad Sci USA. 1997;94:10138–10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zella LA, Kim S, Shevde NK, Pike JW. Enhancers located within two introns of the vitamin D receptor gene mediate transcriptional autoregulation by 1,25-dihydroxyvitamin D3. Mol Endocrinol. 2006;20:1231–1247. [DOI] [PubMed] [Google Scholar]
- 40. Zella LA, Meyer MB, Nerenz RD, Lee SM, Martowicz ML, Pike JW. Multifunctional enhancers regulate mouse and human vitamin D receptor gene transcription. Mol Endocrinol. 2010;24:128–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee SM, Bishop KA, Goellner JJ, O'Brien CA, Pike JW. Mouse and human BAC transgenes recapitulate tissue-specific expression of the vitamin D receptor in mice and rescue the VDR-null phenotype. Endocrinology. 2014;155:2064–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meyer MB, Goetsch PD, Pike JW. A downstream intergenic cluster of regulatory enhancers contributes to the induction of CYP24A1 expression by 1α,25-dihydroxyvitamin D3. J Biol Chem. 2010;285:15599–15610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ignar-Trowbridge DM, Nelson KG, Bidwell MC, et al. Coupling of dual signaling pathways: epidermal growth factor action involves the estrogen receptor. Proc Natl Acad Sci USA. 1992;89:4658–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ma ZQ, Santagati S, Patrone C, Pollio G, Vegeto E, Maggi A. Insulin-like growth factors activate estrogen receptor to control the growth and differentiation of the human neuroblastoma cell line SK-ER3. Mol Endocrinol. 1994;8:910–918. [DOI] [PubMed] [Google Scholar]
- 45. Arnold SF, Obourn JD, Jaffe H, Notides AC. Phosphorylation of the human estrogen receptor by mitogen-activated protein kinase and casein kinase II: consequence on DNA binding. J Steroid Biochem Mol Biol. 1995;55:163–172. [DOI] [PubMed] [Google Scholar]
- 46. Patrone C, Gianazza E, Santagati S, Agrati P, Maggi A. Divergent pathways regulate ligand-independent activation of ERα in SK-N-BE neuroblastoma and COS-1 renal carcinoma cells. Mol Endocrinol. 1998;12:835–841. [DOI] [PubMed] [Google Scholar]
- 47. Wang Q, Li W, Zhang Y, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Malloy PJ, Feldman BJ. Cell-autonomous regulation of brown fat identity gene UCP1 by unliganded vitamin D receptor. Mol Endocrinol. 2013;27:1632–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pike JW, Lee SM, Meyer MB. Regulation of gene expression by 1,25-dihydroxyvitamin D3 in bone cells: exploiting new approaches and defining new mechanisms. Bonekey Rep. 2014;3:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Makishima M, Lu TT, Xie W, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–1316. [DOI] [PubMed] [Google Scholar]
- 51. Su MJ, Bikle DD, Mancianti ML, Pillai S. 1,25-Dihydroxyvitamin D3 potentiates the keratinocyte response to calcium. J Biol Chem. 1994;269:14723–14729. [PubMed] [Google Scholar]
- 52. Pillai S, Bikle DD, Su MJ, Ratnam A, Abe J. 1,25-Dihydroxyvitamin D3 upregulates the phosphatidylinositol signaling pathway in human keratinocytes by increasing phospholipase C levels. J Clin Invest. 1995;96:602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xie Z, Bikle DD. Cloning of the human phospholipase C-γ1 promoter and identification of a DR6-type vitamin D-responsive element. J Biol Chem. 1997;272:6573–6577. [DOI] [PubMed] [Google Scholar]
- 54. Bikle DD, Chang S, Crumrine D, et al. 25 Hydroxyvitamin D 1α-hydroxylase is required for optimal epidermal differentiation and permeability barrier homeostasis. J Invest Dermatol. 2004;122:984–992. [DOI] [PubMed] [Google Scholar]
- 55. Zinser GM, Sundberg JP, Welsh J. Vitamin D3 receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis. 2002;23:2103–2109. [DOI] [PubMed] [Google Scholar]
- 56. Teichert AE, Elalieh H, Elias PM, Welsh J, Bikle DD. Overexpression of hedgehog signaling is associated with epidermal tumor formation in vitamin D receptor-null mice. J Invest Dermatol. 2011;131:2289–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ellison TI, Smith MK, Gilliam AC, MacDonald PN. Inactivation of the vitamin D receptor enhances susceptibility of murine skin to UV-induced tumorigenesis. J Invest Dermatol. 2008;128:2508–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Holick MF, MacLaughlin JA, Clark MB, et al. Photosynthesis of previtamin D3 in human skin and the physiologic consequences. Science. 1980;210:203–205. [DOI] [PubMed] [Google Scholar]
- 59. Zehnder D, Bland R, Williams M, et al. Extrarenal expression of 25-hydroxyvitamin D3-1α-hydroxylase. J Clin Endocrinol Metab. 2001;86:888–894. [DOI] [PubMed] [Google Scholar]
- 60. Ellfolk M, Norlin M, Gyllensten K, Wikvall K. Regulation of human vitamin D3 25-hydroxylases in dermal fibroblasts and prostate cancer LNCaP cells. Mol Pharmacol. 2009;75:1392–1399. [DOI] [PubMed] [Google Scholar]
- 61. Hunziker W, Walters MR, Norman AW. 1,25-Dihydroxyvitamin D3 receptors. Differential quantitation of endogenously occupied and unoccupied sites. J Biol Chem. 1980;255:9534–9537. [PubMed] [Google Scholar]
- 62. Dokoh S, Haussler MR, Pike JW. Development of a radioligand immunoassay for 1,25-dihydroxycholecalciferol receptors utilizing monoclonal antibody. Biochem J. 1984;221:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zarach JM, Beaudoin GM, 3rd, Coulombe PA, Thompson CC. The co-repressor hairless has a role in epithelial cell differentiation in the skin. Development. 2004;131:4189–4200. [DOI] [PubMed] [Google Scholar]