

Abstract
The steroid hormone aldosterone (aldo) contributes to cardiovascular disease in animal models and in humans. Aldo activates the mineralocorticoid receptor (MR), a hormone-activated transcription factor, and indeed, pharmacological MR inhibition improves cardiovascular outcomes. Because the incidence of cardiovascular disease is lower in premenopausal women, we hypothesized that estrogen (E2) signaling through the estrogen receptor (ER) may protect the vasculature by inhibiting the detrimental effects of aldo signaling through the MR. We demonstrate that E2-activated ER inhibits MR-mediated gene transcription from the mouse mammary tumor virus reporter in human embryonic kidney-293 cells. In contrast, aldo-activated MR does not affect ER-mediated gene transcription. The ERα N terminus (amino acids 1–253) containing part of the DNA-binding domain is sufficient to inhibit MR genomic function, although point mutations reveal that DNA binding, ligand-independent activation, and rapid nongenomic ERα signaling are not required for this effect. Furthermore, ERα and MR are part of a complex in cell lysates, with amino acids 1–233 of the ERα N terminus being sufficient to complex with the MR. Overall, the ability of ERα to inhibit MR-mediated gene transcription correlates with the ability of ERα segments to both localize to the nucleus and complex with the MR. In cultured vascular endothelial cells expressing ERα, E2 inhibits aldo induction of the vascular MR target gene intercellular adhesion molecule-1 (ICAM-1). ICAM-1 induction by endothelial MR is known to promote vascular inflammation that could contribute to the mechanism of aldo-induced atherosclerosis. E2 also inhibits aldo induction of ICAM-1 protein and prevents aldo-enhanced leukocyte adhesion to endothelial cells. These studies support a new model in which E2-activated ER in endothelial cells forms a complex with MR in the nucleus to modulate MR regulation of the proinflammatory gene ICAM-1. Estrogen inhibition of MR regulation of genes that contribute to cardiovascular disease may be a new mechanism by which premenopausal women are protected from cardiovascular disease.
Cardiovascular disease is the leading cause of death in both men and women in the developed world (1). Epidemiological studies have consistently demonstrated a relative protection from myocardial infarction and cardiovascular mortality in premenopausal women compared with men of the same age. This protection is lost after the age of menopause (1), yet the mechanisms for it remain poorly understood. Much evidence supports a role for endogenous estrogen in mediating this protective effect (2, 3). The biologically active estrogen, estradiol (E2), is a steroid hormone that acts by binding to intracellular estrogen receptors (ER), of which there are two isoforms, ERα and ERβ. ERs mediate the cardiovascular protective effects of E2 in mouse models (4–7).
The mineralocorticoid receptor (MR) is another member of the nuclear receptor family of hormone-activated transcription factors that is responsive to the steroid hormone aldosterone (aldo). Aldo activation of MR in the kidney regulates sodium and fluid balance thereby regulating blood pressure. MR also is expressed in the vasculature (8–11) and in many other tissues (11–14). As members of the nuclear receptor family, MR and ER share a common domain structure including an N-terminal AB domain that mediates ligand-independent activation, a DNA-binding domain that interacts with specific hormone-responsive DNA sequences, a hinge region that includes a nuclear localization sequence, and a C-terminal ligand-binding domain that recruits cofactors to regulate gene transcription upon ligand binding (15). Accordingly, nuclear receptors can act through multiple mechanisms including genomic mechanisms in which they bind hormone, translocate to the nucleus, bind to specific DNA sequences, and act as transcription factors to regulate gene expression. Steroid receptors can also be activated in a ligand-independent manner by signaling events from transmembrane and G protein-coupled receptors (8, 16–18). In addition, both ER and MR act through rapid signaling (often termed nongenomic) mechanisms in which receptors bind hormone outside the nucleus and participate in signaling cascades to exert rapid effects on cellular functions (19, 20).
In contrast to the many beneficial cardiovascular effects seen with activation of ER by E2, activation of MR by aldo exacerbates cardiovascular disease (21–24). In humans, elevated serum aldo levels correlate with increased incidence of myocardial infarction, stroke, and cardiovascular death in patients with hypertension or other cardiovascular risk factors (25, 26). Conversely, MR antagonists prevent cardiovascular mortality (27–30) by unclear mechanisms. In animal models of hyperlipidemia-induced atherosclerosis or wire-induced vascular endothelial damage, aldo infusion promotes atherosclerosis and vascular remodeling after injury (22, 31), whereas estrogen is protective in the same animal models (6, 32, 33).
Endothelial dysfunction represents an early stage of vessel wall damage and is a marker of subclinical atherosclerosis (34, 35). Human studies demonstrate that endothelium-dependent vasodilation and subsequent progression to atherosclerosis correlate with plasma aldosterone levels, suggesting that aldosterone action specifically in the endothelium may be important in the pathogenesis of atherosclerosis (36). Likewise, multiple human studies reveal an association of serum estrogen levels with preservation of endothelial function in woman (37–39), and even in men, estradiol level is positively associated with flow-mediated vasodilation independent of other cardiovascular risk factors (40). Based on these data, we hypothesized that estrogen signaling through the ER may protect the vasculature by inhibiting the detrimental effects of aldo and MR on the regulation of genes in endothelial cells that contribute to cardiovascular disease. To test this hypothesis, we investigated the interaction between ER and MR transcriptional function using a heterologous promoter system in vitro, examined interactions between the receptors by biochemical assays, determined the impact of ER on MR regulation of intercellular adhesion molecule-1 (ICAM-1) expression in vascular endothelial cells, and assessed the relevance of our findings using an in vitro functional assay of immune cell adhesion to the endothelium.
Materials and Methods
Reagents and cell lines
Aldo and E2 (Sigma-Aldrich) were used as described (8, 41) with dimethylsulfoxide (Sigma-Aldrich) and ethanol (Fisher Scientific) controls, respectively. Human embryonic kidney-293 (HEK293) cells (American Type Culture Collection) were maintained in phenol-containing DMEM (Gibco) with 10% fetal bovine serum (FBS; Atlantic Biologicals). HEK293 cells were switched to phenol-free 1% charcoal-dextran-stripped FBS (S-FBS) for 24 hours prior to any experimental treatment. For protein overexpression experiments, cells were transiently transfected with MR or ERα expression plasmids (described below) by PolyFect transfection reagent (QIAGEN). Endothelial cell line EAhy926 cells [a human umbilical vein endothelial cell hybrid line, a kind gift of C. J. Edgell, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina (32)] were grown in phenol-containing DMEM with 10% FBS and switched to 10% S-FBS prior to experimental treatment. EAhy926 cells were stably transfected with the pcDNA 3.1 ERα. Twenty-four hours after transfection, cells were placed in selective media with puromycin at 5 μg/mL (Sigma) for 2–3 weeks. Single colonies were selected from 96-well plates that stably express moderate levels of ERα and maintained in the presence of 2 μg/mL puromycin. The expression of functional ERα was confirmed by ERα immunoblotting and estrogen response element (ERE)-luciferase reporter assays (data not shown).
Expression and reporter plasmids
The MR expression plasmid contains the full-length human MR cDNA [a generous gift of R. Evans (42)] cloned into the CMX expression vector with an N-terminal hemagluttanin (HA) tag. The MR reporter plasmid contains the mouse mammary tumor virus (MMTV) long-terminal repeat (43) cloned into the PGL2 luciferase reporter vector (Promega). The ERE reporter contains three copies of the Xenopus vitellogenin ERE24 (consensus sequence 5′-GGTCAnnnTGACC-3′) proximal to the thymidine kinase promoter-driving expression of luciferase as described (16).The construction of human full-length wild-type human ERα, ERα-S118A with an alanine-for-serine substitution at amino acid 118, pCMV3 ERα1–271, and pCMV3 ER 176–595 have been described previously (16). ERα 176–271 and 254–370 constructs were made by cloning PCR-derived ERα fragments into a pC2 vector (CLONTECH) by EcoRI and SmaI sites, respectively. The His-tagged ERα plasmids were made by cloning ERα full-length and fragments into pet28a(+) (Novagen) by EcoRI and SmaI sites. The following plasmids were made by a QuikChange II XL site-directed mutagenesis kit (Stratagene). ER DNA binding domain (DBD) mutants were made by alanine-for-cystine substitution in the DBD domain at amino acids 220 and 227; the ER KRR mutant was made by alanine for 231K, 233R, and 234R substitution; ERα 1–263, 1–253, 1–233, and 1–176 were made by the insertion of a stop codon at ERα amino acids 263, 253, 233, and 176, respectively.
Transfections and luciferase assay
Luciferase reporter assays were performed as described (16) by cotransfection of plasmids for receptors (MR, ERα, ERα mutants), a plasmid for MR- or ERα-response element-driving luciferase expression (43), and a β-galactosidase plasmid to normalize for transfection efficiency. Twenty-four hours later, cells were treated with aldo and/or E2 for 18 hours. Quantification of luciferase activity with a luciferase assay kit (Promega) was normalized to β-galactosidase activity assessed with Tropic accelerator (Applied Biosystems). Each treatment was carried out in triplicate and was performed in a minimum of three independent experiments.
Coimmunoprecipitation and immunodetection
HEK293 cells were cotransfected with the human HA-MR and ERα expression plasmids described above and switched to DMEM phenol red free medium with 10% S-FBS for 16 hours and then switched to serum-free medium for 24 hours, followed by treatment with aldo, E2, or aldo plus E2 as indicated. Cells were harvested in lysis buffer (20 mM Tris-Cl, pH 7.5; 0.137 M NaCl; 2 mm EDTA, pH 7.4; 1% Triton X-100; 10% glycerol; 25 mM glycerol phosphate; and in the presence of phenylmethylsulfonyl fluoride and protease inhibitor mixture). The coimmunoprecipitations were performed as described (41, 44) with the incubation of cell lysates with 5 μg of nonimmune rabbit IgG or rabbit anti-HA antibody (Santa Cruz Biotechnology Inc) overnight at 4°C. Protein G beads (Amersham Biosciences) were then added and a further incubation carried out at 4°C for 2 hours. The pellets obtained after centrifugation were washed five times with wash buffer (50 mM Tris, pH 7.5; 7 mM MgCl2; 2 mM EDTA; and 1 mM phenylmethylsulfonyl fluoride). The immunopellets were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and then probed with appropriate primary antibodies. Antibodies used for immunoblotting (Supplemental Table 1) include rabbit polyclonal anti-ERα HC20 (Santa Cruz Biotechnology) and mouse monoclonal anti-MR [a generous gift from Dr Celso Gomez-Sanchez (45)]. Membranes were washed three times with wash buffer followed by incubation with antimouse or antirabbit-horseradish peroxidase secondary antibody (GE Healthcare UK Ltd) and developed with enhanced chemiluminescence reagent (Amersham Biosciences).
His pull-down assay
His-ERα plasmids were transformed into Escherichia coli BL21(DE3) competent cells. His-fusion protein expression was induced by 1 mM isopropyl β-D-1-thiogalactopyranoside for 3 hours. The fusion proteins were purified using Nickel-NTA agarose beads (Invitrogen). Expression of the fusions proteins was confirmed by SDS-PAGE and Coomassie Blue staining. The HEK293 cell lysates expressing human MR were incubated with 50 μL of each His fusion protein beads. The samples were rocked at 4°C overnight, washed five times to remove nonspecific binding, and boiled in sodium dodecyl sulfate sample buffer. Associated proteins were resolved by SDS-PAGE and immunoblotted as described above.
Immunoflourescent staining
HEK293 cells were transfected with flag-tagged ERα, ERα mutants, or ERα fragments. The cells were fixed in 3.7% paraformaldehyde for 10 minutes and permeabilized with 0.3% Triton X-100 for 15 minutes. After blocking with 10% donkey serum for 1 hour, immunostaining was performed as described (41, 44) by incubating the cells with anti-Flag-tag (M2; Sigma) antibody for 1 hour (Supplemental Table 1), washing three times with PBS followed by incubation with CY3-conjugated donkey antirabbit secondary antibody (1:1000) or Cy3-labeled donkey antimouse secondary antibody (1:1000) (Jackson ImmunoResearch). Cells were then washed three times with PBS, and the nuclei were stained with 4′,6′-diamino-2-phenylindole (DAPI; Sigma) for 15 minutes. Cells were analyzed using fluorescence microscopy.
Quantitative RT-PCR
RNA was isolated from endothelial cells and reverse transcribed, and quantitative RT-PCR was performed by methods that have been previously described (9, 46). Cycle threshold values were normalized to glyceraldehyde-3-phosphate dehydrogenase, and mRNA levels in hormone-treated samples were expressed as a fold change relative to the expression level in vehicle-treated samples. Primer sequences were as follows: ICAM-1, forward, GGC CTC AGT CAG TGT GA and reverse, AAC CCC ATT CAG CGT CA; glyceraldehyde-3-phosphate dehydrogenase forward, GAG CCA AAA GGG TCA TCA TCT CT and reverse, GAG CCA AAA GGG TCA TCA TCT CT.
Leukocyte adhesion assay
EAhy926 cells stably expressing ERα were grown in DMEM with 10% S-FBS until 80% confluent and switched to serum-free media for 24 hours. The cells were treated with aldo, E2, aldo plus E2, or vehicle for 18 hours. The leukocytes (U937 cell) were harvested, washed twice with PBS, and labeled with 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein-acetoxymethyl (2 μM) (Molecular Probes) in PBS for 30 minutes at 37°C. The fluorescently labeled U937 cells were incubated with EAhy926 cells for 2 hours. The cells were gently washed with PBS three times to remove the nonadherent cells. The adherent fluorescent cells were counted by a reader blinded to cell treatments as described (9).
Statistical analysis
All values are reported as mean fold change compared with vehicle control ± SEM. Within-group differences were assessed with one-factor ANOVA. Post hoc comparisons were tested with the Student-Newman-Keuls test. P < .05 was considered significant.
Results
The ER inhibits MR-mediated transcriptional activation
To test whether an interaction exists between the transcriptional activity of ER and MR, transcriptional reporter assays were performed in HEK293 cells using a luciferase reporter containing the MMTV MR-responsive element (MRE-luciferase; Figure 1A) or containing the Xenopus vitellogenin ERE24, an ERE (ERE-luciferase; Figure 1B). Because these HEK293 cells do not express endogenous MR [or glucocorticoid receptor (GR)], aldo (and cortisol) did not affect MRE-luciferase reporter activity in the absence of cotransfected MR (Supplemental Figure 1). Expression of MR increased MRE-luciferase reporter activity, likely due to ligand-independent MR activation. MRE activity was further augmented by addition of aldo in a dose-dependent manner. We began with 10 nM because this is a concentration consistent with that found in the serum of patients with cardiovascular disease (47). Cotransfection of ERα-expressing plasmid substantially attenuated MR-dependent reporter activity, and the further addition of estrogen completely inhibited MRE reporter activity (Figure 1A). ERE-luciferase reporter activity was unaffected by cotransfection with MR alone or with aldo at 10 nM (data not shown) or 100 nM (Figure 1B), even with higher levels of MR expression (data not shown) as confirmed by immunoblotting (Supplemental Figure 2A). These data demonstrate that the ER, particularly in the presence of estrogen, attenuates MR transcriptional activity but that the converse is not true because MR does not influence ER genomic activity under the same conditions.
Figure 1.

Estrogen inhibits MR-mediated transcriptional activity. A, ER inhibits MR-mediated transcriptional activation by aldosterone. HEK293 cells were transfected with the MR-responsive MMTV-luciferase reporter. Aldo stimulation (10 nM or 100 nM, as indicated) of MR transcriptional activity is significantly attenuated by the expression of the ER and completely inhibited by E2 (10 nM) with ER. B, No effect of MR on ER-stimulated transcription. HEK293 cells were transfected with a luciferase reporter driven by an ERE. E2 (10 nM) induction of ER-mediated reporter activity was unaffected by the cotransfection with MR with or without aldo (100 nM) (n = 3 experiments). *, P < .001 vs no MR (A) or no ER (B); #, P < .01 vs MR or ER + vehicle; ^, P < .05 vs MR + vehicle; ^^, P < .01 vs MR + 100 nM aldo; ^^^, P < .001 vs MR + 10 nM aldo.
MR transcriptional inhibition by ER is mediated by the ER N terminus and does not require ER DNA binding or nongenomic signaling
To determine the ERα functional domains that are necessary for the inhibition of MR-induced transcription, MRE-luciferase reporter assays were performed in the presence of ERα with mutated functional domains (Figure 2A, top panel). ERα with mutations that delete the C terminus (ABC) or the N terminus (CDEF) or that specifically inactivate DNA binding (DBDmut), ligand-independent activation (S118A), or rapid nongenomic signaling (KRR), all maintain the ability to inhibit MR-mediated transcription. However, a mutant lacking amino acids 254–370, including the ERα hinge region and the nuclear localization signal, is insufficient to inhibit MR transcriptional activity (Figure 2B) despite relatively equal expression of all ER mutants (Supplemental Figure 2B). This domain alone (253–370) is also insufficient to significantly inhibit MR transcriptional activity (Figure 2C), suggesting that additional domains are also needed.
Figure 2.

ERα functional domains that contribute to MR transcriptional inhibition. A, Schematic of full-length ERα alongside functional and deletion mutants. The depicted ERα constructs were cotransfected with MR into HEK293 cells, and MRE-luciferase reporter activity was quantified. B, ERα mutants inhibited MR transcriptional activity despite lacking the AB domain (CDEF) or the ligand binding domain (ABC) or with point mutations that inactivate DNA binding (DBDmut), ligand-independent activation (S118A), or nongenomic signaling (KRR). A deletion mutant lacking the nuclear localization signal (NLS) and hinge region (Δ254–370) did not inhibit MR transcription; however, this domain alone (253–370) was insufficient to significantly inhibit MR transcriptional activity (see panel C). C, ERα amino acids 1–253 without the ligand binding domain was sufficient to inhibit MR transcriptional activity (n = 5 experiments). *, P < .05 vs MR + aldo + E2.
Because the ERα N-terminal ABC domain (amino acids 1–271) was sufficient to inhibit MRE-luciferase activity, additional deletion mutants of the ERα N terminus were generated to identify the portion of ERα that is necessary to inhibit MR-mediated transcription (Figure 2A, bottom panel). MRE-luciferase reporter assays in Figure 2C reveal that ERα amino acids 1–253 is sufficient to mediate MR transcriptional inhibition, whereas the deletion of 20 additional ERα C-terminal amino acids from 253 to 233 completely prevented ERα inhibition of MR function despite similar expression of each ERα deletion mutant (Supplemental Figure 2C).
MR and ERα are part of a protein complex in cells
To further explore the mechanism by which ER inhibits MR activity, we examined whether ER and MR are part of a common protein complex in cells. MR was immunoprecipitated from cell lysates from HEK293 cells transfected with full-length human MR and ERα, and the presence of ERα in the complex was determined by immunoblotting for ERα. Indeed, abundant ERα protein is present in the MR immunoprecipitated complex (Figure 3A). When MR-expressing HEK293 cell lysate was incubated with His-tagged ERα, the ERα-containing His beads pull down a complex that contains MR (Figure 3B). Although this experiment does not test for a direct interaction between the two receptors, the data support the conclusion that MR and ERα are simultaneously part of a protein complex in cells.
Figure 3.

ER and MR are part of a protein complex in cells. A, Cell lysates from HEK293 cells expressing full-length human MR and ERα were immunoprecipitated (IP) with anti-MR antibody or control nonimmune (NI) serum, and the complex was immunoblotted (IB) with anti-MR or anti-ERα antibody. B, His-ERα beads or His beads alone were incubated with MR-expressing HEK293 cell lysate and the bound complex was immunoblotted (IB) to detect the presence of MR. C, Striatin binding is not required for ER to form a complex with MR. Beads with wild-type ERα, His-ERα-KRR (the mutant that cannot bind to striatin, thereby preventing rapid nongenomic ER signaling), or His beads alone were incubated with MR-containing HEK293 cell lysates followed by immunoblotting for MR or striatin. D, E2 treatment increases ERα complexing with MR. The role of the ligand in the ER-MR complex formation was examined by treating HEK293 cells expressing MR and ER with 10 nM E2 and/or 100 nM aldo followed by IP of MR from the lysate and IB for MR or ER (n = 4 experiments). *, P < .01 vs vehicle.
In addition to their role in transcriptional regulation in the nucleus, both ER and MR have been shown to function in a rapid, nongenomic manner in the cytoplasm tethered to the membrane by the scaffolding protein striatin (41, 48). To test whether the ER-MR complex is mediated by interaction of the two receptors with striatin, the His-ERα pull-down was repeated using a His-tagged ERα with mutations in three amino acids critical for striatin binding and rapid signaling [KRR mutant (49)]. ERα with the KRR mutation does not pull down striatin as expected, but it is still able to pull down a complex containing the MR (Figure 3C), suggesting that striatin is not necessary for ER-MR complex formation. The role of the hormonal ligands in the ER-MR complex formation was also explored using lysates from HEK293 cells expressing ERα and MR and treated with the indicated hormones. ER and MR are present in a common complex in the absence and presence of either aldo or E2, and the ratio of the receptors in the complex is not altered by treatment with aldo. Treatment with E2 alone significantly increased the amount of ER in the complex with the MR, although this difference is not present in the cells treated with E2 together with aldo (Figure 3D).
Both complex formation with MR and nuclear localization may be necessary for ERα inhibition of MR-mediated transcription
We next examined the domains required for ER-MR complex formation. His-tagged ERα domains corresponding to the mutants that determined MR transcriptional inhibition in Figure 2 were incubated with lysate from HEK293 cells expressing MR followed by immunoblotting of the bound proteins for MR (Figure 4A). As in the reporter assays, ERα lacking the AB domain (amino acids 176–595) or with the ligand-binding domain deleted (amino acids 1–271) were each able to form a complex containing the MR. Interestingly, deletion of the hinge region containing the NLS (Δ254–370) did not prevent ER interaction with MR in a whole-cell lysate despite the finding that this deletion mutant did not inhibit MR transcriptional activity in cells in vitro. However, this ERα domain alone (254–370) was insufficient to complex with the MR (Figure 4A). Further deletions of the ERα N terminus demonstrated that amino acids 1–253 and 1–233 are sufficient for MR complex formation but that further deletion to 1–176 prevents complex formation with the MR. Comparison with the reporter assay reveals some similarities but also some differences in the domains involved in the ER-MR complex formation (Figure 4C, ER-MR Complex panel) and transcriptional inhibition (Figure 4C, Inhibits MRE Reporter panel).
Figure 4.
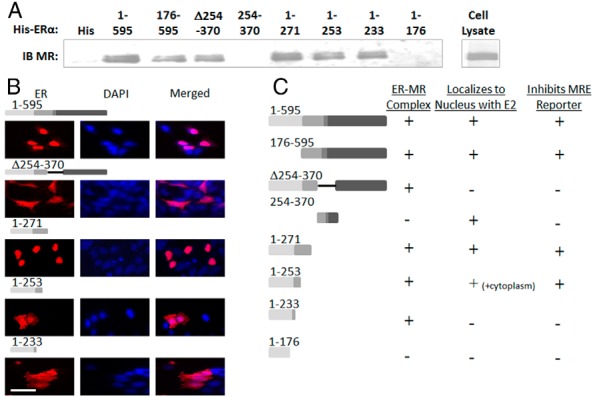
ERα domains that bind MR and also localize to the nucleus with E2 treatment are sufficient to inhibit MR transcriptional activity. A, His beads containing ERα deletion mutants of varying lengths were mixed with lysate from HEK293 cells expressing MR and the bound proteins were immunoblotted (IB) with anti-MR antibody. B, HEK293 cells transfected with flag-tagged ERα mutants were treated with 10 nM E2 followed by immunofluorescent microscopy with antiflag antibody (red) and DAPI to indicate the nuclei (blue). Merged images show nuclear vs cytoplasmic localization. Scale bars, 25 μM. C, Summary of ERα domains capable of complexing with MR (from Figure 4A), localizing to the nucleus with E2 (from Figure 4B and Supplemental Figure 3), and inhibiting MRE reporter activity (from Figure 2). Only mutants that form a complex with MR and also localize in the nucleus in which MR genomic activity takes place are able to inhibit MRE-luciferase reporter activity.
Because MR transcriptional activity occurs in the nucleus, we next investigated which of the ERα mutants can localize to the nucleus in the presence of E2. HEK293 cells transfected with flagged-tagged ERα deletion mutants were treated with E2 and the localization of ERα determined by immunfluorescent staining with antibody to the ERα epitope tag and DAPI to mark the nuclei. As expected, in the presence of E2, full-length ERα (1–595) localizes to the nucleus, whereas cells expressing ERα with the NLS deleted (Δ254–370) showed diffuse cytoplasmic ERα signal that does not colocalize with the DAPI-stained nucleus (Figure 4B). ERα 1–271 is nuclear, whereas cells expressing ERα 1–253 showed both nuclear and cytoplasmic localization of the receptor and cells expressing ERα 1–233 had predominantly cytoplasmic staining (Figure 4B). The observations that ERα 1–233 binds to MR in lysed cells but lacks strong nuclear localization and does not inhibit MR-mediated transcription supports the possibility that ERα inhibition of MR-mediated transcription requires the concentration of ERα in the nucleus in which it can subsequently form a complex with the MR. Indeed, further exploration of the subcellular localization of ERα mutants (Supplemental Figure 3) combined with the MR binding and reporter activity data reveals that the ability of ERα to inhibit MR-mediated gene transcription correlates with the ability of ERα segments to both localize to the nucleus and to form a complex with MR (Figure 4C).
In human endothelial cells, estrogen inhibits aldo induction of MR transcriptional activity and of ICAM-1 expression
Because E2 and aldo appear to have opposing effects on endothelial function, we next explored whether E2 modulates MR transcriptional function in human vascular endothelial cells. Because ER expression is rapidly down-regulated in cultured endothelial cells (ECs), a human endothelial cell line (EAHy926 cells) was generated that stably expresses full-length ERα. These ECs were transfected with the same MMTV MRE-luciferase reporter plasmid and treated with aldo (10 nM) in the absence or presence of E2 (10 nM). This concentration of aldo does not activate the GR (Supplemental Figure 1C). As in human coronary ECs (9), aldo enhances MRE-luciferase reporter activity in EAHy ECs (Figure 5A), although the magnitude is more modest than in HEK293 cells overexpressing the MR, likely due to the lower levels of endogenous MR expression in ECs. Aldo induction of luciferase activity is prevented by E2, supporting the concept that E2 interferes with aldo activation of endogenous MR transcriptional activity in human ECs.
Figure 5.

Estrogen inhibits aldo induction of MR transcriptional activity and ICAM-1 expression in endothelial cells. A, Aldo-activated MRE reporter activity is attenuated by E2 in ECs. Human endothelial cells (EAHy) stably expressing ERα were transfected with the MMTV MR-responsive reporter and luciferase activity was quantified in the presence of vehicle, 10 nM aldosterone (Aldo), or aldo with 10 nM estradiol (Aldo + E2). B, Estrogen prevents aldo regulation of ICAM-1 in ECs. EAHy cells stably expressing ERα were treated with vehicle, aldo, or aldo + E2 for 18 hours and expression of ICAM-1 mRNA was quantified by quantitative RT-PCR (n = 3 or 4 experiments). *, P < .05 vs vehicle; #, P < .05 vs aldo.
We next tested whether E2 modulates aldo induction of a known MR target gene in ECs. We previously demonstrated that the cell adhesion molecule ICAM-1 is an MR transcriptional target gene in human coronary endothelial cells (9). Treatment of EAHy926 ECs expressing ERα with aldo at 10 nM, a concentration found in patients with cardiovascular disease (47), increases expression of ICAM-1 mRNA. E2 (10 nM) prevents aldo induction of ICAM-1 mRNA in these cells (Figure 5B). These data offer evidence that E2 inhibits aldo-stimulated transcription in ECs and impacts mRNA abundance of a physiologically important MR target gene.
Estrogen inhibits aldo induction of endothelial ICAM-1 protein expression and leukocyte-EC adhesion
To explore the functional significance of estrogen inhibition of MR regulation of ICAM-1 mRNA expression, we assessed the effect of E2 on aldo induction of EC ICAM-1 protein expression and on aldo-stimulated immune cell adhesion to ECs. Aldo (10 nM) increases ICAM-1 protein expression in ECs as quantified by immunoblotting with anti ICAM-1 antibody, and this is prevented by E2 (10 nM) (Figure 6, A and B). We have previously demonstrated that aldo enhances leukocyte adhesion to human coronary ECs and that this requires ICAM-1 (9). To quantify leukocyte adhesion to EAHy ECs, endothelial monolayers were treated for 18 hours with 10 nM aldo in the presence or absence of 10 nM E2 and then incubated with fluorescent labeled human monocytic cells (U937 cells). After washing away nonadherent leukocytes, adherent fluorescent cells were visualized (Figure 6C) and quantified (Figure 6D). Here we demonstrate that aldo enhances leukocyte adhesion to the EAHy cells. Although E2 alone does not affect leukocyte adhesion to the ERα-expressing EAHy cells, E2 prevents aldo-enhanced leukocyte-EC adhesion (Figure 6D). These data support the concept that by preventing the aldo induction of MR target genes in ECs, estrogen may prevent the expression of genes that contribute to cardiovascular dysfunction and disease.
Figure 6.
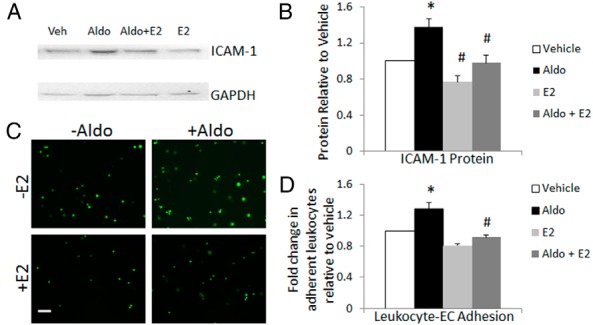
Estrogen inhibits aldo induction of ICAM-1 protein and leukocyte adhesion to endothelial cells. Endothelial cells (EAHy) stably expressing ERα were treated with aldosterone (Aldo, 10 nM), E2 (10 nM), or both for 18 hours, and cell lysates were immunoblotted to quantify ICAM-1 protein (A and B) and incubated with fluorescently labeled human monocytic cells (U937) to quantify leukocyte-EC adhesion (C and D). A, A representative ICAM-1 immunoblot is shown. Veh, vehicle. B, Quantification of ICAM-1 protein shows a significant increase with aldo that is inhibited by E2 (n = 3). C, Representative images of leukocyte adhesion assay. Scale bar, 25 μM. D, Quantification of adherent fluorescent leukocytes reveals that E2 attenuates aldo-stimulated leukocyte adhesion (n = 4). *, P < .05 vs vehicle; #, P < .05 vs aldo.
Discussion
In summary, we have demonstrated the following: 1) ERα in the presence of estrogen prevents aldosterone stimulation of MR transcriptional activity; 2) ER and MR can associate as part of a common protein complex in cells; and 3) estrogen inhibits aldo induction of the endothelial MR target gene ICAM-1 and prevents aldo-induced leukocyte-endothelial cell adhesion. The minimum portion of ERα required for MR transcriptional inhibition is the N-terminal amino acids 1–253 and the capacity of ERα to inhibit MR genomic function appears to require ERα localization to the nucleus and formation of a complex containing the two receptors. Inhibition of MR transcriptional activity does not require ERα to bind to DNA or to striatin, nor is ligand-independent ERα activation necessary. We have previously shown that aldo activation of EC MR induces ICAM-1 mRNA expression in human ECs by modulating ICAM-1 transcription and that enhanced endothelial ICAM-1 surface protein expression mediates aldosterone induction of leukocyte-EC adhesion (9). Furthermore, aldo increases vascular leukocyte infiltration and atherosclerosis progression in animal models (22). Here we demonstrated that estrogen prevents aldo induction of endothelial ICAM-1 expression and leukocyte-endothelial cell adhesion. These findings support the new concept that estrogen inhibition of MR-regulated transcription of proatherogenic genes in the endothelium could be a novel mechanism by which gender modulates cardiovascular risk.
Although further studies are needed to clarify the detailed molecular mechanism(s) by which ER abrogates MR-mediated gene regulation, our findings are consistent with several possible mechanisms. First, the data support that the mechanism requires ERα to form a complex with MR in the nucleus, either through direct binding or indirectly via other proteins, and that this interaction blocks MR function as a transcription factor. This possibility is supported by our immunoprecipitation and nuclear localization experiments (Figures 3 and 4). However, although we identified ERα segment 1–253 as being the smallest portion able to inhibit MR transcriptional activity, the smaller segment 1–233 was able to complex with MR. The explanation for this discrepancy may come from our finding that nuclear localization of 1–233 was much weaker than 1–253, suggesting nuclear localization of ERα may also be critical for its interference with MR-mediated transcriptional activity. This mechanism appears to be distinct from the well-characterized dimerization interaction of the retinoid-X-receptor with nonsteroid nuclear receptors including the thyroid, retinoid and vitamin D receptors (reviewed in reference 50) because this does not require DNA binding by the ER.
Once the ER forms a complex with MR in the nucleus, multiple potential mechanisms are consistent with the data. ER may prevent MR from interacting with a cofactor that is required for aldo-induced transcriptional activation but is not needed for ER transcriptional activation. This is consistent with the finding that MR does not mutually inhibit transcriptional activation of ER (Figure 1B). Recently posttranslational modification of the MR by phosphorylation has been shown to modulate MR transcriptional function in specific renal cell types (51). Thus, another possibility could be that complexing with ER in the nucleus facilitates MR modifications that alter its transcriptional function. The N-terminus of MR has also been shown to interact with the ligand-binding C-terminal domain of MR and this N-C interaction is enhanced by aldo and modulates MR transcriptional function (52, 53). Although we have not investigated which domain of MR mediates complex formation with ER because the ERα N terminus appears to be required to modulate MR function, it is possible that the ERα N terminus affects the MR N-C interaction to modulate MR transcriptional function. Further studies are needed to differentiate between these mechanisms.
There are important limitations of this study that should be considered. The initial in vitro studies exploring interactions between ER and MR transcriptional function use a transformed human embryonic kidney cell line (HEK293 cells) with overexpressed ER and MR proteins and a transfected supercoiled plasmid containing the MMTV MR-responsive element as a transcriptional reporter. This system, although highly artificial, was chosen because these HEK293 cells lack endogenous ER, MR, and GR, and hence, the expression of receptors and ER mutants could be carefully controlled to specifically and independently examine transcriptional function of the ER and the MR (in the absence of GR). The MMTV reporter was used because much of what is known about MR transcriptional function has been determined using this promoter (54, 55), and there are no vascular MR target genes for which the MR DNA-binding site has been determined. The lack of known vascular MR target gene DNA binding sites and an adequate MR antibody for chromatin immunoprecipitation has prevented the use of more modern methods to confirm endogenous MR binding to specific target promoters within intact chromatin (56). To address these limitations, we confirmed the MMTV reporter studies in more physiologically relevant vascular endothelial cells with endogenously expressed MR and also examined the effects of estrogen on aldo regulation of an endogenous vascular MR target gene in the context of intact chromatin.
Another important limitation is that ER expression is rapidly down-regulated in cultured cells; thus, the ER in all of these studies is exogenously expressed. Future studies in whole vessels or in vivo will be needed to interrogate this novel mechanism in the setting of physiological levels of MR and ER. Whether such a mechanism also applies to other combinations of steroid receptors also remains to be determined. Other considerations limit the interpretation of the effects of E2 on aldo regulation of endogenous ICAM-1 in human ECs. In this system, one cannot exclude an independent effect of ER on ICAM-1 transcription. This is less likely because estrogen alone did not affect ICAM-1 expression or leukocyte adhesion but rather, estrogen attenuated the effects of aldo on ICAM-1 expression and function (Figure 6, C and D). Also, because ICAM-1 mRNA induction by aldo is very modest after short-term treatment [only 20% increase after 3 h (9)], the studies here were performed after 18 hours of aldo treatment, leaving open the possibility of secondary effects beyond transcriptional activation. We have previously demonstrated that aldo induction of ICAM-1 mRNA in human coronary endothelial cells is completely inhibited by actinomycin-D, even after 24 hours (9), supporting a transcriptional mechanism; however, that does not rule out the possibility that ER might also interfere with longer-term effects of the MR on ICAM-1 regulation.
Despite these limitations, the findings presented here are important in the larger context of substantial evidence for opposing roles for E2-activated ER and aldo-activated MR in human cardiovascular disease. The decreased incidence of cardiovascular disease in premenopausal women compared with age-match men, which equalizes after menopause, is well known (57), yet the mechanism is still not clear. In addition, serum aldo levels are lower, specifically in premenopausal women compared with age-matched men (58–60). Conversely, elevated serum aldo levels correlate with increased incidence of cardiovascular disease and poor outcomes (25, 26). Gender may also be an important factor in the cardiovascular response to aldo, as suggested by clinical evidence that serum aldo directly correlates with left ventricular remodeling in women, whereas no such correlation was found in men (61). The finding that higher serum aldo levels correlate not only with atherosclerotic progression but specifically with endothelial dysfunction in humans more specifically implicates aldo activation of endothelial MR in cardiovascular disease (36).
Experimental studies in animal models of cardiovascular disease further support the concept that estrogen and aldo act in opposition in the cardiovascular system. Although E2 treatment inhibits the pathological vascular remodeling response to carotid injury in mice (62), aldo infusion exacerbates remodeling in that same vascular injury model (31). Similarly in the apolipoprotein E knockout mouse model of atherosclerosis, E2 treatment decreases atherosclerotic plaque size and immune cell infiltration in the plaques, whereas aldo treatment increases plaque size and vascular inflammation (6, 22). Moreover, treatment with ER agonists attenuate MR-mediated hypertension and vascular fibrosis in rats infused with aldo (63). Estrogen via ERβ has also been found to inhibit angiotensin II-induced hypertension and cardiac fibrosis (7). Because many of the cardiovascular consequences of angiotensin II are mediated by aldo and MR (64), these data could also provide a mechanism for the interaction between E2 and angiotensin II. One important limitation of many of these animal studies is that they were performed in only one gender, and thus, additional studies are required to directly compare the effects of E2 and aldo in males vs females. However, one unifying aspect of these diverse cardiovascular disease models is the disruption of endothelial cell function, whether through mechanical injury of the endothelium in the carotid injury models, oxidative stress in the angiotensin II hypertension model, or inflammation in the atherosclerosis model. The ubiquitous involvement of the endothelium in models of cardiovascular disease that are differentially affected by aldo and E2 implicate the endothelium as a tissue in which E2 modulation of MR function may have profound clinical implications. Indeed, aldo-induced vascular inflammation is thought to be mediated at least in part by the up-regulation of EC ICAM-1 (9). E2 inhibition of the aldo induction of EC ICAM-1 expression and leukocyte adhesion would be expected to decrease plaque inflammation, thereby preventing plaque rupture, the cause of most heart attacks and strokes (65). Thus, these findings may represent an important new mechanism explaining the lower incidence of cardiovascular ischemic events in premenopausal women.
In conclusion, this study identifies a novel mechanism in which E2 signaling through the ERα inhibits transcriptional regulation by aldo and the MR in the vascular endothelium. This new model may offer critical insight into the mechanism of gender differences in cardiovascular pathologies. Ultimately, greater understanding of this pathway may offer new therapeutic opportunities for the prevention and treatment of cardiovascular disease in both men and women.
Acknowledgments
This work was supported by the US National Institutes of Health Grant NIH HL095590 (to I.Z.J.) and the American Heart Association Grant AHA EIA18290005 (to I.Z.J.) and Grant PRE16920014 (to K.B.M.).
Disclosure Summary: The authors have nothing to disclose.
- aldo
- aldosterone
- DAPI
- 4′,6′-diamino-2-phenylindole
- DBD
- DNA-binding domain
- E2
- estradiol
- EC
- endothelial cell
- ER
- estrogen receptor
- ERE
- estrogen response element
- FBS
- fetal bovine serum
- GR
- glucocorticoid receptor
- HA
- hemagluttanin
- HEK293
- human embryonic kidney-293
- ICAM-1
- intercellular adhesion molecule-1
- MMTV
- mouse mammary tumor virus
- MR
- mineralocorticoid receptor
- MRE
- MR-responsive element
- S-FBS
- charcoal-dextran-stripped FBS.
References
- 1. Go AS, Mozaffarian D, Roger VL, et al. American Heart Association Statistics Committee, Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ossewaarde ME, Bots ML, Verbeek AL, et al. Age at menopause, cause-specific mortality and total life expectancy. Epidemiology. 2005;16:556–562. [DOI] [PubMed] [Google Scholar]
- 3. Bairey Merz CN, Johnson BD, Sharaf BL, et al. Hypoestrogenemia of hypothalamic origin and coronary artery disease in premenopausal women: a report from the NHLBI-sponsored WISE study. J Am Coll Cardiol. 2003;41:413–419. [DOI] [PubMed] [Google Scholar]
- 4. Karas RH, Schulten H, Pare G, et al. Effects of estrogen on the vascular injury response in estrogen receptor α, β (double) knockout mice. Circ Res. 2001;89:534–539. [DOI] [PubMed] [Google Scholar]
- 5. Wang M, Wang Y, Weil B, et al. Estrogen receptor β mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integr Comp Physiol. 2009;296:R972–R978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hodgin JB, Krege JH, Reddick RL, Korach KS, Smithies O, Maeda N. Estrogen receptor alpha is a major mediator of 17β-estradiol's atheroprotective effects on lesion size in Apoe−/− mice. J Clin Invest. 2001;107:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pedram A, Razandi M, Korach KS, Narayanan R, Dalton JT, Levin ER. ERβ selective agonist inhibits angiotensin-induced cardiovascular pathology in female mice. Endocrinology. 2013;154:4352–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocorticoid receptors in human coronary artery smooth muscle cells. Circ Res. 2005;96:643–650. [DOI] [PubMed] [Google Scholar]
- 9. Caprio M, Newfell BG, la Sala A, et al. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ Res. 2008;102:1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lombes M, Oblin ME, Gasc JM, Baulieu EE, Farman N, Bonvalet JP. Immunohistochemical and biochemical evidence for a cardiovascular mineralocorticoid receptor. Circ Res. 1992;71:503–510. [DOI] [PubMed] [Google Scholar]
- 11. Barrett KV, McCurley AT, Jaffe IZ. Direct contribution of vascular mineralocorticoid receptors to blood pressure regulation. Clin Exp Pharmacol Physiol. 2013;40:902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Caprio M, Feve B, Claes A, Viengchareun S, Lombes M, Zennaro MC. Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. FASEB J. 2007;21:2185–2194. [DOI] [PubMed] [Google Scholar]
- 13. Lother A, Berger S, Gilsbach R, et al. Ablation of mineralocorticoid receptors in myocytes but not in fibroblasts preserves cardiac function. Hypertension. 2011;57:746–754. [DOI] [PubMed] [Google Scholar]
- 14. Usher MG, Duan SZ, Ivaschenko CY, et al. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. 2010;120:3350–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Greschik H, Moras D. Structure-activity relationship of nuclear receptor-ligand interactions. Curr Topics Med Chem. 2003;3:1573–1599. [DOI] [PubMed] [Google Scholar]
- 16. Karas RH, Gauer EA, Bieber HE, Baur WE, Mendelsohn ME. Growth factor activation of the estrogen receptor in vascular cells occurs via a mitogen-activated protein kinase-independent pathway. J Clin Invest. 1998;101:2851–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nagase M, Fujita T. Mineralocorticoid receptor activation in obesity hypertension. Hypertens Res. 2009;32:649–657. [DOI] [PubMed] [Google Scholar]
- 18. Nagase M, Ayuzawa N, Kawarazaki W, et al. Oxidative stress causes mineralocorticoid receptor activation in rat cardiomyocytes: role of small GTPase Rac1. Hypertension. 2012;59:500–506. [DOI] [PubMed] [Google Scholar]
- 19. Dooley R, Harvey BJ, Thomas W. Non-genomic actions of aldosterone: from receptors and signals to membrane targets. Mol Cell Endocrinol. 2012;350:223–234. [DOI] [PubMed] [Google Scholar]
- 20. Levin ER. Extra-nuclear estrogen receptors roles in physiology: lessons from mouse models. Am J Physiol Endocrinol Metab. 2014;307(2):E133–E140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luther JM, Luo P, Wang Z, et al. Aldosterone deficiency and mineralocorticoid receptor antagonism prevent angiotensin II-induced cardiac, renal, and vascular injury. Kidney Int. 2012;82:643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McGraw AP, Bagley J, Chen WS, et al. Aldosterone increases early atherosclerosis and promotes plaque inflammation through a placental growth factor-dependent mechanism. J Am Heart Assoc. 2013;2:e000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Azibani F, Fazal L, Chatziantoniou C, Samuel JL, Delcayre C. Aldosterone mediates cardiac fibrosis in the setting of hypertension. Curr Hypertens Rep. 2013;15:395–400. [DOI] [PubMed] [Google Scholar]
- 24. Pruthi D, McCurley A, Aronovitz M, Galayda C, Karumanchi SA, Jaffe IZ. Aldosterone promotes vascular remodeling by direct effects on smooth muscle cell mineralocorticoid receptors. Arterioscleros Thromb Vasc Biol. 2014;34:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ivanes F, Susen S, Mouquet F, et al. Aldosterone, mortality, and acute ischaemic events in coronary artery disease patients outside the setting of acute myocardial infarction or heart failure. Eur Heart J. 2012;33:191–202. [DOI] [PubMed] [Google Scholar]
- 26. Milliez P, Girerd X, Plouin PF, Blacher J, Safar ME, Mourad JJ. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol. 2005;45:1243–1248. [DOI] [PubMed] [Google Scholar]
- 27. Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–717. [DOI] [PubMed] [Google Scholar]
- 28. Pitt B, Remme W, Zannad F, et al. Eplerenone Post-Acute Myocardial Infarction Heart Failure E, Survival Study I. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321. [DOI] [PubMed] [Google Scholar]
- 29. Pitt B, Reichek N, Willenbrock R, et al. Effects of eplerenone, enalapril, and eplerenone/enalapril in patients with essential hypertension and left ventricular hypertrophy: the 4E-left ventricular hypertrophy study. Circulation. 2003;108:1831–1838. [DOI] [PubMed] [Google Scholar]
- 30. Zannad F, McMurray JJ, Krum H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. The New England Journal Of Medicine. 2011;364:11–21. [DOI] [PubMed] [Google Scholar]
- 31. Jaffe IZ, Newfell BG, Aronovitz M, et al. Placental growth factor mediates aldosterone-dependent vascular injury in mice. J Clin Invest. 2010;120:3891–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bernelot Moens SJ, Schnitzler GR, Nickerson M, et al. Rapid estrogen receptor signaling is essential for the protective effects of estrogen against vascular injury. Circulation. 2012;126:1993–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hodgin JB, Maeda N. Minireview: estrogen and mouse models of atherosclerosis. Endocrinology. 2002;143:4495–4501. [DOI] [PubMed] [Google Scholar]
- 34. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115:1285–1295. [DOI] [PubMed] [Google Scholar]
- 35. Giannotti G, Landmesser U. Endothelial dysfunction as an early sign of atherosclerosis. Herz. 2007;32:568–572. [DOI] [PubMed] [Google Scholar]
- 36. Hannemann A, Wallaschofski H, Ludemann J, et al. Plasma aldosterone levels and aldosterone-to-renin ratios are associated with endothelial dysfunction in young to middle-aged subjects. Atherosclerosis. 2011;219:875–879. [DOI] [PubMed] [Google Scholar]
- 37. Bechlioulis A, Kalantaridou SN, Naka KK, et al. Endothelial function, but not carotid intima-media thickness, is affected early in menopause and is associated with severity of hot flushes. J Clin Endocrinol Metab. 2010;95:1199–1206. [DOI] [PubMed] [Google Scholar]
- 38. Clapauch R, Mecenas AS, Maranhao PA, Bouskela E. Early postmenopausal women with cardiovascular risk factors improve microvascular dysfunction after acute estradiol administration. Menopause. 2012;19:672–679. [DOI] [PubMed] [Google Scholar]
- 39. Kalantaridou SN, Naka KK, Papanikolaou E, et al. Impaired endothelial function in young women with premature ovarian failure: normalization with hormone therapy. J Clin Endocrinol Metab. 2004;89:3907–3913. [DOI] [PubMed] [Google Scholar]
- 40. Saltiki K, Papageorgiou G, Voidonikola P, et al. Endogenous estrogen levels are associated with endothelial function in males independently of lipid levels. Endocrine. 2010;37:329–335. [DOI] [PubMed] [Google Scholar]
- 41. Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH. Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor α. Proc Nat Acad Sci USA. 2004;101:17126–17131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arriza JL, Weinberger C, Cerelli G, et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237:268–275. [DOI] [PubMed] [Google Scholar]
- 43. Lombes M, Binart N, Oblin ME, Joulin V, Baulieu EE. Characterization of the interaction of the human mineralocorticosteroid receptor with hormone response elements. Biochem J. 1993;292( Pt 2):577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu Q, Surks HK, Ebling H, et al. Regulation of estrogen receptor alpha-mediated transcription by a direct interaction with protein phosphatase 2A. J Biol Chem. 2003;278:4639–4645. [DOI] [PubMed] [Google Scholar]
- 45. Gomez-Sanchez CE, de Rodriguez AF, Romero DG, et al. Development of a panel of monoclonal antibodies against the mineralocorticoid receptor. Endocrinology. 2006;147:1343–1348. [DOI] [PubMed] [Google Scholar]
- 46. Newfell BG, Iyer LK, Mohammad NN, et al. Aldosterone regulates vascular gene transcription via oxidative stress-dependent and -independent pathways. Arterioscler Thromb Vasc Biol. 2011;31:1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Weber KT. Aldosterone in congestive heart failure. N Engl J Med. 2001;345:1689–1697. [DOI] [PubMed] [Google Scholar]
- 48. Pojoga LH, Coutinho P, Rivera A, et al. Activation of the mineralocorticoid receptor increases striatin levels. Am J Hypertens 2012;25:243–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ueda K, Lu Q, Baur W, Aronovitz MJ, Karas RH. Rapid estrogen receptor signaling mediates estrogen-induced inhibition of vascular smooth muscle cell proliferation. Arterioscler Thromb Vasc Biol. 2013;33:1837–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. [DOI] [PubMed] [Google Scholar]
- 51. Shibata S, Rinehart J, Zhang J, et al. Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab. 2013;18:660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pippal JB, Yao Y, Rogerson FM, Fuller PJ. Structural and functional characterization of the interdomain interaction in the mineralocorticoid receptor. Mol Endocrinol. 2009;23:1360–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fuller PJ, Yao Y, Yang J, Young MJ. Mechanisms of ligand specificity of the mineralocorticoid receptor. J Endocrinol. 2012;213:15–24. [DOI] [PubMed] [Google Scholar]
- 54. Clyne CD, Chang CY, Safi R, Fuller PJ, McDonnell DP, Young MJ. Purification and characterization of recombinant human mineralocorticoid receptor. Mol Cell Endocrinol. 2009;302:81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mettu NB, Stanley TB, Dwyer MA, et al. The nuclear receptor-coactivator interaction surface as a target for peptide antagonists of the peroxisome proliferator-activated receptors. Mol Endocrinol. 2007;21:2361–2377. [DOI] [PubMed] [Google Scholar]
- 56. Ueda K, Fujiki K, Shirahige K, et al. Genome-wide analysis of murine renal distal convoluted tubular cells for the target genes of mineralocorticoid receptor. Biochem Biophys Res Commun. 2014;445:132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kannel WB, Hjortland MC, McNamara PM, Gordon T. Menopause and risk of cardiovascular disease: the Framingham study. Ann Intern Med. 1976;85:447–452. [DOI] [PubMed] [Google Scholar]
- 58. Danser AH, Derkx FH, Schalekamp MA, Hense HW, Riegger GA, Schunkert H. Determinants of interindividual variation of renin and prorenin concentrations: evidence for a sexual dimorphism of (pro)renin levels in humans. J Hypertens. 1998;16:853–862. [DOI] [PubMed] [Google Scholar]
- 59. Schunkert H, Danser AH, Hense HW, Derkx FH, Kurzinger S, Riegger GA. Effects of estrogen replacement therapy on the renin-angiotensin system in postmenopausal women. Circulation. 1997;95:39–45. [DOI] [PubMed] [Google Scholar]
- 60. Miller JA, Anacta LA, Cattran DC. Impact of gender on the renal response to angiotensin II. Kidney Int. 1999;55:278–285. [DOI] [PubMed] [Google Scholar]
- 61. Vasan RS, Evans JC, Benjamin EJ, et al. Relations of serum aldosterone to cardiac structure: gender-related differences in the Framingham Heart Study. Hypertension. 2004;43:957–962. [DOI] [PubMed] [Google Scholar]
- 62. Sullivan TR, Jr, Karas RH, Aronovitz M, et al. Estrogen inhibits the response-to-injury in a mouse carotid artery model. J Clin Invest. 1995;96:2482–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Arias-Loza PA, Hu K, Dienesch C, et al. Both estrogen receptor subtypes, α and β, attenuate cardiovascular remodeling in aldosterone salt-treated rats. Hypertension. 2007;50:432–438. [DOI] [PubMed] [Google Scholar]
- 64. Rocha R, Martin-Berger CL, Yang P, Scherrer R, Delyani J, McMahon E. Selective aldosterone blockade prevents angiotensin II/salt-induced vascular inflammation in the rat heart. Endocrinology. 2002;143:4828–4836. [DOI] [PubMed] [Google Scholar]
- 65. McGraw AP, McCurley A, Preston IR, Jaffe IZ. Mineralocorticoid receptors in vascular disease: connecting molecular pathways to clinical implications. Curr Atheroscler Rep. 2013;15:340. [DOI] [PMC free article] [PubMed] [Google Scholar]