

Abstract
The regulation of serum phosphate, an acknowledged risk factor for chronic kidney disease and cardiovascular mortality, is poorly understood. The discovery of fibroblast growth factor 23 (FGF23) as a key regulator of renal phosphate handling and activation of vitamin D has revolutionized our comprehension of phosphate homeostasis. Through as yet undetermined mechanisms, circulating and dietary phosphate appear to have a direct effect on FGF23 release by bone cells that, in turn, causes renal phosphate excretion and decreases intestinal phosphate absorption through a decrease in vitamin D production. Thus, the two major phosphaturic hormones, PTH and FGF23, have opposing effects on vitamin D production, placing vitamin D at the nexus of phosphate homeostasis. While our understanding of phosphate homeostasis has advanced, the factors determining regulation of serum phosphate level remain enigmatic. Diet, time of day, season, gender, age and genetics have all been identified as significant contributors to serum phosphate level. The effects of these factors on serum phosphate have major implications for what is understood as ‘normal’ and for studies of phosphate homeostasis and metabolism. Moreover, other hormonal mediators such as dopamine, insulin-like growth factor, and angiotensin II also affect renal handling of phosphate. How the major hormone effects on phosphate handling are regulated and how the effect of these other factors are integrated to yield the measurable serum phosphate are only now beginning to be studied.
 |
Dr Eleanor Lederer completed medical school and her residency in Internal Medicine from Baylor College of Medicine. She continued on to complete a Nephrology Fellowship at Baylor College of Medicine. She is currently Professor of Medicine and Chief of the Division of Nephrology at the University of Louisville School of Medicine. She has maintained an active research program in the field of renal phosphate transport for the past two decades with funding from the Department of Veterans Affairs, the American Heart Association and the National Kidney Foundation. Dr. Lederer served in various administrative roles including as the Director of Outpatient Clinics for the Kidney Disease Program, the Director of the Nephrology Fellowship Training Program, the Interim Vice Dean for Research for the School of Medicine, and the Acting Chief of Medical Services at the Robley Rex VA Medical Center. She is currently a Councilor for the American Society of Nephrology and will be President of the Society in 2017.
Introduction
Maintenance of phosphate homeostasis centres on the regulation of phosphate handling by bone, intestine, and kidney. Diseases affecting these organs frequently are accompanied by significant hyper- or hypophosphataemia. The major identifiable regulatory substances are parathyroid hormone (PTH), vitamin D, and fibroblast growth factor 23 (FGF23) with its required cofactor, the glucuronidase klotho. Disorders of these three circulating hormones result in defined syndromes of hyper- or hypophosphataemia and phosphate overload or phosphate deficiency. However, physiological regulation of serum phosphate remains largely enigmatic. Interest in understanding regulation of serum phosphate has exploded with the recognition that serum phosphate correlates with cardiovascular risk (Kendrick et al. 2011; Ellam et al. 2011; Ellam & Chico, 2012). A recent genome-wide association study identified single nucleotide polymorphisms in regions of the genome that included some proteins previously known to play a role in phosphate homeostasis but also some unsuspected proteins as well (Kestenbaum et al. 2010). Serum phosphate is a function of phosphate homeostasis as well as the balanced movement of phosphate between the intracellular and extracellular spaces. The factors regulating this latter process have not been identified.
This review will cover briefly our current level of understanding of physiological phosphate homeostasis (see Fig. 1), highlighting the gaps in our knowledge. This will be followed by a discussion of aspects of phosphate homeostasis that are not considered often but that are likely to play a greater role in physiological phosphate homeostasis and regulation of serum phosphate than recognized. The review will conclude with a summary of key questions about regulation of phosphate homeostasis and serum phosphate concentration.
Figure 1. Determinants of serum phosphate.
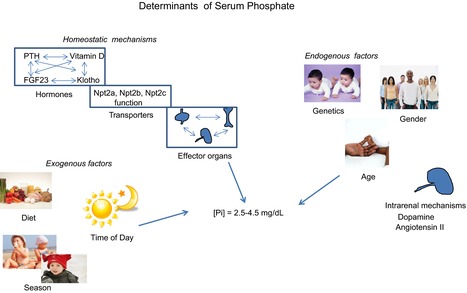
Serum phosphate is a function of multiple factors including those involved in maintaining phosphate homeostasis (hormones, transporters, effector organs), exogenous factors (time of day, season, diet), and endogenous factors (age, gender, genetics). The mechanisms for interactions among these many factors leading to the final level of phosphate measured in the blood have not been identified.
Hormones
The main hormones implicated in regulation of serum phosphate are PTH, vitamin D, and FGF23 with its cofactor klotho. The hypophosphataemic disorders resulting from abnormal function of these hormones include primary hyperparathyroidism; severe vitamin D deficiency; and oncogenic osteomalacia, as well as several forms of congenital hypophosphataemic rickets caused by overexpression or overactivity of FGF23. Conversely, loss of PTH activity through surgical ablation, autoimmune processes, or PTH resistance, as is seen with pseudohypoparathyroidism or vitamin D intoxication, and FGF23 deficiency, as occurs with familial humoral calcinosis, are disorders characterized by hyperphosphataemia (see Table 1).
Table 1.
Disorders of phosphate homeostasis
| Hypophosphataemia | PTH | FGF23 | 1,25 Vit D | Calcium | Phosphate |
|---|---|---|---|---|---|
| Primary hyperparathyroidism | High | Normal | High | High | Normal or low |
| Familial hypocalciuric hypocalcaemia | Normal or high | Unknown | High | High | Normal or low |
| X-linked hypophosphataemic rickets | Normal | High | Low | Normal | Very low |
| Tumor induced osteomalacia | Normal | High | Low | Normal | Very low |
| Sarcoidosis | Low | (?) Low | High | Normal or high | Normal |
| Hyperphosphataemia | |||||
| Hypoparathyroidism | Low | Normal to low | Low | Low | High |
| Pseudohypoparathyroidism | High | Normal to low | Low | High | High |
| Calcitriol Deficiency | High | (?) High | Low | Normal to low | Normal to low |
This table shows the patterns of hormonal alterations in multiple hypophosphataemic and hyperphosphataemic syndromes.
PTH
PTH is an 84 amino acid peptide hormone with receptors widely expressed in bone on cells of osteoblast lineage and in kidney proximal and distal tubule cells. While the major physiological regulator of PTH secretion is the ionized calcium level through interaction with the calcium sensing receptor, PTH secretion is also modulated by serum phosphate, FGF23, and 1,25-dihydroxyvitamin D3 (Krajisnik et al. 2007; Nissenson and Juppner, 2013). PTH has ostensibly conflicting effects on phosphate homeostasis. Through bone breakdown and enhanced gastrointestinal absorption of phosphate mediated by PTH-stimulated vitamin D production, more phosphate is released into the serum (Christensen et al. 2008). These actions, which would be expected to increase serum phosphate, are more than offset by the phosphaturic effect of PTH such that hyperparathyroidism is associated with either low normal or frankly low serum phosphate. PTH decreases the expression of the type 2a sodium–phosphate cotransporter Npt2a at the protein and mRNA levels in renal proximal tubule, which would be expected to produce a long-term decrease in phosphate transport (Blaine et al. 2011; Murray et al. 2013). Patients with primary hyperparathyroidism also have elevated FGF23 which would enhance the phosphaturia (Kawata et al. 2007; Lavi-Moshayoff et al. 2009, 2010; Witteveen et al. 2012). The hypercalciuric effects of PTH may also indirectly affect phosphate transport through the calcium sensing receptor expressed on the luminal membrane of the renal proximal tubule, but the studies investigating the effect of higher serum calcium on proximal tubule phosphate transport are conflicting (Riccardi et al. 2000; Ba & Friedman, 2004; Capasso et al. 2013). Some have suggested that high proximal tubule luminal calcium blunts the phosphaturic effect of PTH while other studies have suggested that direct stimulation of the calcium sensing receptor in proximal tubule decreases expression of Npt2a, which would be expected to enhance the phosphaturic effect of PTH. Not every patient with primary hyperparathyroidism has hypophosphataemia, particularly mild cases. These observations suggest a proposed clinical evolution in the natural history of primary hyperparathyroidism. In early primary hyperparathyroidism, PTH stimulates bone release of calcium and phosphate, enhances gut absorption of calcium and phosphate, and increases renal calcium absorption while it decreases renal phosphate absorption. At this stage, the bone and gut effects offset the renal losses of phosphate to maintain a relatively normal serum phosphate concentration. As the hyperparathyroidism worsens and serum calcium increases, one could envision a sequence of events similar to the following. The higher filtered calcium decreases the expression of the renal sodium–phosphate cotransporters through activation of the proximal tubule calcium sensing receptor, resulting in enhanced phosphaturia. The activation of the renal calcium sensing receptor may also decrease production of 1,25-dihydroxyvitamin D3 thus limiting intestinal phosphate absorption. High PTH will increase FGF23 production, contributing to decreased intestinal phosphate absorption and increased renal excretion of phosphate. The balance would then be tipped, resulting in hypophosphataemia.
FGF23
FGF23, a member of the fibroblast growth factor family, is a recently described regulator of phosphate homeostasis, discovered through the investigation of congenital forms of hypophosphataemic rickets and the syndrome of oncogenic osteomalacia (ADHR Consortium 2000, Martin et al. 2012). FGF23 is produced predominantly by osteocytes and osteoblasts, regulated by serum phosphate levels and dietary phosphate intake. The activity of the hormone is regulated through its cleavage and inactivation by an as yet unidentified enzyme. Identification of mutations in PHEX, the gene for phosphate regulating endopeptidase homologue, X-linked, in individuals with the genetic syndrome of X-linked hypophosphataemic rickets (a hereditary form of hypophosphataemic rickets characterized by abnormally high circulating levels of FGF23 and hypophosphataemia) led early investigators to conclude that PHEX was this FGF23-cleaving enzyme; however, no direct interaction between the two proteins has been demonstrated (Ichikawa et al. 2012). In kidney, FGF23 signals through FGF receptors in conjunction with the glucuronidase klotho and is phosphaturic through a reduction in the expression of Npt2a and Npt2c. FGF23 also decreases intestinal absorption of phosphate, at least in part due to its inhibitory effect on the production of 1,25-dihydroxyvitamin D3. FGF23 decreases PTH mRNA in parathyroid glands, an effect that may be both direct and due to enhanced expression of vitamin D and the calcium sensing receptor (Silver & Naveh-Many, 2012). Of note, the effect of FGF23 on PTH is also dependent on the presence of klotho, similar to what is seen in kidney. Unlike the conflicting effects of PTH on phosphate homeostasis, the effects of FGF23 all promote a decrease in serum phosphate. The combination of a direct phosphaturic effect, decreased intestinal phosphate transport due to diminished vitamin D production, and decreased bone resorption due to decreased PTH production all contribute to the hypophosphataemic effect of FGF23. The end result is frequently quite striking hypophosphataemia, significantly more pronounced than what is seen with primary hyperparathyroidism. These contrasting effects of PTH and FGF23 also are apparent in the resulting bone disease, that is, the high turnover seen with primary hyperparathyroidism vs. the low turnover picture of osteomalacia with high FGF23. Interestingly, although PTH does not require FGF23 for its phosphaturic effect, the opposite is not true (Yuan et al. 2011; Silver & Naveh-Many, 2012). Several lines of evidence suggest that PTH plays a permissive role in the phosphaturic effect of FGF23, suggesting new approaches to the treatment of the syndromes of elevated FGF23 (Bhadada et al. 2013). The mechanism remains unclear.
Vitamin D
Arguably, the effect of vitamin D on phosphate metabolism is the least understood of the three hormonal systems discussed here. Circulating active vitamin D metabolites are largely a result of renal production of the hormone, which is regulated by a number of factors including PTH, FGF23, phosphate, calcium, acid–base balance, and the hormone itself (Maiti & Beckman, 2007; Bikle et al. 2013). 1,25-dihydroxyvitamin D3 activates both osteoblast and osteoclast differentiation suggesting the potential for decreasing or increasing serum phosphate. Likewise, in the kidney, studies have produced conflicting results suggesting either an increase or a decrease in phosphate transport. The genes for both Npt2a and Npt2c express potential vitamin D receptor (VDR) binding elements, suggesting the potential for regulation at the mRNA level; however, no clear-cut evidence for vitamin D-dependent regulation of these proteins has been produced (Kido et al. 2013). Vitamin D receptor null animals fail to develop expression ofNpt2a and Npt2c in the kidney (Capuano et al. 2005); however, this defect can be ameliorated by feeding the animals a high calcium diet, suggesting that the absence of the vitamin D receptor is not responsible for failure to express the renal phosphate transporters. The VDR deficient animals have high PTH levels that decrease with the high calcium diet, providing the most likely explanation for the absence of expression of Npt2a and Npt2c. The effect of vitamin D on the intestine is clearly absorptive; however, the mechanism remains unclear with studies suggesting that vitamin D increases Npt2b expression, but that this effect is specific to both the intestinal segment and species. The upregulation of Npt2b seen with a low phosphate diet is vitamin D receptor independent (Brown et al. 2012). Additionally, the effect of vitamin D on Npt2b expression appears to be age dependent, in that very young animals show a significant increase in Npt2b mRNA and protein expression in response to vitamin D administration, while older animals do not. Studies of intestinal phosphate transport reveals significant species variability in phosphate handling over the several segments of the small intestine, preventing generalization of findings over different model systems (Marks et al. 2010). Overall, high active vitamin D levels are associated with hyperphosphataemia while low active vitamin D levels are associated with hypophosphataemia, but generally only at extremes of vitamin D expression. In contrast, serum calcium levels appear to be much more sensitive to vitamin D levels, as disorders such as sarcoidosis, which is characterized by extra-renal active vitamin D production, exhibit hypercalcaemia but not hyperphosphataemia (Motoyama, 2002). How much of the effect of vitamin D on serum phosphate is due to the direct action of vitamin D and how much is due to secondary effects on PTH and FGF23 are unanswered questions (Prie & Friedlander, 2010). Higher vitamin D levels will suppress PTH secretion directly and through elevation of serum calcium, thus decreasing renal excretion of phosphate. High vitamin D may also decrease FGF23 levels, further limiting phosphate excretion. Conversely, lower vitamin D levels are associated with higher PTH and FGF23 levels, enhanced renal phosphate excretion and lower serum phosphate levels.
The relative strengths of the effects of PTH and FGF23 on vitamin D are not clear. In the pathological state of chronic kidney disease, both PTH and FGF23 are elevated. FGF23 levels increase at the early stages, resulting in the early decrease in 1,25-dihydroxyvitamin D3 levels. PTH levels rise later in the course of the disease but do not restore 1,25 levels. Whether this is due to end organ resistance, even higher FGF23 levels resulting in greater suppression, or reduced kidney mass in the later stages of kidney disease has not been determined.
Klotho
Klotho is a glucuronidase that exists in tissue bound and circulating forms (Hu et al. 2010). This single transmembrane protein associates with FGF receptors in the kidney, enabling FGF23 binding and inhibition of phosphate transport. In parathyroid gland, tissue bound klotho plays a similar role in mediating FGF23 regulation of PTH mRNA and protein synthesis. The circulating forms of klotho include an alternatively spliced form of klotho as well as a cleaved form from the tissue bound protein. The absence of klotho leads to a syndrome similar to FGF23 deficiency, vis à vis, phosphate homeostasis, i.e. hyperphosphataemia with an absence of phosphaturia (Sakan et al. 2014). Klotho, however, has effects independent of FGF23 including salutary effects on lipid and glucose metabolism, oxidative stress, and other manifestations of ageing (Streicher et al. 2012; Kuro-o, 2013). The relative contributions of tissue bound and circulating forms of klotho have not been determined. The isolated effects of either FGF23 or klotho on phosphate metabolism are also debated. The most convincing data suggesting that FGF23 requires klotho as a cofactor for its phosphaturic effects stems from the findings in chronic kidney disease where, early in the course of the illness, klotho levels diminish while FGF23 levels increase. Serum phosphate levels are maintained within the normal range in early chronic kidney disease through elevated FGF23 levels. In isolated kidney proximal tubule cells, FGF23 decreases phosphate transport only in the presence of klotho or other enzymatic manipulations, suggesting an absolute requirement. Isolated cell culture experiments suggest that klotho can cause phosphaturia, through modification of glycosyl groups on Npt2a to decrease surface expression, and thus may exert independent phosphaturic actions.
How are the signals integrated?
All three hormones affect both phosphate and calcium metabolism independently but are highly interrelated. PTH functions predominantly as a calcium regulating hormone with secondary effects on phosphate homeostasis (Nissenson & Juppner, 2013). FGF23 functions predominantly as a phosphate regulating hormone with secondary effects on calcium homeostasis (Rodriguez-Ortiz et al. 2012; Quinn et al. 2013). In this scenario, the contrasting effects on vitamin D metabolism appear to be the wild card, the critical determinant of the differences in the effect on phosphate homeostasis (Prie & Friedlander, 2010). Phosphate itself has effects on the hormone pathways (Ito et al. 2013). The effects of this intrinsic feedback system can be difficult to predict.
Primary disease of the kidney, gut or bone affects serum phosphate through both direct and indirect actions on the major hormonal regulators. Chronic kidney disease is associated with global dysregulation of phosphate metabolism, culminating in the development of sustained hyperphosphataemia in the final stages of kidney disease. The original ‘trade-off hypothesis’ articulated by Neal Bricker (1972) in the 1970s suggested that a reduction in the number of nephrons in early kidney failure necessitated enhanced phosphate excretion by the remaining nephrons, accomplished by increasing levels of PTH (Loghman-Adham, 1997). We know now that primary kidney failure leads to abnormalities in all hormones involved in phosphate homeostasis, starting first with a reduction in klotho expression, followed by an increase in FGF23 and simultaneous decrease in the expression of 1,25-dihydroxyvitamin D3 and finally by an increase in PTH at which time serum phosphate begins to rise, escaping all regulatory processes (Gutierrez, 2010; Pavik et al. 2013). There have been many excellent reviews of this subject. A major unanswered question is why klotho expression decreases so early in the disease, when one might anticipate that its expression would increase to enhance phosphaturia. This unfortunate response is also likely to play a significant role in the accelerated ageing phenotype of chronic kidney disease (Kuro-O, 2013). The profound effects of kidney failure on phosphate homeostasis serve to reinforce the critical role of the kidney in maintenance of phosphate homeostasis.
Other factors regulating serum phosphate level
PTH, vitamin D, and the FGF23–klotho system have preeminent roles in the regulation of phosphate homeostasis at a whole organism level, determining total intake, distribution, and excretion primarily through effects on epithelial transport. Several other factors play a role in the regulation of serum phosphate concentration, but how these factors interact with the classic PTH–vitamin D–FGF23 axis is an almost untouched area of investigation.
Intrinsic factors
Serum phosphate shows a clear circadian rhythm with peaks at mid-afternoon and shortly after midnight in human beings and a nadir at 08.00–10.00 h. This diurnal rhythm has no discernible relation to the phosphaturic effects of PTH or FGF23A as the decrease in serum phosphate between 04.00 and 08.00 h is not associated with an increase in the fractional excretion of phosphate (Bielesz et al. 2006. Dietary phosphate has minimal effect on this circadian rhythm. A study from Portale and coworkers (1987) demonstrated that baseline 08.00 h phosphate level was relatively similar between individuals on a normal or a high phosphate diet but the swings in serum phosphate concentration were wider though qualitatively the same in both groups. A group of individuals ingesting a low phosphate diet demonstrated a lower baseline serum phosphate level at 08.00 h and significantly blunted serum phosphate changes over the day. Interestingly, the decrease in 08.00 h phosphate level required more than 24 h of low phosphate ingestion to occur. Diurnal rhythms for all of the hormones associated with mineral metabolism have been demonstrated (McElderry et al. 2013). A recent study in rodents demonstrated a nearly circadian rhythm for bone mineralization (25h cycle) following fluctuations in the activity of the Clock gene Per1. The physiological and clinical significance of this rhythmic variability in all facets of mineral metabolism is unknown but suggests that variations in this process could produce variations in individual serum phosphate, perhaps linking bone mineralization and cardiovascular risk.
Serum phosphate varies with age, the highest levels occurring in the neonatal period and early infancy, falling rapidly through puberty and adulthood (Keating et al. 1969; Cirillo et al. 2010). The high serum phosphate in childhood is critical for normal bone formation, as evidenced by the fact that human forms of hypophosphataemic rickets result in significant bone disease if untreated (Tiosano & Hochberg, 2009). Phosphate supplementation in people as well as in animal models of rickets rescues the bone phenotype. The high serum phosphate seen in children corresponds to high kidney expression of sodium–phosphate cotransporters and robust bone formation. Obviously, the elevated phosphate level seen in babies does not confer a cardiovascular risk as it does in adults. The explanation for this discrepancy is not known but again suggests a link between bone metabolism and cardiovascular risk.
Sex differences in serum phosphate have been noted for decades and recent studies have shed light on the mechanisms and potential implications for bone metabolism and cardiovascular disease. As early as the 1960s it was recognized that serum phosphorus levels in men and women differed with progressive ageing. From age 20 to 60+, men experience a progressive decline in serum phosphate. In contrast, women show a similar decline in serum phosphate when pre-menopausal but after menopause show an increase (Keating, 1969). That this post-menopausal increase in phosphate is related to the loss of oestrogen effect is supported by the observation that post-menopausal women taking oestrogen replacement therapy have lower serum phosphate than women who are not taking oestrogens (Winkelman et al. 1973; Zhang et al. 2014). Experimental animal studies have demonstrated that oestrogen decreases the expression of kidney sodium–phosphate cotransporters (Guttmann-Rubinstein, 2010). These findings spark several speculations. Is the oestrogen effect in women responsible for both the lower bone mass and the lower cardiovascular risk in younger pre-menopausal women? Is the high serum phosphate in women after menopause a marker of cardiovascular risk as a consequence to low oestrogen or is the higher serum phosphate actually causative? The absence of salutary cardiovascular effects of oestrogen in post-menopausal women would argue against a cause and effect relationship in this setting.
Genetic contributions
A recent genome-wide association study identified several regions of the genome where single nucleotide polymorphisms (SNPs) correlated with serum phosphate in individuals of European ancestry (Kestenbaum et al. 2010). These sites showed no gender differences and remained significant in individuals with a normal epidermal growth factor receptor (eGFR). The contribution of each SNP was additive but in toto accounted for only about 1.5% of the variability in serum phosphate. Interestingly, although the SNPs fell in genomic regions encompassing genes encoding for proteins that might be expected to play a role in phosphate homeostasis such as alkaline phosphatase, Npt2a, the calcium sensing receptor, and FGF23, the SNPs were not in those genes but in nearby regions. The significance of these findings remains completely unknown.
Mutations in the sodium–phosphate cotransporters have been associated with disease and with alterations in serum phosphate (Lederer & Miyamoto, 2012). Most striking is the syndrome of hereditary hypophosphataemic rickets with hypercalciuria for which mutations in Npt2c, the type IIc sodium–phosphate cotransporter, have been identified. Mutations in Npt2a have been associated with osteoporosis and nephrolithiasis in the setting of significant phosphate wasting, but, with the exception of a severe mutation causing Fanconi syndrome, have not been associated with significant changes in serum phosphate. These observations in human subjects contrast significantly with what is seen in rodent models where Npt2a is responsible for 70% of the proximal tubule phosphate reabsorption, while Npt2c exerts a much lesser influence (Iwaki et al. 2008; Segawa et al. 2009). Mice deficient in Npt2c have in essence no distinct phenotype and are able to maintain phosphate homeostasis relatively normally. Mice deficient in Npt2a, however, exhibit persistent hypophosphataemia, urine phosphate wasting, hypercalciuria, and kidney stone formation (Beck et al. 1998). The clear differences between rodents and humans in the phenotypic presentation in the setting of the two transporters suggest that Npt2c may play a greater role in phosphate homeostasis in humans than in rodents. Mutations in the sodium hydrogen exchanger regulatory factor isoform 1, NHERF1, a critical scaffolding protein for Npt2a, have also been associated with abnormal phosphate handling and clinical syndromes similar to Npt2a mutations (Karim et al. 2008; Courbebaisse et al. 2012). Mutations in Npt2b, the type II sodium–phosphate cotransporter expressed in the small intestine, has been associated with calcifications in several organs but not with low serum phosphate (Lederer & Miyamoto, 2012). The effect on phosphate handling overall has not been studied in humans but intestinal Npt2b deficiency in animals causes a 50% decrease in intestinal phosphate absorption. Whether variations in the function of these transporters play a significant role in regulation of serum phosphate under normal physiological conditions remains unknown.
Diet
Dietary phosphate content affects serum phosphate, but most obviously at extremes. In the previously referenced Portale study (1987) comparing diurnal variation in phosphate in individuals ingesting normal, high, and low phosphate diet, the 08.00 h serum phosphate was similar between individuals on the normal and high phosphate diets but substantially lower in those taking the low phosphate diet, 500 mg daily. Many case reports also document a decrease in serum phosphate in individuals ingesting large quantities of calcium supplements, presumably through intestinal phosphate binding. In contrast, fasting does not cause a decrease in phosphate and individuals with very low calorie ingestion or frank malnutrition such as alcoholics or prisoners of war may have a normal serum phosphate (Felsenfeld et al. 2000). Why selective decrease in phosphate reabsorption with phosphate binders would cause severe hypophosphataemia but global nutrient deficiency would not is not entirely understood but may have to do with stimulation of insulin secretion in the former but not in the latter, which would cause phosphate entry into cells, as well as the discrete effect of the phosphate binders specifically on intestinal phosphate transport. Severely malnourished individuals may exhibit profound hypophosphataemia with feeding, the so-called refeeding syndrome (Knochel, 1980; Skipper, 2012).
Not only is total dietary phosphate important, but so is the form of phosphate. Phosphate contained in plant-derived foods appears to be significantly less bioavailable than that in animal-derived foods or heavily processed foods. In a recent study, individuals ingesting a largely vegetarian diet had lower serum phosphate and excreted less phosphate than those on an animal product diet, despite the fact that both diets contained similar phosphate content, suggesting less intestinal absorption of phosphate (Moe et al. 2011). An alternative explanation could be the contrasting effect of these diets on acid–base balance. Vegetarian diets yield significantly less net acid load than animal-based diets. Alkalosis tends to cause phosphate uptake into cells and decreased renal phosphate excretion, a factor that might have contributed to the results of this study. The effect of dietary phosphate and other nutrients on phosphate homeostasis has not been extensively evaluated.
A key unanswered question regarding dietary phosphate is: is dietary phosphate content a contributor to progressive kidney disease or accelerated ageing or is the hormonal/metabolic response to dietary phosphate metabolically more harmful than the diet itself?
Other hormones
Dietary phosphate stimulates renal dopamine synthesis, which in turn decreases phosphate reabsorption by proximal tubule (Perrichot et al. 1995; Bansal et al. 2012). The signal initiating the dopamine synthesis is unknown (Sizova et al. 2013). Abnormalities in proximal tubule dopamine signalling have been implicated in some forms of hypertension and in ageing. Whether these changes translate into altered renal proximal tubule phosphate handling has not been examined. Oestrogen, as indicated previously in the discussion of gender differences in phosphate metabolism, also decreases renal phosphate reabsorption. Angiotensin II stimulates phosphate absorption by proximal tubule (Xu et al. 2004). Likewise insulin like growth factor stimulates phosphate transport (Hammerman et al. 1984; Caverzasio & Bonjour, 1989; Abraham et al. 1990; Caverzasio et al. 1990). In diabetes, phosphaturia has been observed and may contribute to some of the complications of this disease (Hough, 1987). Thyroid hormone plays an important role in the development of the expression of phosphate transporters in the young as well as in regulation of phosphate transport in the adult animal (Alcalde et al. 1999; Beers & Dousa, 1993). The contribution of these other hormones to phosphate homeostasis and serum phosphate concentration has not been adequately studied.
Miscellaneous
Shifts of phosphate from the intracellular to the extracellular space may occur under a variety of conditions. Rhabdomyolysis and tumour lysis are two examples, resulting from massive cell destruction. On the other hand a shift of phosphate from the extracellular to the intracellular space can be seen with endogenous or exogenous insulin, as alluded to above, or with respiratory alkalosis (Hoppe et al. 1982).
Conclusion
Many questions regarding phosphate homeostasis and the regulation of serum phosphate are raised in this review and many have not even been addressed (see Table 2). How are the functions of the gut, kidney, bone, and parathyroid regulated in an integrated fashion on a day-to-day basis? Evidence from studies of phosphate homeostasis in acute liver injury suggest that there may be additional hormonal factors regulating phosphate homeostasis (Nomura et al. 2014). How is tissue, cell, or serum phosphate sensed? What is responsible for the diurnal, age- and gender-related differences in serum phosphate concentration? A phosphate sensor has not been identified, either for intracellular or extracellular phosphate, though many candidate proteins have been suggested, including some of the known sodium–phosphate cotransporters and intracellular metabolites. Some have even suggested that it is not phosphate that is sensed but the calcium × phosphate product. These considerations go well beyond the theoretical. How do we define a ‘normal’ serum phosphate level, if serum phosphate is influenced by endogenous factors such as age, gender, and the status of multiple hormonal factors as well as exogenous factors such as time of day, season, and diet. In clinical practice, how should we account for these factors? In clinical trials, how are we controlling for them? These and many other issues remain challenges for scientists and clinicians alike in attempting to understand the significance of the serum phosphate level in health and disease.
Table 2.
Unanswered questions in phosphate homeostasis
| Questions | Commentary |
|---|---|
| Is there a phosphate sensor that regulates serum phosphate level? | If so, how does it accommodate changes in age, gender, time of year, time of day? |
| What drives the circadian rhythm of serum phosphate? | How much of the cycling is diet driven? How much is a reflection of circadian rhythms in hormones? Can it be disrupted, and if so, what are the consequences? |
| How does serum phosphate relate to bone metabolism? | Serum phosphate is highest in infancy and lowest in old age, reflecting bone metabolism. Is serum phosphate, in fact, simply a reflection of bone metabolism? |
| How do the mechanisms for total body phosphate homeostasis and the mechanisms regulating the intracellular–extracellular balance of phosphate interact with each other? | To what extent do homeostatic processes influence serum phosphate? Why does serum phosphate at times not reflect total body phosphate stores? |
| To what extent does serum phosphate reflect dietary phosphate? | Can we prescribe appropriate parameters for dietary phosphate? |
| How are the functions of the major hormones that regulate phosphate homeostasis integrated? | What are the signals between organ systems and is there a ‘master’ regulator? |
This table presents a list of some of the more important areas of uncertainty in our understanding of phosphate homeostasis and regulation of serum phosphate concentration.
Glossary
- EGFR
epidermal growth factor receptor
- FGF23
fibroblast growth factor 23
- NHERF1
sodium–hydrogen exchanger regulatory factor isoform 1
- PTH
parathyroid hormone
- SNP
single nucleotide polymorphism
- VDR
vitamin D receptor
Additional information
Competing interests
None to declare.
Funding
This work was funded by a grant from the VA Merit Review Board to Dr Lederer, 2 IO1 BX000610-05A1.
References
- Abraham MI, McAteer JA, Kempson SA. Insulin stimulates phosphate transport in opossum kidney epithelial cells. Am J Physiol Renal Physiol. 1990;258:F114–F121. doi: 10.1152/ajprenal.1990.258.6.F1592. [DOI] [PubMed] [Google Scholar]
- ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- Alcalde AI, Sarasa M, Raldua D, Aramayona J, Morales R, Biber J, Murer H, Levi M, Sorribas V. Role of thyroid hormone in regulation of renal phosphate transport in young and aged rats. Endocrinology. 1999;140:1544–1551. doi: 10.1210/endo.140.4.6658. [DOI] [PubMed] [Google Scholar]
- Ba J, Friedman PA. Calcium-sensing receptor regulation of renal mineral ion transport. Cell Calcium. 2004;35:229–237. doi: 10.1016/j.ceca.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Bansal N, Hsu C-y, Whooley M, Berg AH, Ix JH. Relationship of urine dopamine with phosphorus homeostasis in humans: the Heart and Soul Study. Am J Nephrol. 2012;35:483–490. doi: 10.1159/000338483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci U S A. 1998;95:5372–5377. doi: 10.1073/pnas.95.9.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers KW, Dousa TP. Thyroid hormone stimulates the Na+–PO4 symporter but not the Na+–SO4 symporter in renal brush border. Am J Physiol Renal Physiol. 1993;265:F323–F326. doi: 10.1152/ajprenal.1993.265.2.F323. [DOI] [PubMed] [Google Scholar]
- Bhadada SK, Palnitkar SJ, Qiu S, Parikh N, Talpos GB, Rao SD. Deliberate total parathyroidectomy: a potentially novel therapy for tumor-induced hypophosphatemic osteomalacia. J Clin Endocrinol Metab. 2013;98:4273–4278. doi: 10.1210/jc.2013-2705. [DOI] [PubMed] [Google Scholar]
- Bielesz B, Bacic D, Honegger K, Biber J, Murer H, Wagner CA. Unchanged expression of the sodium-dependent phosphate cotransporter NaPi-IIa despite diurnal changes in renal phosphate excretion. Pflugers Arch. 2006;452:683–689. doi: 10.1007/s00424-006-0087-0. [DOI] [PubMed] [Google Scholar]
- Bikle D, Adams JS. Vitamin D: production, metabolism, mechanism of action and clinical requirements. In: Rosen CJ, Christakos S, editors. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th edn. Washington, DC, USA: American Society for Bone and Mineral Research; 2013. pp. 235–248. [Google Scholar]
- Blaine J, Weinman EJ, Cunningham R. The regulation of renal phosphate transport. Adv Chronic Kidney Dis. 2011;18:77–84. doi: 10.1053/j.ackd.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Bricker N. On the pathogenesis of the uremic state. An exposition of the ‘trade off hypothesis’. N Engl J Med. 1972;286:1093–1099. doi: 10.1056/NEJM197205182862009. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Zhang F, Ritter CS. The vitamin D analog ED-71 is a potent regulator of intestinal phosphate absorption and NaPi-IIb. Endocrinology. 2012;153:5150–5156. doi: 10.1210/en.2012-1587. [DOI] [PubMed] [Google Scholar]
- Capasso G, Geibel PJ, Damiano S, Jaeger P, Richards WG, Geibel JP. The calcium sensing receptor modulates fluid reabsorption and acid secretion in the proximal tubule. Kidney Int. 2013;84:277–284. doi: 10.1038/ki.2013.137. [DOI] [PubMed] [Google Scholar]
- Capuano P, Radanovic T, Wagner CA, Bacic D, Kato S, Uchiyama Y, St-Arnoud R, Murer H, Biber J. Intestinal and renal adaptation to a low-Pi diet of type II NaPi cotransporters in vitamin D receptor- and 1αOHase-deficient mice. Am J Physiol Cell Physiol. 2005;288:C429–C434. doi: 10.1152/ajpcell.00331.2004. [DOI] [PubMed] [Google Scholar]
- Caverzasio J, Bonjour JP. Insulin-like growth factor I stimulates Na-dependent Pi transport in cultured kidney cells. Am J Physiol Renal Physiol. 1989;257:F712–F717. doi: 10.1152/ajprenal.1989.257.5.F712. [DOI] [PubMed] [Google Scholar]
- Caverzasio J, Montessuit C, Bonjour JP. Stimulatory effect of insulin-like growth factor-1 on renal Pi transport and plasma 1,25-dihydroxyvitamin D3. Endocrinology. 1990;127:453–459. doi: 10.1210/endo-127-1-453. [DOI] [PubMed] [Google Scholar]
- Christensen SE, Nissen PH, Vestergaard P, Heickendorff L, Rejnmark L, Brixen K, Mosekilde L. Plasma 25-hydroxyvitamin D, 1,25-dihydroxyvitamin D, and parathyroid hormone in familial hypocalciuric hypercalcemia and primary hyperparathyroidism. Eur J Endocrinol. 2008;159:719–727. doi: 10.1530/EJE-08-0440. [DOI] [PubMed] [Google Scholar]
- Cirillo M, Bilancio G, Marcarelli F. Ageing and changes in phosphate transport: clinical implications. J Nephrol. 2010;23(Suppl. 16):S152–S157. [PubMed] [Google Scholar]
- Courbebaisse M, Leroy C, Bakouh N, Salaun C, Beck L, Grandchamp B, Planelles G, Hall RA, Friedlander G, Prie D. A new human NHERF1 mutation decreases renal phosphate transporter NpT2a expression by a PTH-independent mechanism. PLoS One. 2012;7:e34764. doi: 10.1371/journal.pone.0034764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellam TJ, Chico TJ. Phosphate: the new cholesterol? The role of the phosphate axis in nonuremic vascular disease. Atherosclerosis. 2012;220:310–318. doi: 10.1016/j.atherosclerosis.2011.09.002. [DOI] [PubMed] [Google Scholar]
- Ellam T, Wilkie M, Chamberlain J, Crossman D, Eastman R, Francis S, Chico TJ. Dietary phosphate modulates atherogenesis and insulin resistance in apolipoprotein E knockout mice – brief report. Arterioscler Thromb Vasc Biol. 2011;31:1988–1990. doi: 10.1161/ATVBAHA.111.231001. [DOI] [PubMed] [Google Scholar]
- Felsenfeld AJ, Jara A, Avedian G, Kleeman CR. Effects of fasting, feeding, and bisphosphonate administration on serum calcitriol levels in phosphate-deprived rats. Kidney Int. 2000;58:1016–1022. doi: 10.1046/j.1523-1755.2000.00259.x. [DOI] [PubMed] [Google Scholar]
- Gutierrez OM. Fibroblast growth factor 23 and disordered vitamin D metabolism in chronic kidney disease: updating the ‘trade-off’ hypothesis. Clin J Am Soc Nephrol. 2010;5:1710–1716. doi: 10.2215/CJN.02640310. [DOI] [PubMed] [Google Scholar]
- Guttmann-Rubinstein L, Lichtstein D, Ilani A, Gal-Moscovici A, Scherzer P, Rubinger D. Evidence of a parathyroid hormone-independent chronic effect of estrogen on renal phosphate handling and sodium-dependent phosphate cotransporter type IIa expression. Horm Metab Res. 2010;42:230–236. doi: 10.1055/s-0029-1246182. [DOI] [PubMed] [Google Scholar]
- Hammerman MR, Rogers S, Hansen VA, Gavin JR., 3rd Insulin stimulates Pi transport in brush border vesicles from proximal tubular segments. Am J Physiol Endocrinol Metab. 1984;247:E616–E624. doi: 10.1152/ajpendo.1984.247.5.E616. [DOI] [PubMed] [Google Scholar]
- Hoppe A, Metler M, Berndt TJ, Knox FG, Angielski S. Effect of respiratory alkalosis on renal phosphate excretion. Am J Physiol Renal Physiol. 1982;243:F471–F475. doi: 10.1152/ajprenal.1982.243.5.F471. [DOI] [PubMed] [Google Scholar]
- Hough FS. Alterations of bone and mineral metabolism in diabetes mellitus. Part II. Clinical studies in 206 pateints with Type I diabetes mellitus. S Afr Med J. 1987;72:120–126. [PubMed] [Google Scholar]
- Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, Kuro-o M, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010;24:3438–3450. doi: 10.1096/fj.10-154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa S, Austin AM, Gray AK, Econs MJ. A Phex mutation in a murine model of X-linked hypophosphatemia alters phosphate responsiveness of bone cells. J Bone Miner Res. 2012;27:453–460. doi: 10.1002/jbmr.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa T, Sandoval-Cooper MJ, Tenenhouse HS, Castellino FJ. A missense mutation in the sodium phosphate co-transporter Slc34a1 impairs phosphate homeostasis. J Amer Soc Nephrol. 2008;19:1753–1762. doi: 10.1681/ASN.2007121360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito N, Findlay DM, Anderson PH, Bonewald LF, Atkins GJ. Extracellular phosphate modulates the effect of 1α,25-dihydroxy vitamin D3 (1,25D) on osteocyte like cells. J Steroid Biochem Mol Biol. 2013;136:183–186. doi: 10.1016/j.jsbmb.2012.09.029. [DOI] [PubMed] [Google Scholar]
- Karim Z, Gerard B, Bakouh N, Alili R, Leroy C, Beck L, Silve C, Planelles G, Urena-Torres P, Grandchamp B, et al. NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med. 2008;359:1128–1135. doi: 10.1056/NEJMoa0802836. [DOI] [PubMed] [Google Scholar]
- Kawata T, Imanishi Y, Kobayashi K, Miki T, Arnold A, Inaba M, Nishizawa Y. Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol. 2007;18:2683–2688. doi: 10.1681/ASN.2006070783. [DOI] [PubMed] [Google Scholar]
- Keating FR, Jr, Jones JD, Elveback LR, Randall RV. The relation of age and sex to distribution of values in healthy adults of serum calcium, inorganic phosphorus, magnesium, alkaline phosphatase, total proteins, albumin and blood urea. J Lab Clin Med. 1969;73:825–834. [PubMed] [Google Scholar]
- Kendrick J, Kestenbaum B, Chonchol M. Phosphate and cardiovascular disease. Adv Chronic Kidney Dis. 2011;18:113–119. doi: 10.1053/j.ackd.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kestenbaum B, Glazer NL, Köttgen A, Felix JF, Hwang SJ, Liu Y, Lohman K, Kritchevsky SB, Hausman DB, Petersen AK, et al. Common genetic variants associate with serum phosphorus concentration. J Am Soc Nephrol. 2010;21:1223–1232. doi: 10.1681/ASN.2009111104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kido S, Kaneko I, Tatsumi S, Segawa H, Miyamoto K-I. Vitamin D and type II sodium-dependent phosphate cotransporters. Contrib Nephrol. 2013;180:86–97. doi: 10.1159/000346786. [DOI] [PubMed] [Google Scholar]
- Knochel JP. Hypophosphatemia in the alcoholic. Arch Int Med. 1980;140:613–615. [PubMed] [Google Scholar]
- Krajisnik T, Bjorkland P, Marsell R, Ljunggren O, Akerstrom G, Jonsson KB, Westin G, Larssen TE. 1,25. J Endocrinol. 2007;195:125–131. doi: 10.1677/JOE-07-0267. [DOI] [PubMed] [Google Scholar]
- Kuro-o M. Klotho, phosphate and FGF-23 in ageing and disturbed mineral metabolism. Nat Rev Nephrol. 2013;9:650–660. doi: 10.1038/nrneph.2013.111. [DOI] [PubMed] [Google Scholar]
- Lavi-Moshayoff V, Silver J, Naveh-Many T. Human PTH gene regulation in vivo using transgenic mice. Am J Physiol Renal Physiol. 2009;297:F713–F719. doi: 10.1152/ajprenal.00161.2009. [DOI] [PubMed] [Google Scholar]
- Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol. 2010;299:F882–F889. doi: 10.1152/ajprenal.00360.2010. [DOI] [PubMed] [Google Scholar]
- Lederer E, Miyamoto K. Clinical consequences of mutations in sodium phosphate cotransporters. Clin J Am Soc Nephrol. 2012;7:1179–1187. doi: 10.2215/CJN.09090911. [DOI] [PubMed] [Google Scholar]
- Loghman-Adham M. Adaptations to changes in dietary phosphorus in health and in renal failure. J Lab Clin Med. 1997;129:176–188. doi: 10.1016/s0022-2143(97)90137-2. [DOI] [PubMed] [Google Scholar]
- McElderry J-DP, Zhao G, Khmaladze A, Wilson CG, Franceschi RT, Morris MD. Tracking circadian rhythms of bone mineral deposition in murine calvarial organ cultures. J Bone Miner Res. 2013;28:1846–1854. doi: 10.1002/jbmr.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti A, Beckman MJ. Extracellular calcium is a direct effector of VDR levels in proximal tubule epithelial cells that counterbalances effects of PTH on renal vitamin D metabolism. J Steroid Biochem Mol Biol. 2007;107:504–508. doi: 10.1016/j.jsbmb.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Marks J, Debnam ES, Unwin RJ. Phosphate homeostasis and the renal–gastrointestinal axis. Am J Physiol Renal Physiol. 2010;299:F285–F296. doi: 10.1152/ajprenal.00508.2009. [DOI] [PubMed] [Google Scholar]
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev. 2012;92:131–155. doi: 10.1152/physrev.00002.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moe SM, Zidehsarai MP, Chambers MA, Jackman LA, Radcliffe JS, Trevino LL, Donahue SE, Asplin JR. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Amer Soc Nephrol. 2011;6:257–264. doi: 10.2215/CJN.05040610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama K, Inaba M, Emoto M, Morii H, Nishizawa Y. Sarcoidosis initially manifesting as symptomatic hypercalcemia with the absence of organic involvement. Intern Med. 2002;41:449–452. doi: 10.2169/internalmedicine.41.449. [DOI] [PubMed] [Google Scholar]
- Murray RD, Holthouser K, Clark BJ, Salyer SA, Barati MT, Khundmiri SJ, Lederer ED. Parathyroid hormone (PTH) decreases type IIa sodium phosphate cotransporter (Npt2a) mRNA stability. Am J Physiol Renal Physiol. 2013;304:F1076–F1084. doi: 10.1152/ajprenal.00632.2012. [DOI] [PubMed] [Google Scholar]
- Nissenson RA. Parathyroid hormone. In: Rosen CJ, Juppner H, editors. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th edn. Washington, DC, USA: American Society for Bone and Mineral Research; 2013. pp. 208–214. [Google Scholar]
- Nomura K, Tatsumi S, Miyagawa A, Shiozaki Y, Sasaki S, Kaneko I, Ito M, Kido S, Segawa H, Sano M, Fukuwatari T, Shibata K, Miyamoto K-I. Hepatectomy-related hypophosphatemia: a novel phosphaturic factor in the liver–kidney axis. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2013060569. DOI: 10.1681/ASN.2013060569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavik I, Jaeger P, Ebner L, Wagner CA, Petzold K, Spichtig D, Poster D, Wuthrich RP, Russman S, Serra AL. Secreted klotho and FGF23 in chronic kidney disease stage 1 to 5: a sequence suggested from a cross-sectional study. Nephrol Dial Transplant. 2013;28:352–359. doi: 10.1093/ndt/gfs460. [DOI] [PubMed] [Google Scholar]
- Perrichot R, Garcia-Ocana A, Couette S, Comoy E, Amiel C, Friedlander G. Locally formed dopamine regulates renal Na–Pi co-transport through DA1 and DA2 receptors. Biochem J. 1995;312:433–437. doi: 10.1042/bj3120433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portale AA, Halloran BP, Morris RC., Jr Dietary intake of phosphorus modulates the circadian rhythm in serum concentration of phosphorus. implications for the renal production of 1,25 dihydroxyvitamin D. J Clin Invest. 1987;80:1147–1154. doi: 10.1172/JCI113172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prie D, Friedlander G. Reciprocal control of 1,25-dihydroxyvitamin D and FGF23 formation involving the FGF23/Klotho system. Clin J Am Soc Nephrol. 2010;5:1717–1722. doi: 10.2215/CJN.02680310. [DOI] [PubMed] [Google Scholar]
- Quinn SJ, Thomsen ARB, Pang JL, Kantham L, Brauner-Osborne H, Pollak M, Goltzman D, Brown EM. Interactions between calcium and phosphorus in the regulation of the production of fibroblast growth factor 23 in vivo. Am J Physiol Endocrinol Metab. 2013;304:E310–E320. doi: 10.1152/ajpendo.00460.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi D, Traebert M, Ward DT, Kaissling B, Biber J, Hebert SC, Murer H. Dietary phosphate and parathyroid hormone alter the expression of the calcium-sensing receptor (CaR) and the Na+-dependent Pi transporter (NaPi-2) in the rat proximal tubule. Pflugers Archiv. 2000;441:379–387. doi: 10.1007/s004240000436. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Ortiz ME, Lopez I, Munoz-Castaneda JU, Martinez-Moreno JM, Ramirez AP, Pineda C, Canalejo A, Jaeger P, Aguilera-Tejero E, Rodriguez M, Felsenfeld A, Almaden Y. Calcium deficiency reduces circulating levels of FGF23. J Am Soc Nephrol. 2012;23:1190–1197. doi: 10.1681/ASN.2011101006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakan H, Nakatani K, Asai O, Imura A, Tanaka T, Yoshimoto S, Iwamoto N, Kurumatani N, Iwano M, Nabeshima Y, Konishi N, Saito Y. Reduced renal α-Klotho expression in CKD patients and its effect on renal phosphate handling and vitamin D metabolism. PLoS One. 2014;9:e86301. doi: 10.1371/journal.pone.0086301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa H, Onitsuka A, Furutani J, Kaneko I, Aranami F, Matsumoto N, Tomoe Y, Kuwahata M, Ito M, Matsumoto M, Li M, Amizuka N, Miyamoto K. Npt2a and Npt2c in mice play distinct and synergistic roles in inorganic phosphate metabolism and skeletal development. Am J Physiol Renal Physiol. 2009;297:F671–F678. doi: 10.1152/ajprenal.00156.2009. [DOI] [PubMed] [Google Scholar]
- Sizova D, Velasquez H, Sampaio-Maia B, Quelhas-Santos J, Pestana M, Desir G. Renalase regulates renal dopamine and phosphate metabolism. Am J Physiol Renal Physiol. 2013;305:F839–F844. doi: 10.1152/ajprenal.00616.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J. FGF23 and the parathyroid. In: Kuro-o M, Naveh-Many T, editors. Endocrine FGFs and Klothos. Springer; 2012. pp. 92–99. [Google Scholar]
- Skipper A. Refeeding syndrome or refeeding hypophosphatemia: a systematic review of cases. Nutr Clin Pract. 2012;27:34–40. doi: 10.1177/0884533611427916. [DOI] [PubMed] [Google Scholar]
- Streicher C, Zeitz U, Andrukhova O, Rupprecht A, Polh E, Larsson TE, Windisch W, Lanske B, Erben RG. Long-term FGF23 deficiency does not influence aging, glucose homeostasis, or fat metabolism in mice with a nonfunctioning vitamin D receptor. Endocrinology. 2012;153:1795–1805. doi: 10.1210/en.2011-1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiosano T, Hochberg Z. Hypophosphatemia: the common denominator of all rickets. J Bone Miner Metab. 2009;27:392–401. doi: 10.1007/s00774-009-0079-1. [DOI] [PubMed] [Google Scholar]
- Winkelman JW, Cannon DC, Pileggi VJ, Reed AH. Estimation of norms from a controlled sample survey. II. influence of body habitus, oral contraceptives, and other factors on values for the normal range derived from the SMA 12/60 screening group of tests. Clin Chem. 1973;19:488–491. [PubMed] [Google Scholar]
- Witteveen JE, van Lierop AH, Papapoulos SE, Hamdy NAT. Increased circulating levels of FGF23: an adaptive response in primary hyperparathyroidism. Eur J Endocrinol. 2012;166:55–60. doi: 10.1530/EJE-11-0523. [DOI] [PubMed] [Google Scholar]
- Xu L, Dixit MP, Chen R, Dixit NM, Collins JF, Ghishan FK. Effects of angiotensin II on NaPi-IIa cotransporter expression and activity in rat renal cortex. Biochim Biophys Acta. 2004;1667:114–121. doi: 10.1016/j.bbamem.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Yuan Q, Sato T, Densmore M, Saito H, Schuler C, Erben RG, Lanske B. FGF23/Klotho signalling is not essential for the phosphaturic or anabolic effects of PTH. J Bone Miner Res. 2011;26:2026–2035. doi: 10.1002/jbmr.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Maalouf NM, Adams-Huet B, Moe OW, Sakhaee K. Effects of sex and postmenopausal estrogen use on serum phosphorus levels: a cross-sectional study of the National Health and Nutrition Examination Survey (NHANES) 2003–2006. Am J Kidney Dis. 2014;63:198–205. doi: 10.1053/j.ajkd.2013.07.012. [DOI] [PubMed] [Google Scholar]