

Abstract
Hypothalamic GnRH is the master regulator of the neuroendocrine reproductive axis, and its secretion is regulated by many factors. Among these is kisspeptin (Kp), a potent trigger of GnRH secretion. Kp signals via the Kp receptor (KISS1R), a Gαq/11-coupled 7-transmembrane–spanning receptor. Until this study, it was understood that KISS1R mediates GnRH secretion via the Gαq/11-coupled pathway in an ERK1/2-dependent manner. We recently demonstrated that KISS1R also signals independently of Gαq/11 via β-arrestin and that this pathway also mediates ERK1/2 activation. Because GnRH secretion is ERK1/2-dependent, we hypothesized that KISS1R regulates GnRH secretion via both the Gαq/11- and β-arrestin–coupled pathways. To test this hypothesis, we measured LH secretion, a surrogate marker of GnRH secretion, in mice lacking either β-arrestin-1 or β-arrestin-2. Results revealed that Kp-dependent LH secretion was significantly diminished relative to wild-type mice (P < .001), thus supporting that β-arrestin mediates Kp-induced GnRH secretion. Based on this, we hypothesized that Gαq/11-uncoupled KISS1R mutants, like L148S, will display Gαq/11-independent signaling. To test this hypothesis, L148S was expressed in HEK 293 cells. and results confirmed that, although strongly uncoupled from Gαq/11, L148S retained the ability to trigger significant Kp-dependent ERK1/2 phosphorylation (P < .05). Furthermore, using mouse embryonic fibroblasts lacking β-arrestin-1 and -2, we demonstrated that L148S-mediated ERK1/2 phosphorylation is β-arrestin–dependent. Overall, we conclude that KISS1R signals via Gαq/11 and β-arrestin to regulate GnRH secretion. This novel and important finding could explain why patients bearing some types of Gαq/11-uncoupled KISS1R mutants display partial gonadotropic deficiency and even a reversal of the condition, idiopathic hypogonadotropic hypogonadism.
Neurons containing GnRH are neuroendocrine cells that originate in the olfactory placode during embryonic development and migrate along olfactory/vomeronasal nerves into the forebrain (1, 2). They migrate caudally to final sites within the hypothalamus where they send projections to and secrete GnRH into the median eminence. GnRH is transported by the hypophyseal portal vessels to reach anterior pituitary gonadotropes, where it regulates LH and FSH synthesis and secretion and thereby reproduction (3–10).
To date, several factors have been identified that positively or negatively regulate GnRH secretion. Among the most potent positive regulators of secretion are the peptides collectively referred to as kisspeptins (Kps) (11–15). Kps signal via the Kp receptor (KISS1R), a Gαq/11-coupled 7-transmembrane–spanning receptor (16, 17). KISS1R is expressed on the surface of GnRH neurons, couples to Gαq/11 and triggers the activation of phospholipase C (PLC) which in turn hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) and results in inositol 1,4,5-trisphosphate (InsP3) and diacylglycerol formation, calcium release, protein kinase C mobilization, membrane depolarization, and GnRH secretion (11–13, 18, 19). Gαq/11-dependent KISS1R signaling is terminated rapidly through the actions of G protein-coupled receptor kinase 2 and β-arrestin-1 and -2 (20). Two independent studies using heterologous cell systems demonstrated that, in addition to signaling via Gαq/11, KISS1R also signals via β-arrestin-1 and -2 to regulate the activation of ERK1/2 (20, 21). Thus, β-arrestins regulate Gαq/11-dependent KISS1R signaling as well as mediate KISS1R signaling in their own right.
Because ERK1/2 regulate Kp-dependent GnRH secretion (22), we hypothesized that in the GnRH neuron, in addition to using the Gαq/11 signaling pathway (22, 18), KISS1R also signals via β-arrestin to trigger GnRH secretion. To test this hypothesis, we measured LH secretion, a surrogate marker of GnRH secretion, in mice lacking either β-arrestin-1 or β-arrestin-2. The results revealed that Kp-dependent LH secretion is significantly diminished relative to wild-type (WT) mice, thus supporting that β-arrestin mediates Kp-induced GnRH secretion.
To date, a small but growing number of naturally occurring KISS1R loss-of-function or inactivating mutations (reviewed in Refs. 23 and 24) and 1 gain-of-function mutation (25) have been identified. At the physiological level, the term loss of function reflects the association with idiopathic hypogonadotropic hypogonadism (IHH) (15), a disorder of the hypothalamus or pituitary that is characterized by the absence of spontaneous sexual maturation in the presence of low or inappropriately normal gonadotropin levels in humans. At the cellular level, KISS1R loss of function is characterized by its diminished capacity to activate the Gαq/11 signaling pathway. Based on our finding that, in addition to using Gαq/11, Kiss1r also employs the β-arrestin pathway to mediate LH secretion, patients bearing some KISS1R mutations might continue to secrete LH, albeit at diminished levels. If that is so, such patients might display a less severe form of IHH and may even experience a reversal of IHH.
One of the first loss-of-function KISS1R mutants identified was L148S. L148S was isolated from a consanguineous Saudi Arabian family in whom members bearing the mutated receptor gene in the homozygous state were diagnosed with IHH and infertility (15, 26). In an in vitro assay, L148S was shown to be strongly defective in triggering InsP3 formation and therefore functionally uncoupled from Gαq/11 at Kp concentrations that triggered a maximum response with WT KISS1R (15). A subsequent study showed that whereas L148S was unable to trigger the ligand-induced catalytic activation of Gαq/11 and signal via this pathway, it remained physically coupled to Gαq/11 (27). That study also showed L148S was expressed at WT levels, localized to the cell surface, bound Kp at levels equivalent to WT KISS1R, and interacted with a large number of proteins with which WT KISS1R also interacted. Consistent with this finding was our observation that, like WT KISS1R, L148S also physically associated with β-arrestins (21).
In 2007, Tenenbaum-Rakover et al (28) reported on another naturally occurring KISS1R mutant, L102P. The mutation is associated with IHH, but based on a detailed phenotypic and endocrine profiling of 5 patients bearing the mutation, the authors observed that gonadotropic deficiency was highly variable, ranging from partial to complete. Yet the mutant receptor was shown to be strongly uncoupled from InsP3 formation at the supraphysiological concentration of 1μM Kp (29), leading the authors to conclude that KISS1R loss-of-function mutations do not necessarily cause complete gonadotropic deficiency (14, 24, 28). Although it was clearly recognized that, based on InsP3 formation, L102P was a loss-of-function mutant that only delayed pubertal development, the mechanism by which this was possible was not understood.
We hypothesize that some Gαq/11-uncoupled KISS1R mutants like L148S and L102P retain some ability to signal, although such signaling would be diminished. Such signaling should be sufficient to trigger some GnRH secretion and activation of the gonadotropic axis. Based on the L102P study, activation may be variable and likely developmentally regulated. To test this hypothesis, L148S was expressed in HEK 293 cells, and it was confirmed that although it was strongly uncoupled from the Gαq/11 pathway, it retained the ability to trigger significant ERK1/2 phosphorylation after Kp treatment. Taken together with the β-arrestin–knockout results, we conclude that KISS1R signals via both Gαq/11 and β-arrestin to regulate GnRH secretion and that some KISS1R mutants that are uncoupled from Gαq/11 retain the ability to signal independently of Gαq/11, likely via β-arrestin, to mediate GnRH secretion. The current study identifies the β-arrestin pathway as a novel pathway mediating GnRH secretion downstream of KISS1R and highlights the need to reassess the activity of some Gαq/11-uncoupled KISS1R mutants and patients that bear such mutations.
Materials and Methods
Animal husbandry and genotyping
Arrb1−/− and Arrb2−/− mice were generated as previously described (30, 31). Homozygous Arrb1−/− and heterozygous Arrb2+/− mice were obtained from R. J. Lefkowitz (Duke University Medical Center, Durham, NC). Animals were housed at the London Regional Cancer Program Animal Facility (London, Ontario, Canada) at controlled temperature and a 12-hour light, 12-hour dark cycle. Animal care and handling were done according to the guidelines of the University of Western Ontario (Canada) Animal Care Committee approved by the Canadian Council on Animal Care. The Arrb1−/− and Arrb2−/− lines were in the C57BL/6J and 129/S1/SvImJ backgrounds, respectively. Arrb2−/− mice and their WT littermates (Arrb2+/+) were generated by breeding heterozygous animals. Mouse genotyping was performed by PCR using Arrb2-WT forward GATCAAAGCCCTCGATGATC (intron 2) and Arrb2-knockout (KO) forward GCTAAAGCGCATGCTCCAGA (Neo), which are specific for WT and knockout (KO), respectively, and the primer Arrb2 reverse ACAGGGTCCACTTTGTCCA (exon 3), which is in common for both WT and KO. PCR products are 605 and 300 bp for Arrb2+/+ and Arrb2−/− alleles, respectively. Homozygous Arrb1−/− mice were crossed to WT C57BL/6J mice to generate a segregating colony. Due to the design of the targeting sequences, Arrb1−/− mice could not be distinguished from Arrb1+/− mice by PCR analysis as was done for Arrb2 segregating populations. Instead, segregating Arrb1 colonies were genotyped at Transnetyx, Inc, based on the determination of Neo copy numbers (30) and data were verified by Western blotting. All experiments were performed using 8-week-old postpubertal male mice to preclude the effects of the estrous cycle on GnRH secretion in females.
Reagents
Kp-10 was purchased from Tocris Cookson Inc; all other biochemical reagents were acquired from Sigma, Fisher, and VWR Scientific. The following plasmid constructs were described previously: untagged WT KISS1R (formerly referred to as GPR54) and untagged L148S (15); FLAG-KISS1R (WT) (formerly referred to as FLAG-GPR54) (20); WT KISS1R-YFP (21); GFP-PHPLC-δ1 (19); and myc-KISS1R (WT) (32). FLAG-L148S and L148S-YFP were created by conducting site-directed mutagenesis (QuikChange kit; Stratagene) to introduce the mutation in the corresponding epitope-tagged WT receptor. Myc-L148S was subcloned from the previously constructed myc-L148S-PCS2+ (33) into the pcDNA3 expression vector.
Cell culture and electroporation, confocal microscopy, Western blot analysis of ERK1/2 phosphorylation, and inositol phosphate assay
Analysis of GnRH neurons by immunohistochemistry
Mice were killed at 8 weeks of age. Mice were transcardially perfused with 0.08% heparinized saline followed by 4% paraformaldehyde. Brains were removed and postfixed overnight in 4% paraformaldehyde and then embedded in 5% agarose and cut into 50-μm-thick coronal sections using a vibrating microtome (Leica VT1000S). Tissue sections were then blocked in 5% normal goat serum for 1 hour at 4°C and incubated over 2 nights at 4°C in primary antisera with antibodies directed against the GnRH peptide. The polyclonal anti-GnRH primary antiserum EL14 (34) (generously provided by Dr Oline Rønnekleiv, Oregon Health & Science University, Portland, OR) was used at a 1:5000 dilution, and alternate sections were incubated in either a second polyclonal anti-GnRH antiserum (PA1-122; Affinity BioReagents) used at a 1:300 dilution or a monoclonal anti-GnRH antibody (19304; QED Bioscience) used at a 1:5000 dilution. After primary antisera, tissue sections were incubated at room temperature in secondary antisera for 2 hours. For sections incubated in EL14 or PA1-122, a biotin-conjugated donkey antirabbit secondary antibody (Jackson Immunoresearch) was used at 1:2500. For sections incubated in the monoclonal antibody 19304, a biotin-conjugated donkey antimouse secondary antibody (Jackson Immunoresearch) was used at 1:5000. Secondary antibody labeling was amplified using a Vectastain ABC Elite kit (Vector Laboratories) for 1 hour at room temperature. Reaction product was visualized after 5 minutes incubation in Tris-buffered saline (pH 7.5) containing 0.025% diaminobenzidine, 0.02% nickel, and 0.02% H2O2. Free-floating tissue sections were washed in Tris-buffered saline mounted on gelatin-coated glass microscope slides and allowed to dry before being coverslipped with Permount (Fisher Scientific). Sections were viewed and images were captured using an Olympus BH2 microscope with an Insight QE digital camera with Spot Advanced Software. Coronal brain slices along the rostral-caudal axis were ordered and aligned relative to the organum vasculosum of the lamina terminalis (OVLT), and an investigator blind to genotype manually counted GnRH neuronal cell bodies using a ×40 objective (35). The effect of genotype on the number and location of GnRH neurons was determined by 2-way ANOVA as a repeated measure using SPSS software. Values are reported as mean ± SEM, and P < .05 was considered statistically significant.
Buserelin- (a GnRH receptor agonist) and Kp-54–triggered LH assay
Eight-week-old male mice (Arrb1−/−, Arrb2−/− and their respective WT littermate controls) were administered 3 ng buserelin/g body weight (for the buserelin challenge) or 100 nmol Kp-54/kg body weight (for the Kp challenge) ip. For the buserelin- and Kp-challenged mice, 20 and 60 minutes later, respectively, mice were anesthetized by CO2 exposure followed by cervical dislocation, and blood was collected immediately after by cardiac puncture. Blood was allowed to clot at room temperature for 120 minutes. The clot was then removed with a wooden applicator stick, and the remaining serum centrifuged at 2000g for 20 minutes at room temperature. Serum LH was measured by the Endocrine Technology and Support Lab, Oregon National Primate Research Center (Beaverton, OR). LH was analyzed by a traditional double-antibody RIA procedure similar to that described previously (36). The detection limit of the assay was 0.1 to 0.2 ng/mL. The interassay variation was <12%, and the intra-assay variation was <8%. The means ± SE are shown for values obtained for the number of independent experiments indicated in the figure legends. GraphPad Prism software was used to analyze data for statistical significance. Statistical significance was determined by Student's t test.
Statistical analysis
Statistical significance was validated when P < .05. The specific analysis performed is discussed in the relevant sections of Materials and Methods.
Results
KISS1R triggers LH secretion via β-arrestin
We have previously reported that WT KISS1R signals via the Gαq/11- and β-arrestin–dependent pathways to phosphorylate ERK1/2 (20, 21). Because ERK1/2 regulate Kp-dependent GnRH secretion (22), we hypothesized that KISS1R stimulates GnRH secretion via the β-arrestin pathway. Because the Arrb1−/−;Arrb2−/− double-KO mice die around birth (37), we used the single-KO mice lacking either Arrb1 (encodes for β-arrestin-1) or Arrb2 (encodes for β-arrestin-2) to test our hypothesis. Based on brood size, although the single-KO mice are as fertile as their WT littermates, we reasoned that because the reproductive axis can withstand significant insults without having an effect on fertility, again as measured by brood size (reviewed in Ref. 38), we might instead be able to detect differences at the endocrine level that would support our hypothesis. We therefore proposed measuring LH secretion, a well-established surrogate for GnRH secretion in several vertebrate species (15, 39, 40), to test whether KISS1R stimulates GnRH secretion via the β-arrestin pathway. Although this strategy is possible because GnRH receptor activity on the anterior pituitary is not regulated by β-arrestin (41–45), it still depends on first demonstrating that the loss of Arrb1 or Arrb2 did not affect the size and distribution of the hypothalamic GnRH neuronal population in the anterior hypothalamus, nor did it affect LH secretion from the pituitary gonadotropes due to developmental or other defects resulting from the whole-body loss of Arrb1 and Arrb2.
Loss of either β-arrestin-1 or -2 has no effect on the GnRH immunoreactive neuronal population size and distribution in the hypothalamus
The effect of β-arrestin-1 or -2 on the number and location of the adult GnRH cell population was analyzed by comparing 8-week-old male KO mice with to their respective WT littermates. Representative coronal images from sections at the level of the OVLT are shown (Figure 1, A and E). The mean number of GnRH-immunoreactive neurons in each section was plotted as a histogram (Arrb1−/− [n = 6] and Arrb1+/+ littermates [n = 6]; Arrb2−/− [n = 5] and Arrb2+/+ littermates [n = 5]) (Figure 1, B and F). As expected, the highest number of GnRH-immunoreactive neurons was generally found at or near the OVLT with fewer cells in sections rostral and caudal to the OVLT (35) (Figure 1, B and F). Genotype (KO vs WT) had no significant effect on the total GnRH-immunoreactive cell counts (P > .1) (Figure 1, C and G). This finding, based on the EL14 GnRH antiserum, was corroborated using 2 other GnRH antisera, PA1-122 and QED. PA1-122 data are shown for the β-arrestin-1 WT and KO mice (n = 3 for WT and KO) (Figure 1, C and D), and QED data are shown for the β-arrestin-2 WT and KO mice (n = 5 for WT and KO) (Figure 1, G and H). With respect to the location of GnRH-immunoreactive neurons, genotype (KO vs WT) also had no significant effect in the Arrb1−/− vs Arrb1+/+ mice (P > .1) (Figure 1, B, D, F, and H) and Arrb2−/− vs Arrb2+/+ mice (P > .1) (Figure 1B, D, F and H). Overall, these findings suggest that the β-arrestin-1 and -2 single KO mice could be used to test whether KISS1R stimulates GnRH secretion via the β-arrestin pathway because the GnRH neuronal population was fundamentally intact.
Figure 1.

Loss of either Arrb1−/− or Arrb2−/− has no effect on the GnRH-immunoreactive neuronal population size and distribution in the hypothalamus. A and E, Photomicrographs of coronal brain sections at the level of the OVLT from 8-week-old Arrb1−/− and Arrb2−/− mice and their WT littermate. In photomicrographs, GnRH neurons were revealed in the brain slices by immunohistochemistry using the EL14 antibody, and the OVLT was used as a neuroanatomical reference point for rostral to caudal alignment of coronal sections. B, D, F, and H, Histograms displaying the rostral-caudal distribution of GnRH-immunoreactive neurons from WT and KO mice aligned at the OVLT. The plot represents average GnRH-immunoreactive neurons per 50-μm coronal section. GnRH-immunoreactive neurons were identified with the EL14 antibody (B, Arrb1−/− mice; F, Arrb2−/− mice), PA1–122 antibody (D, Arrb1−/− mice), and QED antibody (H, Arrb2−/− mice). C and G, Total number of GnRH-immunoreactive neurons identified with the EL14 and PA1–122 antibody (C, Arrb1−/− mice) and EL14 and QED antibody (G, Arrb2−/− mice). Error bars represent SEM.
Loss of either β-arrestin-1 or -2 has no effect on buserelin-triggered LH secretion
Next, we determined whether the loss of β-arrestin in the pituitary gonadotropes affected LH secretion due to developmental or other defects along the hypothalamic-pituitary axis. To do this, 8-week-old male Arrb1−/− and Arrb2−/− mice and their WT littermates were administered buserelin (a GnRH receptor agonist) ip, and serum LH was measured 20 minutes later. There was no difference in buserelin-triggered LH secretion in the single-KO mice relative to their WT littermates (P > .02 for Arrb1−/− [3.8 ± 0.2 ng/mL LH; n = 11] vs WT [3.3 ± 0.4 ng/mL LH; n = 13]; P > .02 for Arrb2−/− [2.7 ± 10.3 ng/mL LH; n = 15] vs WT [2.7 ± 0.1 ng/mL LH; n = 9]) (Figure 2, A and B). Thus, the loss of either β-arrestin isoform in pituitary gonadotropes did not affect buserelin-stimulated LH secretion, and these single-KO mice could be used to test whether KISS1R stimulates GnRH secretion via the β-arrestin pathway.
Figure 2.

Loss of either Arrb1−/− or Arrb2−/− does not affect buserelin-triggered LH secretion. Serum LH levels were measured by RIA. Mice were either left untreated or treated with saline or 3 ng buserelin/g body weight. Twenty minutes later, blood was collected and serum prepared. Buserelin-triggered LH levels in Arrb1−/− (A) and Arrb2−/− (B) mice were significantly higher (P < .001) than levels from their respective untreated and saline-treated controls. Buserelin-triggered LH levels were not different (P > .2) between Arrb1−/− (A) and Arrb2−/− (B) mice and their WT littermate controls, respectively. Only male mice were used to preclude the effects of estrous cycle variation on GnRH secretion in females. Error bars represent SEM.
Loss of either β-arrestin-1 or -2 significantly reduces Kp-triggered LH secretion
To determine what effect a loss of either β-arrestin-1 or -2 has on LH secretion, 8-week-old male Arrb1−/− and Arrb2−/− mice and their WT littermates were administered Kp-54 ip, and serum LH was measured 1 hour later. There was significant reduction in Kp-triggered LH secretion in each of the single-KO mice relative to their WT littermates (P < .001 for Arrb1−/− [12.1 ± 0.4 ng/mL LH; n = 14] vs WT [15.4 ± 0.8 ng/mL LH; n = 10]; P < .001 for Arrb2−/− [13.2 ± 0.8 ng/mL LH; n = 14] vs WT [18.4 ± 1.1 ng/mL LH; n = 12]) (Figure 3, A and B). Thus, the loss of either β-arrestin isoform diminished Kp-stimulated LH secretion, and β-arrestin-1 and -2 regulate LH secretion in young adult male mice.
Figure 3.
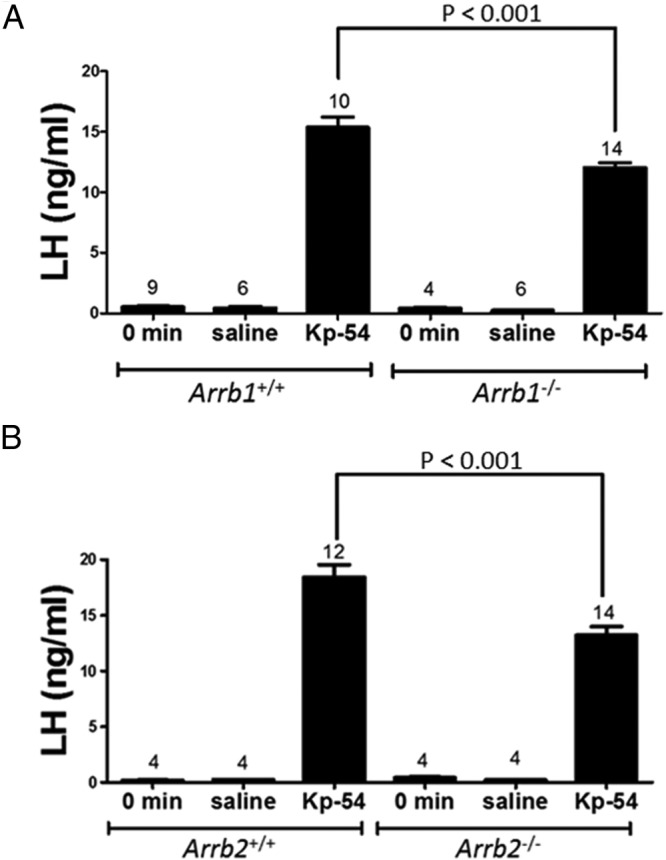
Loss of either Arrb1−/− or Arrb2−/− significantly diminishes Kp-54–triggered LH secretion. Serum LH levels were measured by RIA. Mice were either left untreated or treated with saline or 100 nmol Kp-54/kg body weight. Sixty minutes later, blood was collected and serum prepared. Kp-54–triggered LH levels in Arrb1−/− (A) and Arrb2−/− (B) mice were significantly higher (P < .001) than levels from their respective untreated and saline-treated controls. Kp-54–triggered LH levels were significantly lower (P < .001) in Arrb1−/− (A) and Arrb2−/− (B) mice compared with their respective WT littermate controls. Only male mice were used to preclude the effects of estrous cycle variation on GnRH secretion in females. Error bars represent SEM.
L148S displays Gαq/11-independent signaling
WT KISS1R displays Gαq/11-independent signaling via β-arrestin (20, 21). We subsequently determined that, like WT KISS1R, L148S also physically associates with β-arrestin-1 and -2. Additionally, it was reported that L148S is expressed at WT levels, localizes to the cell surface, binds Kp, and continues to interact with many signaling and regulatory molecules identical to WT KISS1R (27). These observations led to the hypothesis that although L148S is uncoupled from InsP3 formation, it has the capacity to undergo Kp-triggered Gαq/11-independent signaling.
L148S triggers significant ERK1/2 phosphorylation
Relative to Kp-treated WT KISS1R-expressing cells, Kp-treated L148S-expressing cells triggered about 2.5-fold less (P < .05) ERK1/2 phosphorylation (Figure 4, A, B, and D). However, after 0.1μM and 3.0μM Kp-10 stimulation, L148S-triggered ERK1/2 phosphorylation was approximately 2-fold greater (P < .05) than unstimulated L148S-expressing cells (Figure 4, B and D) and both unstimulated and stimulated nontransfected cells (Figure 4, B–D). All these studies were conducted with the untagged WT and L148S receptor.
Figure 4.
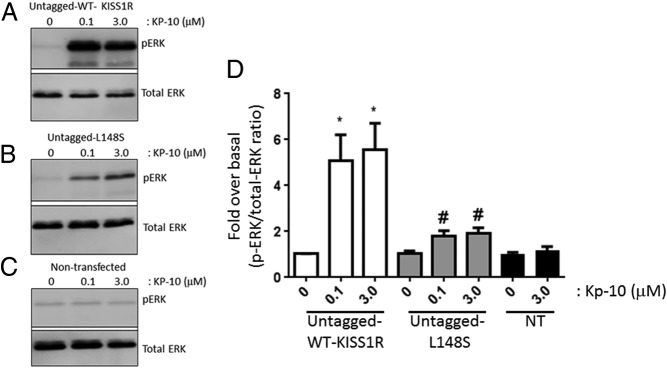
L148S triggers ERK1/2 phosphorylation. A–D, Representative autoradiographs (A–C) and densitometric analysis (D) showing the expression of total and phosphorylated ERK1/2 in WT untagged KISS1R- and untagged L148S-overexpressing HEK 293 cells and nontransfected (NT) HEK 293 cells after a 10-minute Kp-10 treatment (0μM, 0.1μM, and 3.0μM). Western blot analyses were done using monoclonal anti-ERK1/2 and anti–phospho-ERK1/2 antibodies. The data represent the mean of 3 independent experiments. *, P < .05, pERK1/2/total ERK1/2 ratio compared with 0μM in WT group; #, P < .05, pERK1/2/total ERK1/2 ratio compared with 0μM in L148S group. Error bars represent SEM.
Because we routinely use tagged receptors to study KISS1R signaling, we determined how tagged-WT and -L148S receptors compared with the untagged receptors with respect to ERK1/2 phosphorylation. Tagged (YFP, FLAG, and myc) WT and L148S receptors triggered similar levels of ERK1/2 phosphorylation (P > .05) compared with the untagged WT and L148S receptors, respectively, after 0.1μM and 3.0μM Kp-10 stimulation (Figure 5, A–F vs Figure 4 A, C, and D). These results indicate that the presence of epitopes YFP, FLAG, and myc did not alter the signaling potential of WT and mutant KISS1R. Kp-treated L148S-expressing cells triggered significant ERK1/2 phosphorylation (P < .05) compared with both unstimulated L148S-expressing cells and stimulated nontransfected cells (Figures 4 and 5). These findings were verified independently in both the Babwah and Kaiser laboratories, and expression constructs were routinely sequenced to verify their nucleotide composition.
Figure 5.

L148S triggers ERK1/2 phosphorylation. Representative autoradiographs (A, C, and E) and densitometric analysis (B, D, and F) showing the expression of total and phosphorylated ERK1/2 in WT KISS1R (YFP-, FLAG-, and myc-tagged), L148S (YFP-, FLAG-, and myc-tagged) and nontransfected (NT) overexpressing HEK 293 cells after a 10-minute Kp-10 treatment (0μM, 0.1μM, and 3.0μM). Western blot analyses were done using monoclonal anti-ERK1/2 and anti–phospho-ERK1/2 antibodies. The data represent the mean of 3 independent experiments. *, P < .05, pERK1/2/total ERK1/2 ratio compared with 0μM in WT group; #, P < .05, pERK1/2/total ERK1/2 ratio compared with 0μM in L148S group. Error bars represent SEM.
L148S triggers ERK1/2 phosphorylation via the β-arrestin pathway
Because L148S physically associates with β-arrestin-1 and -2 (21), we hypothesized that L148S-dependent ERK1/2 phosphorylation (Figures 4 and 5) is β-arrestin–dependent. This was tested by assessing L148S-dependent ERK1/2 phosphorylation after Kp-10 treatment in the presence and absence of β-arrestin-1 and -2. As seen, when L148S was expressed in WT mouse embryonic fibroblast (MEFs), after Kp-10 treatment, it triggered significant ERK1/2 phosphorylation (P < .05) (Figure 6). However, when L148S was expressed in MEFs lacking β-arrestin-1 and -2, the ability to trigger Kp-dependent ERK1/2 phosphorylation was almost completely abolished (Figure 6), thus supporting our hypothesis that L148S-triggered ERK1/2 phosphorylation is β-arrestin–dependent.
Figure 6.
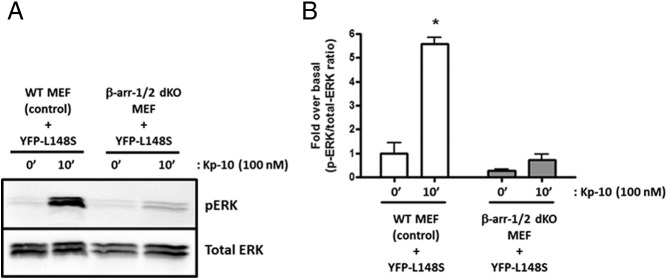
L148S triggers ERK1/2 phosphorylation in a β-arrestin–dependent manner. A and B, A representative autoradiograph (A) and densitometric analysis (B) showing the expression of total and phosphorylated ERK1/2 in YFP-L148S–overexpressing WT and β-arrestin-1 and -2 double-KO (dKO) MEFs after 0- and 10-minute Kp-10 treatment (100nM). Western blot analyses were done using monoclonal anti-ERK1/2 and anti-phospho–ERK1/2 antibodies. The data represent the mean of 3 independent experiments. *, P < .05, pERK1/2/total ERK1/2 ratio of Kp-10–stimulated L148S-expressing WT MEFs compared with unstimulated L148S-expressing WT MEFs. Error bars represent SEM.
L148S is strongly uncoupled from Gαq/11 signaling at Kp-10 concentrations that trigger robust signaling via WT KISS1R
L148S is impaired in G protein activation by inhibiting the dissociation of Gα and Gβγ subunits (27) and, as a result, is uncoupled from InsP3 formation (15, 27). In the present study, we also show that, whereas WT KISS1R triggered InsP3 formation over a wide range of Kp-10 concentrations (1.0 × 10−9M to 3 × 10−6M Kp-10) with an EC50 of 1.5 × 10−9M, L148S was significantly diminished (P < .05) in its capacity to do so, displaying an EC50 of 1.7 × 10−8M that was not significantly different (P > .05) from the InsP3-generating capacity of nontransfected cells (Figure 7). Although these findings confirm previous reports (15, 27) that L148S is strongly uncoupled from Gαq/11 signaling, it was important to confirm these findings in parallel with the evidence that ERK1/2 phosphorylation was still occurring (Figures 4 and 5). Thus, the small but significant L148S-dependent ERK1/2 phosphorylation that we observed was unlikely due to differences such as in expression constructs, experimental techniques and cell lines that may exist between the current and previous studies (15, 27).
Figure 7.
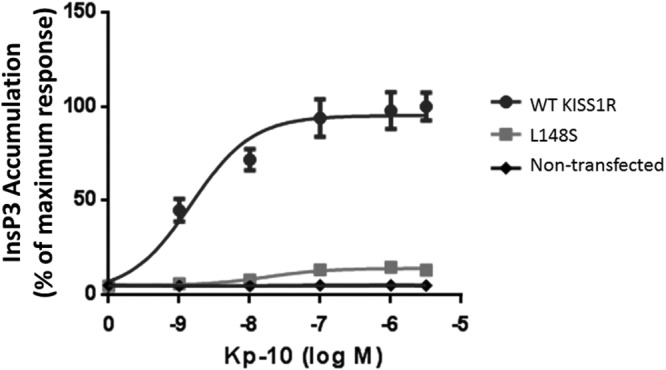
L148S is strongly uncoupled from Gαq/11 signaling. COS7 cells were transiently transfected with WT KISS1R or L148S. Kp-10–stimulated InsP3 accumulation was measured by conversion of prelabeled [2-3H]myoinositol to [3H]IP. Data are percentage of maximum response. The results are from pooled data from 3 independent experiments. All assays were performed in triplicate. Error bars represent SEM. IP, inositol trisphosphate.
WT KISS1R and L148S differentially regulate PIP2 hydrolysis
Although L148S-expressing cells were not significantly different (P > .05) from nontransfected cells in their InsP3-generating capacity, to further strengthen the finding that L148S is functionally uncoupled from the Gαq/11 signaling pathway, we monitored the spatial distribution of GFP-PH-PLC-δ1 after Kp treatment in cells coexpressing either WT KISS1R or L148S. The spatial localization of GFP-PH-PLC-δ1 in the cell reflects the activation state of Gαq/11. In the unstimulated cell, GFP-PH-PLC-δ1 is localized to the plasma membrane, but upon stimulation, GFP-PH-PLC-δ1 is released from the plasma membrane as a consequence of both PIP2 hydrolysis and the high affinity of the PLC-δ1 PH domain for InsP3 (46). In cells expressing GFP-PH-PLC-δ, but lacking KISS1R, GFP-PH-PLC-δ1 was localized at the plasma membrane, even after Kp-10 treatment, due to the interaction of the PLC-δ1 PH domain with membrane PIP2 (Figure 8, A–C). However, in cells coexpressing GFP-PH-PLC-δ1 and FLAG-KISS1R (WT), in response to Kp-10 treatment, GFP-PH-PLC-δ1 was released from the plasma membrane and accumulated in the cytosol of the cell (Figure 8, D–G).
Figure 8.

L148S is strongly uncoupled from Gαq/11 signaling. A–M, representative images selected from a time series of laser scanning confocal microscopic images showing the plasma membrane and cytosolic localization of GFP-PH-PLC-δ1 in nontransfected HEK 293 cells (A–C) and in HEK 293 cells expressing WT KISS1R or L148S (D–G, FLAG-WT-KISS1R-expressing cells; H–J, FLAG-L148S-expressing cells; K-M: myc-L148S-expressing cells) in the absence of agonist (0 seconds) and in response to 3μM Kp-10 treatment (added gently and in a drop-wise manner to cells cultured on a confocal dish) approximately 1 minute later (see solid white arrowheads on x-axis of graphs C, G, J, and M). Cytosolic regions of interest in cells (shown by small white circle in panels A, B, D–F, H, I, K, and L) were analyzed quantitatively by determining the changes in GFP fluorescence over time. These changes are presented graphically (C, G, J, and M). These studies were conducted 5 independent times, and representative images from 1 independent experiment are shown. A–C, Approximately 14 minutes after Kp-10 treatment, translocation of GFP-PH-PLC-δ1 from the plasma membrane to the cytosol was visually undetectable. This confirms that translocation is dependent on KP/KISS1R signaling. D–G, At approximately 440 seconds (7.5 minutes) after adding Kp-10 (open arrowhead) GFP-PH-PLC-δ1 began translocating from the plasma membrane to the cytosol. Thus, the KISS1R-coupled Gαq/11 pathway is activated. H–J, Translocation of GFP-PH-PLC-δ1 from the plasma membrane to the cytosol was visually undetectable after 15 minutes of Kp-10 treatment. K–M, Translocation of GFP-PH-PLC-δ1 from the plasma membrane to the cytosol was visually undetectable after 16 minutes of Kp-10 treatment.
Our previously published study (19) demonstrated that 100nM Kp-10 was sufficient to trigger a robust redistribution of GFP-PH-PLC-δ1 in cells coexpressing WT KISS1R. However, the present study was conducted with 3μM Kp-10 to determine whether a high concentration of Kp-10, which triggers a robust L148S-distal signaling response (that is, ERK1/2 phosphorylation) (Figures 4 and 5), would stimulate a Gαq/11-coupled L148S-proximal signaling event (that is, PIP2 hydrolysis). If it did not, we would conclude that L148S is strongly uncoupled from the Gαq/11 signaling pathway and that L148S-dependent ERK1/2 phosphorylation is primarily the consequence of Gαq/11-independent signaling. As seen, when either FLAG-L148S (Figure 8, H–J) or myc-L148S (Figure 8, K–M) were coexpressed with GFP-PH-PLC-δ1, 3 μM Kp-10 completely failed to stimulate the cellular redistribution of GFP-PH-PLC-δ1 (Figure 8H–M) and was indistinguishable from Kp-treated cells expressing GFP-PH-PLC-δ1 alone (Figure 8A–C). Cells coexpressing GFP-PH-PLC-δ with either FLAG-L148S (Figure 8H–J) or myc-L148S (Figure 8K–M) were imaged for up to about 17 minutes after Kp-10 treatment. Even if there were weak L148S-dependent InsP3 formation over this period, transient cellular accumulation of InsP3 was still insufficient to trigger detectable levels of PIP2 hydrolysis as determined by a cellular redistribution of GFP-PH-PLC-δ1. Based on the GFP-PH-PLC-δ1cellular redistribution data, it appears that at up to 3μM Kp-10, L148S, is strongly uncoupled from the Gαq/11 signaling pathway.
Discussion
One of the most significant findings in the field of reproductive neuroendocrinology is that the hypothalamic Kp/KISS1R signaling system is a potent trigger of GnRH release (15). Since this discovery in 2003, a large number of studies have provided significant insight into how this signaling system is regulated and affects fertility under normal and adverse conditions (reviewed in Ref. 47). In comparison, a much smaller number of studies have provided substantial mechanistic insight into how KISS1R signals intracellularly to regulate biological functions and how receptor-based mutations affect these signaling events (20, 21, 23–25, 27, 33, 48, 49). Based on these studies, it is now well established that KISS1R regulates GnRH secretion in a Gαq/11-dependent manner and patients bearing Gαq/11-uncoupling mutations display IHH (15). Based on the current study and previously published data (20, 21), we now recognize and report that, in addition to coupling and signaling via Gαq/11 in the GnRH neuron to trigger GnRH secretion, KISS1R also couples to and signals via β-arrestin in the GnRH neuron to mediate GnRH secretion (as assessed indirectly by measuring LH secretion from the pituitary). β-Arrestin has been reported to mediate insulin secretion (50, 51), thus it is likely that β-arrestin plays a more widespread role in regulating peptide secretion. In this study, we demonstrated that a loss of either β-arrestin-1 or -2 results in about 20% to 25% reduction in LH secretion. This had no effect on fertility based on brood size and is consistent with what has been previously reported regarding how much secretion is required for fertility (reviewed in Ref. 38). Because both β-arrestin-1 and -2 mediate LH secretion, we predict that their combined loss would result in a much larger reduction in LH secretion, reflecting an even greater role for β-arrestin in mediating GnRH secretion. It is also possible that a combined loss of both isoforms would result in a subfertile phenotype, but unlikely an infertile phenotype, given that KISS1R-coupled Gαq/11 signaling would still be intact. However, because the β-arrestin-1 and -2 double-KO mouse dies around birth (37) and floxed Arrb mice are unavailable, we could not test this prediction.
Based on an earlier in vitro study from our laboratory (21), we determined that in MEFs, β-arrestin-2 positively regulates ERK1/2 phosphorylation downstream of KISS1R, whereas β-arrestin-1 negatively regulates it. Thus, we expected to find that β-arrestin-1 and -2 would have opposite effects on Kp-triggered GnRH secretion; instead, we found that they both mediate Kp-dependent secretion. Although the in vivo and in vitro studies both clearly demonstrate a role for β-arrestin in regulating Kp-dependent signaling, it is not surprising to encounter some differences given differences in the cell types and in the in vivo and in vitro environments. Thus, although the in vitro study establishes a role for β-arrestin in regulating Kp-dependent signaling, the in vivo study establishes the precise nature of that role for a given cellular process, such as Kp-dependent GnRH secretion.
A number of recent studies have determined that the pituitary from various species, including the mouse, express both Kiss1 and Kiss1r (reviewed in Refs. 52 and 53). Several studies have also demonstrated a direct action of Kp on pituitary cells in regulating gonadotropin gene expression and secretion (54–56). However, several lines of evidence (reviewed in Ref. 52), such as those that have shown that the Kp-induced increase in gonadotropin secretion can be blocked with GnRH antagonists (39, 58–61), suggest that Kp-dependent gonadotropin secretion largely reflects direct activation of GnRH neurons and not pituitary gonadotropes. Nevertheless, because Kp-54 was administered systemically in our study, the possibility remains that the attenuated LH response in the β-arrestin–KO mice was at least in a very small part due to the loss of β-arrestin signaling by Kiss1r in the pituitary, rather than in GnRH neurons.
The finding that KISS1R employs the β-arrestin pathway to mediate GnRH secretion led us to consider that some Gαq/11-uncoupled KISS1R mutants might maintain the ability to couple to and signal via Gαq/11-independent mechanisms to regulate GnRH secretion. To do so, a given mutant must fulfill the following criteria. It must localize to the cell surface, bind Kp, and interact with signaling molecules capable of mediating G protein-independent signaling. Because KISS1R mutations such as R331X and F272S (20, 23) block receptor trafficking to the cell surface and consequently preclude KISS1R/Gαq/11 coupling, we would predict that such mutants would not display Gαq/11-independent signaling. However, we predicted that KISS1R mutations such as L148S (27) would show Gαq/11-independent signaling and indeed demonstrated that it did. Furthermore, we demonstrated that the β-arrestin pathway represents a major conduit for L148S signaling. The finding that L148S displays Gαq/11-independent signaling is important because it reveals that some mutant KISS1Rs that appeared as loss-of-function receptors based on InsP3 formation might, in fact, be signaling competent and need to be examined more closely.
An important question that emerges from our findings is whether KISS1R-coupled Gαq/11-independent signaling is physiologically relevant in the human population. There is evidence among human patients to suggest that it is. This might in part or perhaps even fully explain why patients bearing the L102P KISS1R mutation, which we predict to meet the criteria for Gαq/11-independent signaling based on its pharmacological and molecular characterization (28), displayed variable gonadotropic deficiency. Interestingly, an L102P-bearing male patient who was examined from ages 12 to 21 was observed to undergo progressive changes in pituitary response from an early pubertal to an almost full pubertal pattern (28). This led the authors to propose that KISS1R inactivation, as seen with L102P, does not impede pubertal onset but instead delays pubertal maturation of the gonadotropic axis. Based on the current study, we suggest that, mechanistically, gradual pubertal maturation among patients bearing KISS1R mutations like L102P is in part due to Gαq/11-independent, and in particular β-arrestin–dependent, GnRH secretion.
IHH was once considered a permanent condition, but a growing number of studies over the last 2 decades have reported on the reversal of this condition among some patients. In their study, Sidhoum et al (62) present clinical, laboratory, neuroendocrine, and neuroradiologic characteristics of 308 patients with IHH. Of these, 44 patients, even some with severe IHH, underwent reversal. Patients exhibiting reversal had a higher frequency of mutations affecting neurokinin B (NKB) signaling compared with patients with no reversal. Perturbations in NKB activity or signaling have been implicated in GnRH deficiency (63–65). The NKB receptor (NK3R) is a Gαq/11-coupled receptor that appears to signal via β-arrestin (66) in addition to protein kinase C (67). Thus, it is possible that NK3R, like Kiss1r, regulates GnRH secretion via β-arrestin, and therefore, some NK3R mutants that are uncoupled from Gαq/11 might continue to regulate GnRH secretion via β-arrestin (65).
In developing novel molecular strategies to treat diseases, it is critical to develop a detailed mechanistic understanding of the signaling pathways that underlie normal and pathological responses. For example, drugs that trigger β-arrestin–coupled KISS1R signaling with great efficacy may prove useful in treating IHH patients who bear Gαq/11-uncoupled KISS1R mutants that localize to the cell surface and bind Kp. Such treatment may even be extended to patients bearing KISS1R mutants such as R331X that do not localize efficiently to the cell surface (20) by developing and using other drugs that can rescue mislocalized receptors (57, 68). The future development of β-arrestin superagonists that can trigger KISS1R-dependent GnRH secretion is possible only by first recognizing that KISS1R displays Gαq/11-independent signaling.
Given that reproductive success is paramount to the survival of any species, it is not surprising to find that there is more than one pathway significantly coregulating GnRH secretion. Thus, reproductive success, although it might be diminished, can still be achieved in animals bearing certain types of inactivating mutations. The current study describes an important and novel discovery that KISS1R signals and triggers LH secretion via the β-arrestin pathway in the male mouse. We suggest that other 7-transmembrane–spanning receptors such as NK3R employ β-arrestin to regulate the reproductive endocrine axis at several levels and that the β-arrestin signaling pathway likely represents a major regulator of fertility. Finally, we propose that for some patients, Gαq/11-independent signaling could account for the phenomenon of IHH reversal. Further investigations into this may eventually lead to new approaches in the treatment of IHH and several other reproductive endocrine disorders.
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council of Canada (RGPIN/327334-2011) and Lawson Health Research Institute (London, Ontario) (to A.V.B.); the Canadian Institutes of Health Research (MOP 107972 to M.B.); the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health (NIH), through cooperative agreement U54 HD28138 (to U.B.K.) and HD012303 (to P.L.M.) as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research; RO1 HD019938 (to U.B.K.), NIDDK/NIH T32 DK007529 and K08 HD070957 (to L.M.), R01 MH061376 (to S.A.T.), NIH R01 DK044838, R01 HD072754, and R01 HD020377 (to P.L.M.). P.L.M. was also partially supported by P30 DK063491, P30 CA023100, and P42 ES101337. C.G.K. was partially supported by the Hartwell Foundation, T32 HD007203 and T32 DK007044.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- IHH
- idiopathic hypogonadotropic hypogonadism
- InsP3
- inositol 1,4,5-trisphosphate
- KISS1R
- Kp receptor
- KO
- knockout
- Kp
- kisspeptin
- MEF
- mouse embryonic fibroblast
- NKB
- neurokinin B
- NK3R
- NKB receptor
- OVLT
- organum vasculosum of the lamina terminalis
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- WT
- wild-type.
References
- 1. Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989;338:161–164. [DOI] [PubMed] [Google Scholar]
- 2. Wray S. Development of luteinizing hormone releasing hormone neurones. J Neuroendocrinol. 2001;13:3–11. [DOI] [PubMed] [Google Scholar]
- 3. Belchetz PE, Plant TM, Nakai Y, Keogh EJ, Knobil E. Hypophysial responses to continuous and intermittent delivery of hypopthalamic gonadotropin-releasing hormone. Science. 1978;202:631–633. [DOI] [PubMed] [Google Scholar]
- 4. Clayton RN. Gonadotrophin releasing hormone: from physiology to pharmacology. Clin Endocrinol (Oxf). 1987;26:361–384. [DOI] [PubMed] [Google Scholar]
- 5. Clayton RN. Mechanism of GnRH action in gonadotrophs. Hum Reprod. 1988;3:479–483. [DOI] [PubMed] [Google Scholar]
- 6. Savoy-Moore RT, Swartz KH. Several GnRH stimulation frequencies differentially release FSH and LH from isolated, perfused rat anterior pituitary cells. Adv Exp Med Biol. 1987;219:641–645. [DOI] [PubMed] [Google Scholar]
- 7. Wildt L, Häusler A, Marshall G, et al. Frequency and amplitude of gonadotropin-releasing hormone stimulation and gonadotropin secretion in the rhesus monkey. Endocrinology. 1981;109:376–385. [DOI] [PubMed] [Google Scholar]
- 8. Kaiser UB, Conn PM, Chin WW. Studies of gonadotropin-releasing hormone (GnRH) action using GnRH receptor-expressing pituitary cell lines. Endocr Rev. 1997;18:46–70. [DOI] [PubMed] [Google Scholar]
- 9. Thompson IR, Ciccone NA, Xu S, Zaytseva S, Carroll RS, Kaiser UB. GnRH pulse frequency-dependent stimulation of FSHβ transcription is mediated via activation of PKA and CREB. Mol Endocrinol. 2013;27:606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thompson IR, Kaiser UB. GnRH pulse frequency-dependent differential regulation of LH and FSH gene expression. Mol Cell Endocrinol. 2014;385:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kotani M, Detheux M, Vandenbogaerde A, et al. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem. 2001;276:34631–34636. [DOI] [PubMed] [Google Scholar]
- 12. Muir AI, Chamberlain L, Elshourbagy NA, et al. AXOR12, a novel human G protein-coupled receptor, activated by the peptide KiSS-1. J Biol Chem. 2001;276:28969–28975. [DOI] [PubMed] [Google Scholar]
- 13. Ohtaki T, Shintani Y, Honda S, et al. Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001;411:613–617. [DOI] [PubMed] [Google Scholar]
- 14. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100:10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627. [DOI] [PubMed] [Google Scholar]
- 16. Lee DK, Nguyen T, O'Neill GP, et al. Discovery of a receptor related to the galanin receptors. FEBS Lett. 1999;446:103–107. [DOI] [PubMed] [Google Scholar]
- 17. Kirby HR, Maguire JJ, Colledge WH, Davenport AP. International Union of Basic and Clinical Pharmacology. LXXVII. Kisspeptin receptor nomenclature, distribution, and function. Pharmacol Rev. 2010;62:565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu X, Lee K, Herbison AE. Kisspeptin excites gonadotropin-releasing hormone neurons through a phospholipase C/calcium-dependent pathway regulating multiple ion channels. Endocrinology. 2008;149:4605–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Babwah AV, Pampillo M, Min L, Kaiser UB, Bhattacharya M. Single-cell analyses reveal that KISS1R-expressing cells undergo sustained kisspeptin-induced signaling that is dependent upon an influx of extracellular Ca2+. Endocrinology. 2012;153:5875–5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pampillo M, Camuso N, Taylor JE, et al. Regulation of GPR54 signaling by GRK2 and β-arrestin. Mol Endocrinol. 2009;23:2060–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Szereszewski JM, Pampillo M, Ahow MR, Offermanns S, Bhattacharya M, Babwah AV. GPR54 regulates ERK1/2 activity and hypothalamic gene expression in a Gαq/11 and β-arrestin-dependent manner. PLoS One. 2010;5:e12964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Castellano JM, Navarro VM, Fernández-Fernández R, et al. Ontogeny and mechanisms of action for the stimulatory effect of kisspeptin on gonadotropin-releasing hormone system of the rat. Mol Cell Endocrinol. 2006;257–258:75–83. [DOI] [PubMed] [Google Scholar]
- 23. Nimri R, Lebenthal Y, Lazar L, et al. A novel loss-of-function mutation in GPR54/KISS1R leads to hypogonadotropic hypogonadism in a highly consanguineous family. J Clin Endocrinol Metab. 2011;96:E536–E545. [DOI] [PubMed] [Google Scholar]
- 24. Chevrier L, de Brevern A, Hernandez E, et al. PRR repeats in the intracellular domain of KISS1R are important for its export to cell membrane. Mol Endocrinol. 2013;27:1004–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Teles MG, Bianco SD, Brito VN, et al. A GPR54-activating mutation in a patient with central precocious puberty. N Engl J Med. 2008;358:709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bo-Abbas Y, Acierno JS, Jr, Shagoury JK, Crowley WF, Jr, Seminara SB. Autosomal recessive idiopathic hypogonadotropic hypogonadism: genetic analysis excludes mutations in the gonadotropin-releasing hormone (GnRH) and GnRH receptor genes. J Clin Endocrinol Metab. 2003;88:2730–2737. [DOI] [PubMed] [Google Scholar]
- 27. Wacker JL, Feller DB, Tang XB, et al. Disease-causing mutation in GPR54 reveals the importance of the second intracellular loop for class A G-protein-coupled receptor function. J Biol Chem. 2008;283:31068–31078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tenenbaum-Rakover Y, Commenges-Ducos M, Iovane A, Aumas C, Admoni O, de Roux N. Neuroendocrine phenotype analysis in five patients with isolated hypogonadotropic hypogonadism due to a L102P inactivating mutation of GPR54. J Clin Endocrinol Metab. 2007;92:1137–1144. [DOI] [PubMed] [Google Scholar]
- 29. Horikoshi Y, Matsumoto H, Takatsu Y, et al. Dramatic elevation of plasma metastin concentrations in human pregnancy: metastin as a novel placenta-derived hormone in humans. J Clin Endocrinol Metab. 2003;88:914–919. [DOI] [PubMed] [Google Scholar]
- 30. Conner DA, Mathier MA, Mortensen RM, et al. β-Arrestin1 knockout mice appear normal but demonstrate altered cardiac responses to β-adrenergic stimulation. Circ Res. 1997;81:1021–1026. [DOI] [PubMed] [Google Scholar]
- 31. Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science. 1999;286:2495–2498. [DOI] [PubMed] [Google Scholar]
- 32. Min L, Soltis K, Reis AC, et al. Dynamic kisspeptin receptor trafficking modulates kisspeptin-mediated calcium signaling. Mol Endocrinol. 2014;28:16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bianco SD, Vandepas L, Correa-Medina M, et al. KISS1R intracellular trafficking and degradation: effect of the Arg386Pro disease-associated mutation. Endocrinology. 2011;152:1616–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ellinwood WE, Ronnekleiv OK, Kelly MJ, Resko JA. A new antiserum with conformational specificity for LHRH: usefulness for radioimmunoassay and immunocytochemistry. Peptides. 1985;6:45–52. [DOI] [PubMed] [Google Scholar]
- 35. Gill JC, Wadas B, Chen P, et al. The gonadotropin-releasing hormone (GnRH) neuronal population is normal in size and distribution in GnRH-deficient and GnRH receptor-mutant hypogonadal mice. Endocrinology. 2008;149:4596–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pau KY, Orstead KM, Hess DL, Spies HG. Feedback effects of ovarian steroids on the hypothalamic-hypophyseal axis in the rabbit. Biol Reprod. 1986;35:1009–1023. [DOI] [PubMed] [Google Scholar]
- 37. Zhang M, Teng H, Shi J, Zhang Y. Disruption of β-arrestins blocks glucocorticoid receptor and severely retards lung and liver development in mice. Mech Dev. 2011;128:368–375. [DOI] [PubMed] [Google Scholar]
- 38. Popa SM, Moriyama RM, Caligioni CS, et al. Redundancy in Kiss1 expression safeguards reproduction in the mouse. Endocrinology. 2013;154:2784–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gottsch ML, Cunningham MJ, Smith JT, et al. A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology. 2004;145:4073–4077. [DOI] [PubMed] [Google Scholar]
- 40. Ramaswamy S, Dwarki K, Ali B, Gibbs RB, Plant TM. The decline in pulsatile GnRH release, as reflected by circulating LH concentrations, during the infant-juvenile transition in the agonadal male rhesus monkey (Macaca mulatta) is associated with a reduction in kisspeptin content of KNDy neurons of the arcuate nucleus in the hypothalamus. Endocrinology. 2013;154:1845–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vrecl M, Anderson L, Hanyaloglu A, et al. Agonist-induced endocytosis and recycling of the gonadotropin-releasing hormone receptor: effect of beta-arrestin on internalization kinetics. Mol Endocrinol. 1998;12:1818–1829. [DOI] [PubMed] [Google Scholar]
- 42. McArdle CA, Davidson JS, Willars GB. The tail of the gonadotrophin-releasing hormone receptor: desensitization at, and distal to, G protein-coupled receptors. Mol Cell Endocrinol. 1999;151:129–136. [DOI] [PubMed] [Google Scholar]
- 43. Heding A, Vrecl M, Hanyaloglu AC, Sellar R, Taylor PL, Eidne KA. The rat gonadotropin-releasing hormone receptor internalizes via a beta-arrestin-independent, but dynamin-dependent, pathway: addition of a carboxyl-terminal tail confers beta-arrestin dependency. Endocrinology. 2000;141:299–306. [DOI] [PubMed] [Google Scholar]
- 44. Hislop JN, Everest HM, Flynn A, et al. Differential internalization of mammalian and non-mammalian gonadotropin-releasing hormone receptors. Uncoupling of dynamin-dependent internalization from mitogen-activated protein kinase signaling. J Biol Chem. 2001;276:39685–39694. [DOI] [PubMed] [Google Scholar]
- 45. Pawson AJ, Faccenda E, Maudsley S, Lu ZL, Naor Z, Millar RP. Mammalian type I gonadotropin-releasing hormone receptors undergo slow, constitutive, agonist-independent internalization. Endocrinology. 2008;149:1415–1422. [DOI] [PubMed] [Google Scholar]
- 46. Hirose K, Kadowaki S, Tanabe M, Takeshima H, Iino M. Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. Science. 1999;284:1527–1530. [DOI] [PubMed] [Google Scholar]
- 47. Pinilla L, Aguilar E, Dieguez C, Millar RP, Tena-Sempere M. Kisspeptins and reproduction: physiological roles and regulatory mechanisms. Physiol Rev. 2012;92:1235–1316. [DOI] [PubMed] [Google Scholar]
- 48. Zajac M, Law J, Cvetkovic DD, et al. GPR54 (KISS1R) transactivates EGFR to promote breast cancer cell invasiveness. PLoS One. 2011;6:e21599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cvetkovic D, Dragan M, Pape C, et al. KISS1R induces invasiveness of estrogen receptor-negative human mammary epithelial and breast cancer cells. Endocrinology. 2013;154:1999–2014. [DOI] [PubMed] [Google Scholar]
- 50. Sonoda N, Imamura T, Yoshizaki T, Babendure JL, Lu JC, Olefsky JM. β-Arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic beta cells. Proc Natl Acad Sci U S A. 2008;105:6614–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ravier MA, Leduc M, Richard J, et al. β-Arrestin2 plays a key role in the modulation of the pancreatic β cell mass in mice. Diabetologia. 2014;57:532–541. [DOI] [PubMed] [Google Scholar]
- 52. Kauffman AS, Clifton DK, Steiner RA. Emerging ideas about kisspeptin- GPR54 signaling in the neuroendocrine regulation of reproduction. Trends Neurosci. 2007;30:504–511. [DOI] [PubMed] [Google Scholar]
- 53. Richard N, Corvaisier S, Camacho E, Kottler ML. KiSS-1 and GPR54 at the pituitary level: overview and recent insights. Peptides. 2009;30:123–129. [DOI] [PubMed] [Google Scholar]
- 54. Gutiérrez-Pascual E, Martínez-Fuentes AJ, Pinilla L, Tena-Sempere M, Malagón MM, Castaño JP. Direct pituitary effects of kisspeptin: activation of gonadotrophs and somatotrophs and stimulation of luteinising hormone and growth hormone secretion. J Neuroendocrinol. 2007;19:521–530. [DOI] [PubMed] [Google Scholar]
- 55. Suzuki S, Kadokawa H, Hashizume T. Direct kisspeptin-10 stimulation on luteinizing hormone secretion from bovine and porcine anterior pituitary cells. Anim Reprod Sci. 2008;103:360–365. [DOI] [PubMed] [Google Scholar]
- 56. Witham EA, Meadows JD, Hoffmann HM, Shojaei S, Coss D, Kauffman AS, Mellon PL. Kisspeptin regulates gonadotropin genes via immediate early gene induction in pituitary gonadotropes. Mol Endocrinol. 2013;27:1283–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Janovick JA, Stewart MD, Jacob D, et al. Restoration of testis function in hypogonadotropic hypogonadal mice harboring a misfolded GnRHR mutant by pharmacoperone drug therapy. Proc Natl Acad Sci U S A. 2013;110:21030–21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Irwig MS, Fraley GS, Smith JT, et al. Kisspeptin activation of gonadotropin releasing hormone neurons and regulation of KiSS-1 mRNA in the male rat. Neuroendocrinology. 2004;80:264–272. [DOI] [PubMed] [Google Scholar]
- 59. Matsui H, Takatsu Y, Kumano S, Matsumoto H, Ohtaki T. Peripheral administration of metastin induces marked gonadotropin release and ovulation in the rat. Biochem Biophys Res Commun. 2004;320:383–388. [DOI] [PubMed] [Google Scholar]
- 60. Shahab M1, Mastronardi C, Seminara SB, Crowley WF, Ojeda SR, Plant TM. Increased hypothalamic GPR54 signaling: a potential mechanism for initiation of puberty in primates. Proc Natl Acad Sci U S A. 2005;102:2129–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Navarro VM, Castellano JM, Fernández Fernández R, et al. Effects of KiSS1 peptide, the natural ligand of GPR54, on follicle-stimulating hormone secretion in the rat. Endocrinology. 2005;146:1689–1697. [DOI] [PubMed] [Google Scholar]
- 62. Sidhoum VF, Chan YM, Lippincott MF, et al. Reversal and relapse of hypogonadotropic hypogonadism: resilience and fragility of the reproductive neuroendocrine system. J Clin Endocrinol Metab. 2014;99:861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Topaloglu AK, Reimann F, Guclu M, et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet. 2009;41:354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gianetti E, Tusset C, Noel SD, et al. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J Clin Endocrinol Metab. 2010;95:2857–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Noel SD, Abreu AP, Xu S, et al. TACR3 mutations disrupt NK3R function through distinct mechanisms in GnRH-deficient patients. FASEB J. 2014;28:1924–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schmidlin F, Roosterman D, Bunnett NW. The third intracellular loop and carboxyl tail of neurokinin 1 and 3 receptors determine interactions with beta-arrestins. Am J Physiol Cell Physiol. 2003;285:C945–C958. [DOI] [PubMed] [Google Scholar]
- 67. Glidewell-Kenney CA, Shao PP, Iyer AK, Grove AM, Meadows JD, Mellon PL. Neurokinin B causes acute GnRH secretion and repression of GnRH transcription in GT1–7 GnRH neurons. Mol Endocrinol. 2013;27:437–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Re M, Pampillo M, Savard M, et al. The human gonadotropin releasing hormone type I receptor is a functional intracellular GPCR expressed on the nuclear membrane. PLoS One. 2010;5:e11489. [DOI] [PMC free article] [PubMed] [Google Scholar]