

Abstract
Leptin is a cytokine produced by white fat cells, skeletal muscle, the placenta, and the pituitary gland among other tissues. Best known for its role in regulating appetite and energy expenditure, leptin is produced largely by and in proportion to white fat cells. Leptin is also important to the maintenance and function of the GH cells of the pituitary. This was shown when the deletion of leptin receptors on somatotropes caused decreased numbers of GH cells, decreased circulating GH, and adult-onset obesity. To determine the source of leptin most vital to GH cells and other pituitary cell types, we compared two different leptin knockout models with Cre-lox technology. The global Lep-null model is like the ob/ob mouse, whereby only the entire exon 3 is deleted. The selective adipocyte-Lep-null model lacks adipocyte leptin but retains pituitary leptin, allowing us to investigate the pituitary as a potential source of circulating leptin. Male and female mice lacking adipocyte leptin (Adipocyte-lep-null) did not produce any detectable circulating leptin and were infertile, suggesting that the pituitary does not contribute to serum levels. In the presence of only pituitary leptin, however, these same mutants were able to maintain somatotrope numbers and GH mRNA levels. Serum GH trended low, but values were not significant. However, hypothalamic GHRH mRNA was significantly reduced in these animals. Other serum hormone and pituitary mRNA differences were observed, some of which varied from previous results reported in ob/ob animals. Whereas pituitary leptin is capable of maintaining somatotrope numbers and GH mRNA production, the decreased hypothalamic GHRH mRNA and low (but not significant) serum GH levels indicate an important role for adipocyte leptin in the regulation of GH secretion in the mouse. Thus, normal GH secretion may require the coordinated actions of both adipocyte and pituitary leptin.
Leptin is an anorexigenic protein first isolated and identified in murine white adipose tissue in the mid-1990s in the laboratory of Friedman and colleagues (1). However, due to the appearance of the ob/ob mouse at the Jackson Laboratory in the 1950s, scientists had been collecting data on leptin for over 4 decades before the gene product was identified (2). Leptin is known for its ability to control appetite and energy expenditure by way of long-form leptin receptors (LepRbs) in the hypothalamus (3–8). Serum leptin is produced and secreted in proportion to fat mass, and there exists an apparent lack of appetite suppression and energy control in obese individuals despite their high serum levels (9–13). Some investigators therefore propose a model of leptin resistance in obesity, a theory enforced by the underwhelming effects of systemic leptin administration in obese individuals (10, 14). After the initial studies highlighting adipose tissue as the main source of leptin production in humans and animals, several studies revealed extraadipocyte sources of leptin. These sources include the skeletal muscle, the placenta, the fundus of the stomach, the brain, and the pituitary (15–19). Researchers are now working to decipher the specific roles for leptin in each of these tissues, although until now, a tissue-specific leptin knockout animal model has not been available.
The ob/ob mouse model, a model that arose due to a spontaneous point mutation in the lep gene, has given researchers valuable information concerning the effects of total leptin deficiency on various endocrine systems, especially concerning the hypothalamic-pituitary axis. These mice have multiple pituitary hormone deficiencies, and these deficiencies are manifest in several systemic abnormalities.
The ob/ob mice have decreased gonadotropin secretion and are infertile, and these deficits can be corrected (at least to some degree) with exogenous administration of leptin (2, 20–23). The GH axis is also impaired in ob/ob animals, evident in the low GH serum protein levels (24, 25) as well as reduced levels of pituitary GH mRNA (26). A study by Luque et al (27) demonstrated that leptin replacement in ob/ob mice enhanced circulating GH levels (although not to significance) while significantly increasing pituitary GH and GHRH receptor (GHRHR) mRNA expression in vivo. Skeletal growth and bone mass in ob/ob animals are severely impaired but can be restored upon leptin administration (28). Prolactin, another pituitary-derived hormone, is abnormally low in ob/ob mice (25). These mice also exhibit abnormally high levels of pituitary ACTH (29).
Our laboratory has previously shown that the leptin signal, specifically through the long form of the leptin receptor, regulates both the number of GH cells and the secretion of GH because the deletion of LepRb on somatotropes results in decreased numbers of GH cells and decreased serum GH levels (30). Additionally, these somatotrope LepRb-null mice develop adult-onset obesity. Knowing that the leptin signal is an important regulator of somatotrope maintenance and function, we sought to determine whether the pituitary itself was the source of the leptin signaling to the somatotrope population.
All pituitary cell types produce some amount of leptin, although somatotropes produce most of the hormone. Upward of 60%–70% of leptin-producing pituitary cells are somatotropes in male and nonpregnant female rats (31, 32). Although gonadotropes represent only 10%–20% of leptin-bearing cells in male and (cycling) female rats, as much as 70% of leptin-bearing cells in the pituitary coexpress gonadotropins during pregnancy and lactation (33). Fasting drastically reduces the number of pituitary cells with leptin mRNA (31). The effect that obesity has on the expression of pituitary leptin, however, is not known.
Given the wealth of information connecting the regulation of the various pituitary hormones and cell types to leptin, we propose that pituitary leptin is an important paracrine and/or autocrine regulator of pituitary development and function. Whereas several tissues are known to produce leptin, it is not known whether these extraadipocyte tissues, including the pituitary, are sources of circulating serum leptin. Additionally, there has not been an in vivo method of knocking out leptin in specific cell types or organs. Our laboratory has created two novel leptin knockout models with the use of Cre-lox technology: one in which leptin is knocked out in all cell types and one in which leptin is knocked out only in white adipose tissue. This report will detail the phenotype of these mice along with two new discoveries. First, adipose tissue is the only source of circulating leptin in mice; other sources of leptin do not appear to contribute to serum leptin. Second, pituitary leptin is not dependent on adipocyte leptin and may be capable of supporting the development of GH cells. We also report interesting results that contrast with the global leptin knockouts (ob/ob) and may reflect the more extensive deletion of leptinexon 3 in our knockout models.
Materials and Methods
Creation of targeting vector and chimera lines
Using genomic FVB mouse DNA and standard molecular cloning, a targeting vector that contained 5′ and 3′ homology arms and loxP sequences flanking exon 3 of leptin (Taconic)was created. Exon 3 is the largest coding portion of the lep gene, coding 119 amino acids compared with exon 2, which encodes only 48 amino acids (34). It is known that a single-point mutation in exon 3 is known to cause obesity, thus making the exon an excellent target for our model (35). The vector also included two expression cassettes: a Diphtheria toxin A expression cassette for negative selection of embryonic stem cells and a neomycin expression cassette flanked by Flippase recombination target sequences for the positive selection of embryonic stem cells. After the electroporation and selection of embryonic stem cells, two potential target clones were expanded and injected into blastocysts, creating chimeras for both a B12 and a C5 line. The male chimeras from the B12 line were used to create Leptinfl/fl founders.
Experimental animals
All animal care protocols have been approved by the Institutional Animal Care and Use Committee. After weaning, mice were fed a diet of 22% crude protein, 5% crude fat, and 4.5% crude fiber (Teklad 8640 diet; Harlan) and water ad libitum. Experimental deletion mutants and littermate controls were bred in-house and maintained in a room with a 14-hour light, 10-hour dark cycle at 23°C.
Leptinfl/fl mice (FVB/Ntac; Taconic) from the B12 line were bred to produce homozygous founders. To remove the neocassette, these founders were bred to mice expressing Flippase recombination enzyme [129S4/SvJaeSor-Gt(ROSA)26Sortm1(FLP1)DYM/J; Jackson Laboratory], an enzyme that causes a recombination event at Flippase recombination target sites. Heterozygous floxed leptin mice from both lines (50% FVB/NTac, 50% 129S4) that had no neocassette were then selected, and these were bred to create neocassette-null Leptinfl/fl founders. Because they were bred in the FVB background, the floxed leptin mice carried the retinal degeneration gene, which made them blind by the time they reached puberty. However, after breeding with the Flippase mice, we selected for mice that were null for the retinal degeneration gene to found the lines.
Leptinfl/fl mice were subsequently crossed with a line of mice bearing Cre-recombinase behind the adiponectin promoter [B6.FVB-Tg(Adipoq-Cre)1Evdr/J; stock number 010803; Jackson Laboratory]. The use of this promoter allows the deletion of leptin only in white and brown adipose tissue (36). Adipoq-creTg/0 Leptinfl/Δ males and littermate Leptinfl/fl females were mated to produce subsequent generations because the Adipocyte-lep-null homozygous deletion mutants (bearing Adipoq-creTg/0 LeptinΔ/Δ) were infertile.
Leptinfl/fl mice were also bred to a line of mice expressing Cre-recombinase under the control of the adenovirus EIIa promoter [FVB/N-Tg(EIIa-Cre)C5397Lmgd/J, Jackson Laboratory]. This promoter initiates Cre expression in the preimplanted embryo in all cell types, leading to global recombination (37). Heterozygous deletion mutants were then crossed to create homozygous leptin deletion mutants. As was the case with the Adipocyte-lep-null deletion mutants, both male and female Global-lep-null (EIIa-cre) deletion mutants are infertile.
All animals were weighed weekly from weaning until death or 6 months of age.
PCR design and validation of tissue-specific knockouts
Genotypes of 21-day-old mice were determined by genomic DNA extracts of tail snip DNA. Cre-recombinase is detected as previously described (11). For the detection of floxed lep, the forward sequence is 5′-TGAGCAGTTCTGCAAACCAGCCT-3′, and the reverse sequence is 5′-AAGGGATGACTGTTCTGTGACTGC-3′ (327 bp). Detection of the lep gene in which exon 3 is deleted (535 bp) requires the forward sequence 5′-TCCTTTACAACCAGTCCTTGTGTAGC-3′ and the floxed lep reverse sequence.
Female and male deletion mutants and controls from each line were used for extensive organ genotyping to validate the specificity of each knockout. The purposes of the genotyping were to determine whether there was Cre-recombinase activity outside adipose tissue in the Adipocyte-lep-null line and to ensure the complete excision of the lep gene in all tissues in the Global-lep-null line. The activity of the Cre would be detected by the presence of the excised lep alleles and the absence of floxed lep alleles. Sixteen organs (fat, muscle, heart, lung, liver, pancreas, spleen, adrenal, kidney, stomach, ovaries/testes, uterus/uterine tubes, cerebrum, cerebellum, hypothalamus, and pituitary) were collected from each mouse and genotyped using the primer sets mentioned previously. Sample genotyping from the male experiments are included in this report.
Quantitative real-time PCR (qRT-PCR) mRNA analysis of hypothalami and pituitaries
Whole pituitaries and hypothalami were collected from male control and deletion mutant animals from both lines at 6 months of age. Samples were stored in RNAlater solution (Ambion) at −20°C.
Sample tissue was homogenized with sterile pestles, and RNA was isolated using the Maxwell 16 LEV simplyRNA tissue kit (Promega; AS1280). Recovered sample RNA was measured (Nanodrop 2000c; Thermo Fisher Scientific), concentrated at 1 μg/mL for cDNA preparation with iQ Supermix (Bio-Rad Laboratories; 170–8891), and amplified for qRT-PCR. Samples and primers were combined with Power SYBR Green PCR master mix (Applied Biosystems; 4367659) for amplification and detection.
All qRT-PCRs were performed using the QuantStudio 12K Flex system (Applied Biosystems, Life Technologies) with the following protocol in three stages: incubation/denaturation stage, 50°C for 2 minutes and 95°C for 10 minutes; PCR amplification stage (50 cycles), 95°C for 15 seconds, 55°C for 15 seconds, and 72°C for 1 minute; and melt curve stage, 95°C for 15 seconds, 60°C for 1 minute, and 95°C for 15 seconds. Data collection occurred after each of the 72°C steps in the amplification stage. Samples were normalized to cyclophilin expression, and relative expression values were determined by the QuantStudio 12K Flex Software version 1.0 using the Δ-Δ-cycle threshold method (38).
Information on the quantitative PCR primer sets can be found in Supplemental Table 1. Primer sets identified included cyclophilin, FSHβ, GH, GHRH, GHRHR, GH secretagogue receptor (GHS-R), LHβ, proopiomelanocortin (POMC), prolactin, and TSHβ, sequences taken from Luque et al (39), somatostatin (40), leptin receptors (all isoforms) (41), and ghrelin (42).
Primary pituitary cell culture and serum enzyme immunoassays (EIAs)
After isoflurane anesthesia and decapitation, trunk blood was collected and serum stored at −20°C. All sera were collected between 9:00 and 10:00 am to prevent variation in hormone levels across samples. Primary pituitary cell dispersion and fixation were performed on 6-month-old animals from males and females, control and deletion, of both lines as described previously (30). Pituitaries were dissected, washed in DMEM, and incubated with trypsin and deoxyribonuclease in DMEM at 37°C. After mechanical dispersion of individual pituitaries in fresh DMEM with deoxyribonuclease and trypsin inhibitor using a 1-mL syringe with 26-G needle, cells were resuspended in fresh DMEM and insulin/transferrin/sodium selenite supplement (1:100; Sigma). The number of cells per pituitary was determined using a hemocytometer, and cells were plated on glass coverslips in 24-well trays at 12 000 pituitary cells/well. After 3 hours of incubation at 37°C, cells were fixed in 2% glutaraldehyde, washed in a phosphate sucrose glycine solution, and stored at 4°C until used for immunocytochemistry.
Abdominal fat samples from adult animals of both transgenic lines were collected and stored at −20°C until protein extraction. Protein from fat samples was extracted and quantified in a method previously described by our laboratory (30). Protein from fat samples, along with serum from 21-day-old and 6-month-old animals, was assayed with a solid-phase ELISA designed to detect recombinant mouse leptin (R&D Systems; mouse/rat leptin Quantikine ELISA, MOB00). The antibody used in this system has been shown to work well with both mouse serum and protein extracted from mouse adipose tissue (43–46).
Pituitary hormones (ACTH, FSH, GH, prolactin, TSH, and LH) in serum from 6-month-old animals were quantified using the Milliplex mouse pituitary magnetic bead panel (EMD Millipore; MPTMAG-49K) run on a Luminex 100/200 System (Luminex Corp).
Immunocytochemistry on dispersed pituitary cells
Adult pituitary cells from both lines were immunolabeled using an avidin-peroxidase protocol as previously described by our laboratory for GH (47) and leptin (32, 33). The affinity-isolated polyclonal antibody to leptin (Sigma; L3410) was raised in rabbit and is specific only for the N-terminal residues of human and mouse leptin (amino acids 22–40). This leptin antibody was used at a 1:10 000 dilution, whereas the antirat GH was used at a dilution of 1:200 000 (Hormone Distribution Program, Torrance, California) and has been previously used by our laboratory (30).
Statistical analysis
For all serum and fat hormone assays and qPCR (pituitary and fat), samples from at least five animals were run for each experimental group. For the immunocytochemistry study, cells from 10 fields each on two coverslips per animal were analyzed, with at least three animals in every group (at least 60 fields per sex per genotype). Female and male samples were always analyzed separately. A one-way ANOVA and Student's t tests were performed with Welch's correction using PRISM software (GraphPad). A post hoc power analysis was performed with the use of pilot data from Adipocyte-lep-null animals to determine sample size as described previously (31).
Results
Creation of transgenic leptin knockout animals
To create two models of Cre-mediated leptin excision, we developed a floxed leptin animal line in which exon 3 of the lep gene is targeted (Figure 1). Exon 3 was chosen because it comprises most of the coding portion of the lep gene (34). Homozygous deletion mutants from the EIIa line were produced from two Cre-negative Leptinfl/Δ mice because the EIIa-cre transgene always sorted with the wild-type leptin allele. These homozygous mutants are referred to as Global-lep-null mutants throughout the report. Even without the presence of Cre-recombinase, the inherited deletion of both alleles of leptin appears to be complete, as indicated in the organ genotyping (Figure 2A) and serum leptin levels (Figure 3A).
Figure 1.
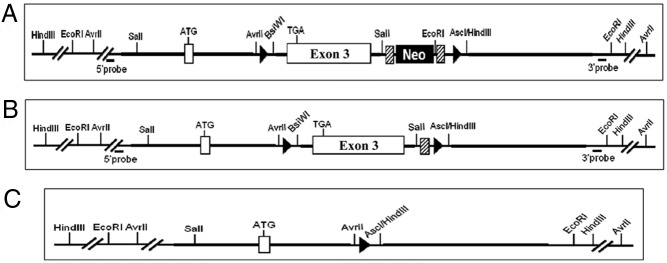
A diagram of the floxed leptin gene. A, Representation of the floxed leptin exon 3 (Taconic) used to create the tissue-specific leptin knockout mice. Leptin exon 3 is flanked by two loxP sites (represented by black arrowheads). Also contained within these loxP sites is the selection neocassette, flanked by two Flippase recognition target sites (diagonal striped boxes). B, Floxed leptin gene after the neocassette has been deleted, by crossing with an animal bearing the Flippase recombination enzyme. One Flippase recognition target site remains, along with lep exon 3 and both loxP sites. C, Final product of recombination after the floxed leptin gene has been exposed to Cre-recombinase. Exon 3 has been deleted, and only a single loxP site remains.
Figure 2.

Sample genotyping of Global-lep-null and Adipocyte-lep-null mutant and control organs. Figure 2A contains a sample of the extensive organ genotyping done on the Global-lep-null animals and littermate Leptinfl/fl controls. The first panel demonstrates the genotyping for detection of Cre-recombinase (166 bp), which is not present in either the three control samples or the three Global-lep-null samples of adipose tissue. Because the EIIa-cre always sorted with the wild-type lep gene, we used Cre-negative Leptinfl/Δ males and females to create Cre-negative LeptinΔ/Δ animals. The second panel shows genotyping for the floxed lep gene (327 bp), which is present in the control pituitaries but absent in the Global-lep-null pituitaries. This is because both alleles of lep have been excised. The final three panels indicate the excision of lep (535 bp band) in deletion pituitaries, fat samples, and stomachs but not in any of the control samples. B, Sample organ genotyping in the Adipocyte-lep-null line. All Adipocyte-lep-null samples (but none of the control samples) are Cre positive, as indicated by the adipose samples in the first panel. Floxed lep alleles are present in both control and deletion pituitaries because the Adipoq-cre is specific only to adipose tissue. The final three panels show no lep excision in control or deletion pituitaries or stomach samples but strong excision in Adipocyte-lep-null adipose tissue (but not control tissue).
Figure 3.

Analysis of serum and fat leptin protein levels. Panel A contains serum leptin levels for control and Global-lep-null males (left) and females (right). A one-tailed Student's t test was used to compare values between groups within sexes. Values are expressed as average leptin levels (picograms per milliliter) ± SEM. Significance is set at P < .05. B, Serum leptin levels (picograms per milliliter ± SEM) for control and Adipocyte-lep-null males (left) and females (right). A two-tailed Student's t test was used to determine differences. C, Control and deletion Adipocyte-lep-null male (left) and female (right) adipose leptin protein levels. Values are displayed as picograms per milliliter per microgram of protein ± SEM, and differences were determined with one-tailed Student's t tests. Significance (for this figure and all others) is indicated as follows: *, P = .01 to P = .05; **, P = .01 to P = .001; ***, P = .001 to P = .0001; ****, P < .0001.
Adipocyte-lep-null males and females were produced from heterozygous deletion males and Leptinfl/fl females because neither male nor female homozygotes were fertile (data not shown). Organ genotyping from the male mutants and controls show that floxed leptin is expressed in every organ, but the excised gene (lepΔ/Δ) occurs only in adipose tissue (Figure 2B).
Serum, fat leptin content, and body weight measurements
To determine whether the deletion of lep exon 3 is a functional, translatable deletion, we measured serum leptin from Global-lep-null mutants and controls. As predicted, neither male nor female LeptinΔ/Δ animals had any detectable circulating serum leptin (Figure 3A). As should be expected from an ob/ob-like animal, male mutants are significantly more obese than controls at 3 weeks of age (P < .05) and continue to diverge from controls throughout life. Female Global-lep-null mutants are significantly heavier than controls at week 5 (P < .05) and continue to gain weight (Figure 4A).
Figure 4.

Characterization of weight over time in the Global- and Adipocyte-lep-null mutants and controls. A, Weekly average weights from weaning to 5 months of age in male (top) and female (bottom) Global-lep-null mutants (squares) and controls (circles). Values are expressed as averages of at least five animals per group per sex at each time point ± SEM. Student's t tests were used to determine the differences, with significance set as P < .05. Weights for Adipocyte-lep-null mutants are shown in a similar manner in panel B.
To ensure that our Adipocyte-lep-null mutants truly did not produce any adipocyte leptin, we measured adipose leptin protein (Figure 3C). Adipose leptin protein was also not detectable in the Adipoq-cre adult deletion mutants. To determine whether circulating leptin is produced only by adipocytes or whether extraadipocyte sources contribute to the circulating leptin pool, we measured serum leptin from Adipocyte-lep-null mutants and Leptinfl/fl controls. The Adipocyte-lep-null males and females showed no detectable serum leptin (Figure 3B), confirming for the first time that circulating leptin must be produced only by adipocytes. The male and female mutants from the Adipocyte-lep-null line, much like those of the Global-lep-null line, were significantly more obese than controls by postnatal day 21 (P < .05, Figure 4B).
Pituitary and hypothalamic qRT-PCR
We hypothesized that pituitary leptin is sufficient to support the development and maintenance of the pituitary in the absence of adipocyte leptin. We analyzed adult male and female Adipocyte-lep-null mutant and Leptinfl/fl control pituitaries for mRNA content (Figure 5A). Adult male mutants had significantly higher GHRHR (P < .05) and POMC transcripts (P < .01) and significantly decreased prolactin transcripts (P < .005), as compared with controls. Female mutants had significant decreases in the following transcripts: GHRH (P < .005), GHS-R (P < .05), LH (P < .01), and prolactin (P < .05). The female mutants also had significantly increased GHRHR (P < .01), POMC (P < .01), and TSH (P < .05) transcripts. Neither male nor female mutants had significant changes in FSH, GH, or ghrelin transcripts. The male mutants also had no change in GHRH, GHS-R, LH, or TSH transcript levels.
Figure 5.

Pituitary and hypothalamic mRNA analysis. A, Male (left) and female (right) control and deletion Adipocyte-lep-null pituitary mRNA levels. For all mRNA results, levels were determined using qRT-PCR and Δ-Δ-cycle threshold analysis, with cyclophilin A as an internal control. Relative quantification values are expressed with ± SEM, with control mRNA levels set at 1. A Student's t test (two tailed) was used to determine differences within sexes, with significance set at P < .05. Target pituitary mRNAs included FSH, GH, ghrelin, GHRH, GHS-R, LH, POMC, prolactin, and TSH. At least five pituitaries were used per experimental group per sex. B, mRNA analysis for male (left) and female (right) control and Adipocyte-lep-null mutant hypothalami, and at least five whole hypothalamus samples were analyzed per group per sex. Target hypothalamus mRNAs were GHRH, ghrelin, leptin receptor (all isoforms), and somatostatin. C, mRNA analysis for male (left) and female (right) control and Global-lep-null hypothalami, and n is at least five per group per sex.
To determine whether the hypothalamus was protected by local production of leptin (17, 48), we measured GHRH, ghrelin, leptin receptor, and somatostatin mRNA levels (Figure 5B). Males and female mutants from the Adipocyte-lep-null (Adipoq-cre) line had significantly lower GHRH mRNA levels (P < .05) than controls. This differs from the deletion mutants of the Global-lep-null (EIIa-cre) line, which had no significant differences in any of the hypothalamic mRNAs tested (Figure 5C).
Pituitary GH and leptin content
We hypothesized that pituitary leptin may be important for the development and maintenance of the somatotrope cell population, even in the absence of adipocyte leptin. Immunolabeling of dispersed adult male pituitary cells reveals no significant difference in the percentage of cells labeled for GH between Leptinfl/fl controls (27.00% ± 1.343%, n = 12) and Adipocyte-lep-null deletion mutants (27.58% ± 2.466%, n = 12) (Figure 6, A and B). Similar results were found when GH cells from deletion mutant females were counted (33.00% ± 3.8%, n = 3). The results were not different from control females or control or deletion mutant males. To determine whether pituitary leptin is affected by the loss of adipocyte leptin, we also immunolabeled adult male pituitary cells for leptin (Figure 6A). Adipocyte-lep-null deletion mutants showed an increased percentage of cells labeled for leptin (45.53% ± 2.818%, n = 15) when compared with Leptinfl/fl controls (35.25% ± 2.157%, n = 12) (P = .0079). Global-lep-null mutants had very few pituitary cells that were immunopositive for leptin (1.750% ± 0.4787%, n = 4) compared with controls (35.63% ± 1.451%, n = 8) (Figure 6, A and C), and they showed a significant decrease in the percentage of cells labeled for GH (controls: 27.67% ± 0.6667%, n = 3; mutants: 15.00% ± 0.5774%, n = 3) (Figure 6A).
Figure 6.

Immunolabeling of GH and leptin in primary pituitary cells. A, Results from immunolabeling of male Adipocyte-lep-null and Global-lep-null control and mutant primary pituitary cell cultures. At least two coverslips for each of three animals per group were immunolabeled for either GH or leptin (amino acids 22–40). Ten fields per coverslip were analyzed for labeled vs unlabeled pituitary cells. Values are expressed as the average percentage of cells immunolabeled ± SEM. Student's t tests were used to determine differences in GH or leptin labeling. Representative fields of GH-labeled pituitary cells from a control and an Adipocyte-lep-null male are shown in panel B. Fields showing leptin-labeled pituitary cells from a control and a Global-lep-null animal are shown in panel C.
Analysis of circulating pituitary hormones
To determine whether pituitary leptin could sustain the secretory functions of the pituitary in the absence of adipocyte leptin, we collected serum from adult male and female Adipocyte-lep-null (Adipoq-cre) mutants and Leptinfl/fl controls and analyzed the samples for circulating pituitary hormone levels (Figure 7). Male mutants (DEL) had significantly decreased FSH compared with controls (CTL) (CTL: 11062 ± 1335 pg/mL, n = 6; DEL: 7195 ± 495.1 pg/mL, n = 6) (P < .05). Male mutants showed a trend toward a significant reduction in serum GH (CTL: 23630 ± 11823 pg/mL, n = 6; DEL: 3103 ± 1622 pg/mL, n = 6), although the result is not significant. Female mutants have significantly decreased LH (CTL: 2552 ± 663.5 pg/mL, n = 5; DEL: 734.5 ± 181.5 pg/mL, n = 5; P < .05) as well as significantly decreased prolactin (CTL: ± 16659 pg/mL, n = 5; DEL: 5177 ± 2046 pg/mL, n = 5; P < .05). Global-lep-null males had significantly decreased FSH (CTL: 10622 ± 934.9 pg/mL, n = 10; DEL: 5479 ± 964.8 pg/mL, n = 12; P < .005), and females had significantly decreased serum prolactin (CTL: 36801 ± 9818 pg/mL, n = 11; DEL: 4296 ± 1142 pg/mL, n = 9; P < .01).
Figure 7.

Serum levels of pituitary hormones. Male and female pituitary hormone levels from Adipocyte-lep-null and Global-lep-null males and females are shown in a series of graphs. Values for FSH, GH, and LH (top row) and prolactin and TSH (bottom row) are displayed on individual charts. Values are expressed as nanograms per milliliter ± SEM, with significance set at P < .05. Samples from at least five animals per group per sex are represented for each hormone. Student's t tests were used to determine differences.
Discussion
The cytokine leptin inhibits appetite and increases energy expenditure by inhibiting neuropeptide Y neurons and stimulating POMC neurons in the hypothalamus (1, 3, 5, 49, 50). The protein is produced by and in proportion to white fat cells, although leptin is also produced in other tissues in humans, rodents, and other species [see review by Denver et al (51), 2011] (10, 11, 15–19, 51). Leptin receptor expression is even more widespread, found in many tissues in humans, rats, and mice, including the pituitary (4, 7, 52–59). Our laboratory has previously shown that the long-form LepRb is important for the maintenance of the GH-secreting cells of the pituitary (30). The current study was designed to identify the source of leptin that is crucial to the somatotrope cell population.
Creation of an adipocyte leptin knockout mouse
To compare the impact of the local pituitary source of leptin with that from adipocytes, we designed a tissue-specific leptin knockout mouse using a floxed leptin exon 3 model (Taconic) and the Adipoq-cre model that expresses Cre-recombinase under control of the adiponectin promoter (36). Organ genotyping indicates that Cre-recombinase is expressed in adipose tissue of deletion mutants only. The organ genotyping also shows that leptin is deleted only in the adipose tissue and not in other leptin-producing tissues such as the stomach and the pituitary. Additionally, fat assayed from Adipocyte-lep-null males had no detectable leptin protein, indicating that the deletion of exon 3 is sufficient to eliminate the production of the protein.
The pituitaries of Adipocyte-lep-null male mice were found to contain abundant leptin-producing cells, which indicates that pituitary leptin production is not dependent on circulating leptin from adipocytes. This is the first tissue-specific leptin knockout mouse created with the use of Cre-lox technology, and the results indicate that the floxed leptin mouse will prove to be a valuable tool in future studies of other extraadipocyte sources of leptin. There are unique challenges, however. The same EIA used to detect leptin proteins in rat pituitaries contains an antibody that does not detect mouse pituitary leptin either in an EIA or by immunocytochemistry (31). An EIA with the antibodies that detect mouse pituitary leptin is being developed for future studies.
To validate the functionality of the floxed leptin exon 3 model, we also used the EIIa-cre mouse line, which creates a global knockout (37). Organ genotyping of the Global-lep-null line shows that leptin is knocked out in all of the organs, such as adipose tissue, the pituitary, and the stomach. The deletion mutants from this line do not express Cre-recombinase because the EIIa-cre always sorts with the wild-type lep gene. Therefore, it was impossible for us to obtain animals that expressed both the EIIa-cre transgene as well as two floxed lep alleles. We were able to obtain total deletion mutants by crossing Cre-negative heterozygotes that had inherited leptin deletions from their Cre-bearing parents. Even though Cre was not expressed in the experimental animals, the deletion was complete because the Global-lep-null mutants did not have any detectable serum leptin.
Deletion of adipocyte leptin eliminates all detectable circulating leptin
Our results confirm, for the first time, the ongoing assumption that circulating leptin is secreted mainly or completely by adipocytes. In animals in which leptin is deleted only in adipocytes, circulating leptin levels are undetectable. This indicates that, although leptin is produced by several different tissue and cell types, these extraadipocyte sources of leptin do not secrete detectable levels of the hormone into the bloodstream, even during the postnatal leptin surge (Odle A. K., et al, manuscript in preparation) (60, 61). This discovery also points to the significance of extraadipocyte sources of leptin, which may serve as paracrine or autocrine regulators in their respective tissues.
Both Global-lep-null and Adipocyte-lep-null mutants are obese by or shortly after weaning and continue to gain weight into adulthood, mimicking the weight gain observed in the ob/ob mouse (2). We found no significant differences in the weights of the Global-lep-null mutants vs the weights of the Adipocyte-lep-null mutants (within sexes, data not shown). Therefore, the leptin produced by nonadipocyte sources (such as the muscle, the stomach, and the pituitary) were not enough to prevent the massive weight gain in the adipocyte leptin deletion mutants. As in the case of ob/ob males and females, neither male nor female mutants from the Global- or Adipocyte-lep-null lines are fertile. Rarely an ob/ob male is fertile (62); however, our tests of young males from either of our lines resulted in no offspring (data not shown).
Deletion of adipocyte leptin changes pituitary gonadotropin and prolactin secretion
Ob/ob animals, which have a point mutation in the lep gene, have a significantly impaired hypothalamic-pituitary-gonadal axis. Ob/ob males and females at 5–6 months of age have significantly decreased serum FSH values, whereas serum LH values do not differ from controls (21, 22). Leptin treatment of ob/ob animals can restore reproductive ability in both males and females (20, 22). Leptin treatment prevented the age-related decline of LH in ob/ob females while increasing the serum FSH levels of ob/ob males compared with pair-fed ob/ob controls. Although pituitary defects are not the only contributing factors to the infertility of the leptin-deficient mouse, they are the major contributors. Female Adipocyte-lep-null pituitaries have significantly reduced LH mRNA, which correlates with low serum LH values in this group. Male Adipocyte-lep-null animals have no significant differences in the LH or FSH transcript levels, but they do have significantly decreased serum FSH levels. Although female Adipocyte-lep-null animals differ from ob/ob females of the same age, the defects seen in the pituitary and serum are severe enough to contribute to the infertility of the model. The changes in our Adipocyte-lep-null serum gonadotropin levels suggest that circulating leptin is important for gonadotropin release in a sex-dependent manner. Pituitary leptin was not sufficient to maintain LH release in females or FSH release in males. Similarly, our Global-lep-null males had a significant decrease in serum FSH.
Ob/ob males and females both have significantly decreased pituitary prolactin content as well as decreased circulating prolactin levels (25, 63). Both male and female Adipocyte-lep-null mutants had decreased prolactin transcript levels, and females had significantly decreased circulating prolactin levels (male levels trended low, but variation was too great to achieve significance). Global-lep-null females also had significantly decreased prolactin. It is evident that both the production and the release of prolactin appear to be dependent on circulating leptin and that pituitary leptin cannot sustain the functions of this cell population.
Effects of deleting adipocyte leptin on corticotropes and thyrotropes
Ob/ob adult pituitaries contain elevated levels of both ACTH and MSH (64, 65) and the resulting high glucocorticoids may be partially responsible decreasing hypothalamic GHRH mRNA (66). Both male and female Adipocyte-lep-null mutants have high POMC transcript levels, with no significant differences in serum ACTH levels. Although neither male nor female Adipocyte-lep-null had significant changes in serum TSH levels, female mutant pituitaries had significantly higher levels of TSH mRNA. TSH secretion is decreased in the fasted rodent, a phenomenon that can be corrected with leptin administration, suggesting that leptin plays a role in the regulation of thyroid hormone secretion (67)
Effects of deleting adipocyte leptin on the GH axis
There are many studies on the relationship between leptin and GH. Leptin stimulates GH secretion from the pituitaries of several species (68–74), and our laboratory has shown that deletion of leptin receptors on somatotropes leads to decreased secretion of GH (30). In this study, the deletion of leptin from adipocytes did not significantly change serum GH levels. However, this lack of significance may be due to the large SE that is often the result of averaging pulsatile hormone levels (or the relatively low number). This also may be the case for our Global-lep-null males, whose decrease in serum GH is also not statistically significant. Pituitary GH mRNA did not differ significantly in either Adipocyte-lep-null males nor females, and the percentage of pituitary cells that are GH positive in adult males and females did not differ from controls. The fact that our Adipocyte-lep-null males have no change in the percentage of pituitary cells labeled for GH is interesting and suggests that the expansion in somatotropes to adult levels, which occurs during the first week of postnatal life (75), does not depend on circulating leptin. This is further confirmed by the decrease in pituitary cells labeled for GH in our Global-lep-null males. It also suggests that circulating leptin is not needed to maintain somatotrope numbers or GH mRNA in the adult animals. Having recently demonstrated the importance of leptin to optimal somatotrope numbers and function (30), these data collectively suggest that pituitary leptin is capable of sustaining the somatotrope development and numbers, in the absence of circulating adipocyte leptin.
Pituitary GHRHR mRNA was increased in both male and female Adipocyte-lep-null mutants. The high GHRHR mRNA levels in the mutants do not correlate with those of the ob/ob mice as reported by Luque and Kineman (26), in which ob/ob mouse GHRHR transcript levels did not differ from controls. Female Adipocyte-lep-null mutants have significantly decreased GH secretagogue mRNA. This result has also been seen in male ob/ob mice, although male Adipocyte-lep-null mutants did not have a significant change in GHS-R mRNA (26). We also investigated transcription in the governing organ of the pituitary, the hypothalamus. In ob/ob mice, there are no significant changes in hypothalamic GHRH, LepR, or somatostatin transcript levels (26). Like the ob/ob mice, our Global-lep-null male deletion mutants had no significant changes in any of the three transcripts. However, the Adipocyte-lep-null deletion mutant males and females had significantly decreased GHRH mRNA levels. It is interesting to note that the numbers of somatotropes are maintained despite the loss of hypothalamic GHRH mRNA in Adipocyte-lep-null deletion males. The numbers of somatotropes are consistent with the findings that GH mRNA is normal, and GHRHR mRNA is higher in the pituitaries of Adipocyte-lep-null mutant mice. Luque et al (27) reported that in vivo leptin treatment stimulated both GH and GHRHR mRNA in ob/ob mouse pituitaries. Collectively these data point to pituitary leptin as a key factor in the regulation of the somatotrope. Obviously circulating adipocyte leptin is needed to lower neuropeptide Y and maintain hypothalamic GHRH expression, and thus, the two sources of leptin work together to support the hypothalamic-pituitary-GH axis.
Conclusion
We created a tissue-specific leptin knockout model to determine the importance of adipocyte vs pituitary leptin to the development and function of the pituitary. Even though the pituitaries of male Adipocyte-lep-null animals contained significantly higher percentages of cells producing leptin, this leptin could not be detected in the bloodstream. We confirmed for the first time that adipocytes truly are the only source of circulating leptin detected in the mouse. And although extraadipocyte sources of leptin were unable to prevent the Adipocyte-lep-null animals from becoming obese, differences were found between these mutants and results previously found in ob/ob mice. However, we note that ob/ob mice have a single-point mutation in the lep gene, and this is different from our deletion, which eliminates the largest coding exon. The Adipocyte-lep-null mutants (males and females) had significantly decreased hypothalamic GHRH mRNA and significantly increased pituitary GHRHR mRNA. Additionally, the percentage of pituitary cells in the Adipocyte mutant males containing GH did not differ from controls, even though the serum GH levels trended low. This suggests that the pituitary was able to develop the somatotrope cell population, perhaps because of the presence (and increase) of pituitary leptin. However, we hypothesize that the low GHRH and low serum GH levels seen in the males indicate a specific role for adipocyte leptin in the secretion of GH. Future studies will complement this one by deleting leptin only in the pituitary. With a tool as powerful as the floxed leptin mouse, investigators may soon understand the roles of individual sources of leptin.
Acknowledgments
We thank Dr A. F. Parlow and the National Institutes of Health Hormone Pituitary Distribution Program for the antiserum to GH. We also thank Douglas Kessel, Orpa Ali, and Farhan Syed for their excellent technical assistance.
Information from this publication was presented in abstract form at the 95th Annual Meeting of The Endocrine Society as poster MON-146 as well as the 2014 International Congress of Endocrinology/Annual Meeting of The Endocrine Society as poster MON-699.
This paper is submitted in partial fulfillment of the requirements for the PhD degree (A. Odle).
This work was supported by National Institutes of Health Grants R03 HD059066 and 1R01HD059056 (to G.V.C.), the core facilities of the Center for Translational Neuroscience funded by National Institutes of Health NIGMS IDeA Award P20 GM103425, and the Neurosciences Center supported by National Institutes of Health Grant P30 NS047546 at the University of Arkansas for Medical Sciences.
Disclosure Summary: The authors have no conflicts to disclose.
Footnotes
- CTL
- controls
- DEL
- male mutant
- EIA
- enzyme immunoassay
- GHRHR
- GHRH receptor
- GHS-R
- GH secretagogue receptor
- LepRb
- long-form leptin receptor
- POMC
- proopiomelanocortin
- qRT-PCR
- quantitative real-time-PCR.
References
- 1. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. [DOI] [PubMed] [Google Scholar]
- 2. Ingalls AM, Dickie MM, Snell GD. Obese, a new mutation in the house mouse. J Heredity. 1950;41:317–318. [DOI] [PubMed] [Google Scholar]
- 3. Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest. 1996;98:1101–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tartaglia LA, Dembski M, Weng X, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. [DOI] [PubMed] [Google Scholar]
- 5. Schwartz MW, Baskin DG, Bukowski TR, et al. Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes. 1996;45:531–535. [DOI] [PubMed] [Google Scholar]
- 6. Woods AJ, Stock MJ. Leptin activation in hypothalamus. Nature. 1996;381:745. [DOI] [PubMed] [Google Scholar]
- 7. Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 1996;387:113–116. [DOI] [PubMed] [Google Scholar]
- 8. Halaas JL, Gajiwala KS, Maffei M, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. [DOI] [PubMed] [Google Scholar]
- 9. Lonnqvist F, Arner P, Nordfors L, Schalling M. Overexpression of the obese (ob) gene in adipose tissue of human obese subjects. Nat Med. 1995;1:950–953. [DOI] [PubMed] [Google Scholar]
- 10. Hamilton BS, Paglia D, Kwan AY, Deitel M. Increased obese mRNA expression in omental fat cells from massively obese humans. Nat Med. 1995;1:953–956. [DOI] [PubMed] [Google Scholar]
- 11. Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–1314. [DOI] [PubMed] [Google Scholar]
- 12. Maffei M, Fei H, Lee GH, et al. Increased expression in adipocytes of ob RNA in mice with lesions of the hypothalamus and with mutations at the db locus. Proc Natl Acad Sci USA. 1995;92:6957–6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maffei M, Halaas J, Ravussin E, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–1161. [DOI] [PubMed] [Google Scholar]
- 14. Shetty GK, Matarese G, Magkos F, et al. Leptin administration to overweight and obese subjects for 6 months increases free leptin concentrations but does not alter circulating hormones of the thyroid and IGF axes during weight loss induced by a mild hypocaloric diet. Eur J Endocrinol. 2011;165:249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cioffi JA, Van Blerkom J, Antczak M, Shafer A, Wittmer S, Snodgrass HR. The expression of leptin and its receptors in pre-ovulatory human follicles. Mol Hum Reprod. 1997;3:467–472. [DOI] [PubMed] [Google Scholar]
- 16. Masuzaki H, Ogawa Y, Sagawa N, et al. Nonadipose tissue production of leptin: leptin as a novel placenta-derived hormone in humans. Nat Med. 1997;3:1029–1033. [DOI] [PubMed] [Google Scholar]
- 17. Morash B, Li A, Murphy PR, Wilkinson M, Ur E. Leptin gene expression in the brain and pituitary gland. Endocrinology. 1999;140:5995–5998. [DOI] [PubMed] [Google Scholar]
- 18. Sobhani I, Bado A, Vissuzaine C, et al. Leptin secretion and leptin receptor in the human stomach. Gut. 2000;47:178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J, Liu R, Hawkins M, Barzilai N, Rossetti L. A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature. 1998;393:684–688. [DOI] [PubMed] [Google Scholar]
- 20. Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet. 1996;12:318–320. [DOI] [PubMed] [Google Scholar]
- 21. Swerdloff RS, Batt RA, Bray GA. Reproductive hormonal function in the genetically obese (ob/ob) mouse. Endocrinology. 1976;98:1359–1364. [DOI] [PubMed] [Google Scholar]
- 22. Barash IA, Cheung CC, Weigle DS, et al. Leptin is a metabolic signal to the reproductive system. Endocrinology. 1996;137:3144–3147. [DOI] [PubMed] [Google Scholar]
- 23. Yu WH, Kimura M, Walczewska A, Karanth S, McCann SM. Role of leptin in hypothalamic-pituitary function. Proc Natl Acad Sci USA. 1997;94:1023–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roos P, Martin JM, Westman-Naeser S, Hellerstrom C. Immunoreactive growth hormone levels in mice with the obese-hyperglycemic syndrome (genotype obob). Horm Metab Res. 1974;6:125–128. [DOI] [PubMed] [Google Scholar]
- 25. Sinha YN, Salocks CB, Vanderlaan WP. Prolactin and growth hormone secretion in chemically induced and genetically obese mice. Endocrinology. 1975;97:1386–1393. [DOI] [PubMed] [Google Scholar]
- 26. Luque RM, Kineman RD. Impact of obesity on the growth hormone axis: evidence for a direct inhibitory effect of hyperinsulinemia on pituitary function. Endocrinology. 2006;147:2754–2763. [DOI] [PubMed] [Google Scholar]
- 27. Luque RM, Huang ZH, Shah B, Mazzone T, Kineman RD. Effects of leptin replacement on hypothalamic-pituitary growth hormone axis function and circulating ghrelin levels in ob/ob mice. Am J Physiol Endocrinol Metab. 2007;292:E891–E899. [DOI] [PubMed] [Google Scholar]
- 28. Steppan CM, Crawford DT, Chidsey-Frink KL, Ke H, Swick AG. Leptin is a potent stimulator of bone growth in ob/ob mice. Regul Pept. 2000;92:73–78. [DOI] [PubMed] [Google Scholar]
- 29. Garthwaite TL, Kalkhoff RK, Guansing AR, Hagen TC, Menahan LA. Plasma free tryptophan, brain serotonin, and an endocrine profile of the genetically obese hyperglycemic mouse at 4–5 months of age. Endocrinology. 1979;105:1178–1182. [DOI] [PubMed] [Google Scholar]
- 30. Childs GV, Akhter N, Haney A, et al. The somatotrope as a metabolic sensor: deletion of leptin receptors causes obesity. Endocrinology. 2011;152:69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Crane C, Akhter N, Johnson BW, et al. Fasting and glucose effects on pituitary leptin expression: is leptin a local signal for nutrient status? J Histochem Cytochem. 2007;55:1059–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McDuffie IA, Akhter N, Childs GV. Regulation of leptin mRNA and protein expression in pituitary somatotropes. J Histochem Cytochem. 2004;52:263–273. [DOI] [PubMed] [Google Scholar]
- 33. Akhter N, Johnson BW, Crane C, et al. Anterior pituitary leptin expression changes in different reproductive states: stimulation, in vitro, by gonadotropin releasing hormone (GnRH). J Histochem Cytochem. 2007;55:151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Isse N, Ogawa Y, Tamura N, et al. Structural organization and chromosomal assignment of the human obese gene. J Biol Chem. 1995;270:27728–27733. [DOI] [PubMed] [Google Scholar]
- 35. Hong CJ, Tsai PJ, Cheng CY, et al. ENU mutagenesis identifies mice with morbid obesity and severe hyperinsulinemia caused by a novel mutation in leptin. PLoS One. 2010;5:e15333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eguchi J, Wang X, Yu S, et al. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 2011;13:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lakso M, Pichel JG, Gorman JR, et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci USA. 1996;93:5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-δδC(T)] method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 39. Luque RM, Lin Q, Cordoba-Chacon J, et al. Metabolic impact of adult-onset, isolated, growth hormone deficiency (AOiGHD) due to destruction of pituitary somatotropes. PLoS One. 2011;6:e15767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luque RM, Cordoba-Chacon J, Ibanez-Costa A, et al. Obestatin plays an opposite role in the regulation of pituitary somatotrope and corticotrope function in female primates and male/female mice. Endocrinology. 2014;en20131728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Groba C, Mayerl S, van Mullem AA, et al. Hypothyroidism compromises hypothalamic leptin signaling in mice. Mol Endocrinol. 2013;27:586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kineman RD, Gahete MD, Luque RM. Identification of a mouse ghrelin gene transcript that contains intron 2 and is regulated in the pituitary and hypothalamus in response to metabolic stress. J Mol Endocrinol. 2007;38:511–521. [DOI] [PubMed] [Google Scholar]
- 43. Ernst MC, Haidl ID, Zuniga LA, et al. Disruption of the chemokine-like receptor-1 (CMKLR1) gene is associated with reduced adiposity and glucose intolerance. Endocrinology. 2012;153:672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kiefer FW, Zeyda M, Gollinger K, et al. Neutralization of osteopontin inhibits obesity-induced inflammation and insulin resistance. Diabetes. 2010;59:935–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lijnen HR, Christiaens V, Scroyen I, et al. Impaired adipose tissue development in mice with inactivation of placental growth factor function. Diabetes. 2006;55:2698–2704. [DOI] [PubMed] [Google Scholar]
- 46. Shapiro NI, Khankin EV, Van Meurs M, et al. Leptin exacerbates sepsis-mediated morbidity and mortality. J Immunol. 2010;185:517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Childs GV, Unabia G, Miller BT. Cytochemical detection of gonadotropin-releasing hormone-binding sites on rat pituitary cells with luteinizing hormone, follicle-stimulating hormone, and growth hormone antigens during diestrous up-regulation. Endocrinology. 1994;134:1943–1951. [DOI] [PubMed] [Google Scholar]
- 48. Ueno N, Inui A, Kalra PS, Kalra SP. Leptin transgene expression in the hypothalamus enforces euglycemia in diabetic, insulin-deficient nonobese Akita mice and leptin-deficient obese ob/ob mice. Peptides. 2006;27:2332–2342. [DOI] [PubMed] [Google Scholar]
- 49. Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology. 1997;138:4489–4492. [DOI] [PubMed] [Google Scholar]
- 50. Schwartz MW, Seeley RJ, Woods SC, et al. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes. 1997;46:2119–2123. [DOI] [PubMed] [Google Scholar]
- 51. Denver RJ, Bonett RM, Boorse GC. Evolution of leptin structure and function. Neuroendocrinology. 2011;94:21–38. [DOI] [PubMed] [Google Scholar]
- 52. Kieffer TJ, Heller RS, Habener JF. Leptin receptors expressed on pancreatic β-cells. Biochem Biophys Res Commun. 1996;224:522–527. [DOI] [PubMed] [Google Scholar]
- 53. Zamorano PL, Mahesh VB, De Sevilla LM, Chorich LP, Bhat GK, Brann DW. Expression and localization of the leptin receptor in endocrine and neuroendocrine tissues of the rat. Neuroendocrinology. 1997;65:223–228. [DOI] [PubMed] [Google Scholar]
- 54. Serradeil-Le Gal C, Raufaste D, Brossard G, et al. Characterization and localization of leptin receptors in the rat kidney. FEBS Lett. 1997;404:185–191. [DOI] [PubMed] [Google Scholar]
- 55. Hoggard N, Mercer JG, Rayner DV, Moar K, Trayhurn P, Williams LM. Localization of leptin receptor mRNA splice variants in murine peripheral tissues by RT-PCR and in situ hybridization. Biochem Biophys Res Commun. 1997;232:383–387. [DOI] [PubMed] [Google Scholar]
- 56. Hoggard N, Hunter L, Duncan JS, Williams LM, Trayhurn P, Mercer JG. Leptin and leptin receptor mRNA and protein expression in the murine fetus and placenta. Proc Natl Acad Sci USA. 1997;94:11073–11078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Couce ME, Burguera B, Parisi JE, Jensen MD, Lloyd RV. Localization of leptin receptor in the human brain. Neuroendocrinology. 1997;66:145–150. [DOI] [PubMed] [Google Scholar]
- 58. Henson MC, Swan KF, O'Neil JS. Expression of placental leptin and leptin receptor transcripts in early pregnancy and at term. Obstet Gynecol. 1998;92:1020–1028. [DOI] [PubMed] [Google Scholar]
- 59. Breidert M, Miehlke S, Glasow A, et al. Leptin and its receptor in normal human gastric mucosa and in Helicobacter pylori-associated gastritis. Scand J Gastroenterol. 1999;34:954–961. [DOI] [PubMed] [Google Scholar]
- 60. Rayner DV, Dalgliesh GD, Duncan JS, Hardie LJ, Hoggard N, Trayhurn P. Postnatal development of the ob gene system: elevated leptin levels in suckling fa/fa rats. Am J Physiol. 1997;273:R446–R450. [DOI] [PubMed] [Google Scholar]
- 61. Ahima RS, Prabakaran D, Flier JS. Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J Clin Invest. 1998;101:1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Charlton HM. Mouse mutants as models in endocrine research. Q J Exp Physiol. 1984;69:655–676. [DOI] [PubMed] [Google Scholar]
- 63. Larson BA, Sinha YN, Vanderlaan WP. Serum growth hormone and prolactin during and after the development of the obese-hyperglycemic syndrome in mice. Endocrinology. 1976;98:139–145. [DOI] [PubMed] [Google Scholar]
- 64. Edwardson JA, Hough CA. The pituitary-adrenal system of the genetically obese (ob/ob) mouse. J Endocrinol. 1975;65:99–107. [DOI] [PubMed] [Google Scholar]
- 65. Peaslee MH, Moisset B, Shibuya H, Pert CB. Increase in pituitary melanocyte-stimulating hormone activity of genetically obese (ob/ob) mice. Experientia. 1980;36:133–134. [DOI] [PubMed] [Google Scholar]
- 66. Luque RM, Park S, Kineman RD. Severity of the catabolic condition differentially modulates hypothalamic expression of growth hormone-releasing hormone in the fasted mouse: potential role of neuropeptide Y and corticotropin-releasing hormone. Endocrinology. 2007;148:300–309. [DOI] [PubMed] [Google Scholar]
- 67. Ahima RS, Prabakaran D, Mantzoros C, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. [DOI] [PubMed] [Google Scholar]
- 68. Saleri R, Grasselli F, Tamanini C. Effects of different culture conditions and leptin on GH mRNA expression and GH secretion by pig pituitary cells. Horm Metab Res. 2005;37:214–219. [DOI] [PubMed] [Google Scholar]
- 69. Isozaki O, Tsushima T, Miyakawa M, Demura H, Seki H. Interaction between leptin and growth hormone (GH)/IGF-I axis. Endocr J. 1999;46(suppl):S17–S24. [DOI] [PubMed] [Google Scholar]
- 70. Nagatani S, Zeng Y, Keisler DH, Foster DL, Jaffe CA. Leptin regulates pulsatile luteinizing hormone and growth hormone secretion in the sheep. Endocrinology. 2000;141:3965–3975. [DOI] [PubMed] [Google Scholar]
- 71. Zieba DA, Amstalden M, Morton S, et al. Effects of leptin on basal and GHRH-stimulated GH secretion from the bovine adenohypophysis are dependent upon nutritional status. J Endocrinol. 2003;178:83–89. [DOI] [PubMed] [Google Scholar]
- 72. Roh S, Clarke IJ, Xu R, Goding JW, Loneragan K, Chen C. The in vitro effect of leptin on basal and growth hormone-releasing hormone-stimulated growth hormone secretion from the ovine pituitary gland. [DOI] [PubMed] [Google Scholar]
- 73. Roh SG, Nie GY, Loneragan K, Gertler A, Chen C. Direct modification of somatotrope function by long-term leptin treatment of primary cultured ovine pituitary cells. Endocrinology. 2001;142:5167–5171. [DOI] [PubMed] [Google Scholar]
- 74. Shimon I, Yan X, Magoffin DA, Friedman TC, Melmed S. Intact leptin receptor is selectively expressed in human fetal pituitary and pituitary adenomas and signals human fetal pituitary growth hormone secretion. J Clin Endocrinol Metab. 1998;83:4059–4064. [DOI] [PubMed] [Google Scholar]
- 75. Taniguchi Y, Yasutaka S, Kominami R, Shinohara H. Proliferation and differentiation of pituitary somatotrophs and mammotrophs during late fetal and postnatal periods. Anat Embryol (Berl). 2001;204:469–475. [DOI] [PubMed] [Google Scholar]