

Abstract
Cardiovascular disease (CVD) and kidney disease are closely interrelated. Disease of one organ can induce dysfunction of the other, ultimately leading to failure of both. Clinical awareness of synergistic adverse clinical outcomes in patients with coexisting CVD and kidney disease or ‘cardiorenal syndrome (CRS)’ has existed. Renal dysfunction, even mild, is a strong independent predictor for poor prognosis in CVD patients. Developing therapeutic interventions targeting acute kidney injury (AKI) has been limited due mainly to lack of effective tools to accurately detect AKI in a timely manner. Neutrophil gelatinase-associated lipocalin and kidney injury molecule-1 have been recently demonstrated to be potential candidate biomarkers in patients undergoing cardiac surgery. However, further validation of AKI biomarkers is needed in other CVD settings, especially acute decompensated heart failure and acute myocardial infarction where AKI commonly occurs. The other concern with regard to understanding the pathogenesis of renal complications in CVD is that mechanistically oriented studies have been relatively rare. Pre-clininal studies have shown that activation of renal inflammation–fibrosis processes, probably triggered by haemodynamic derangement, underlies CVD-associated renal dysfunction. On the other hand, it is postulated that there still are missing links in the heart–kidney connection in CRS patients who have significant renal dysfunction. At present, non-dialysable protein-bound uraemic toxins (PBUTs) appear to be the main focus in this regard. Evidence of the causal role of PBUTs in CRS has been increasingly demonstrated, mainly focusing on indoxyl sulfate (IS) and p-cresyl sulfate (pCS). Both IS and pCS are derived from colonic microbiotic metabolism of dietary amino acids, and hence the colon has become a target of treatment in addition to efforts to improve dialysis techniques for better removal of PBUTs. Novel therapy targeting the site of toxin production has led to new prospects in early intervention for predialysis patients with chronic kidney disease.
 |
Suree Lekawanvijit is an assistant professor of pathology at Faculty of Medicine, Chiang Mai University, Thailand. She received a PhD scholarship from ‘Prince Doctor Fund’, Thailand, under the Royal Patronage of Her Royal Highness Princess Galyanivadhana. Her research interest area is cardiorenal syndrome focusing on protein-bound uraemic toxins as a missing link in cardiorenal crosstalk and the pathophysiology of renal changes secondary to cardiovascular disease. During her PhD at Centre of Cardiovascular Research & Education in Therapeutics, Monash University, Melbourne, Australia, she with her team leaded by Professor Henry Krum discovered adverse cardiac effects of a uraemic toxin ‘indoxyl sulfate’ and a new application of an existing drug to alleviate uraemic toxin-related cardiac pathology. After resuming her position at Chiang Mai University, she has developed a molecular pathology lab for diagnosis and research and still desires to further explore cardio-renal interrelationship. Her work is currently supported by Ananda Mahidol Foundation (Scholar Support Fund), Thailand Research Fund and Faculty Research Fund. Henry Krum is a recognised leader in cardiovascular therapeutics, particularly in the area of novel cardiovascular drug therapies. This is attested to by his publication record, grant success and invitations to give plenary talks at major international meetings. He is currently Editor-in-Chief, Associate Editor and/or Editorial Board member of many international cardiovascular therapeutics journals and former President of the Asian Pacific Society of Heart Failure. Prof Krum has been Chairman and Executive Committee member of numerous international clinical trials as well as serving on Endpoint Committees and DSMBs for such trials. He was recently appointed Study Chairman of a 7000 patient global heart failure trial. He has in addition led many investigator-initiated trials of cardiovascular therapeutic agents.
Introduction
The common coexistence of cardiovascular disease (CVD) and kidney disease, the so called ‘cardiorenal syndrome (CRS)’ or ‘renocardiac syndrome’, is associated with poor clinical outcomes. A bidirectional heart–kidney interaction in CRS usually leads to accelerated progression of both organ failure. Ronco (2008) has proposed a well-known classification of the CRS by taking the primary diseased organ and the duration of the diseased state defined as either ‘acute’ or ‘chronic’ into account. Type 1 or acute CRS is defined as a rapid worsening of cardiac function, leading to acute kidney injury; type 2 or chronic CRS as chronic abnormalities in cardiac function such as chronic heart failure (HF) causing progressive chronic kidney disease (CKD); type 3 or acute renocardiac syndrome as an abrupt and primary worsening of kidney function such as hypoxic–ischaemic injury, leading to acute cardiac dysfunction such as acute HF, arrhythmia and ischaemia; and type 4 or chronic renocardiac syndrome as a condition of primary CKD contributing to decreased cardiac function, ventricular hypertrophy, diastolic dysfunction, and/or increased risk of adverse cardiovascular (CV) events. An additional subtype of CRS or type 5 or secondary CRS is defined as the presence of combined cardiac and renal dysfunction due to acute or chronic systemic disorders such as sepsis.
Despite advanced progress in management of both CVD and kidney disease, specific interventions for CRS are warranted since CRS is still a burden on global health. Most patients with significant kidney disease have specifically been excluded from most clinical trials on CV interventions, although CV mortality is predominantly responsible for the death of patients with CRS, irrespective of the primary affected organ.
In CRS when the heart is the primary diseased organ, especially in type 1 CRS, acute kidney injury (AKI) is highly incident and predictive of poor clinical outcomes in many serious CV conditions such as cardiac surgery, acute decompensated heart failure (ADHF) and acute myocardial infarction (MI). AKI needs timely diagnosis and management but there currently appear to be no satisfactorily effective tools. Clinical evaluation of AKI still depends on laboratory measurement of serum creatinine concentration. In the past, the major problem of defining AKI was lack of a standard definition, but two serum creatinine-based consensus criteria, the RIFLE (Bellomo et al. 2004) and the Acute Kidney Injury Network or AKIN (Mehta et al. 2007) criteria, were established in 2002 and 2005, respectively, and have been increasingly utilized for AKI diagnosis. However, serum creatinine is not a sensitive marker for detecting minor renal parenchymal damage. Recently, two large scale studies (n = 2322 and 1635) in critically ill patients, predominantly cardiac surgery patients (n = 1452), demonstrated that up to one-fifth of the cohorts having subclinical AKI detected by novel kidney injury biomarkers (more specific to intrinsic AKI) but not fulfilling the RIFLE criteria (more specific to prerenal AKI or severe degree of renal parenchymal damage) are associated with a significantly increased risk for renal replacement therapy and in-hospital mortality (Haase et al. 2011; Nickolas et al. 2012). Many candidate biomarkers for early detection of AKI have been demonstrated in the past decade and have been proposed for incorporation into future updated AKI criteria. However, the performance of individual biomarkers that can be released from other than renal sources and that have a different characteristic metabolism should be validated in any specific CV conditions at high risk of AKI before becoming widely used. In common with AKI detection, mechanistically oriented studies of renal changes secondary to CVD are extremely rare, despite clearly observed clinical evidence of its negative impact.
In CRS when the kidney is the primary diseased organ, especially type 4 CRS, CKD has become a major concern worldwide due to its increasing incidence as well as the associated high CV morbidity and mortality. There are several known CKD-specific/related, non-traditional CV risk factors underlying CV complications in the CKD population such as anaemia, hypertension and abnormal calcium–phosphorus homeostasis; however CV death is still high despite such risk factors being controlled. This suggests that there are still missing links. Protein-bound uremic toxins (PBUTs) are currently the main focus of efforts to find these links due to recently emerging evidence regarding their causal role in the pathogenesis of CRS.
Collectively, at this stage there appear to be several knowledge gaps with regard to CRS that need to be investigated further. These include: (1) lack of effective tools for early detection of AKI in specific CV conditions, (2) incomplete understanding of the pathophysiology of renal changes secondary to CVD, and (3) missing links between heart–kidney crosstalk in CKD patients.
Renal complications in patients with cardiovascular disease
AKI, which frequently accompanies CVD, is one of the strongest predictors of mortality in patients with either MI or with HF. Renal dysfunction, even mild, is associated with increased risk for long-term mortality (Parikh et al. 2008). AKI occurs in 10–20% of hospitalized patients with acute MI (Parikh et al. 2008). Approximately 24–45% of these MI patients with AKI die during hospitalization, a rate that is 4.4–8.8 times higher than those without AKI (Goldberg et al. 2005, 2009). Importantly, decline in renal function post MI tends to progress with time (Hillege et al. 2003).
The incidence of AKI, defined according to the AKIN criteria as increased serum creatinine >0.3 mg dl−1, is 27% in patients hospitalized for worsening HF or ADHF. In HF patients, 21–43% have concomitant renal dysfunction, defined as an estimated glomerular filtration rate (eGFR) or estimated creatinine clearance <60 ml min−1 (equivalent to CKD stage 3 onwards) (Weinfeld et al. 1999; Dries et al. 2000; Shlipak, 2003; Hillege et al. 2006). Absolute serum creatinine levels of greater than 1.5 mg dl−1 have been demonstrated as a risk predictor in HF patients (McClellan et al. 2002; Akhter et al. 2004). One study has reported that every increase in serum creatinine of 0.5 mg dl−1 is associated with a more than 10–15% increased risk of mortality in these patients (Smith et al. 2005).
Common risk factors for AKI in CVD patients include history of MI, angina pectoris, stroke and HF, hypertension, diabetes mellitus, advanced New York Heart Association (NYHA) functional class, previous hospitalizations for HF, old age and male gender (Dries et al. 2000; Forman et al. 2004; Adams et al. 2005; Hillege et al. 2006; Santolucito et al. 2010). Notably, approximately three-quarters of patients with acute MI or ADHF develop AKI within 72 h of admission (Gottlieb et al. 2002; Goldberg et al. 2005; Liang et al. 2008). This is in agreement with the AKIN criteria, indicating that an abrupt decline in renal function within 48 h is associated with adverse outcomes (Mehta et al. 2007).
Although the adverse impact of renal impairment is clear, there are still no detailed management guidelines specific to the CVD population with renal insufficiency. There are three reasons for this. First, 90% of major CV trials fail to provide adequate information on kidney function and only 3% have performed subgroup analysis of the intervention effects stratified by renal function (Coca et al. 2006). Furthermore, approximately 60% of major clinical trials of CVD specifically exclude patients with kidney disease. Second, given that the clinical manifestations of renal insufficiency are a late phenomenon of kidney damage and change in creatinine levels may be influenced by factors other than kidney damage, more accurate and timely detection of kidney injury by biomarkers with better sensitivity and specificity could lead to progression in developing timely therapeutic interventions to combat AKI. Last, there is still much to discover about the basic pathophysiological mechanisms of renal insufficiency secondary to CVD that link to the clinical manifestations.
Acute kidney injury biomarkers in patients with cardiovascular disease
AKI is classified into two types, intrinsic and prerenal. Intrinsic AKI is associated with injured renal cells which may abnormally secrete cellular substances into the urine (such as neutrophil gelatinase-associated lipocalin (NGAL), kidney injury molecule (KIM)-1, interleukin (IL)-18, N-acetyl-β-d-glucodaminidase (NAG) and liver fatty acid-binding protein (LFABP)) or lose the ability to reabsorb filtered substances such as cystatin C. Urinary concentrations of these substances are therefore potential candidate biomarkers for early detection of intrinsic AKI, and may also reflect the degree of renal parenchymal damage. Prerenal AKI is associated with reduced glomerular filtration, mostly contributory to a decompensation response of the kidney to haemodynamic derangements such as reduced renal blood flow and increased venous pressure. Biomarkers for detecting prerenal AKI are usually circulating substances such as creatinine and cystatin C which ideally should be freely filtered through the glomeruli without renal tubular excretion. Increased circulating levels of these substances may reflect renal damage if the degree of damage is high enough to cause defective glomerular filtration.
Serum creatinine is a glomerular filtration marker (but not an ideal one as it is also excreted from the renal tubule) traditionally used to evaluate renal function. However, there has been growing evidence that it is not sensitive and specific enough to detect early renal parenchymal damage/injury. Elevation of serum creatinine levels above the ‘normal range’ occurs after a substantial amount of functioning kidney tissue has been lost. Minor effects on renal parenchyma are difficult to detect due to the functional reserve capacity of the kidney. Furthermore, serum creatinine is not an accurate marker of glomerular filtration rate (GFR) in the non-steady-state condition of AKI (Thadhani et al. 1996). In most CV studies, an increase in serum creatinine >0.3 mg dl−1 is widely used as the criteria for diagnosis of AKI, and this increase has been demonstrated to have satisfactory sensitivity (81%) and specificity (62%) to predict mortality in patients hospitalized for HF (Gottlieb et al. 2002). A study in ADHF patients demonstrated that decline in estimated GFR (eGFR), defined as a >20% reduction of eGFR calculated by the Modified Diet and Renal Disease equation, and a >0.3 mg dl−1 increased serum creatinine level are both predictive of mortality. However, mortality prediction of an increase in creatinine, but not a decline in eGFR, is dependent of baseline renal function (Testani et al. 2010). This study has raised concern about the interpretation of change in creatinine levels in heterogenous populations with a wide range of baseline renal function. In fact, both serum creatinine and creatinine-based eGFR are affected by many confounding factors such as age, gender, race, sex, protein intake, muscle mass and underlying illness.
Serum cystatin C is also a glomerular filtration marker. Cystatin C is produced from all nucleated cells at a constant rate (Filler et al. 2005). It is solely and freely filtered through the glomeruli without tubular excretion and then completely reabsorbed and catabolized by the proximal tubular cells, thereby normally not being excreted into the urine and not returning to the circulation. Thus, circulating cystatin C appears to be a more ideal and reliable filtration marker than serum creatinine. Compared to serum creatinine, serum cystatin C has been demonstrated to detect AKI 6 h to 2 days earlier in various settings (Herget-Rosenthal et al. 2004; Nejat et al. 2010), including acute MI (Fouad & Boraie, 2013), and to be more strongly predictive of mortality and CV events in the elderly (Shlipak et al. 2005). Serum cystatin C levels are associated with left ventricular hypertrophy (LVH) and diastolic dysfunction in patients with known coronary artery disease (Ix et al. 2006). In addition, cystatin C is less likely to be affected by age, gender, height and muscle mass than creatinine (Filler et al. 2005) but its circulating levels may be altered in the inflammatory state (Keller et al. 2007). Estimating GFR from serum cystatin C has been demonstrated to be at least equally as accurate (Tidman et al. 2008) as creatinine-based eGFR estimation, but superior in specific patient groups such as children, the elderly, and patients with reduced muscle mass (Filler et al. 2005). However, cystatin C level is more expensive to measure than creatinine and is not normally available; in addition it is crucial to standardize the test and to validate the cystatin C-based eGFR equation before it becomes widely used (Tidman et al. 2008). It should also be noted that use of serum cystatin C for detecting AKI in the setting of CRS needs special attention because high cystatin C levels have been significantly observed in atherosclerosis-associated CVD, independently of GFR (Salgado et al. 2013).
Urinary concentrations of biomarkers directly released from injured renal cells into the urine are likely to be more sensitive and specific for early detection of intrinsic AKI, as well as quantifying the degree of kidney injury, mainly specific to tubular damage, compared to the filtration markers. Most are enzymes or intracellular or transmembrane proteins. Potential biomarkers include NGAL, KIM-1, IL-18, NAG and LFABP. Given that filtered cystatin C is not normally present in the urine, urinary cystatin C may be also considered as a biomarker for early renal (proximal tubule) dysfunction/injury. Elevated urinary cystatin C levels have been demonstrated to differentiate intrinsic AKI from prerenal AKI and have a graded association with AKI severity (Park et al. 2013b), whilst serum cystatin C can better distinguish between AKI and non-AKI (Soto et al. 2010). Among biomarkers of intrinsic AKI, KIM-1 appears to be the only biomarker solely originating from renal proximal tubular cells whilst cystatin C originates from all nucleated cells; however, filtered cystatin C is reabsorbed by tubular cells as previously mentioned. NGAL, NAG and LFABP are also produced from specific sources other than renal tubular cells such as leukocytes (NGAL), liver cells (NAG, LFABP) and brain (NAG) (Vanmassenhove et al. 2013). IL-18 is mainly derived from monocytes/macrophages but can be secreted by epithelial cells including renal proximal tubular cells (Charlton et al. 2014).
Study of these novel AKI biomarkers, predominantly utilizing their urinary concentrations, in CRS is usually conducted in specific CV conditions as discussed below.
Cardiac surgery
Post-cardiac surgery appears to be the most common CV setting in which biomarkers of AKI have been investigated. The incidence of post-operative AKI defined by the RIFLE criteria is 15–23% in adults and 35–49% in children undergoing cardiac surgery (Haase et al. 2011). NGAL and KIM-1 are the two biomarkers which perform best as an AKI diagnostic marker after cardiac surgery compared to cystatin C, IL-18, NAG and LFABP (Thurman & Parikh, 2008; McIlroy et al. 2010), where AKI is defined by a 50% increase in serum creatinine. Urinary, and serum to a lesser extent, NGAL has high accuracy early post-cardiac surgery, especially at 2–4 h, and sharply declines after 12 h, whilst urinary KIM-1 has high accuracy after 12 h. NGAL is particularly advantageous in pediatric cardiac surgery patients for highly accurate prediction of AKI. Urinary NGAL performs better than serum NGAL in most studies (Mishra et al. 2005; Koyner et al. 2008; Tuladhar et al. 2009; Parikh et al. 2011b), but this observation is less pronounced (Parikh et al. 2011a) or even reversed (Koyner et al. 2012) in some studies. Interestingly, elevated urine and/or plasma NGAL levels in the absence of AKI diagnosed by RIFLE criteria, so-called subclinical AKI, is associated with a 16.4 times higher risk of subsequent renal replacement therapy compared to normal NGAL levels in a study of 2322 critically ill patients, predominantly (63%) cardiac surgery patients (Haase et al. 2011).
Whether a marked increase in circulating NGAL concentration post-cardiac surgery is mainly due to back-leakage from renal tubular reabsorption of urinary NGAL is not known. To our knowledge, none of studies investigating both circulating and urinary NGAL concentrations in cardiac surgery populations report a correlation between both parameters (Mishra et al. 2005; Koyner et al. 2008, 2012; Tuladhar et al. 2009; Krawczeski et al. 2011; Parikh et al. 2011a). Interestingly, the duration of cardiopulmonary bypass during cardiac surgery is not only a risk factor for post-operative AKI (Mishra et al. 2005; Parikh et al. 2011a,b2011b; Koyner et al. 2012) but also induces leukocyte activation (Edmunds, 1998). Activated leukocytes can be a source of massive NGAL release into the circulation which may have effects (yet unknown) on the performance of serum NGAL in post-cardiac surgery AKI prediction.
ADHF
In contrast to cardiac surgery patients, the use of urinary NGAL in early detection of AKI does not appear to be very promising in patients with ADHF. One study reported that both urine and serum NGAL levels are predictive of AKI defined as a serum creatinine increase >0.3 mg dl−1 within 5 monitoring days in hospitalized ADHF patients (n = 93) (Shrestha et al. 2012). A modest correlation between urinary and serum NGAL levels has been demonstrated and high urinary NGAL levels are associated with impaired natriuresis and diuresis during the 24 h period after initiation of diuretic therapy, reflecting distal tubular injury/dysfunction in ADHF patients (Shrestha et al. 2012). Another study (n = 141) demonstrated a significant difference in urinary NGAL levels between ADHF patients with and without AKI only on day 3, which is later than detection by the AKIN criteria (Dupont et al. 2012). However, a marginal rise in urinary NGAL preceding AKI was observed.
No association between urinary NGAL and KIM-1 concentrations and recovery from AKI has been recently reported (Park et al. 2013a). A further study in 83 patients hospitalized for ADHF showed that urinary NGAL, KIM-1 and IL-18 are all inadequately sensitive/specific to predict AKI when AKI is defined as a ≥25% decrease in eGFR during days 1–5 (Verbrugge et al. 2013). In contrast, urinary IL-18 is predictive of persistent renal impairment (defined as persistently increased serum creatinine >0.3 mg dl−1 for 6 months) and all-cause mortality (average follow-up 7 ± 4 months) (Verbrugge et al. 2013).
With the limited number of studies that utilize a variety of referenced AKI criteria and tested biomarkers, currently available data appear insufficient for pooled analysis of (intrinsic AKI) biomarker performance in ADHF patients. Notably, urine concentrations of NGAL, the most extensively investigated biomarker, seem to be only slightly higher in ADHF patients with AKI than in healthy subjects but rather low compared with AKI due to other causes such as post-cardiac surgery. Therefore, considering the reported performance of intrinsic AKI biomarkers in these patients, it is suggesting that prerenal AKI may be predominant in ADHF and possibly related to either reduced renal blood flow or increased systemic venous pressure. In addition, baseline urinary and serum NGAL levels in ADHF patients can be increased in association with common co-morbidities such as diabetes, hypertension and old age (Shrestha et al. 2012).
Chronic HF
Studies on kidney injury biomarkers in patients with stable chronic HF mainly evaluate their ability to predict clinical outcomes rather than AKI. Urinary NAG, KIM-1 and NGAL concentrations are increased in chronic HF patients (Damman et al. 2010). One study (n = 2130) demonstrated an independent association between these biomarkers and the composite of all-cause mortality and HF hospitalization (Damman et al. 2011), while two smaller-scale studies reported this association for urinary KIM-1 and NAG, but not NGAL (Damman et al. 2010; Jungbauer et al. 2011). High urinary KIM-1 and NAG concentrations are also significantly correlated with decreased LV function and worsened NYHA functional class (Jungbauer et al. 2011). In addition, one study demonstrated a significant elevation of serum NGAL concentrations in stable HF patients (n = 150) with normal serum creatinine levels and an increase in NGAL protein expression within failing myocardial tissue, suggesting that increased serum NGAL concentrations may not only reflect impaired renal function in chronic HF (Yndestad et al. 2009). Only NAG is correlated with measured GFR ([125I]iothalamate) and effective renal plasma flow (Damman et al. 2010).
MI
To our knowledge, there have been no clinical studies comparing performance on AKI prediction among novel kidney injury biomarkers in patients with acute MI. However, a few studies have examined kidney injury biomarkers in post-MI animal models. An in vivo time course (3 days and 4 and 8 weeks) study in a rat MI model demonstrated a significant increase in serum and urinary NGAL at day 3 and week 2, but not at weeks 4 and 8, post MI (Cho et al. 2013). However, a significant increase in circulating activated monocytes observed on day 3 post MI, as well as an inflammatory reaction in infarcted myocardium, may be also sources of serum NGAL (Cho et al. 2013). In a different time course in vivo (1, 4, 8, 12 and 16 weeks) MI study, renal KIM-1 protein expression significantly increased at 1 week post MI. KIM-1 expression declined at 4 weeks and then gradually increased to reach significance at 12 and 16 weeks in association with development of renal fibrosis (Lekawanvijit et al. 2012c). An increase in renal tubular KIM-1 protein expression (1 and 12–16 weeks) occurred earlier but was associated with a biphasic pattern of reduced GFR post MI (1–4 weeks and 16 weeks). Interestingly, an obvious increase in renal KIM-1 expression at 1 week post MI was associated with a significant decrease in systolic blood pressure (BP) and measured GFR, suggesting that KIM-1 may reflect a contribution by haemodynamic derangements post acute MI to renal tubular injury. An association between renal dysfunction and increased renal KIM-1 expression has been confirmed in a further 16 week post-MI animal study (Lekawanvijit et al. 2013). A significant increase in both urinary NGAL and KIM-1 concentrations measured at day 5 post acute MI has been demonstrated in a rat MI model which reverted to the sham level on follow-up day 30 (Entin-Meer et al. 2012). In the same study, a significantly higher-than-sham level of both urinary biomarkers was persistently observed in a rat CKD model until the study endpoint (9 weeks post subtotal nephrectomy) but was significantly less than the level on day 5 post insult, suggesting that urinary NGAL and KIM-1 may be useful in assessing renal damage in the CKD setting.
The currently used criteria for AKI were initially established based on simple markers for the prime purpose of generating a universal consensus, despite awareness of their clinical weakness. Increasing evidence of novel early kidney injury biomarkers is promising, especially in AKI caused by primary kidney disease. However, use of these kidney injury biomarkers for early detection of AKI in common CV conditions needs to be further validated.
Pre-clinical study of renal changes following cardiovascular disease
Pre-clinical study of the mechanisms of renal impairment secondary to CVD is mainly conducted in an experimental model of either cardiac dysfunction alone, for investigating incipient renal changes, or combined cardiorenal dysfunction, for investigating further worsening of the kidney.
Study of incipient renal changes is mostly conducted in a rat MI model induced by left anterior descending coronary artery ligation. Renal interstitial fibrosis with a mild reduction in GFR has been demonstrated in association with systolic dysfunction without changes in BP at 3 weeks post MI (Martin et al. 2007). Significant renal gene dysregulation related to cell proliferation, metabolic processes, and cell communication was detected by microarray analysis. The mechanisms underlying such changes, however, were not described.
A time course study of post-MI renal changes was systematically investigated at 1, 4, 8, 12 and 16 weeks in a rat MI model (Lekawanvijit et al. 2012c). Renal dysfunction determined by measured GFR was observed in a biphasic pattern, initially at 1 and 4 weeks and again at 16 weeks, with a function restored in between. Reduced GFR at 1 week post MI was associated with a significant decrease in systolic BP, suggesting a contributing role of haemodynamic disturbance to early renal dysfunction. Inflammatory activation in the kidney was detected at 1 week by an increase in renal macrophage infiltration, IL-6 mRNA expression and transforming growth factor-β (TGF-β) protein expression. Renal interstitial fibrosis, predominantly at the cortex-to-corticomedullary junction area, mediated by the Smad-associated pathway, was demonstrated at 4 weeks onwards, reaching a maximum at 16 weeks when the second decline in GFR was observed, suggesting that progressive renal fibrosis may contribute to late renal dysfunction. Progressively increased albuminuria following MI despite no significant difference from sham levels may reflect intrinsic renal injury. Similarly, early decline in renal function (3 days post MI), in association with decreased systolic BP and increased renal expression of pro-inflammatory cytokines (TGF-β, IL-1β and vascular cell adhesion molecule 1), followed by progressive renal fibrosis, has been demonstrated in a 3 week MI mouse study (Lu et al. 2012).
A recent time-course (3 days and 4 and 8 weeks) MI rat study has demonstrated a significant decrease in systolic BP 1 day post MI and a significant increase in circulating activated monocytes on day 3 post MI, accompanied by an increase in renal macrophage infiltration, renal expression of nuclear factor-κB (NF-κB) and renal inflammatory cytokine gene (IL-1, IL-6 and tumour necrosis factor-α) expression. Renal interstitial fibrosis, mainly at the corticomedullary junction was observed at 4 and 8 weeks post MI that was significantly reversed by use of liposomal clodronate to deplete circulating activated monocytes (Cho et al. 2013). Interestingly, post-MI (3 and 9 weeks) renal inflammation and tubular necrosis and fibrosis in association with increased blood cystatin C levels are all attenuated by the angiotensin II type 1 receptor blocker losartan (Wen et al. 2013).
Whether infarct size, usually determined by a fraction of the infarcted and non-infarcted endocardial and epicardial lengths at mid-left ventricular cross-section and expressed as a percentage, has effects on timing of incipient renal changes is not clear. With an equal average MI size of 34%, Windt et al. (2008) did not see any renal structural pathology while Bongartz et al. (2012) reported mild glomerulosclerosis at 6 weeks in post-MI rats. The 1, 4, 8, 12 and 16 weeks time-course study referred to above, with larger average infarct sizes of 46, 44, 45, 42 and 41%, respectively, reported significant tubulointerstitial inflammation or fibrosis at all time points post MI (Lekawanvijit et al. 2012c). In contrast, 12 weeks post-MI rats with a large average MI size of 54.2% demonstrated significant renal dysfunction without significant renal structural changes in both glomeruli and tubulointerstitium (Bauersachs et al. 2000), whereas renal structural change only, without renal dysfunction, was observed in 12 and 14 weeks post-MI rats with smaller infarct sizes (42% and 35%) (Lekawanvijit et al. 2012c; Liu et al. 2013). However, one study with a modest MI size (18.1%) showed significant renal dysfunction in 16 weeks post-MI rats, despite using plasma creatinine levels for assessment, in association with significant renal interstitial damage and an increased level of glomerular macrophage infiltration (van Dokkum et al. 2004). Methods of renal structural assessment vary across studies, from mainly semiquantitative assessment (Bauersachs et al. 2000; Windt et al. 2008; Bongartz et al. 2012), to quantitative assessment and digital image analysis (van Dokkum et al. 2004; Lekawanvijit et al. 2012c; Liu et al. 2013). Renal changes following MI are generally minimal and therefore are likely to be more sensitive to detection by quantitative methods.
In summary, early renal dysfunction post MI can be reversed due at least in part to transient haemodynamic derangement (decreased systolic BP). Whilst this transient functional impairment is probably a reflection of prerenal AKI, the process of intrinsic renal damage associated with inflammatory activation occurs simultaneously and tends to be persistent and to progress to fibrogenesis. Renal structural changes following acute MI are predominantly located around the corticomedullary junction, the region most vulnerable to ischaemic injury due to reduced renal blood flow. Renal fibrosis tends to progress over time, ultimately contributing to late renal dysfunction. Renal inflammation and fibrosis might be mediated via the TGF-β–Smad–NF-κB pathway in association with activation of the renin–angiotensin–aldosterone system (RAAS). Thus, interventions with an anti-inflammatory effect or RAAS blockade may prevent progression of renal fibrosis and late renal dysfunction.
Further renal changes following CV events superimposed on existing CKD have also been investigated in combined cardiorenal dysfunction models mostly induced by either unilateral or 5/6-subtotal nephrectomy (5/6-STNx) followed by left anterior descending coronary artery ligation. These models represent all stages of CKD patients who are considered at high risk for CV death irrespective of the severity of renal dysfunction (Levey, 1998).
In unilaterally nephrectomized rats, MI further impairs renal function, determined by plasma creatinine and proteinuria, and renal structural changes including glomerular inflammation, sclerosis and interstitial damage, compared with unilaterally nephrectomized rats without MI (van Dokkum et al. 2004). Larger infarct size is associated with a higher degree of additional proteinuria and sclerotic glomeruli. Another study demonstrated that the rapidly progressive proteinuria and striking renal interstitial injury (mainly confined to the cortex and outer medulla) observed in rats undergoing unilateral nephrectomy plus MI is mediated via an angiotensin II type 1a receptor and alleviated by blockade of this receptor (Homma et al. 2012).
In a more severe renal failure model of 5/6-STNx, superimposed MI results in a significant decrease in systolic BP from 2 weeks post MI onwards but does not further reduce creatinine clearance (Bongartz et al. 2012) Compared with 5/6-STNx alone, in rats with 5/6-STNx + MI only worsened glomerulosclerosis and lower urinary excretion of nitric oxide metabolites were observed in this study (Bongartz et al. 2012). By contrast, another study showed a further decline in creatinine clearance without further renal damage (proteinuria and glomerulosclerosis), and the renoprotective effects of angiotensin-converting enzyme inhibitor observed in 5/6-STNx rats did not have additional beneficial renal effects in 5/6-STNx + MI animals (Windt et al. 2008). Both studies, Bongartz et al. (2012) and Windt et al. (2008), have comparable follow-up periods (both 6 weeks post MI), infarct size (34% vs. 39%) and renal structural assessment based mainly on semiquantitative methods. The only obvious difference is the interval between 5/6-STNx and MI surgery. In the former study with MI 2 weeks after 5/6-STNx, smaller renal changes secondary to superimposed MI is possibly obscured by massive renal damage from 5/6-STNx which may not have developed fully within 2 weeks, but may be evident in the latter study with a 9 week interval between the two insults.
Cardiovascular complications in patients with kidney disease: uraemic toxins as a potential ‘missing link’ in cardiorenal connection
A high prevalence of CVD is responsible for approximately half of all deaths in the renal failure population (USRDS, 2007). CV mortality is 10–30 times higher in dialysis patients than in the general population (Sarnak et al. 2003). In addition, CKD patients have a greater risk of CV events and mortality than progression to end-stage renal disease (ESRD) (Keith et al. 2004). In the CKD population, HF is the most common clinical presentation of CVD whilst left ventricular hypertrophy (LVH), a surrogate of uraemic cardiomyopathy, is predictive of CV mortality. Sudden cardiac death, HF and MI are the three most common causes of cardiac death (USRDS, 2012). Despite the high incidence of accelerated atherosclerosis and MI, as well as high mortality post acute MI (Lindner et al. 1974), only 15–25% of cardiac deaths in the CKD population are attributable to ischaemic heart disease, half of which are angiographically negative for significant coronary atherosclerosis (Rostand et al. 1984). Cardiac interstitial fibrosis (a crucial component of uraemic cardiomyopathy) and non-obstructive vascular diseases (such as vascular stiffness, calcification and ossification) are the predominant CV pathology in CKD patients; both are independent of hypertension (Amann et al. 1995; Glassock et al. 2009) and may contribute to the high incidence of sudden cardiac death and ischaemic heart disease in the absence of significant coronary atherosclerosis. Importantly, traditional CV risk factors are insufficient to explain the extraordinarily high prevalence of CVD in the CKD population. CKD-specific/related risk factors such as anaemia, hypertension and abnormal calcium–phosphorus homeostasis have been demonstrated to be closely associated with CV pathology and mortality; however death due to CV complications is still predominant even when these risk factors have been corrected. This strongly suggests that there are still ‘missing links’ in the connection between heart and kidney (Fig. 1) in the setting of CRS.
Figure 1. Pathophysiology of cardiorenal syndrome.
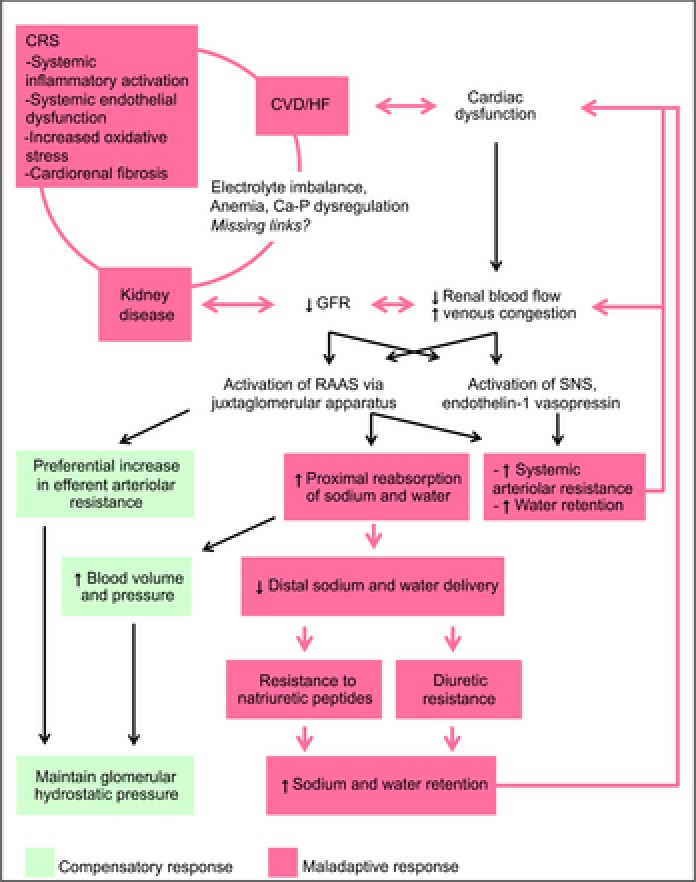
CRS, cardiorenal syndrome; CVD, cardiovascular disease; HF, heart failure; GFR, glomerular filtration rate; Ca-P, calcium and phosphorus; RAAS, renin–angiotensin–aldosterone system; SNS, sympathetic nervous system.
Despite tremendous advances in the development of dialysis technology, CV mortality is still unacceptably high in dialysis patients compared with renal transplantation patients (USRDS, 2012). Whilst LVH gradually regresses after transplantation (Gross & Ritz, 2008), progression of LVH persists in long-term haemodialysis patients (London et al. 2001; Zoccali et al. 2004) even if hypertension and anaemia are controlled (London et al. 2001). Such collective data have drawn attention to non-dialysable uraemic toxins as a potential further link in kidney–heart interactions. Protein-bound uraemic toxins (PBUTs) are currently the main focus due to recent emerging evidence regarding their causal role in the pathogenesis of CRS (for review, see Lekawanvijit et al. 2012b).
Role of protein-bound uraemic toxins in CRS
Indoxyl sulfate (IS) and p-cresyl sulfate (pCS) are the two PBUTs most extensively studied with regard to their contribution to the pathogenesis and progression of CRS. Both are associated with renal progression, as well as increased CV and all-cause mortality (Barreto et al. 2009; Liabeuf et al. 2010; Wu et al. 2011, 2012). Pre-clinical studies have demonstrated their causative effects on CV and renal pathology, which include renal inflammation and fibrosis, increased cardiac protein and collagen synthesis and endothelial dysfunction (Dou et al. 2007; Meijers et al. 2009; Lekawanvijit et al. 2012b; Neirynck et al. 2012). IS is implicated in vascular calcification and ossification (Muteliefu et al. 2009; Adijiang et al. 2010) which is commonly found in advanced-stage CKD patients and is associated with poor CV outcomes (Aoki et al. 2005; Guerin et al. 2006). In addition, IS also enhances oxidative stress in kidney (Owada et al. 2008; Tumur & Niwa, 2008), heart (Fujii et al. 2009; Lekawanvijit et al. 2012a) and endothelial cells (Dou et al. 2007; Yu et al. 2011). Cardiorenal fibrosis induced by IS is potentially mediated via the ROS–NF-κB–TGF-β1 pathway. Recently, serum IS levels have been demonstrated in vivo to be positively correlated with renal KIM-1 expression in a chronic MI model, with consequent renal dysfunction and fibrosis (Lekawanvijit et al. 2013). However, no reports of direct evidence of renal and/or CV dysfunction induced by PBUTs have yet been published. Most studies on PBUTs in CRS merely document an association.
Serum IS, but not pCS, levels have recently been demonstrated to be independently correlated with serum fibroblast growth factor 23 (FGF23) levels in stages 3–5 CKD patients (Lin et al. 2013). Elevated serum levels of FGF23 are usually observed in CKD patients. FGF23 normally acts on the kidney and parathyroid gland through FGF receptors in the presence of Klotho co-receptors to enhance phosphaturia, to inhibit renal 1-α hydroxylase, thereby reducing vitamin D production, and to suppress parathyroid hormone secretion. A decline in Klotho expression usually observed with the progression of CKD contributes to a compensatory elevation of FGF23 levels; however, FGF23 fails to regulate phosphorus, vitamin D and parathyroid hormone without Klotho (Kuro-o, 2012), leading to typical CKD manifestations such as hyperphosphataemia, secondary hyperparathyroidism and bone disease. Elevated serum FGF23 levels are associated with not only progression to ESRD (Isakova et al. 2011) but also increasing risk of new-onset LVH and mortality in predialysis CKD patients (Faul et al. 2011). Although FGF23-associated CV pathology in CKD can be explained by cardiovascular toxicity due to hyperphosphataemia (Amann et al. 2003) and hyperparathyroidism (Gross & Ritz, 2008), FGF23 itself has been demonstrated in vivo and in vitro to directly induce LVH through the FGF receptor-dependent calcineurin–NFAT signalling pathway which is independent of Klotho co-receptors (Faul et al. 2011). This indicates that the normal mineral regulatory function of FGF23 shifts towards cardiovascular toxicity in the event of Klotho deficiency.
Whether IS and FGF23 are mechanistically interrelated in the pathophysiology of CRS is not known. However, recent data indicate that PBUTs are implicated in Klotho deficiency. Exogenous IS and pCS can directly induce renal fibrosis, suppress renal Klotho protein and mRNA expression, and promote DNA hypermethylation of the Klotho gene in association with increased expression of DNA methyltransferase 1 in the absence of a reduction in GFR in a mildly function-compromised CKD model (Sun et al. 2012a). Reduced Klotho protein expression by IS or pCS is prevented by administration of an oral adsorbent, AST-120 (Adijiang & Niwa, 2010). ROS and NF-κB have been demonstrated to be involved in the mechanism of IS-reduced Klotho protein and mRNA expression in the kidney (Shimizu et al. 2011). This suggests that Klotho might be involved in PBUT-induced renal fibrosis mediated via the ROS–NF-κB–TGF-β1 pathway. In addition, renal fibrosis induced by IS or pCS is associated with activation of the renal RAAS and epithelial-to-mesenchymal transition of renal tubular cells. Inhibition with losartan significantly reduces renal fibrosis, suppresses activation of the Smad-dependent TFG-β1 pathway and restores the epithelial phenotype of renal tubular cells in mice with unilateral nephrectomy treated with either IS or pCS (Sun et al. 2012b). Thus, the potential pathway of PBUT-induced (more specific to IS) renal fibrosis is likely to be the ROS–NF-κB–Klotho/Smad-dependent TGF-β1 pathway. Further investigation of the effects of RAAS blockade on PBUT-associated CV pathology is needed, which could be performed in cultured cardiac cells stimulated with PBUTs, experimental CRS models, or in CRS patients with established renal dysfunction by analysis of CV endpoints associated with IS concentrations.
Klotho deficiency also plays a role in IS-induced endothelial cell dysfunction. Increased ROS production, reduced nitric oxide (NO) and decreased cell viability observed in endothelial cells stimulated with IS are attenuated by Klotho protein (Yang et al. 2012). Recently, aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor mediating cellular adaptive and toxic responses, has been demonstrated to be activated by IS (Schroeder et al. 2010). Stimulation of human umbilical vein endothelial cells with IS results in enhanced oxidative stress and increased expression of AhR target genes and monocyte chemoattractant protein-1 (MCP-1) (Watanabe et al. 2013). Administration of AhR antagonists inhibits the IS-induced increase in MCP-1 expression in a dose dependent manner (Watanabe et al. 2013).
Both IS and pCS are derived from colonic microbiotic metabolism of dietary amino acids tryptophan and tyrosine, respectively (Aronov et al. 2011). Novel therapy targeting the site of toxin production has opened the prospect of reducing the levels of such PBUTs in predialysis CKD patients. Given that CKD patients are at greater risk of CV mortality than progression to dialysis-dependent ESRD (Keith et al. 2004), such interventions may be beneficial in improving IS/pCS-associated CV pathology and outcomes. Strategies include low protein diet, probiotic and prebiotic treatment and use of oral adsorbents. Trials of probiotic and/or prebiotic treatments to maintain normal colonic microbiotic homeostasis demonstrate promising results in improving renal function and quality of life in predialysis CKD patients (Ranganathan et al. 2010). However, beneficial CV endpoints have never been studied. A strict low protein diet decreases serum IS levels in predialysis CKD patients (Marzocco et al. 2013) but has raised concerns regarding protein malnutrition in this setting.
The therapeutic role of an oral adsorbent, AST-120, in reducing colon-derived PBUTs has been widely investigated. Renoprotective effects of AST-120 are observed at various stages of CKD in both CKD patients and in experimental CKD models, mostly demonstrated in association with an IS-lowering effect. A multicentre, randomized, controlled trial has demonstrated a beneficial effect of AST-120 on preservation of renal function in predialysis CKD patients (n = 460) (Akizawa et al. 2009), although there was no difference in incipient dialysis or survival between treated and control groups due mainly to infrequent events over the 1 year treatment duration. Preliminary results from two large-scale trials (Evaluating Prevention of Progression In Chronic Kidney Disease, EPPIC) showed no benefits on study endpoints comprising times to initiation of dialysis, doubling serum creatinine levels and death; however, a reduction in CKD progression was observed in high risk (proteinuria and haematuria) patients with good compliance (Schulman et al. 2014). The potential benefit of AST-120 on CV protection has recently been demonstrated, for example, in preventing cardiac fibrosis in CKD rats (Fujii et al. 2009; Lekawanvijit et al. 2012a) and limiting or stabilizing atherosclerotic plaque in CKD mice with apolipoprotein E deficiency (Yamamoto et al. 2011). In predialysis CKD patients, AST-120 improves parameters of endothelial dysfunction (Yu et al. 2011), vascular stiffness (Nakamura et al. 2004) and calcification (Goto et al. 2013), and may be beneficial in delaying progression of LVH (Nakai et al. 2011). Studies of the effects of AST-120 on both CV and renal endpoints are, however, very rare.
Conclusions
Renal impairment is a strong independent risk factor associated with poor prognosis in CVD patients. Renal dysfunction is probably caused by progressive renal structural damage. Accurate detection of kidney parenchymal injury in a timely manner, as well as increased knowledge of the pathophysiology and mechanisms underlying this injury, is of great importance in developing therapeutic interventions for combating renal complications at an early stage.
Regarding the role of uraemic solutes in the pathophysiology of CRS, a number of further studies are warranted. Uraemic solutes may be identified from proteomics studies that have not yet been chemically identified or tested for biological activity. Beyond PBUTs, uraemic solutes in other classes (according to the European Uraemic Toxin Work Group (EUTox) classification (Vanholder et al. 2003) may have adverse cardiorenal effects. Although, most small water-soluble solutes and middle molecules can be satisfactorily removed by either conventional or newly developed dialysis strategies, targeting uraemic toxins with cardiorenal toxicity at predialysis-stage CKD may delay or prevent incident dialysis as well as the initiation/progression of CRS.
Glossary
- ADHF
acute decompensated heart failure
- AhR
aryl hydrocarbon receptor
- AKI
acute kidney injury
- BP
blood pressure
- CKD
chronic kidney disease
- CRS
cardiorenal syndrome
- CV
cardiovascular
- CVD
cardiovascular disease
- eGFR
estimated glomerular filtration rate
- ESRD
end-stage renal disease
- FGF23
fibroblast growth factor 23
- GFR
glomerular filtration rate
- HF
heart failure
- IL
interleukin
- IS
indoxyl sulfate
- KIM-1
kidney injury molecule-1
- LVH
left ventricular hypertrophy
- MCP-1
monocyte chemoattractant protein-1
- MI
myocardial infarction
- NAG
N-acetyl-β-d-glucodaminidase
- NF-κB
nuclear factor-κB
- NGAL
neutrophil gelatinase-associated lipocalin
- NYHA
New York Heart Association
- PBUTs
protein-bound uraemic toxins
- pCS
p-cresyl sulfate
- RAAS
renin–angiotensin–aldosterone system
- STNx
subtotal nephrectomy
- TGF-β
transforming growth factor-β
Additional information
Completing interests
There are no competing interests.
Funding
This work was supported by National Health and Medical Research Council of Australia (Program Grants 334008 and 546272) and Thailand Research Fund (MRG568079).
References
- Adams KF, Jr, Fonarow GC, Emerman CL, LeJemtel TH, Costanzo MR, Abraham WT, Berkowitz RL, Galvao M, Horton DP. Characteristics and outcomes of patients hospitalized for heart failure in the United States: rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE) Am Heart J. 2005;149:209–216. doi: 10.1016/j.ahj.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Adijiang A, Higuchi Y, Nishijima F, Shimizu H, Niwa T. Indoxyl sulfate, a uremic toxin, promotes cell senescence in aorta of hypertensive rats. Biochem Biophys Res Commun. 2010;399:637–641. doi: 10.1016/j.bbrc.2010.07.130. [DOI] [PubMed] [Google Scholar]
- Adijiang A, Niwa T. An oral sorbent, AST-120, increases Klotho expression and inhibits cell senescence in the kidney of uremic rats. Am J Nephrol. 2010;31:160–164. doi: 10.1159/000264634. [DOI] [PubMed] [Google Scholar]
- Akhter MW, Aronson D, Bitar F, Khan S, Singh H, Singh RP, Burger AJ, Elkayam U. Effect of elevated admission serum creatinine and its worsening on outcome in hospitalized patients with decompensated heart failure. Am J Cardiol. 2004;94:957–960. doi: 10.1016/j.amjcard.2004.06.041. [DOI] [PubMed] [Google Scholar]
- Akizawa T, Asano Y, Morita S, Wakita T, Onishi Y, Fukuhara S, Gejyo F, Matsuo S, Yorioka N, Kurokawa K. Effect of a carbonaceous oral adsorbent on the progression of CKD: a multicenter, randomized, controlled trial. Am J Kidney Dis. 2009;54:459–467. doi: 10.1053/j.ajkd.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Amann K, Neususs R, Ritz E, Irzyniec T, Wiest G, Mall G. Changes of vascular architecture independent of blood pressure in experimental uremia. Am J Hypertens. 1995;8:409–417. doi: 10.1016/0895-7061(94)00248-a. [DOI] [PubMed] [Google Scholar]
- Amann K, Tornig J, Kugel B, Gross ML, Tyralla K, El-Shakmak A, Szabo A, Ritz E. Hyperphosphatemia aggravates cardiac fibrosis and microvascular disease in experimental uremia. Kidney Int. 2003;63:1296–1301. doi: 10.1046/j.1523-1755.2003.00864.x. [DOI] [PubMed] [Google Scholar]
- Aoki J, Ikari Y, Nakajima H, Mori M, Sugimoto T, Hatori M, Tanimoto S, Amiya E, Hara K. Clinical and pathologic characteristics of dilated cardiomyopathy in hemodialysis patients. Kidney Int. 2005;67:333–340. doi: 10.1111/j.1523-1755.2005.00086.x. [DOI] [PubMed] [Google Scholar]
- Aronov PA, Luo FJ, Plummer NS, Quan Z, Holmes S, Hostetter TH, Meyer TW. Colonic contribution to uremic solutes. J Am Soc Nephrol. 2011;22:1769–1776. doi: 10.1681/ASN.2010121220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto FC, Barreto DV, Liabeuf S, Meert N, Glorieux G, Temmar M, Choukroun G, Vanholder R, Massy ZA. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol. 2009;4:1551–1558. doi: 10.2215/CJN.03980609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauersachs J, Braun C, Fraccarollo D, Widder J, Ertl G, Schilling L, Kirchengast M, Rohmeiss P. Improvement of renal dysfunction in rats with chronic heart failure after myocardial infarction by treatment with the endothelin A receptor antagonist, LU 135252. J Hypertens. 2000;18:1507–1514. doi: 10.1097/00004872-200018100-00020. [DOI] [PubMed] [Google Scholar]
- Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P. Acute renal failure – definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8:R204–212. doi: 10.1186/cc2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongartz LG, Joles JA, Verhaar MC, Cramer MJ, Goldschmeding R, Tilburgs C, Gaillard CA, Doevendans PA, Braam B. Subtotal nephrectomy plus coronary ligation leads to more pronounced damage in both organs than either nephrectomy or coronary ligation. Am J Physiol Heart Circ Physiol. 2012;302:H845–H854. doi: 10.1152/ajpheart.00261.2011. [DOI] [PubMed] [Google Scholar]
- Charlton JR, Portilla D, Okusa MD. A basic science view of acute kidney injury biomarkers. Nephrol Dial Transplant. 2014 doi: 10.1093/ndt/gft510. doi: 10.1093/ndt/gft510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E, Kim M, Ko YS, Lee HY, Song M, Kim MG, Kim HK, Cho WY, Jo SK. Role of inflammation in the pathogenesis of cardiorenal syndrome in a rat myocardial infarction model. Nephrol Dial Transplant. 2013;28:2766–2778. doi: 10.1093/ndt/gft376. [DOI] [PubMed] [Google Scholar]
- Coca SG, Krumholz HM, Garg AX, Parikh CR. Underrepresentation of renal disease in randomized controlled trials of cardiovascular disease. JAMA. 2006;296:1377–1384. doi: 10.1001/jama.296.11.1377. [DOI] [PubMed] [Google Scholar]
- Damman K, Masson S, Hillege HL, Maggioni AP, Voors AA, Opasich C, van Veldhuisen DJ, Montagna L, Cosmi F, Tognoni G, Tavazzi L, Latini R. Clinical outcome of renal tubular damage in chronic heart failure. Eur Heart J. 2011;32:2705–2712. doi: 10.1093/eurheartj/ehr190. [DOI] [PubMed] [Google Scholar]
- Damman K, Van Veldhuisen DJ, Navis G, Vaidya VS, Smilde TD, Westenbrink BD, Bonventre JV, Voors AA, Hillege HL. Tubular damage in chronic systolic heart failure is associated with reduced survival independent of glomerular filtration rate. Heart. 2010;96:1297–1302. doi: 10.1136/hrt.2010.194878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou L, Jourde-Chiche N, Faure V, Cerini C, Berland Y, Dignat-George F, Brunet P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J Thromb Haemost. 2007;5:1302–1308. doi: 10.1111/j.1538-7836.2007.02540.x. [DOI] [PubMed] [Google Scholar]
- Dries DL, Exner DV, Domanski MJ, Greenberg B, Stevenson LW. The prognostic implications of renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dysfunction. J Am Coll Cardiol. 2000;35:681–689. doi: 10.1016/s0735-1097(99)00608-7. [DOI] [PubMed] [Google Scholar]
- Dupont M, Shrestha K, Singh D, Awad A, Kovach C, Scarcipino M, Maroo AP, Tang WH. Lack of significant renal tubular injury despite acute kidney injury in acute decompensated heart failure. Eur J Heart Fail. 2012;14:597–604. doi: 10.1093/eurjhf/hfs039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds LH., Jr Inflammatory response to cardiopulmonary bypass. Ann Thorac Surg. 1998;66:S12–16. doi: 10.1016/s0003-4975(98)00967-9. Discussion S25–18. [DOI] [PubMed] [Google Scholar]
- Entin-Meer M, Ben-Shoshan J, Maysel-Auslender S, Levy R, Goryainov P, Schwartz I, Barshack I, Avivi C, Sharir R, Keren G. Accelerated renal fibrosis in cardiorenal syndrome is associated with long-term increase in urine neutrophil gelatinase-associated lipocalin levels. Am J Nephrol. 2012;36:190–200. doi: 10.1159/000341651. [DOI] [PubMed] [Google Scholar]
- Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutierrez OM, Aguillon-Prada R, Lincoln J, Hare JM, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filler G, Bokenkamp A, Hofmann W, Le Bricon T, Martinez-Bru C, Grubb A. Cystatin C as a marker of GFR – history, indications, and future research. Clin Biochem. 2005;38:1–8. doi: 10.1016/j.clinbiochem.2004.09.025. [DOI] [PubMed] [Google Scholar]
- Forman DE, Butler J, Wang Y, Abraham WT, O'Connor CM, Gottlieb SS, Loh E, Massie BM, Rich MW, Stevenson LW, Young JB, Krumholz HM. Incidence, predictors at admission, and impact of worsening renal function among patients hospitalized with heart failure. J Am Coll Cardiol. 2004;43:61–67. doi: 10.1016/j.jacc.2003.07.031. [DOI] [PubMed] [Google Scholar]
- Fouad M, Boraie M. Cystatin C as an early marker of acute kidney injury and predictor of mortality in the intensive care unit after acute myocardial infarction. Arab J Nephrol Transplant. 2013;6:21–26. [PubMed] [Google Scholar]
- Fujii H, Nishijima F, Goto S, Sugano M, Yamato H, Kitazawa R, Kitazawa S, Fukagawa M. Oral charcoal adsorbent (AST-120) prevents progression of cardiac damage in chronic kidney disease through suppression of oxidative stress. Nephrol Dial Transplant. 2009;24:2089–2095. doi: 10.1093/ndt/gfp007. [DOI] [PubMed] [Google Scholar]
- Glassock RJ, Pecoits-Filho R, Barberato SH. Left ventricular mass in chronic kidney disease and ESRD. Clin J Am Soc Nephrol. 2009;4(Suppl. 1):S79–S91. doi: 10.2215/CJN.04860709. [DOI] [PubMed] [Google Scholar]
- Goldberg A, Hammerman H, Petcherski S, Zdorovyak A, Yalonetsky S, Kapeliovich M, Agmon Y, Markiewicz W, Aronson D. Inhospital and 1-year mortality of patients who develop worsening renal function following acute ST-elevation myocardial infarction. Am Heart J. 2005;150:330–337. doi: 10.1016/j.ahj.2004.09.055. [DOI] [PubMed] [Google Scholar]
- Goldberg A, Kogan E, Hammerman H, Markiewicz W, Aronson D. The impact of transient and persistent acute kidney injury on long-term outcomes after acute myocardial infarction. Kidney Int. 2009;76:900–906. doi: 10.1038/ki.2009.295. [DOI] [PubMed] [Google Scholar]
- Goto S, Kitamura K, Kono K, Nakai K, Fujii H, Nishi S. Association between AST-120 and abdominal aortic calcification in predialysis patients with chronic kidney disease. Clin Exp Nephrol. 2013;17:365–371. doi: 10.1007/s10157-012-0717-0. [DOI] [PubMed] [Google Scholar]
- Gottlieb SS, Abraham W, Butler J, Forman DE, Loh E, Massie BM, O'Connor CM, Rich MW, Stevenson LW, Young J, Krumholz HM. The prognostic importance of different definitions of worsening renal function in congestive heart failure. J Card Fail. 2002;8:136–141. doi: 10.1054/jcaf.2002.125289. [DOI] [PubMed] [Google Scholar]
- Gross ML, Ritz E. Hypertrophy and fibrosis in the cardiomyopathy of uremia – beyond coronary heart disease. Semin Dial. 2008;21:308–318. doi: 10.1111/j.1525-139X.2008.00454.x. [DOI] [PubMed] [Google Scholar]
- Guerin AP, Pannier B, Marchais SJ, London GM. Cardiovascular disease in the dialysis population: prognostic significance of arterial disorders. Curr Opin Nephrol Hypertens. 2006;15:105–110. doi: 10.1097/01.mnh.0000203186.11772.21. [DOI] [PubMed] [Google Scholar]
- Haase M, Devarajan P, Haase-Fielitz A, Bellomo R, Cruz DN, Wagener G, Krawczeski CD, Koyner JL, Murray P, Zappitelli M, et al. The outcome of neutrophil gelatinase-associated lipocalin-positive subclinical acute kidney injury: a multicenter pooled analysis of prospective studies. J Am Coll Cardiol. 2011;57:1752–1761. doi: 10.1016/j.jacc.2010.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herget-Rosenthal S, Marggraf G, Husing J, Goring F, Pietruck F, Janssen O, Philipp T, Kribben A. Early detection of acute renal failure by serum cystatin C. Kidney Int. 2004;66:1115–1122. doi: 10.1111/j.1523-1755.2004.00861.x. [DOI] [PubMed] [Google Scholar]
- Hillege HL, Nitsch D, Pfeffer MA, Swedberg K, McMurray JJ, Yusuf S, Granger CB, Michelson EL, Ostergren J, Cornel JH, de Zeeuw D, Pocock S, van Veldhuisen DJ. Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation. 2006;113:671–678. doi: 10.1161/CIRCULATIONAHA.105.580506. [DOI] [PubMed] [Google Scholar]
- Hillege HL, van Gilst WH, van Veldhuisen DJ, Navis G, Grobbee DE, de Graeff PA, de Zeeuw D. Accelerated decline and prognostic impact of renal function after myocardial infarction and the benefits of ACE inhibition: the CATS randomized trial. Eur Heart J. 2003;24:412–420. doi: 10.1016/s0195-668x(02)00526-2. [DOI] [PubMed] [Google Scholar]
- Homma T, Sonoda H, Manabe K, Arai K, Mizuno M, Sada T, Ikeda M. Activation of renal angiotensin type 1 receptor contributes to the pathogenesis of progressive renal injury in a rat model of chronic cardiorenal syndrome. Am J Physiol Renal Physiol. 2012;302:F750–F761. doi: 10.1152/ajprenal.00494.2011. [DOI] [PubMed] [Google Scholar]
- Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, Wahl P, Gutierrez OM, Steigerwalt S, He J, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011;305:2432–2439. doi: 10.1001/jama.2011.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ix JH, Shlipak MG, Chertow GM, Ali S, Schiller NB, Whooley MA. Cystatin C, left ventricular hypertrophy, and diastolic dysfunction: data from the Heart and Soul Study. J Card Fail. 2006;12:601–607. doi: 10.1016/j.cardfail.2006.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungbauer CG, Birner C, Jung B, Buchner S, Lubnow M, von Bary C, Endemann D, Banas B, Mack M, Boger CA, Riegger G, Luchner A. Kidney injury molecule-1 and N-acetyl-ß-d-glucosaminidase in chronic heart failure: possible biomarkers of cardiorenal syndrome. Eur J Heart Fail. 2011;13:1104–1110. doi: 10.1093/eurjhf/hfr102. [DOI] [PubMed] [Google Scholar]
- Keith DS, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med. 2004;164:659–663. doi: 10.1001/archinte.164.6.659. [DOI] [PubMed] [Google Scholar]
- Keller CR, Odden MC, Fried LF, Newman AB, Angleman S, Green CA, Cummings SR, Harris TB, Shlipak MG. Kidney function and markers of inflammation in elderly persons without chronic kidney disease: the health, aging, and body composition study. Kidney Int. 2007;71:239–244. doi: 10.1038/sj.ki.5002042. [DOI] [PubMed] [Google Scholar]
- Koyner JL, Bennett MR, Worcester EM, Ma Q, Raman J, Jeevanandam V, Kasza KE, O'Connor MF, Konczal DJ, Trevino S, Devarajan P, Murray PT. Urinary cystatin C as an early biomarker of acute kidney injury following adult cardiothoracic surgery. Kidney Int. 2008;74:1059–1069. doi: 10.1038/ki.2008.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyner JL, Garg AX, Coca SG, Sint K, Thiessen-Philbrook H, Patel UD, Shlipak MG, Parikh CR. Biomarkers predict progression of acute kidney injury after cardiac surgery. J Am Soc Nephrol. 2012;23:905–914. doi: 10.1681/ASN.2011090907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczeski CD, Woo JG, Wang Y, Bennett MR, Ma Q, Devarajan P. Neutrophil gelatinase-associated lipocalin concentrations predict development of acute kidney injury in neonates and children after cardiopulmonary bypass. J Pediatr. 2011;158:1009–1015 e1. doi: 10.1016/j.jpeds.2010.12.057. [DOI] [PubMed] [Google Scholar]
- Kuro-o M. Klotho in health and disease. Curr Opin Nephrol Hypertens. 2012;21:362–368. doi: 10.1097/MNH.0b013e32835422ad. [DOI] [PubMed] [Google Scholar]
- Lekawanvijit S, Kompa AR, Manabe M, Wang BH, Langham RG, Nishijima F, Kelly DJ, Krum H. Chronic kidney disease-induced cardiac fibrosis is ameliorated by reducing circulating levels of a non-dialysable uremic toxin, indoxyl sulfate. PLoS One. 2012a;7:e41281. doi: 10.1371/journal.pone.0041281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekawanvijit S, Kompa AR, Wang BH, Kelly DJ, Krum H. Cardiorenal syndrome: the emerging role of protein-bound uremic toxins. Circ Res. 2012b;111:1470–1483. doi: 10.1161/CIRCRESAHA.112.278457. [DOI] [PubMed] [Google Scholar]
- Lekawanvijit S, Kompa AR, Zhang Y, Wang BH, Kelly DJ, Krum H. Myocardial infarction impairs renal function, induces renal interstitial fibrosis, and increases renal KIM-1 expression: implications for cardiorenal syndrome. Am J Physiol Heart Circ Physiol. 2012c;302:H1884–H1893. doi: 10.1152/ajpheart.00967.2011. [DOI] [PubMed] [Google Scholar]
- Lekawanvijit S, Kumfu S, Wang BH, Manabe M, Nishijima F, Kelly DJ, Krum H, Kompa AR. The uremic toxin adsorbent AST-120 abrogates cardiorenal injury following myocardial infarction. PLoS One. 2013;8:e83687. doi: 10.1371/journal.pone.0083687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey AS. Controlling the epidemic of cardiovascular disease in chronic renal disease: where do we start? Am J Kidney Dis. 1998;32:S5–S13. doi: 10.1053/ajkd.1998.v32.pm9820463. [DOI] [PubMed] [Google Scholar]
- Liabeuf S, Barreto DV, Barreto FC, Meert N, Glorieux G, Schepers E, Temmar M, Choukroun G, Vanholder R, Massy ZA. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol Dial Transplant. 2010;25:1183–1191. doi: 10.1093/ndt/gfp592. [DOI] [PubMed] [Google Scholar]
- Liang KV, Williams AW, Greene EL, Redfield MM. Acute decompensated heart failure and the cardiorenal syndrome. Crit Care Med. 2008;36:S75–88. doi: 10.1097/01.CCM.0000296270.41256.5C. [DOI] [PubMed] [Google Scholar]
- Lin CJ, Pan CF, Chuang CK, Liu HL, Sun FJ, Wang TJ, Chen HH, Wu CJ. Association of indoxyl sulfate with fibroblast growth factor 23 in patients with advanced chronic kidney disease. Am J Med Sci. 2013;347:370–376. doi: 10.1097/MAJ.0b013e3182989f26. [DOI] [PubMed] [Google Scholar]
- Lindner A, Charra B, Sherrard DJ, Scribner BH. Accelerated atherosclerosis in prolonged maintenance hemodialysis. New Eng J Med. 1974;290:697–701. doi: 10.1056/NEJM197403282901301. [DOI] [PubMed] [Google Scholar]
- Liu S, Kompa AR, Kumfu S, Nishijima F, Kelly DJ, Krum H, Wang BH. Subtotal nephrectomy accelerates pathological cardiac remodeling post-myocardial infarction: implications for cardiorenal syndrome. Int J Cardiol. 2013;168:1866–1880. doi: 10.1016/j.ijcard.2012.12.065. [DOI] [PubMed] [Google Scholar]
- London GM, Pannier B, Guerin AP, Blacher J, Marchais SJ, Darne B, Metivier F, Adda H, Safar ME. Alterations of left ventricular hypertrophy in and survival of patients receiving hemodialysis: follow-up of an interventional study. J Am Soc Nephrol. 2001;12:2759–2767. doi: 10.1681/ASN.V12122759. [DOI] [PubMed] [Google Scholar]
- Lu J, Wang X, Wang W, Muniyappa H, Deshmukh A, Hu C, Das K, Mehta JL. Abrogation of lectin-like oxidized LDL receptor-1 attenuates acute myocardial ischemia-induced renal dysfunction by modulating systemic and local inflammation. Kidney Int. 2012;82:436–444. doi: 10.1038/ki.2012.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan WM, Flanders WD, Langston RD, Jurkovitz C, Presley R. Anemia and renal insufficiency are independent risk factors for death among patients with congestive heart failure admitted to community hospitals: a population-based study. J Am Soc Nephrol. 2002;13:1928–1936. doi: 10.1097/01.asn.0000018409.45834.fa. [DOI] [PubMed] [Google Scholar]
- McIlroy DR, Wagener G, Lee HT. Biomarkers of acute kidney injury: an evolving domain. Anesthesiology. 2010;112:998–1004. doi: 10.1097/ALN.0b013e3181cded3f. [DOI] [PubMed] [Google Scholar]
- Martin FL, Huntley BK, Harders GE, Sandberg SM, Chen HH, Burnett JC., Jr Myocardial infarction mediates renal fibrosis and activates renal molecular remodeling in the absence of heart failure. Circulation. 2007;116:II_60. (abstract) [Google Scholar]
- Marzocco S, Dal Piaz F, Di Micco L, Torraca S, Sirico ML, Tartaglia D, Autore G, Di Iorio B. Very low protein diet reduces indoxyl sulfate levels in chronic kidney disease. Blood Purif. 2013;35:196–201. doi: 10.1159/000346628. [DOI] [PubMed] [Google Scholar]
- Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11:R31. doi: 10.1186/cc5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijers BK, Van Kerckhoven S, Verbeke K, Dehaen W, Vanrenterghem Y, Hoylaerts MF, Evenepoel P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am J Kidney Dis. 2009;54:891–901. doi: 10.1053/j.ajkd.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Mishra J, Dent C, Tarabishi R, Mitsnefes MM, Ma Q, Kelly C, Ruff SM, Zahedi K, Shao M, Bean J, Mori K, Barasch J, Devarajan P. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet. 2005;365:1231–1238. doi: 10.1016/S0140-6736(05)74811-X. [DOI] [PubMed] [Google Scholar]
- Muteliefu G, Enomoto A, Jiang P, Takahashi M, Niwa T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol Dial Transplant. 2009;24:2051–2058. doi: 10.1093/ndt/gfn757. [DOI] [PubMed] [Google Scholar]
- Nakai K, Fujii H, Kono K, Goto S, Fukagawa M, Nishi S. Effects of AST-120 on left ventricular mass in predialysis patients. Am J Nephrol. 2011;33:218–223. doi: 10.1159/000324354. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Kawagoe Y, Matsuda T, Ueda Y, Shimada N, Ebihara I, Koide H. Oral ADSORBENT AST-120 decreases carotid intima-media thickness and arterial stiffness in patients with chronic renal failure. Kidney Blood Press Res. 2004;27:121–126. doi: 10.1159/000077536. [DOI] [PubMed] [Google Scholar]
- Neirynck N, Vanholder R, Schepers E, Eloot S, Pletinck A, Glorieux G. An update on uremic toxins. Int Urol Nephrol. 2012;45:139–150. doi: 10.1007/s11255-012-0258-1. [DOI] [PubMed] [Google Scholar]
- Nejat M, Pickering JW, Walker RJ, Endre ZH. Rapid detection of acute kidney injury by plasma cystatin C in the intensive care unit. Nephrol Dial Transplant. 2010;25:3283–3289. doi: 10.1093/ndt/gfq176. [DOI] [PubMed] [Google Scholar]
- Nickolas TL, Schmidt-Ott KM, Canetta P, Forster C, Singer E, Sise M, Elger A, Maarouf O, Sola-Del Valle DA, O'Rourke M, et al. Diagnostic and prognostic stratification in the emergency department using urinary biomarkers of nephron damage: a multicenter prospective cohort study. J Am Coll Cardiol. 2012;59:246–255. doi: 10.1016/j.jacc.2011.10.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owada S, Goto S, Bannai K, Hayashi H, Nishijima F, Niwa T. Indoxyl sulfate reduces superoxide scavenging activity in the kidneys of normal and uremic rats. Am J Nephrol. 2008;28:446–454. doi: 10.1159/000112823. [DOI] [PubMed] [Google Scholar]
- Parikh CR, Coca SG, Thiessen-Philbrook H, Shlipak MG, Koyner JL, Wang Z, Edelstein CL, Devarajan P, Patel UD, Zappitelli M, Krawczeski CD, Passik CS, Swaminathan M, Garg AX. Postoperative biomarkers predict acute kidney injury and poor outcomes after adult cardiac surgery. J Am Soc Nephrol. 2011a;22:1748–1757. doi: 10.1681/ASN.2010121302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh CR, Coca SG, Wang Y, Masoudi FA, Krumholz HM. Long-term prognosis of acute kidney injury after acute myocardial infarction. Arch Intern Med. 2008;168:987–995. doi: 10.1001/archinte.168.9.987. [DOI] [PubMed] [Google Scholar]
- Parikh CR, Devarajan P, Zappitelli M, Sint K, Thiessen-Philbrook H, Li S, Kim RW, Koyner JL, Coca SG, Edelstein CL, Shlipak MG, Garg AX, Krawczeski CD. Postoperative biomarkers predict acute kidney injury and poor outcomes after pediatric cardiac surgery. J Am Soc Nephrol. 2011b;22:1737–1747. doi: 10.1681/ASN.2010111163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Vittinghoff E, Liu KD, Shlipak MG, Hsu CY. Urine biomarkers neutrophil gelatinase-associated lipocalin (ngal) and kidney injury molecule-1 (KIM-1) have different patterns in heart failure exacerbation. Biomark Insights. 2013a;8:15–18. doi: 10.4137/BMI.S11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MY, Choi SJ, Kim JK, Hwang SD, Lee YW. Urinary cystatin C levels as a diagnostic and prognostic biomarker in patients with acute kidney injury. Nephrology (Carlton) 2013b;18:256–262. doi: 10.1111/nep.12037. [DOI] [PubMed] [Google Scholar]
- Ranganathan N, Ranganathan P, Friedman EA, Joseph A, Delano B, Goldfarb DS, Tam P, Rao AV, Anteyi E, Musso CG. Pilot study of probiotic dietary supplementation for promoting healthy kidney function in patients with chronic kidney disease. Adv Ther. 2010;27:634–647. doi: 10.1007/s12325-010-0059-9. [DOI] [PubMed] [Google Scholar]
- Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52:1527–1539. doi: 10.1016/j.jacc.2008.07.051. [DOI] [PubMed] [Google Scholar]
- Rostand SG, Kirk KA, Rutsky EA. Dialysis-associated ischemic heart disease: insights from coronary angiography. Kidney Int. 1984;25:653–659. doi: 10.1038/ki.1984.70. [DOI] [PubMed] [Google Scholar]
- Salgado JV, Souza FL, Salgado BJ. How to understand the association between cystatin C levels and cardiovascular disease: Imbalance, counterbalance, or consequence. J Cardiol. 2013;62:331–335. doi: 10.1016/j.jjcc.2013.05.015. [DOI] [PubMed] [Google Scholar]
- Santolucito PA, Tighe DA, McManus DD, Yarzebski J, Lessard D, Gore JM, Goldberg RJ. Management and outcomes of renal disease and acute myocardial infarction. Am J Med. 2010;123:847–855. doi: 10.1016/j.amjmed.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL, McCullough PA, Kasiske BL, Kelepouris E, Klag MJ, Parfrey P, Pfeffer M, Raij L, Spinosa DJ, Wilson PW. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation. 2003;108:2154–2169. doi: 10.1161/01.CIR.0000095676.90936.80. [DOI] [PubMed] [Google Scholar]
- Schroeder JC, Dinatale BC, Murray IA, Flaveny CA, Liu Q, Laurenzana EM, Lin JM, Strom SC, Omiecinski CJ, Amin S, Perdew GH. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry. 2010;49:393–400. doi: 10.1021/bi901786x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman G, Vanholder R, Niwa T. AST-120 for the management of progression of chronic kidney disease. Int J Nephrol Renovasc Dis. 2014;7:49–56. doi: 10.2147/IJNRD.S41339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Bolati D, Adijiang A, Adelibieke Y, Muteliefu G, Enomoto A, Higashiyama Y, Higuchi Y, Nishijima F, Niwa T. Indoxyl sulfate downregulates renal expression of Klotho through production of ROS and activation of nuclear factor-kB. AmJ Nephrol. 2011;33:319–324. doi: 10.1159/000324885. [DOI] [PubMed] [Google Scholar]
- Shlipak MG. Pharmacotherapy for heart failure in patients with renal insufficiency. Ann Intern Med. 2003;138:917–924. doi: 10.7326/0003-4819-138-11-200306030-00013. [DOI] [PubMed] [Google Scholar]
- Shlipak MG, Sarnak MJ, Katz R, Fried LF, Seliger SL, Newman AB, Siscovick DS, Stehman-Breen C. Cystatin C and the risk of death and cardiovascular events among elderly persons. New Eng J Med. 2005;352:2049–2060. doi: 10.1056/NEJMoa043161. [DOI] [PubMed] [Google Scholar]
- Shrestha K, Shao Z, Singh D, Dupont M, Tang WH. Relation of systemic and urinary neutrophil gelatinase-associated lipocalin levels to different aspects of impaired renal function in patients with acute decompensated heart failure. Am J Cardiol. 2012;110:1329–1335. doi: 10.1016/j.amjcard.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GL, Shlipak MG, Havranek EP, Masoudi FA, McClellan WM, Foody JM, Rathore SS, Krumholz HM. Race and renal impairment in heart failure: mortality in blacks versus whites. Circulation. 2005;111:1270–1277. doi: 10.1161/01.CIR.0000158131.78881.D5. [DOI] [PubMed] [Google Scholar]
- Soto K, Coelho S, Rodrigues B, Martins H, Frade F, Lopes S, Cunha L, Papoila AL, Devarajan P. Cystatin C as a marker of acute kidney injury in the emergency department. Clin J Am Soc Nephrol. 2010;5:1745–1754. doi: 10.2215/CJN.00690110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CY, Chang SC, Wu MS. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012a;81:640–650. doi: 10.1038/ki.2011.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CY, Chang SC, Wu MS. Uremic toxins induce kidney fibrosis by activating intrarenal rennin–angiotensin–aldosterone system associated epithelial-to-mesenchymal transition. PLoS One. 2012b;7:e34026. doi: 10.1371/journal.pone.0034026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testani JM, McCauley BD, Chen J, Shumski M, Shannon RP. Worsening renal function defined as an absolute increase in serum creatinine is a biased metric for the study of cardio-renal interactions. Cardiology. 2010;116:206–212. doi: 10.1159/000316038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thadhani R, Pascual M, Bonventre JV. Acute renal failure. New Eng J Med. 1996;334:1448–1460. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- Thurman JM, Parikh CR. Peeking into the black box: new biomarkers for acute kidney injury. Kidney Int. 2008;73:379–381. doi: 10.1038/sj.ki.5002739. [DOI] [PubMed] [Google Scholar]
- Tidman M, Sjostrom P, Jones I. A comparison of GFR estimating formulae based upon s-cystatin C and s-creatinine and a combination of the two. Nephrol Dial Transplant. 2008;23:154–160. doi: 10.1093/ndt/gfm661. [DOI] [PubMed] [Google Scholar]
- Tuladhar SM, Puntmann VO, Soni M, Punjabi PP, Bogle RG. Rapid detection of acute kidney injury by plasma and urinary neutrophil gelatinase-associated lipocalin after cardiopulmonary bypass. J Cardiovasc Pharmacol. 2009;53:261–266. doi: 10.1097/FJC.0b013e31819d6139. [DOI] [PubMed] [Google Scholar]
- Tumur Z, Niwa T. Oral sorbent AST-120 increases renal NO synthesis in uremic rats. J Ren Nutr. 2008;18:60–64. doi: 10.1053/j.jrn.2007.10.013. [DOI] [PubMed] [Google Scholar]
- U.S. Renal Data System (USRDS) 2007. Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2007.
- U.S. Renal Data System (USRDS) 2012. Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2012.
- van Dokkum RP, Eijkelkamp WB, Kluppel AC, Henning RH, van Goor H, Citgez M, Windt WA, van Veldhuisen DJ, de Graeff PA, de Zeeuw D. Myocardial infarction enhances progressive renal damage in an experimental model for cardio-renal interaction. Am J Kidney Dis. 2004;15:3103–3110. doi: 10.1097/01.ASN.0000145895.62896.98. [DOI] [PubMed] [Google Scholar]
- Vanholder R, De Smet R, Glorieux G, Argiles A, Baurmeister U, Brunet P, Clark W, Cohen G, De Deyn PP, Deppisch R, et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 2003;63:1934–1943. doi: 10.1046/j.1523-1755.2003.00924.x. [DOI] [PubMed] [Google Scholar]
- Vanmassenhove J, Vanholder R, Nagler E, Van Biesen W. Urinary and serum biomarkers for the diagnosis of acute kidney injury: an in-depth review of the literature. Nephrol Dial Transplant. 2013;28:254–273. doi: 10.1093/ndt/gfs380. [DOI] [PubMed] [Google Scholar]
- Verbrugge FH, Dupont M, Shao Z, Shrestha K, Singh D, Finucan M, Mullens W, Tang WH. Novel urinary biomarkers in detecting acute kidney injury, persistent renal impairment, and all-cause mortality following decongestive therapy in acute decompensated heart failure. J Card Fail. 2013;19:621–628. doi: 10.1016/j.cardfail.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe I, Tatebe J, Namba S, Koizumi M, Yamazaki J, Morita T. Activation of aryl hydrocarbon receptor mediates indoxyl sulfate-induced monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells. Circ J. 2013;77:224–230. doi: 10.1253/circj.cj-12-0647. [DOI] [PubMed] [Google Scholar]
- Weinfeld MS, Chertow GM, Stevenson LW. Aggravated renal dysfunction during intensive therapy for advanced chronic heart failure. Am Heart J. 1999;138:285–290. doi: 10.1016/s0002-8703(99)70113-4. [DOI] [PubMed] [Google Scholar]
- Wen Z, Cai M, Mai Z, Chen Y, Geng D, Wang J. Protection of renal impairment by angiotensin II type 1 receptor blocker in rats with post-infarction heart failure. Ren Fail. 2013;35:766–775. doi: 10.3109/0886022X.2013.780561. [DOI] [PubMed] [Google Scholar]
- Windt WA, Henning RH, Kluppel AC, Xu Y, de Zeeuw D, van Dokkum RP. Myocardial infarction does not further impair renal damage in 5/6 nephrectomized rats. Nephrol Dial Transplant. 2008;23:3103–3110. doi: 10.1093/ndt/gfn233. [DOI] [PubMed] [Google Scholar]
- Wu IW, Hsu KH, Hsu HJ, Lee CC, Sun CY, Tsai CJ, Wu MS. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients – a prospective cohort study. Nephrol Dial Transplant. 2012;27:1169–1175. doi: 10.1093/ndt/gfr453. [DOI] [PubMed] [Google Scholar]
- Wu IW, Hsu KH, Lee CC, Sun CY, Hsu HJ, Tsai CJ, Tzen CY, Wang YC, Lin CY, Wu MS. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol Dial Transplant. 2011;26:938–947. doi: 10.1093/ndt/gfq580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Zuo Y, Ma J, Yancey PG, Hunley TE, Motojima M, Fogo AB, Linton MF, Fazio S, Ichikawa I, Kon V. Oral activated charcoal adsorbent (AST-120) ameliorates extent and instability of atherosclerosis accelerated by kidney disease in apolipoprotein E-deficient mice. Nephrol Dial Transplant. 2011;26:2491–2497. doi: 10.1093/ndt/gfq759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Nie L, Huang Y, Zhang J, Xiao T, Guan X, Zhao J. Amelioration of uremic toxin indoxyl sulfate-induced endothelial cell dysfunction by Klotho protein. Toxicol Lett. 2012;215:77–83. doi: 10.1016/j.toxlet.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Yndestad A, Landro L, Ueland T, Dahl CP, Flo TH, Vinge LE, Espevik T, Froland SS, Husberg C, Christensen G, Dickstein K, Kjekshus J, Oie E, Gullestad L, Aukrust P. Increased systemic and myocardial expression of neutrophil gelatinase-associated lipocalin in clinical and experimental heart failure. Eur Heart J. 2009;30:1229–1236. doi: 10.1093/eurheartj/ehp088. [DOI] [PubMed] [Google Scholar]
- Yu M, Kim YJ, Kang DH. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin J Am Soc Nephrol. 2011;6:30–39. doi: 10.2215/CJN.05340610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoccali C, Benedetto FA, Mallamaci F, Tripepi G, Giacone G, Stancanelli B, Cataliotti A, Malatino LS. Left ventricular mass monitoring in the follow-up of dialysis patients: prognostic value of left ventricular hypertrophy progression. Kidney Int. 2004;65:1492–1498. doi: 10.1111/j.1523-1755.2004.00530.x. [DOI] [PubMed] [Google Scholar]