

Abstract
Primate sequence comparisons are difficult to interpret due to the high degree of sequence similarity shared between such closely related species. Recently, a novel method, phylogenetic shadowing, has been pioneered for predicting functional elements in the human genome through the analysis of multiple primate sequence alignments. We have expanded this theoretical approach to create a computational tool, eShadow, for the identification of elements under selective pressure in multiple sequence alignments of closely related genomes, such as in comparisons of human-to-primate or mouse-to-rat DNA. This tool integrates two different statistical methods and allows for the dynamic visualization of the resulting conservation profile. eShadow also includes a versatile optimization module capable of training the underlying Hidden Markov Model to differentially predict functional sequences. This module grants the tool high flexibility in the analysis of multiple sequence alignments and in comparing sequences with different divergence rates. Here, we describe the eShadow comparative tool and its potential uses for analyzing both multiple nucleotide and protein alignments to predict putative functional elements.
Cross-species sequence comparisons between distantly related genomes, such as those of humans and rodents, have been instrumental in defining evolutionarily conserved elements with critical biological roles, whether they function as coding exons (Pennacchio et al. 2001; Gilligan et al. 2002), regulatory elements (Loots et al. 2000; Ghanem et al. 2003; Nobrega et al. 2003), or microRNAs (Lim et al. 2003). Four widely used tools intended for generating genomic alignments are web accessible and augmented by easy-to-use graphical interfaces, Blast2Seq (Tatusova and Madden 1999), PipMaker (Schwartz et al. 2000; http://bio.cse.psu.edu/pipmaker/), Vista (Mayor et al. 2000; http://www-gsd.lbl.gov/VISTA/), and zPicture (Ovcharenko et al. 2004; http://zpicture.dcode.org). Also, several genome browsers provide an effortless access to precomputed genome-scale sequence alignments for any region in the human genome. These include Ensembl (Hubbard et al. 2002; http://www.ensembl.org/), the ECR Browser (http://ecrbrowser.dcode.org/), Pip Dispenser (Schwartz et al. 2003; http://bio.cse.psu.edu/genome/hummus/), Vista Browser (Couronne et al. 2003; http://pipeline.lbl.gov), and the Genome Browser at UCSC (Karolchik et al. 2003; http://genome.ucsc.edu).
However, a deeper understanding of the biology and the evolution of Homo sapiens will require comparisons not only to distantly related genomes, such as rodents and fishes, but also to our closest relatives, the great apes. In such comparisons, it is very challenging to extract statistically significant differences, as the genomes of humans and their primate relatives are very similar at the nucleotide level (>90%) (Britten 2002; Silva and Kondrashov 2002; Anzai et al. 2003; Hellmann et al. 2003). Available comparative sequence analysis tools and methodologies have, in general, been developed to analyze more distant evolutionary relationships and are not fine-tuned to analyze recent evolutionary events. Such tools are not sensitive enough to allow for meaningful comparisons involving recent segmental duplications in the human genome (Bailey et al. 2002), dynamically evolving clusters of paralogous genes such as the zinc-finger transcription factor families (Shannon et al. 2003), or slowly diverging genomic intervals such as the Hox gene clusters (Balavoine et al. 2002). But most importantly, in light of the emerging sequence of the chimpanzee genome, we require new resources that will permit meaningful comparisons between humans and other primates. Such tools will advance our ability to extract functional information from primate comparisons and expand our current understanding of human health and biology.
Recently, a novel approach, phylogenetic shadowing, was developed to compute and statistically evaluate conservation profiles of multiple sequence alignments from closely related species. This statistical method permitted the accurate prediction of exons and transcriptional regulatory elements in human–primate comparisons, and validated the use of this approach for deciphering primate-specific functional DNA sequences (Boffelli et al. 2003). On the basis of the successful use of the phylogenetic shadowing method, we have created a publicly accessible automated tool, eShadow (http://eshadow.dcode.org/), that applies this strategy to the analysis of any closely related sequences. Also, eShadow extends the phylogenetic shadowing approach to include the analysis of multiple protein alignments, and to reduce the number of species required for the identification of functional elements. The eShadow tool incorporates two distinct approaches for finding functional elements—Hidden Markov Model Islands (HMMI) and Divergence Threshold (DT) scans of multiple (or pairwise) sequence alignments. Here, we report the computational design and algorithms underlining the eShadow tool, and we suggest several applications, including the analysis of (1) coding exons, (2) noncoding elements, and (3) protein domains. We show that by overlapping HMMI predictions with a distribution of ORFs and fully conserved splice sites, eShadow can also be used to highlight regions with coding potential. Finally, we demonstrate how this program is trainable, and highly flexible to be generalized to human/baboon pairwise comparisons.
RESULTS
eShadow Computational Design and Visualization Scheme
eShadow is an interactive computational tool for aligning, visualizing, and evaluating evolutionarily conservation profiles in multiple and pairwise nucleotide or protein alignments of closely related sequences. The tool works best in alignments between sequences characterized by divergence rates less than the average human versus mouse neutral substitution rate of 0.46 substitutions per site (Waterston et al. 2002). eShadow analysis proceeds in three major steps as follows: (1) generating multiple sequence alignments (MSAs), (2) visualizing MSAs as percent variation plots, and (3) statistically evaluating MSAs to detect regions of high conservation (Fig. 1). eShadow generates MSAs using the multiple aligner program ClustalW (http://www.ebi.ac.uk/clustalw/). This tool creates alignments by first constructing a phylogenetic tree on the basis of sequence similarity, and then follows with successive pairwise alignments in the order provided by the tree. ClustalW is able to align multiple closely related sequences (such as human/baboon/chimp or mouse/rat genomic sequences, which are ≥80% identical) up to 200 kb in length. While performing protein alignments, ClustalW weighs amino acid substitutions according to the divergence rates of the sequences being aligned, and assigns residue-specific gap penalties that are adjusted locally, resulting in increasing or decreasing the score, depending on the potential secondary structure of the protein (Thompson et al. 1994; Chenna et al. 2003).
Figure 1.

Schematic dataflow of the eShadow program.
eShadow visualizes MSAs as percent variation (or mismatch) plots. The percent of mismatched nucleotides or amino acids is calculated in a sliding window of user-defined length; where the percent identity y, in a window size w, centered at a given position x, is plotted at the (x,y) coordinate. The peaks and valleys of the conservation plot correspond to regions of low and high variation, respectively, and 0% variation signifies 100% sequence identity in the MSA. To increase plasticity for the visual representation of the data, eShadow uniquely allows users to interactively choose the base organism, modify parameters, and annotation files—features absent from all other available multiple or pairwise sequence alignment tools.
The eShadow analytical module implements two different statistical methods of scanning MSAs to detect slow-mutating regions as follows: (1) HMMI, and (2) DT. Whereas Hidden Markov Model methods are used extensively to detect functional elements in raw genomic and protein sequences as well as in sequence alignments, DT methods are typically used for the analysis of conservation plots. HMMI implementations include gene prediction (Burge and Karlin 1997; Krogh 1997), CpG islands localization (Takai and Jones 2002), noncoding RNAs identification (Rivas and Eddy 2001), protein domain (Truong and Ikura 2002), and protein fold predictions (Bienkowska et al. 2000). We used a two-state HMMI to analyze conservation profiles and to predict conserved (slowly diverging) regions. This method does not utilize a sliding window, rather it analyzes the underlying distribution of matches and mismatches in the alignments. The DT method distinguishes conserved elements on the basis of their length and the level of complete nucleotide/amino acid identity. In contrast to the HHMI approach, DT analysis is performed by scanning the alignment summary for regions corresponding to the number of matches x in a sliding window of predefined length y. The DT method is used by most pairwise alignment programs and is probably the most commonly used approach for biologists to define evolutionary conservation (Schwartz et al. 2000).
The analytical component of the eShadow tool also contains several optional features as follows: (1) an ORF detection block and (2) an optimization module to assist during the characterization of conservation patterns across alignments. By superimposing ORF predictions, eShadow's HMMI detection module identifies nucleotide regions with high potential to code for proteins, possibly differentiating coding from noncoding conserved elements. The optimization module allows the user to train the program to identify parameters that would distinguish features similar in nature to a set of known elements (exons or regulatory elements) from the background noise. We have implemented three complementary optimization methods into the eShadow tool as follows: (1) Baum-Welch (Durbin et al. 1998), (2) Maximum Likelihood (Durbin et al. 1998), and (3) Golden Section Search (Press et al. 1988).
Applications for the eShadow Tool
eShadow has been designed for comparing sequences with relatively small interspecies divergence rates, preferably classified to the same class or order. The detection of such elements is very difficult by currently available computational means, and is critical for molecularly distinguishing biological functions unique to small clades of organisms (Cooper et al. 2003). Unlike most available comparative sequence analysis tools, eShadow can be dynamically adjusted and trained to accommodate evolutionary relationships as distant as human and mouse or as close as two primates. However, other tools such as PipMaker, zPicture or VISTA are more effective in pairwise analysis of distantly related sequences and are specifically designed for such applications (Mayor et al. 2000; Schwartz et al. 2000). The particular analytical scope of the eShadow tool is to assist in the discovery of elements distinctly shared by classes of organisms tightly clustered on the same branch of the evolutionary tree. Here, we illustrate three major applications for the eShadow tool as follows: (1) identification of coding exons, (2) prediction of conserved noncoding elements, and (3) evaluation of protein domains in alignments.
Detecting Coding Exons
The two statistical methods implemented into the eShadow tool were tested for the ability to detect conserved elements across four genomic intervals and to accurately predict the known coding regions corresponding to five exons from four different genes (ApoB; Plasminogen; LXR-α; CETP). These four genomic intervals have been sampled extensively across multiple primate species and currently represent the only data set for which there are from 13 to 16 unique primate sequences available (Boffelli et al. 2003). Even though the set of sequenced primates varied slightly for each tested region, the available sequences for each genomic interval spanned the primate phylogeny evenly, resulting in similar substitution rates per human base pair in all tested cases (Supplemental Table S1). We optimized the parameters for each analytical method to detect regions with slow mutation rates, and found a set of common parameters that identified all of the exons present in these genomic intervals. The HMMI method detected all exons (parameters: eS = 0.85; eF = 0.77; T = 0.1 [details in Supplemental materials; Fig. S1]), whereas the DT method missed the shortest exon (parameters: percent variation = 15; length = 100 bp; Fig. 2).
Figure 2.

Predicting exons in multiple primate alignments. eShadow exon prediction and conservation plots for ApoB (1.4 kb) (A), Plasminogen (1.2 kb) (B), LXR-α (1.35 kb) (C), and CETP (1 kb) (D) loci using multiple primate alignments. HMMI predictions (0.85/0.77/0.1) are in beige and DT regions (15/100) are in green. The locations of known exons are depicted as red bars. Sequences are available at NCBI (accession nos. AY190030–AY190042, AY192729–AY192785).
Despite the tremendous advances achieved in DNA-sequencing technologies, obtaining the sequence of dozens of closely related vertebrate-sized genomes is still not a practical goal. We therefore asked whether single human/primate pairwise alignments might contain enough information to distinguish conserved (slow-mutating) from neutral (fast-mutating) regions. This task is highly intricate, as the substitution rate decreases significantly when switching from an MSA to a pairwise alignment (Supplemental Fig. S2). Figure 3 illustrates eShadow's ability to predict the ApoB exon from a single human/primate (Allouatta seniculus) pairwise alignment (Fig. 3C) as accurately as it can be predicted from a human/mouse (Fig. 3A) or a primate MSA (Fig. 3B). Similar results were obtained in 54 other exons analyzed in human/baboon alignments (Table 1). These results suggest that if properly analyzed, single human/primate pairwise alignments have the potential to be as informative for exon identification as human/rodent alignments are.
Figure 3.

Predicting exons in single primate pairwise alignments. eShadow analysis for ApoB (1.4 kb) region using human/mouse (A), human and 14 primate sequences (B), and human/Allouatta seniculus sequences (C); 50-bp sliding window was utilized to smoothen the conservation profile. (Right) Human/mouse (D) and human/Allouatta seniculus (E) sequence comparisons plotted with standard 50%–100% thresholds and 100-bp sliding window. Exons (blue) and evolutionarily conserved regions (ECRs) (red) are indicated in D and E zplots (http://zpicture.dcode.org). HMMI predictions (beige), ORF predictions (gray), exon annotations (red), and exon predictions (yellow) are visualized in eShadow plots (A–C). Percent variation: y-axis, size in bp: x-axis (A–C). Percent identity: y-axis, size in kb: x-axis (D,E).
Table 1.
Evaluating eShadow's Performance on Recapitulating Human/Mouse Conservation Patterns in Human/Baboon Alignments
|
Detected coding exons
|
|||||
|---|---|---|---|---|---|
| Gene locus | Sensitivity | Specificity | Common region lengtha | All | >150 bp |
| WNT2 | 67.9% | 61.1% | 52.8 kb | 5/5 | 4/4 |
| CECR | 55.7% | 82.5% | 63.4 kb | 7/20 | 5/7 |
| PCQAP | 51.1% | 89.8% | 123.4 kb | 13/20 | 8/8 |
| SNAP29 | 46.0% | 89.6% | 125.1 kb | 19/27 | 7/10 |
| TCF4 | 61.4% | 56.7% | 137.8 kb | 10/11 | 1/1 |
| Average | 59.3% | 77.6% | 100.5 kb | 54/87 (62%) | 25/30 (83%) |
Sequences for the WNT2 region were obtained by excising ∼53 kb from the ∼2 Mb region [NISC Comparative Sequencing Program has sequenced for the CFTR locus sequencing project (Thomas et al., 2003)]. A total of four baboon BACs spanning CECR, PCQAP, SNAP29, and TCF4 gene loci were downloaded from NCBI (Accession nos. AC091672, AC128639, AC129881, AC113267). Human and mouse sequences were obtained from UCSC database.
Common region lengths are defined as regions of homology (kb) flanked by at least one ECR and one HMMI prediction. HMMI parameters used: 0.98/0.90/0.005.
Detecting Conserved Noncoding Elements
Because humans and rodents share the majority of their protein-coding genes (Waterston et al. 2002), it has been hypothesized that most of the phenotypic differences between clades of mammals are attributed to differences in noncoding sequences. In some cases, these differences may involve substantial changes in regulatory sequences that have occurred during the ∼80 Myrs separating the rodent and human lineages (Dermitzakis and Clark 2002; Scemama et al. 2002), limiting H/M comparisons to functions that are more globally shared by mammals and vertebrates. The phylogenetic shadowing approach has been shown to be suited for the identification of lineage-specific noncoding conserved elements through comparisons of several closely related primate genomic regions (Boffelli et al. 2003). Because this approach is limited by the amount of sequence data available for species tightly clustered on the same branch of the phylogenetic tree, we tested eShadow's ability to recapitulate conservation patterns when the number of input sequences is reduced from >10 different species to only two or three. We also limited our analysis to organisms evolutionarily close to humans and in the primate lineage—such as baboon and chimp (genome sequences expected to be generated in the foreseeable future).
We analyzed 53 kb from the Wingless-type MMTV Integration Site Family, Member 2 gene locus (WNT2), which has been deeply sequenced in many species, including chimps and baboons in addition to humans and mice (Thomas et al. 2003). H/M comparisons identified all translated and untranslated (UTR) exons included in a collection of 62 evolutionarily conserved regions (ECRs; ≥100 bp and ≥70%; Fig. 4). We addressed whether eShadow primate-specific comparisons can possibly recapitulate the H/M conservation patterns, as well as identify additional primate-specific conserved elements through the use of human/baboon/chimp (H/B/C) comparisons. The eShadow HMMI module was first trained on the WNT2 exons, and the optimized parameters were used to analyze the conservation pattern across the entire 53-kb genomic interval.
Figure 4.

eShadow analysis for the WNT2 region. Human/mouse conservation plot (A) compared with human/baboon/chimp eShadow conservation plot (B). Human/mouse alignments were generated and visualized by the zPicture program (http://zpicture.dcode.org), using standard parameters (≥ 100 bp; ≥ 70%) and conserved elements corresponding to exons (blue), UTRs (yellow), intronic (pink), and intergenic (red) elements are indicated (A). Human/baboon/chimp alignment plot (B) depicting regions of conservation (purple) and HMMI predictions (beige). y-axis corresponds to percent identity (A) and percent variation (B). x-axis corresponds to size in base pairs (A,B).
Whereas the H/B/C alignment demonstrated ∼12% nucleotide variation, the conservation pattern clearly distinguished regions with different evolutionary rates. HMMI predictions (0.98/0.90/0.005) identified 26 ECRs, including all the WNT2 coding exons. Although this number (26) is approximately three times less then the corresponding number for H/M ECRs (62), in general, strings of smaller H/M ECRs were incorporated within larger H/B/C ECRs, such that 68% (42) H/M ECRs were recapitulated in H/B/C ECRs. On a per base-pair basis, the sum of all human nucleotides classified as highly conserved either in rodent (H/M) or primate (H/B/C) comparisons were highly similar, spanning 15 kb in H/B/C and 17 kb H/M alignments. Four primate-specific ECRs lacked a highly homologous counterpart in the mouse ortholog. These elements could either represent regions that did not accumulate enough mutations throughout primate evolution due to chance alone, or could be primate-specific elements. Computationally distinguishing between these two possibilities is not yet feasible; rather, the true biological relevance of these lineage-specific elements must be determined experimentally.
To evaluate the specificity and sensitivity by which the eShadow tool is able to recapitulate H/M conservation profiles from a single human/primate alignment, we analyzed a test set of four completely finished baboon BACs spanning ∼677 kb that were syntenic to contiguous regions in both the human and mouse genomes (Table 1). This analysis was performed using fixed HMMI parameters trained on the WNT2 region (0.98/0.90/0.005). These regions exhibited strong correlations between the H/B HMMI predictions and the ECRs present in the H/M alignments. We estimated the sensitivity of recapitulating H/M conservation patterns by HMMI modeling of H/B alignments to be ∼59.3% and the specificity ∼77.6% (Table 1). To provide a measure of H/B exon-detection sensitivity, we calculated the number of exons identified by the HMMI approach across these four baboon BACs. Exons were scored as “detected” if they contained a partial or a full overlap with the HMMI prediction. A total of 62% (54/87) of all exons were detected by this approach. That also corresponds to 83% (25/30) of the long exons (>150 bp), suggesting that the eShadow approach works more efficiently for detecting longer elements (Table 1). These results imply that similar analysis may be used to define primate-specific elements in the human genome in an unbiased manner by comparing a minimum number of different primate sequences, and therefore suggest the use of the eShadow tool for computationally identifying primate-specific elements when the genome sequences of additional primates become available.
Identifying Conserved Protein Domains
Discovering deleterious mutations within candidate genes is fundamental to elucidating the genetic basis of human disorders. For most genes, the significance of any particular amino acid change is mostly unknown and requires comprehensive structural and functional studies. Multispecies protein alignments (MPA) can provide valuable information about the phylogenetic relationships between species and identify evolutionarily constraints in regions that are central to structural and biochemical interactions (Cline et al. 2002). Evaluating evolutionary rates at specific sites through the use of likelihood-ratio tests (LRTs) has been used extensively to characterize amino acid rate changes likely to underlie functional constraints on proteins (Knudsen and Miyamoto 2001). Evolutionary rate analysis complements existing approaches for the identification of conserved residues. Despite the intuitive correlation between conserved residues and functionally significant protein domains, distinguishing conservation associated with genuine biological interactions solely resulting from the shared phylogeny is a very difficult task (Pollock and Taylor 1997).
One application for the eShadow tool includes the analysis of MPAs to detect protein domains under selective pressure using HMMI predictions. This strategy is particularly promising, as HMM profiling is one of the most successful strategies for detecting statistically significant regions of protein homology (Madera and Gough 2002) and is already implemented by homology-based protein motif search tools such as the Pfam database (http://www.sanger.ac.uk/Software/Pfam/). The eShadow tool can also be used to visualize the distribution of nonsynonymous amino acid changes within MPA. To illustrate this, we have mapped all cystic fibrosis (CF) missense mutations documented in the Cystic Fibrosis Mutation Database to the CFTR MPA built from seven different species and correlated their distribution with the eShadow HHMI predictions. A total of 80% of the documented CF missense mutations (32/40) were enclosed by regions of high-protein homology as identified by HMMI (0.77/0.65/0.1; Fig. 5). At the same time, 95% of the most common CF disease-causing alleles (16 CF missense mutations; three single amino acid deletions) were found to be present in HMMI predicted domains.
Figure 5.

eShadow application in multiple protein alignment analysis. Human, baboon, cow, sheep, mouse, rat, and rabbit CFTR proteins were aligned using eShadow, and highly homologous regions were predicted using the HMMI module (0.77/0.65/0.1). Mutations known to cause cystic fibrosis (http://www.genet.sickkids.on.ca/cftr) are mapped to the CFTR MPA and annotated as red tick marks.
DISCUSSION
We have developed a computational Web-based tool, eShadow, that is highly proficient in performing phylogenetic shadowing analysis for closely related nucleotide and protein sequences. eShadow amplifies the information content from pairwise or multiple alignments by combining independent mutations present in each different lineage, and detects regions with the lowest cumulative density of mutations through the use of two different statistical methods, DT and HMMI. This tool also includes a parameters-optimization module for the HMMI model that can be amended to any particular evolutionary history underlying the input sequences, and trains the program to predict conserved elements in a wide variety of alignments. Unlike other available tools that analyze conservation across alignments using static parameters, eShadow allows for dynamic modifications of all parameters and picture settings, creating conservation plots in realtime.
eShadow can be used to detect coding exons, protein domains, and conserved noncoding elements. Whereas eShadow identifies exons, exon-intron boundaries are not exactly delineated; therefore, this tool provides a good starting point for transcript analysis that could benefit from external gene-prediction information. When protein alignments are analyzed, eShadow can be used to highlight protein domains that are conserved across multiple species and may be involved in vital biochemical processes such as protein–protein contacts or DNA binding. We have also indicated how amino acid mutational analysis can be superimposed on HHMI predictions in MPAs, and this analysis can be used to evaluate missense mutations.
We have shown that eShadow can recreate most of the information obtained from human/mouse alignments when human/baboon/chimp or human/baboon alignments are analyzed. A 53-kb three-way primate alignment analysis for the WNT2 locus recovered 68% of the human/mouse conserved elements, as well as identified several primate-specific conserved elements. Whereas the functional significance of these lineage-specific sequence elements is presently unknown, we speculate that they may potentially represent sequences that underlie noncoding functions shared by primates but not by other mammals. Regulatory modifications of conserved genes have been proposed to define the major molecular differences that set different organisms phenotypically apart (Boffelli et al. 2003), suggesting one potentially very interesting application for eShadow that cannot currently be performed by any other publicly available computational tool. Although we have focused on primate sequence alignments, eShadow can be tuned to align closely related sequences from any species. In addition, eShadow may be uniquely applicable to other problems, including alignments between recent segmental duplications. Such duplications can often generate new functional gene copies that do not have true orthologs in other species, and are therefore not amenable to standard cross-species comparative analysis (Bailey et al. 2002; Shannon et al. 2003). eShadow therefore adds an important set of capabilities to the current comparative genomics toolkit, providing unique access to species- and lineage-specific elements throughout sequenced genomes from any evolutionary clade.
METHODS
Hidden Markov Model Islands
We used a two-state HMM method to predict slow diverging regions in MSA. We modeled the distribution of matches and mismatches, assuming that they correspond to two mutation states (slow and fast), with different probabilities of emitting a match (eS and eF in slow- and fast-mutation states, respectively), and under the assumption that the probability T of transferring from state to state is equal in both directions (Supplemental Fig. S1). The emission probability of a state relates to the average sequence conservation of that particular state, whereas the transfer probability is inversely proportional to the average length of regions in the alignment. Viterbi algorithm was used to predict the underlying distribution of slow-mutation states.
Parameters Optimization
Baum-Welch (Durbin et al. 1998), Maximum Likelihood (Durbin et al. 1998), and Golden Section Search (Press et al. 1988) were used for optimizing parameters. Only Maximum Likelihood and Golden Section Search utilize user-provided annotation files (base sequence) as a training data set. Maximum Likelihood scans MSAs and sets emission probabilities equal to the observed number of matches per length of sequence in a particular state; the transition probability is estimated as a number of transitions from state to state, divided by the total length of the base sequence. The guided Baum-Welch optimization tests different paths through the sets of slow- and fast-mutation states and identifies the set that maximizes the likelihood probability for the HMMI. This method does not require annotation (it is used as an initial guess, if available) to calculate HMMI parameters; instead, it scans through all of the possible regions of slow mutation and identifying regions and HMMI parameters that will maximize the log-likelihood of the model, given the distribution of matches and mismatches. Golden Section Search (Brent's method) is a one-dimensional approach for determining the minimum (or optimum) of a nonlinear function, and is used to fine-tune the HMMI parameters by cyclically optimizing the probability parameters until full convergence is achieved. Every parameter optimization is performed, whereas the other two variables are fixed. This is achieved by sampling the surface and identifying the local minimum of the penalty function. We define the scoring function S as a discrepancy in the location of the HMMI predictions and the locations of functional regions (G):
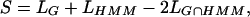 |
where LG, LHMM, and LG∩HMMare the lengths of guiding regions and HMMI predictions, and the length of their overlap, respectively. An exact fit for the HMMI predictions and guiding regions will zero the scoring function S, whereas any discrepancies will increase it. This method iterates and converges to the point of the local minimum, due to the discrete nature of the scoring function. Usually there are several local extremum points in the three-dimensional surface of HMMI probability parameters in Baum-Welch and Golden Section Surface optimizations. The implemented procedure converges to one of the extremum points, depending on the input parameters. Therefore, when HMMI generates no predictions, or there is a large discrepancy between the predicted and the expected elements, parameters need to be modified accordingly.
ORF Detection
ORFs are identified in all six reading frames, excluding the ones <60-bp in length, and are illustrated as gray bars. Stop codons from all sequences are collapsed onto the reference sequence. All ORFs internally spanned by HMMI predictions >75% in length are preserved, identifying the most probable frame of translation for each HMMI. At the final step, every frame is truncated in order to confirm the standard AG-GT exon-intron splice-site (Burset et al. 2000). AG and GT dinucleotides are required to be evolutionarily conserved in all of the species throughout the alignment, and the pair closest to HMMI edge is used to define the exon boundaries. Independently generated ORF and HMM predictions that overlap with each other are demarcated by different colors; red (positive strand); blue (negative strand; Fig. 3C).
Output Option
The text output option of the eShadow tool provides the user with a detailed summary for the alignment and exhaustive information about the detected slow-mutating regions. This module calculates the number of mismatches in all of the pairwise sequence comparisons for the base sequence versus each different sequence, and compares it with the whole MSA. This serves as an estimate of evolutionary divergence between different species, as well as it characterizes the substitution rate in the MSA. The second part of the summary data consists of a module calculating the discriminative power introduced by each additional species used in the multiple-sequence alignment. The program presents the tabulated data identifying the n most distant species, where n varies from two to the maximum number of species in the initial alignment by analyzing all of the possible combinations of n species and extracting the one with the highest substitution rate per base sequence (Supplemental Fig. S2). This is done by a sequential grouping of sequences that introduce the largest number of mutations into the MSA. Every group of n sequences that is presented is the optimal group that has the maximum possible amount of mutations in all of the possible sets of groups of n sequences.
This section also contains a report of all of the slow-mutating regions identified by the selected methods, providing the coordinates of the predicted elements in the base sequence, and indicating their parameters. HMMI predictions also contain scores that reflect confidence (statistically evaluating predicted regions vs. background noise for a given set of parameters). Every predicted slow-mutating interval I of the HMMI collects a score S(I), which reflects a log likelihood probability of this interval not to be a fast-mutating region:
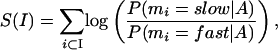 |
where the summation is done across all the base pairs in the interval and P(mi = k | A) is the posterior probability of the observed state k at the position i in the alignment A, which has a known structure of complete matches/mismatches. We prioritized longer conserved intervals over the shorter ones by not normalizing interval scores based on length.
Acknowledgments
We thank Lisa Stubbs and Marcelo Nobrega for critical reading of the manuscript, valuable suggestions, and comments. We thank Eddy Rubin for his support in creating this tool. This work was performed under the auspices of the U.S. Department of Energy by the University of California, Lawrence Livermore National Laboratory Contract No. W-7405-Eng-48 and Lawrence Berkeley National Laboratory Contract No. AC0376SF00098.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1773104.
Footnotes
[Supplemental material is available online at www.genome.org.]
References
- Anzai, T., Shiina, T., Kimura, N., Yanagiya, K., Kohara, S., Shigenari, A., Yamagata, T., Kulski, J.K., Naruse, T. K., Fujimori, Y., et al. 2003. Comparative sequencing of human and chimpanzee MHC class I regions unveils insertions/deletions as the major path to genomic divergence. Proc. Natl. Acad. Sci. 100: 7708–7713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, J.A., Gu, Z., Clark, R.A., Reinert, K., Samonte, R.V., Schwartz, S., Adams, M.D., Myers, E.W., Li, P.W., and Eichler, E.E. 2002. Recent segmental duplications in the human genome. Science 297: 1003–1007. [DOI] [PubMed] [Google Scholar]
- Balavoine, G., de Rosa, R., and Adoutte, A. 2002. Hox clusters and bilaterian phylogeny. Mol. Phylogenet. Evol. 24: 366–373. [DOI] [PubMed] [Google Scholar]
- Bienkowska, J.R., Yu, L., Zarakhovich, S., Rogers Jr., R.G., and Smith, T.F. 2000. Protein fold recognition by total alignment probability. Proteins 40: 451–462. [DOI] [PubMed] [Google Scholar]
- Boffelli, D., McAuliffe, J., Ovcharenko, D., Lewis, K.D., Ovcharenko, I., Pachter, L., and Rubin, E.M. 2003. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 299: 1391–1394. [DOI] [PubMed] [Google Scholar]
- Britten, R.J. 2002. Divergence between samples of chimpanzee and human DNA sequences is 5%, counting indels. Proc. Natl. Acad. Sci. 99: 13633–13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge, C. and Karlin, S. 1997. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268: 78–94. [DOI] [PubMed] [Google Scholar]
- Burset, M., Seledtsov, I.A., and Solovyev, V.V. 2000. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 28: 4364–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T.J., Higgins, D.G., and Thompson, J.D. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31: 3497–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline, M., Hughey, R., and Karplus, K. 2002. Predicting reliable regions in protein sequence alignments. Bioinformatics 18: 306–314. [DOI] [PubMed] [Google Scholar]
- Cooper, G.M., Brudno, M., Green, E.D., Batzoglou, S., Sidow, A., and NISC Comparative Sequencing Program. 2003. Quantitative estimates of sequence divergence for comparative analysis of mammalian genomes. Genome Res. 13: 813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couronne, O., Poliakov, A., Bray, N., Ishkhanov, T., Ryaboy, D., Rubin, E., Pachter, L., and Dubchak, I. 2003. Strategies and tools for whole-genome alignments. Genome Res. 13: 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis, E.T. and Clark, A.G. 2002. Evolution of transcription factor binding sites in Mammalian gene regulatory regions: Conservation and turnover. Mol. Biol. Evol. 19: 1114–1121. [DOI] [PubMed] [Google Scholar]
- Durbin, R.E., Eddy, S.R., Krogh, A., and Mitchison, G. 1998. Biological sequence analysis. Probabilistic models of proteins and nucleic acids. Cambridge University Press, Cambridge, UK.
- Ghanem, N., Jarinova, O., Amores, A., Long, Q., Hatch, G., Park, B.K., Rubenstein, J.L., and Ekker, M. 2003. Regulatory roles of conserved intergenic domains in vertebrate dlx bigene clusters. Genome Res. 13: 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilligan, P., Brenner, S., and Venkatesh, B. 2002. Fugu and human sequence comparison identifies novel human genes and conserved non-coding sequences. Gene 294: 35–44. [DOI] [PubMed] [Google Scholar]
- Hellmann, I., Zollner, S., Enard, W., Ebersberger, I., Nickel, B., and Paabo, S. 2003. Selection on human genes as revealed by comparisons to chimpanzee cDNA. Genome Res. 13: 831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard, T., Barker, D., Birney, E., Cameron, G., Chen, Y., Clark, L., Cox, T., Cuff, J., Curwen, V., Down, T., et al. 2002. The Ensembl genome database project. Nucleic Acids Res. 30: 38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik, D., Baertsch, R., Diekhans, M., Furey, T.S., Hinrichs, A., Lu, Y.T., Roskin, K.M., Schwartz, M., Sugnet, C.W., Thomas, D.J., et al. 2003. The UCSC Genome Browser Database. Nucleic Acids Res. 31: 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen, B. and Miyamoto, M.M. 2001. A likelihood ratio test for evolutionary rate shifts and functional divergence among proteins. Proc. Natl. Acad. Sci. 98: 14512–14517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh, A. 1997. Two methods for improving performance of an HMM and their application for gene finding. Proc. Int. Conf. Intell. Syst. Mol. Biol. 5: 179–186. [PubMed] [Google Scholar]
- Lim, L.P., Glasner, M.E., Yekta, S., Burge, C.B., and Bartel, D.P. 2003. Vertebrate microRNA genes. Science 299: 1540. [DOI] [PubMed] [Google Scholar]
- Loots, G.G., Locksley, R.M., Blankespoor, C.M., Wang, Z.E., Miller, W., Rubin, E.M., and Frazer, K.A. 2000. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science 288: 136–140. [DOI] [PubMed] [Google Scholar]
- Madera, M. and Gough, J. 2002. A comparison of profile hidden Markov model procedures for remote homology detection. Nucleic Acids Res. 30: 4321–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor, C., Brudno, M., Schwartz, J.R., Poliakov, A., Rubin, E.M., Frazer, K.A., Pachter, L.S., and Dubchak, I. 2000. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16: 1046–1047. [DOI] [PubMed] [Google Scholar]
- Nobrega, M.A., Ovcharenko, I., Afzal, V., and Rubin, E.M. 2003. Scanning human gene deserts for long-range enhancers. Science 302: 413. [DOI] [PubMed] [Google Scholar]
- Ovcharenko, I., Loots, G.G., Hardison, R.C., Miller, W., and Stubbs, L. 2004. zPicture: Dynamic alignment and visualization tool for analyzing conservation profiles. Genome Res. 14: 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio, L.A., Olivier, M., Hubacek, J.A., Cohen, J.C., Cox, D.R., Fruchart, J.C., Krauss, R.M., and Rubin, E.M. 2001. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science 294: 169–173. [DOI] [PubMed] [Google Scholar]
- Pollock, D.D. and Taylor, W.R. 1997. Effectiveness of correlation analysis in identifying protein residues undergoing correlated evolution. Protein Eng. 10: 647–657. [DOI] [PubMed] [Google Scholar]
- Press, W.H., Flannery, B.P., Teukolsky, S.A., and Vetterling, W.T. 1988. Numerical Recipes in C. Cambridge University Press, UK.
- Rivas, E. and Eddy, S.R. 2001. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics 2: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scemama, J.L., Hunter, M., McCallum, J., Prince, V., and Stellwag, E. 2002. Evolutionary divergence of vertebrate Hoxb2 expression patterns and transcriptional regulatory loci. J. Exp. Zool. 294: 285–299. [DOI] [PubMed] [Google Scholar]
- Schwartz, S., Zhang, Z., Frazer, K.A., Smit, A., Riemer, C., Bouck, J., Gibbs, R., Hardison, R., and Miller, W. 2000. PipMaker—A web server for aligning two genomic DNA sequences. Genome Res. 10: 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, S., Kent, W.J., Smit, A., Zhang, Z., Baertsch, R., Hardison, R.C., Haussler, D., and Miller, W. 2003. Human—mouse alignments with BLASTZ. Genome Res. 13: 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, M., Hamilton, A.T., Gordon, L., Branscomb, E., and Stubbs, L. 2003. Differential expansion of zinc-finger transcription factor Loci in homologous human and mouse gene clusters. Genome Res. 13: 1097–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, J.C. and Kondrashov, A.S. 2002. Patterns in spontaneous mutation revealed by human—baboon sequence comparison. Trends Genet. 18: 544–547. [DOI] [PubMed] [Google Scholar]
- Takai, D. and Jones, P.A. 2002. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. 99: 3740–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusova, T.A. and Madden, T.L. 1999. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 174: 247–250. [DOI] [PubMed] [Google Scholar]
- Thomas, J.W., Touchman, J.W., Blakesley, R.W., Bouffard, G.G., Beckstrom-Sternberg, S.M., Margulies, E.H., Blanchette, M., Siepel, A.C., Thomas, P.J., McDowell, J.C., et al. 2003. Comparative analyses of multi-species sequences from targeted genomic regions. Nature 424: 788–793. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong, K. and Ikura, M. 2002. Identification and characterization of subfamily-specific signatures in a large protein superfamily by a hidden Markov model approach. BMC Bioinformatics 3: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston, R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.ebi.ac.uk/clustalw/; ClustalW.
- http://www.genet.sickkids.on.ca/cftr/; Cystic Fibrosis Mutation Database.
- http://ecrbrowser.dcode.org/; ECR Browser.
- http://www.ensembl.org/; Ensembl.
- http://eshadow.dcode.org/; eShadow.
- http://genome.ucsc.edu/; Human Genome Browser at UCSC.
- http://bio.cse.psu.edu/genome/hummus/; Pip Dispenser.
- http://bio.cse.psu.edu/pipmaker/; PipMaker.
- http://www.sanger.ac.uk/Software/Pfam/; Pfam database.
- http://www-gsd.lbl.gov/VISTA/; Vista.
- http://pipeline.lbl.gov; Vista Browser.
- http://zpicture.dcode.org/; zPicture.