

Abstract
Diabetic kidney disease (DKD) defines the functional, structural and clinical abnormalities of the kidneys that are caused by diabetes. This complication has become the single most frequent cause of end-stage renal disease. The pathophysiology of DKD comprises the interaction of both genetic and environmental determinants that trigger a complex network of pathophysiological events, which leads to the damage of the glomerular filtration barrier, a highly specialized structure formed by the fenestrated endothelium, the glomerular basement membrane and the epithelial podocytes, that permits a highly selective ultrafiltration of the blood plasma. DKD evolves gradually over years through five progressive stages. Briefly they are: reversible glomerular hyperfiltration, normal glomerular filtration and normoalbuminuria, normal glomerular filtration and microalbuminuria, macroalbuminuria, and renal failure. Approximately 20–40% of diabetic patients develop microalbuminuria within 10–15 years of the diagnosis of diabetes, and about 80–90% of those with microalbuminuria progress to more advanced stages. Thus, after 15–20 years, macroalbuminuria occurs approximately in 20–40% of patients, and around half of them will present renal insufficiency within 5 years. The screening and early diagnosis of DKD is based on the measurement of urinary albumin excretion and the detection of microalbuminuria, the first clinical sign of DKD. The management of DKD is based on the general recommendations in the treatment of patients with diabetes, including optimal glycaemic and blood pressure control, adequate lipid management and abolishing smoking, in addition to the lowering of albuminuria.
 |
Carmen Mora-Fernández is responsible for the Scientific Management of the Research Unit at the University Hospital Nuestra Señora de Candelaria (RU-HUNSC). She obtained her Bachelor of Medicine and Surgery in 1991 from the University of Alcalá de Henares (Madrid, Spain). She is principal associate researcher within the Nephrobiology and Cardiovascular Risk Section at the RU-HUNSC. She is a member of the Clinical Research Ethics Committee of the HUNSC. Dr. Navarro-González is Chair of the Research Division with a joint appointment as Staff Nephrologist at the HUNSC, Assistant Professor of Nephrology at the University of La Laguna (Santa Cruz de Tenerife, Spain), Teaching and Research Coordinator of the Spanish Society of Nephrology, and National Coordinator of the GEENDIAB (Spanish Study Group for the study of Diabetic Nephropathy).
Introduction
New terminology to describe kidney disease attributable to diabetes is being introduced in recent guidelines (National Kidney Foundation, 2007). Thus, the term ‘diabetic nephropathy’ should be replaced by diabetic kidney disease (DKD), a long-term highly prevalent major microvascular complication defined as functional, structural and clinical abnormalities of the kidneys that are caused by diabetes. This complication has become the single most frequent cause of end-stage renal disease (ESRD), and it is considered ‘a medical catastrophe of worldwide dimensions’ (Ritz et al. 1999). Additionally, it is strongly associated with cardiovascular morbidity and mortality. Of the complications of diabetes, DKD may be the most detrimental regarding patients’ quality of life and survival.
This review presents a brief update of DKD, including the physiological aspects of the glomerular filtration barrier, the pathogenic mechanisms and natural history of this complication, and aspects related to screening, diagnosis and management.
Glomerular filtration barrier: a physiological update
The kidneys produce a virtually protein-free primary urine through an apparently simple process: the blood enters the glomerular tuft via the afferent arteriole and perfuses the glomerular capillaries, where filtration across the glomerular filtration barrier into Bowman's space occurs. The primary urine is drained into the renal tubules, and the blood exits the tuft via the efferent arteriole. However, the success of this filtration process is based on the presence of a highly specialized structure: the glomerular filtration barrier (GFB). This filtration barrier permits a highly selective ultrafiltration of the blood plasma: it is freely permeable to water, small- and mid-sized solutes, and low-molecular-weight proteins up to the mass of albumin, but largely precludes the filtration of plasma proteins with a mass of more than 60–70 kDa, especially if they are negatively charged. As an example, the estimated sieving coefficient of albumin is lower than 0.0005, meaning that normally less than 0.05% of plasma albumin passes through the GFB to the urinary space.
This filtration apparatus is formed by three layers: the fenestrated endothelium, the glomerular basement membrane (GBM), and the epithelial podocytes with their ‘slit diaphragms’ (Fig. 1). The GFB functions as a whole, with each layer making an important contribution to selective permeability, and its integrity and functionality is maintained by an interplay of the cell types and constituents (Pavenstädt et al. 2003) (Table 1).
Figure 1.
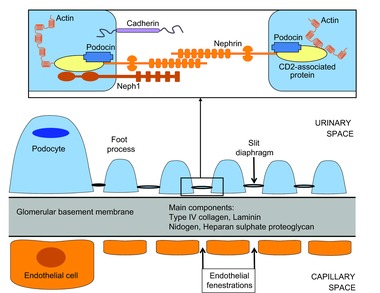
Schematic representation of the kidney filtration apparatus and main protein components of the podocyte slit diaphragm
Table 1.
Key molecules in the glomerular filtration barrier
| Glomerular endothelial cells | ||
| Glycocalyx | ||
| Proteoglycans | ||
| Glycosaminoglycans: heparan sulphate | ||
| Plasma proteins: albumin | ||
| Glomerular basement membrane | ||
| Laminin (laminin-521 (α5β2γ1)) | ||
| Type IV collagen (α3α4α5) | ||
| Heparan sulphate proteoglycans (agrin) | ||
| Nidogen-1 and -2 | ||
| Podocytes and slit diaphragm | ||
| Nephrin | Neph1/Neph3 | Podocin |
| CASK | JAM-A | Zona occludens-1 |
| Catenins | ||
| TrpC6 | CLIC5 | CD2-associated protein |
CASK, calcium–calmodulin-dependent serine protein kinase; JAM-A, junctional adhesion molecule A; TrpC6, transient receptor potential-6 channel; CLIC5, chloride intracellular channel 5; CD2, cluster of differentiation 2.
The glomerular endothelium
Glomerular endothelial cells have special characteristics, such as the presence of fenestrations that appear as transcytoplasmic holes, a structural feature characterized by the absence of the diaphragm and the lack of ‘plasmalemmal vesicle-associated protein-1’ (a type II transmembrane glycoprotein that is an integral part of the diaphragm), retention of the basal lamina, and the presence of surface glycocalyx that extends over the fenestrations.
The fenestrations are transcellular holes of 60–80 nm in diameter that occupy 30–40% of the cell surface. They are mainly concentrated toward the peripheral cytoplasm, and arranged in a cluster of sieves separated by the ridges of cytoplasm. They are thus localized in the region of cytoplasm that is opposite to the podocyte foot processes and filtration slit across the basement membrane (Satchell & Braet, 2009). Fenestrations are an adaptation that facilitates the high water permeability of the glomerular endothelium which is essential for filtration function. The size of the fenestrations is much too large to easily exclude large protein, including albumin, from the glomerular filtrate. Therefore, the glomerular capillary wall is not a perfect barrier for macromolecules and it allows the albumin to move across. However, glomerular filtration of albumin is followed by tubular reabsorption, allowing urine to remain virtually albumin free (Obeidat & Ballermann, 2012).
The glycocalyx is a polyanionic hydrated mesh composed principally of proteoglycans, glycoproteins and sialoproteins, with particular molecular and charge characteristics, on the luminal surface of glomerular endothelial cells. This structure covers the fenestrations and participates in regulating permeability as well as ligand/receptor and cellular interactions (Weinbaum et al. 2007). The biophysical models indicate that glycocalyx contributes to about 50% of the overall hydraulic resistance of the glomerular filtration barrier, and the specific composition of the glycocalyx in the fenestrations is important for maintaining the permeability properties and the glomerular filtration rate (Singh et al. 2007).
The glomerular basement membrane
The GBM is a thin layer (250–400 nm) of extracellular matrix proteins that separates the vasculature from the urinary space. Most of this membrane is located between two cellular beds: the endothelial cells that line the glomerular capillaries, and the podocytes that sit on the opposite side within the urinary space. The remaining portions of the GBM lie between mesangial cells and podocytes at the bases of the capillary loops. Podocytes and glomerular endothelial cells synthesize the components of the GMB (with mesangial cells participating in the turnover), which contains specific protein isoforms, some of them being crucial for glomerular development, structure and function (Miner, 2011, 2012).
Nine major proteins have been found in the GBM, although like other basement membranes, the main four types of extracellular matrix macromolecules are type IV collagen, laminin, heparan sulphate proteoglycan and nidogen (Miner, 2011, 2012). Type IV collagen is the most abundant protein in basement membranes: There are six genetically distinct collagen IV α chains (α1 to α6), and these chains assemble to form three different heterotrimers. In the mature GBM, the composition of collagen IV chains is α3α4α5, although there is a switch of these chains during glomerulogenesis. Regarding laminin, this is a ubiquitous basement membrane component that possesses several isoforms. They are all composed of three different homologous chains (α, β and γ), and secreted as αβγ heterotrimers. Laminin-521 (α5β2γ1) is the major isoform in the normal GBM. However, during the processes of GBM formation and maturation, there is a developmental transition in laminin trimer deposition, which occurs coincidentally with the transition of type IV collagen chains. Concerning the third constituent of the GBM, nidogen-1 and nidogen-2, these are two virtually ubiquitous basement membrane homologous glycoproteins encoded by two different genes. Since nidogen-1 binds to both laminin and type IV collagen, it has been suggested that nidogen functions to connect the separate laminin and type IV collagen networks in basement membranes, providing extra stability under situations of unusual stress. Finally, the last main component is agrin, the major heparan sulphate proteoglycan of the GBM. Agrin is highly negatively charged due to the presence of sulfated glycosaminoglycan with anionic sites that ultimately provide a net negative charge on the GBM. Agrin is important due to its effects on different aspects. Such as the charge selectivity within the glomerular filtration barrier and the linking of the GBM to the adjacent cells.
The GBM participates importantly in the normal homeostasis and function of the GFB through different functions: it provides structural support for the glomerular capillaries, harbours ligands for receptors on the surface of the adjacent endothelial cells, podocytes and mesangial cells, and contributes to glomerular permselectivity. In support of this idea, mutations in genes encoding the four major proteins of the GBM are known to cause human kidney diseases. In addition, environmental conditions can also produce alterations in the composition and structure of the GBM, with DKD as an example in which the GBM is adversely affected by the microenvironment. Finally, primary changes in the GBM may result in structural and functional alterations in the neighbouring cells (podocytes, endothelial cells and mesangial cells), thereby affecting glomerular filtration.
The podocytes and the slit diaphragm
The podocytes are the glomerular visceral epithelial cells, terminally differentiated and highly specialized cells of mesenchymal origin that cover the urinary aspect of the GBM. They have a cell body, long primary processes and a complex network of interdigitating cellular extensions called secondary processes or foot processes. The foot processes of different podocytes interact at specialized intercellular junctions called filtering slit or slit diaphragms (Pavenstädt et al. 2003; Grahammer et al. 2013).
The complex cellular architecture of podocytes, essential to maintain its unique structure and function, is preserved by the presence of an actin cytoskeleton, a particular organization of microtubules and actin microfilaments in the cellular cytoplasm. Actinin-4 and synaptopodin are actin-associated proteins with an important role in the regulation of actin cytoskeleton (Mathieson, 2012). In addition to this architectural aspect of podocytes, increasing interest has been paid to the composition of the slit diaphragm, a dynamic structure with an essential role in the function of GFB (Huber & Benzing, 2005).
The protein components of the slit diaphragm have to fulfil several tasks, including their role as a macromolecular filter: anchor the filter to the GBM, connect the slit diaphragm to the actin cytoskeleton, and be part of a signalling complex that integrates and mediates extracellular and intracellular signals regulating the plasticity of foot processes (Grahammer et al. 2013). A number of proteins that are associated with junctions in other locations are also found at the slit diaphragms, including adherens junction proteins, such as P-cadherin or catenins, and proteins typically found in tight junctions, such as zona occludens-1 and junctional adhesion molecule A. In addition, several integral membrane proteins, including nephrin, podocin and Neph1, not found in other junctions, have been identified as slit diaphragm components. During development, podocytes are initially connected via tight-junction and gap-junction components, which are gradually replaced by nephrin, podocin and Neph1, although tight-junction and adherens-junction molecules remain in the vicinity of the slit diaphragm. Nephrin, podocin and Neph1 form a zipper-like structure that is the hallmark of the mature slit diaphragm (Grahammer et al. 2013).
Nephrin, a type 1 transmembrane protein, is considered the central molecule to bridge the gap between adjacent podocyte foot processes through interactions with other nephrin molecules (homologous) or with Neph1 molecules (heterologous), and therefore, is hypothesized to be the ‘pore’ of the slit diaphragm (Wartiovaara et al. 2004). In biopsy-controlled clinical studies, it has been demonstrated that nephrin expression is reduced by more than 60% in type 2 diabetic patients. Finally, podocin seems to recruit other slit diaphragm proteins and to increase the signalling properties of the nephrin–Neph1 complex.
Whenever podocytes undergo stress or injury, irrespective of the causative stimulus, they undergo a response that ultimately leads to foot process effacement and loss of slit diaphragms, resulting in proteinuria.
Epidemiology of diabetic kidney disease
Diabetes mellitus is one of the most common chronic diseases. The frequency of diabetes, especially type 2, is continuing to be an increasing international health burden. This is related to several factors such as the ageing of the population and the burden of obesity. Importantly, similar trends are also found for type 1 diabetes, with the predictions showing that the number of new cases of type 1 diabetes in European children younger than 5 years is predicted to double between 2005 and 2020, and the prevalent cases younger than 15 years will rise by 70% (Patterson et al. 2012). Therefore, the data from the International Diabetes Federation estimate that the number of people with diabetes will rise from 366 million in 2011 to more than 550 million in 2030, which represents a 50% increase in less than 20 years (Whiting et al. 2011). Thus, ever more subjects are exposed to the risk of ultimately developing diabetic complications which impose a tremendous burden on patients, their families, health-care systems and society.
The frequency of nephropathy in type 1 diabetes has a rather predictable prevalence (around 30–40%), whereas in type 2 diabetes it depends on several factors, such as race and ethnicity, which have a major impact on the risk for the development of kidney disease as well as for progression to renal damage. In the last 10 years, rates of ESRD due to diabetes in the African American, Native American and Hispanic populations are increasing, whereas rates have been unchanged in the white and Asian populations in the United States (Stanton, 2014). Irrespective of these considerations, diabetes mellitus is the most common cause of ESRD, and DKD is the leading specific primary renal diagnosis for patients initiating renal replacement therapy.
However, in the last few years an interesting paradox has been noted: a declining trend in the incidence of ESRD due to diabetes at the same time as diabetes prevalence is increasing in the general population (Burrows et al. 2010). Several factors have been considered to explain this situation, including the success of current efforts in prevention, early detection and management of DKD, the improvement of general treatment and care, the better control of additional ESRD risk factors (i.e. hypertension), or the effectiveness of pharmacological agents as renoprotective instruments. However, it might be premature to state a real decline in ESRD in diabetes, and these initial data must be confirmed by adequate epidemiological studies.
Pathogenic mechanisms of diabetic kidney disease
Although DKD was classically considered the result of the interaction between metabolic and haemodynamic factors, renal damage in diabetes is not solely explained by these elements. Current knowledge indicates that this represents only a partial view of a much more complex scenario with the interaction of both genetic and environmental determinants that trigger a complex network of pathophysiological events (Fig. 2 and Table 2).
Figure 2.
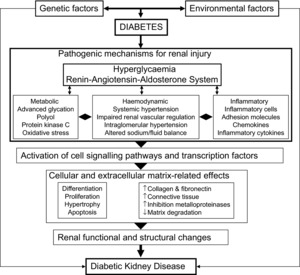
Schematic overview of the pathogenic mechanisms of diabetic kidney disease
Table 2.
Key factors and pathways in the pathogenesis of diabetic kidney disease
| Haemodynamic | ||
| Prostanoids | Nitric oxide | |
| Renin–angiotensin–aldosterone system | ||
| Vascular endothelial growth factor (VEGF) | ||
| Transforming growth factor (TGF)-β1 | ||
| Metabolic | ||
| Glucose transporters: GLUT-1, GLUT-4, glucokinase/hexokinase. | ||
| Advanced glycation end products and their receptors | ||
| Aldose reductase | Protein kinase C | |
| Diacylglyerol | UDP-N-acetylglucosamine | |
| NADPH oxido-reductase | Oxidative stress | |
| TGF-β–Smad–mitogen-activated protein kinase (TGF-β–Smad–MAPK) | ||
| Janus kinase–signal transducer and activator of transcription (JAK-STAT) | ||
| Growth factors | ||
| VEGF | TGF-β1 | Connective tissue growth factor |
| Inflammation | ||
| Chemokines: monocyte chemoattractant protein-1 | ||
| Adhesion molecules: intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1). | ||
| Inflammatory cytokines: interleukin-1, -6, -18, and tumour necrosis factor-α | ||
| Transcription factors: nuclear transcription factor κ-B (NF-κB) | ||
Genetic factors
During the last decades, growing evidence supports the participation of genetic factors in the development and progression of DKD. Both candidate gene approaches and genome-wide linkage analyses have suggested several candidate genes with potential impact on this complication. However, many of these findings have not been robustly replicated, and many genes responsible for susceptibility to diabetic nephropathy remain to be identified. In recent years, several meta-analyses have strongly pointed to some polymorphisms as potential genetic factors for DKD (Table 3), although definitive studies are required to firmly establish the role of these polymorphisms on the risk of DKD.
Table 3.
Association between genetic polymorphisms and diabetic kidney disease in recent meta-analysis
| Gene | Polymorphism | Effect | References |
|---|---|---|---|
| Angiotensin-converting enzyme | Insertion/deletion (I/D) | D or DD associated ESRD susceptibility | Yu et al. 2012 |
| Glucose transporter 1 | Xbal, Enh2, HaeIII | Susceptibility to DKD | Cui et al. 2012 |
| Carnosinase | D18S880 microsatellite | Susceptibility to DKD | Zhu et al. 2013 |
| MTHF reductase | C677T | Increased risk of DKD | Yang et al. 2013; Chang et al. 2013 |
| Tumour necrosis factor-α | -308 G/A | A allele protective against DKD | Zhao et al. 2013 |
| Adiponectin | rs17300539/rs2241766 | Increased risk of DKD | Lin et al. 2014 |
| Transforming growth factor-β1 | T869C | Reduced risk of DKD | Zhou et al. 2014 |
| Endothelial nitric oxide synthase | 4b/4a, T786C, G894T | Increased risk of DKD | Dellamea et al. 2014 |
| Apolipoprotein E | ε2 and ε4 | Increased risk of DKD | Yin et al. 2014 |
D or DD, heterozygous and homozygous for the deletion (D) allele; DKD, diabetic kidney disease; ESRD, end-stage renal disease; MTHF, methylenetetrahydrofolate.
Metabolic factors
The cellular elements of the kidney respond to hyperglycaemia by prompting diverse intracellular processes, including: alteration in cellular energy production; activation of different enzymes, such as aldose reductase and protein kinase C (PKC); enhanced flux of polyols and hexosamines; generation of advanced glycation end-products (AGEs) and reactive oxygen species; activation of transducer signalling pathways; abnormal expression of cyclin kinases and their inhibitors; and dysregulation of factors controlling the extracellular matrix homeostasis (Kanwar et al. 2011; Forbes & Cooper, 2013).
Cells within tissues that are prone to diabetic complications are not able to modulate glucose transport rates to prevent excessive intracellular glucose accumulation.
Increased expression of the glucose transporter-1 has been related to the pathobiology of diabetic complications. In addition, the expression of glucokinase/hexokinase, an important enzyme involved in the transport of glucose into cells, is regulated by glucose-6-phosphate dehydrogenase, which presents significant changes at sites of diabetes complications.
Hence, the highly regulated energy production process in the cells is uncontrolled in the context of diabetes as a result of excess substrate availability, in particular glucose. Abnormalities in energy production are common signs seen in micro- and macrovascular diabetic complications, playing an important role in their development. These include alteration in release of substrates, switching the proportions of cell-specific fuel sources among glucose intermediates, fatty acids and amino acids, changes in respiratory chain protein function, and uncoupling of the respiratory chain.
Once glucose is transported inside the cell, most of this molecule is metabolized via glycolysis, with the conversion of glucose-6-phosphate to fructose-6 phosphate (F6-P), and then to glyceraldehyde 3-phosphate (G3-P). G3-P forms glycerol phosphate, a precursor of diacylglycerol (DAG), a signalling molecule responsible for the recruitment and activation of PKC. This enzyme occupies a central position among signalling kinases in the pathogenesis of DKD. A number of PKC isoforms are expressed in the kidney and are activated in diabetic conditions, with special relevance for PKC-β2. PKC is activated by multiple routes, including DAG, the polyol pathway and AGEs. PKC activation influences a number of downstream pathways (endothelial nitric oxide synthase, endothelin-1, vascular endothelial growth factor, transforming growth factor-β1 (TGF-β1), NADPH oxido-reductase, nuclear factor-κB (NF-κB)), with pathophysiological modifications that result in altered blood flow and capillary permeability, and increased production of extracellular matrix proteins.
Under conditions of elevated intracellular glucose concentrations, glycolysis diverts F6-P stepwise to UDP-N-acetylglucosamine, which is a precursor of extracellular matrix proteins such as the proteoglycans. In addition, UDP-N-acetylglucosamine produces the posttranslational modification of proteins within the cytosol and nucleus. Therefore, these modified sugar residues compete with phosphate groups altering gene expression in diabetic tissues.
It is relevant that the excess glucose is also channelled into the polyol pathway, where it is reduced to sorbitol by aldose reductase, a NADPH-dependent enzyme with a physiological role in detoxification of aldehydes into inert alcohols. Under basal conditions, the polyol pathway has a very low activity, but in a high-glucose environment, around 30% of the glucose is derived to this pathway. This increased activity results in the relative depletion of NADPH and reduced glutathione and glutathione peroxidase activity, an increase in the NADH/NAD+ ratio, and decreased levels of nitric oxide, leading to oxidant and osmotic stress.
Finally, through the non-enzymatic reaction of reducing sugars with free amino groups of proteins, lipids and nucleic acids, AGEs are formed. The initial product of this reaction is called a Schiff base, which spontaneously rearranges itself into an Amadori product. A series of subsequent reactions lead to the formation of AGEs. Under normal conditons, AGEs are produced in small amounts but their concentrations markedly increase under hyperglycaemia, in both the cellular and extracellular compartments. Intracellularly, the AGEs can modulate various events, such as the activation of PKC, mitogen-activated protein kinase (MAPK) and transcription factors such as NF-κB, which, in turn, regulate the expression of diverse cytokines and growth factors, with special relevance for TGF-β1, which is a key contributor to glomerular sclerosis and tubulointerstitial fibrosis.
Regarding extracellular AGEs, these molecules are formed based on a key characteristic of certain reactive or precursor AGEs, their ability for covalent crosslink formation between proteins, which alters their structure and function, as in cellular matrix, basement membranes, and vessel wall components. These modified proteins may have decreased susceptibility to enzymatic hydrolysis by matrix metalloproteinases, which would allow them to accumulate. Thus, the characteristic structural changes of DKD, thickened glomerular basement membrane and mesangial expansion, are accompanied by accumulation of AGEs, leading to glomerulosclerosis and interstitial fibrosis. In addition, glycation of sulfated proteoglycans reduces their electronegativity and thus modifies the charge-selective filtration properties of the basement membrane. Finally, the other major feature of AGEs is related to their interaction with a variety of cell-surface AGE-binding receptors, leading to the modulation of cellular functions, and the activation of pro-oxidant, pro-inflammatory events.
Haemodynamic factors
Haemodynamic dysfunctions in patients with diabetes are represented by intraglomerular hypertension, hyperfiltration and systemic increase in arterial blood pressure (BP).
Early studies demonstrated that the hyperglycaemic state impairs the physiological autoregulatory mechanism that maintains normal glomerular capillary pressure, protecting the intraglomerular structures from changes in systemic BP. The final result is the ready transmission of systemic pressure to the glomerular capillary, with the subsequent elevation of the transcapillary hydraulic pressure and the development of hyperfiltration, which is associated with more serious structural changes (Hostetter et al. 1982). Interestingly, it has been postulated that there is a bidirectional interplay between metabolic and mechanical stimuli at the cellular level. Therefore, the mechanical forces on glomerular cells are able to induce the overexpression of GLUT-1, resulting in greater glucose uptake and activation of intracellular signalling pathways (Gnudi et al. 2007).
One of the most important elements participating in the haemodynamic dysfunction in diabetes and DKD is the renin–angiotensin–aldosterone system (RAAS), which is directly involved in the regulation of BP and fluid volume, and participates in vascular injury and inflammation. Although aldosterone, renin, and several breakdown products of angiotensin I (AI) are also involved, most of the effects of the RAAS on target tissues are mediated by angiotensin II (AII), which is generated both in the circulation and in the tissues. Hyperglycaemia induces the activation of the local renal RAAS (e.g. in the tubular epithelial cells, mesangial cells and podocytes) and increases the production of AII, which exerts a preferential constrictor effect on the efferent glomerular arteriole, resulting in a higher intraglomerular capillary pressure. In addition, the locally produced Ang II activates multiple intracellular signalling pathways and induces inflammation, renal cell growth, mitogenesis, apoptosis, migration and differentiation (Macía-Heras et al. 2012).
In the classic pathway of the RAAS, renin is secreted from the juxtaglomerular apparatus of the kidney and acts on the circulating precursor angiotensinogen to generate AI. Angiotensin-converting enzyme (ACE) present in the endothelium and tissues converts AI to the octapeptide AII. In the heart, kidneys and brain, AII is also produced by non-ACE pathways involving chymases, cathepsin G, kallikrein-like enzymes and endopeptidases, and seems to exert effects on target tissues that are even greater than the effects of centrally generated AII. AII acts on the heart and the kidneys by binding to the G protein-coupled receptors type 1 (ATR1) and type 2 (ATR2). The ATR1 mediates the more deleterious effects of AII, whereas the ATR2 regulates opposing effects.
Recently, a new component of the RAAS has been described, ACE2, a homologue of ACE that act as a negative regulator of the system, counterbalancing the actions of ACE. ACE2 cleaves AI and AII into the inactive angiotensin 1–9, and the vasodilator and anti-proliferative angiotensin 1–7, respectively. ACE2 within the kidneys is largely expressed in tubular epithelial cells and glomerular epithelial cells. In DKD, reduced expression of ACE2 coupled with increased expression of ACE has been described. In addition, both ACE2 genetic ablation and pharmacological ACE2 inhibition have been shown to increase albuminuria and promote glomerular injury (Soler et al. 2008).
Inflammation
Significant advances have been made in relation to the pathogenesis of DKD that have recognized the involvement of inflammatory molecules and pathways in the development and progression of DKD (Navarro-González & Mora-Fernández, 2008; Rivero et al. 2009). The relationships between this inflammatory state and the initiation and evolution of DKD involve very complex network processes. Diverse inflammatory molecules play significant roles in this scenario, including toll-like receptors, chemokines (monocyte chemoattractant protein-1, also known as CCL2), adhesion molecules (intracellular adhesion molecule-1 and vascular adhesion molecule-1), and proinflammatory cytokines (interleukin-1, -6, -18, and tumour necrosis factor-α).
New pathogenic factors from transgenic experimental models
Recent studies with the use of genetically modified animals as experimental models of DKD have reported novel factors with interest from a pathogenic and therapeutic perspective, including tissue inhibitor of metalloproteinase-3 (TIMP-3), matrix metalloproteinase-2 (MMP-2), and CCL2/C-C chemokine receptor 2 (CCR2).
TIMP-3, an endogenous inhibitor of matrix metalloproteinases, is the most highly expressed TIMP in the kidney. It influences the extracellular matrix integrity and the tissue microenvironment. Loss of TIMP-3 in mice has been reported to increase renal damage (Kassiri et al. 2009). A recent study using double mutant mice in which the TIMP-3 gene was deleted in the genetic diabetic Akita mouse (Basu et al. 2012) showed that loss of TIMP-3 leads to the aggravation of diabetic renal injury as exemplified by significantly increased kidney mass, glomerular mesangial matrix score, and urinary albumin excretion, which are all early features of DKD. These changes occurred in the absence of worsening of hypertension or glycaemic control, demonstrating that TIMP-3 plays a key and organ-specific role in diabetic renal injury and is consistent with observations made in human DKD (Ewens et al. 2005).
MMP-2, an endopeptidase responsible for the degradation of type IV collagen and laminin, major components of extracellular matrix proteins, has been showed to have a reduced expression and activity in advanced human DKD. To further elucidate the pathophysiological role of MMP-2, renal injury was assessed in a model of streptozotocin-induced DKD in the context of genetic deletion of MMP-2 (Takamiya et al. 2013). This study showed that the renal expression and activity of MMP-2 are increased as a compensatory mechanism in the early phase of DKD, and that MMP-2 could play a protective role against the progression of renal injury in diabetes.
Finally, a recent study investigated the effect of the blockade of CCL2/CCR2 signalling on the development of experimental DKD (Seok et al. 2013). Administration of the CCR2 antagonist RS102895 to type 2 diabetic db/db mice improved the pathological (e.g. mesangial matrix expansion, GBM thickening and podocyte foot process effacement) and the functional (e.g. increased albuminuria) impairments of DKD. This therapy ameliorated DKD not only by improving blood glucose levels but also by preventing CCL2/CCR2 signalling from altering renal nephrin and vascular endothelial growth factor expression through blocking macrophage infiltration, inflammation and oxidative stress.
Natural history of diabetic kidney disease
The natural history of DKD, a process that advances gradually over years, was defined in the eighties based on longitudinal studies of patients with type 1 and type 2 diabetes, being reported that the general evolution of DKD was comparable in both major types of diabetes. Approximately 20–40% of diabetic patients develop microalbuminuria within 10–15 years of the diagnosis of diabetes, and about 80–90% of those with microalbuminuria progress to more advanced stages. Thus, after 15–20 years, macroalbuminuria occurs in approximately 20–40% of patients, and around half of them will present renal insufficiency within 5 years (Hasslacher et al. 1989; Remuzzi et al. 2002).
Five stages of DKD were defined. (1) Early functional phase with reversible glomerular hyperfiltration. (2) Normal glomerular filtration and normoalbuminuria (urinary albumin excretion (UAE) < 30 mg day–1). (3) Incipient nephropathy, defined by normal glomerular filtration and microalbuminuria (UAE of 30–300 mg day–1), which develops between 5 and 10 years after the diagnosis of diabetes, and it is the first clinical sign of DKD. (4) Macroalbuminuria (UAE higher than 300 mg day–1), after 10–20 years of diabetes evolution. Once macroalbuminuria is present, there is a progressive reduction in glomerular filtration rate (GFR), the rate varying widely from patient to patient, with an average decline of approximately 10 ml min–1 year–1 in untreated subjects. (5) Renal failure, which leads ultimately to ESRD. More precise knowledge about the progression of nephropathy in type 2 diabetes was provided by the United Kingdom Prospective Diabetes Study (UKPDS). Thus, using observed and modelled data, from the diagnosis of diabetes, development of microalbuminuria occurred at 2.0% per year, progression from microalbuminuria to macroalbuminuria at 2.8% per year, and progression from macroalbuminuria to elevated plasma creatinine or renal replacement therapy at 2.3% per year (Adler et al. 2003).
The main pathophysiological changes in DKD include the thickening of the GBM, mesangial expansion, nodular sclerosis (Kimmelstiel–Wilson change), diffuse glomerular sclerosis, tubular interstitial fibrosis, and arteriosclerosis and hyalinosis of kidney blood vessels. The severity of glomerular damage is proportional to GFR value, diabetes mellitus duration, and blood glucose regulation. Recently, the Scientific Committee of the Society for Pathological Anatomy has established the pathological classification of DKD, where diabetic glomerular lesions have been histologically divided into four stages (Table 4).
Table 4.
Classification of glomerular lesions in diabetic kidney disease
| Class | Description | Inclusion criteria |
|---|---|---|
| I | Mild or non-specific light microscopy changes and GBM thickening proved by electron microscopy | Biopsy does not meet any of the criteria for class II, III or IV GBM >395 nm in female and >430 nm in male subjects 9 years of age or older |
| IIa | Mild mesangial expansion | Mild mesangial expansion in 25% of the observed mesangium. Biopsy does not meet criteria for class III or IV |
| IIb | Severe mesangial expansion | Severe mesangial expansion in 25% of the observed mesangia. Biopsy does not meet criteria for class III or IV |
| III | Nodular sclerosis (Kimmelstiel–Wilson lesion) | At least one convincing Kimmesteil–Wilson lesion. Biopsy does not meet criteria for class IV |
| IV | Advanced diabetic glomerulosclerosis | Global glomerular sclerosis in >50% of glomeruli. Lesions from class I–III |
GBM, glomerular basement membrane. Adapted from Tervaert et al. (2010).
Although the largest proportion of patients develop the ‘classical’ natural history of DKD, suffering from proteinuria and diabetic glomerulosclerosis as the underlying pathology, DKD is more heterogeneous than previously thought. Thus, an increasing number of studies have indicated other possibilities regarding the evolution of DKD: a significant initial deterioration of GFR, a spontaneous reduction of microalbuminuria, the presence of reduced renal function accompanied, rather than preceded, by macroalbuminuria, or even present in patients with microalbuminuria, or in those with albuminuria levels that revert to normal, or in subjects whose albuminuria levels remain normal. At the present moment, it is not clear what factors determine this different presentation and natural history (Halimi, 2012). In addition, intervention strategies for the management of DKD have changed the natural history of the disease. As an example, in the seventies, the median time to ESRD from the appearance of proteinuria in type 1 diabetes was less than 7 years, whereas now this time is higher than 14 years.
An important consideration is that the presence of trace amounts of albumin in the urine is not only a predictor of high renal risk, but is also an independent predictor of increased cardiovascular risk, especially in type 2 diabetes. The cardiovascular risk increases progressively even when the UAE rate is within the normal range. Remarkably, the annual death rate increases from less than 1% in the stage of normoalbuminuria to more than 12% in the stage of renal failure. Subjects who present macroalbuminuria are more likely to die than to develop renal failure during the evolution of the disease, highlighting that in these patients, the death rate exceeds the rate of progression to worse nephropathy (Adler et al. 2003).
Detection and management of diabetic kidney disease
Diagnosis and screening
Kidney biopsy is the definitive method to establish the diagnosis of DKD, although in most cases, careful evaluation of patients can identify subjects most likely to have DKD without the need for kidney biopsy.
The screening and early diagnosis of DKD is based on the measurement of UAE. In normal individuals, a small amount of albumin is filtered in the glomeruli, which is almost completely reabsorbed by the tubules. The UAE is considered normal when it is less than 30 mg day–1, a threshold based on the fact that the UAE of 95% of normal subjects falls below this value. A new nomenclature intends to emphasize the continuous nature of albuminuria as a risk factor, thus the terms microalbuminuria (30–299 mg (24 h)–1) and macroalbuminuria (>300 mg (24 h)–1) are rather referred to as persistent albuminuria at levels 30–299 mg (24 h)–1 and levels ≥300 mg (24 h)–1, respectively (American Diabetes Association, 2014).
Historically, detection of macroalbuminuria was the basis of the diagnosis of DKD. However, the development of more sensitive and accurate assays specific for albumin has enabled the detection of very small concentrations of albumin in urine. Clinically, the first sign of DKD is the presence of microalbuminuria, an analysis widely available nowadays. The recommended method for screening is the measurement of the albumin-to-creatinine ratio (ACR) in a spot urine sample (preferably the first morning void), since timed collections are inconvenient and susceptible to inaccuracy, and changes in urinary concentration caused by hydration status may confound the evaluation of tests of albumin concentration alone. Patients with diabetes should be screened annually for DKD. Initial screening should commence 5 years after diagnosis of type 1 diabetes, whereas it must be performed at diagnosis in type 2 diabetes, due to the inability to establish the onset of the disease with certitude. Since urinary albumin excretion rate has a high variability due to diverse factors, including metabolic and haemodynamic perturbations, physical exercise, fever, protein intake, congestive heart failure or urinary tract infection, multiple positive test results are required. Thus, at least two specimens with elevated ACR collected within a 3–6 months period are needed for confirmation (National Kidney Foundation, 2012; American Diabetes Association, 2014).
Although the current screening recommendations are effective for the great majority of patients, they are not sufficient in all cases, since a proportion of patients with diabetes may present a decreased GFR in the absence of increased UAE. Thus, serum creatinine with estimated GFR should be assessed at least annually in all adults with diabetes, regardless of the degree of UAE excretion. Information on UAE and estimated GFR may be used to stage DKD (American Diabetes Association, 2014). In addition, significant diabetic renal lesions may occur in subjects with normoalbuminuria and normal GFR, and furthermore, concomitant conditions may suggest the presence of non-diabetic kidney disease, such as active urinary sediment (mainly haematuria), a short time of diabetes evolution, or the absence of retinopathy (in most people with diabetes, chronic kidney disease should be attributable to DKD in the presence of increased UAE and retinopathy). Therefore, further evaluation, including consideration of kidney biopsy, may be required in some cases to establish the diagnosis of DKD.
Management of diabetic kidney disease
The general suggestions in the treatment of diabetes (glycaemic and BP control, adequate lipid management, and abolishing smoking) must be applied strictly when DKD is present. These recommendations, in addition to the lowering of UAE, are the primary interventions that prevent the development, and slow the progression, of DKD.
Hyperglycaemia is the main initial factor in the development of DKD, and therefore, adequate glycaemic control is the principal target for therapy in patients with potential development of DKD, and the best strategy to reduce the risk of vascular complications (DCCT, 1995; Duckworth et al. 2009). Optimal glycaemic control established as soon as possible after diagnosis of diabetes will reduce the risk of developing diabetic nephropathy, and delay the progression of renal disease. A number of interventions with different anti-diabetic drugs have been demonstrated to reduce the risk and slow the progression of renal disease both in type 1 and type 2 diabetes (DCCT, 1995; UKPDS, 1998a,b1998b; DCCT, 2000; Patel et al. 2008; Ismail-Beigi et al. 2010). While is evident that insulin is the obligate therapy in type 1 diabetes, metformin is the preferred initial pharmacological agent in patients with type 2 diabetes, barring contraindication or intolerance. When metformin fails to achieve or maintain glycaemic goals, another agent should be added. Although there are numerous trials comparing dual therapy to metformin alone, few directly compare drugs as add-on therapy. The effects of oral antidiabetic agents on diabetic nephropathy have been thought to be due to their ability to control blood glucose levels, and few studies have examined whether these agents may also have direct effects on the kidney. A recent systematic review on the ‘Comparative effectiveness and safety of oral diabetes medications for adults with type 2 diabetes’, sponsored by the Agency for Healthcare Research and Quality (US), concluded that overall, each new class of non-insulin agents added to the initial therapy lowers the concentration of glycated haemoglobin around 0.9–1.1%, and that there is insufficient and low-quality evidence on the effectiveness of individual new oral antidiabetic drugs on the development and progression of nephropathy (Bennett et al. 2011).
In addition to metabolic regulation, optimization of BP control is another key element to reduce the risk or slow the progression of DKD (UKPDS, 1998). Hypertension is a common comorbidity of diabetes, affecting the majority of patients, especially when renal involvement is present. There is solid evidence demonstrating the benefit (reduction of cardiac events, stroke and nephropathy) of lowering BP lower than 140 mmHg systolic and less than 80 mmHg diastolic. Patients with blood pressure higher than 120/80 mmHg should be advised on lifestyle changes to reduce BP, consisting, among others, of weight loss, if overweight; reducing sodium intake; moderation of alcohol intake; and increased physical activity. Patients with confirmed BP higher than 140/80 mmHg should, in addition to lifestyle modifications, have prompt initiation and timely subsequent titration of pharmacological therapy to achieve blood pressure goals. Pharmacological therapy should comprise a regimen that includes a blocker of the RAAS, either an ACE inhibitor (ACEI) or an angiotensin receptor blocker (ARB), which is recommended to titrate up to the maximum approved dose in the absence of side effects or adverse events. Diuretics, calcium channel blockers, and β-blockers should be used as additional therapy to further lower BP in patients already treated with ACEIs or ARBs or as alternative therapy in the individual unable to tolerate these drugs. Multiple-drug therapy is generally required to achieve BP targets, and one or more antihypertensive medications should be administered at bedtime since there is an association between increase in sleeptime BP and incidence of cardiovascular events (American Diabetes Association, 2014).
A special multiple-drug strategy is the simultaneous use of two RAAS inhibitors (i.e. dual RAAS blockade). This approach is based on the finding that incomplete blockade of the RAAS with ACEI or ARB monotherapy can cause a lack of negative feedback from end-products of the RAAS, producing a reactive increase in renin and consequent increases in AI and AII concentrations, which overwhelm the pharmacological blockade. Thus, dual blockade would provide more complete blockade of the system by limiting the compensatory responses of AII, aldosterone, renin or their effects (Macía-Heras et al. 2012).
The use of ACEIs plus ARBs represents the most frequent dual RAAS blockade strategy. A recent meta-analysis evaluated the effect of this combination on albuminuria (Kunz et al. 2008). Analysing seven clinical trials of ACEI plus ARB versus placebo plus ARB, the addition of an ACEI reduced albuminuria by 24% (95% confidence interval (CI), 15%, 32% reduction) over 1–4 months follow-up. Analysing seven clinical trials of ARB plus ACEI versus placebo plus ACEI, the addition of an ARB reduced albuminuria by 22% (95% CI, 16%, 28% reduction) over 1–4 months follow-up. These data demonstrate that the combination of an ACEI and ARB reduces albuminuria compared with monotherapy.
In addition to the effect of this strategy on UAE, the recently published VA NEPHRON-D (Veterans Affairs Nephropathy in Diabetes) trial evaluated the efficacy of combination therapy with an ACEI (lisinopril) and an ARB (losartan) as compared with ARB monotherapy in slowing the progression of proteinuric DKD (Fried et al. 2013). This multicentric, double-blind, randomized, controlled study, which is specific to patients with DKD, included 1448 patients with type 2 diabetes and macroalbuminuria, with a median follow-up of 2.2 years. The primary end point was the first occurrence of a decline in the estimated GFR (an absolute decrease of ≥30 ml min–1 (1.73 m2)–1 if the estimated GFR was ≥60 ml min–1 (1.73 m2)–1 at randomization or a relative decrease of ≥50% if the estimated GFR was <60 ml min–1 (1.73 m2)–1), ESRD (defined by the initiation of maintenance dialysis or an estimated GFR of <15 ml min–1 (1.73 m2)–1), or death. The study was prematurely interrupted according to the data and safety monitoring committee, primarily on account of safety concerns due to increased rates of serious adverse events, hyperkalaemia, and acute kidney injury in the combination therapy group as compared with the monotherapy group, along with low conditional power (<5% for the observed trend) to detect a treatment effect on the primary end point. This study demonstrated that combination therapy with an ACEI and an ARB in patients with type 2 diabetes and proteinuric DKD did not provide an overall clinical benefit, being associated with an increased risk of serious adverse events.
The effects of the addition of aldosterone antagonists (spironolactone and eplerenone) to ACEIs or ARBs for the reduction of albuminuria have been recently evaluated. In a systematic review including 14 studies with spironolactone, and one with eplerenone, most of these investigations reported that addition of an aldosterone antagonist reduced albuminuria by 30–40% (Bomback et al. 2008). The antiproteinuric effect of spironolactone was compared with losartan (an ARB) and placebo as add-on therapy to a high-dose of lisinopril (an ACEI) in patients with diabetes (mostly type 2), hypertension and macroalbuminuria. Compared with placebo, the addition of spironolactone to lisinopril reduced albuminuria by 34% at 48 weeks, whereas the addition of losartan to lisinopril reduced albuminuria by 17% (Mehdi et al. 2009).
The newest RAAS inhibitor available for clinical use is the direct renin inhibitor aliskiren. This drug directly blocks the activity of secreted renin. Effects of the dual RAAS blockade with aliskiren and losartan on albuminuria in the setting of type 2 DKD were investigated in the AVOID study. Patients treated with the combination experienced a 20% reduction in albuminuria as compared with participants assigned to placebo (Parving et al. 2008). More recently, a randomized, double-blind, placebo-controlled trial was conducted in more than 8500 patients with type 2 diabetes already taking an ACEI or an ARB as standard practice, to determine the effectiveness and safety of aliskiren, compared with placebo, with respect to fatal and non-fatal renal and cardiovascular events (Parving et al. 2012). The independent data and safety monitoring committee recommended the early termination of the study, on the basis that the excessive risk of adverse events in the aliskiren group could not be offset by a reduction in major cardiovascular and renal events. Therefore, the ALTITUDE (‘Aliskiren trial in type 2 diabetes using cardiorenal endpoints’) trial demonstrated that dual RAAS blockade, in this case with the direct renin inhibitor aliskiren added to standard-of-care renin–angiotensin blockade in high-risk patients with type 2 diabetes, did not reduce cardiovascular or renal outcomes, and resulted in an increased number of adverse events.
An ACEI or ARB for the primary prevention of diabetic kidney disease is not recommended in diabetic patients with normal BP and UAE lower than 30 mg (24 h)–1. Either ACEIs or ARBs are recommended for the treatment of the patient with modestly elevated (30–299 mg (24 h)–1) or higher levels (300 mg (24 h)–1) of UAE (American Diabetes Association, 2014).
Despite the implementation of these strategies, the number of patients with diabetes that ultimately develop DKD and ESRD remains high. Additional studies have focused on the potential of novel therapies that either target various pathways upregulated by hyperglycaemia or other targets believed to promote the progression of renal disease, such as the inhibition of AGEs and PKC, endothelin system, vitamin D and inflammation pathways.
Different approaches (dietary and pharmacological) have been used to reduce the accumulation of AGEs or to block the interaction with their receptors in experimental studies, showing potential beneficial effects on DKD (Zheng et al. 2002; Flyvbjerg et al. 2004; Thallas-Bonke et al. 2004; Coughlan et al. 2007). More importantly, the clinical use of the specific AGE inhibitor aminoguanidine in patients with type 1 diabetes and renal disease showed a significant reduction of proteinuria (Bolton et al. 2004). A similar trial in patients with overt type 2 diabetic nephropathy was initiated (Freedman et al. 1999). However, the development of those studies were discontinued for financial reasons by its pharmaceutical company sponsors. More recently, a 1 year clinical trial with pyridoxamine, a metabolic derivative of pyridoxal phosphate able to inhibit AGEs, failed to significantly alter the progressive loss of renal function in patients with type 2 diabetes mellitus and advanced nephropathy (Lewis et al. 2012).
Ruboxistaurin mesylate is a bisindolylmaleimide that specifically inhibits the β isoform of PKC. In a variety of experimental models of DKD, ruboxistaurin normalized glomerular hyperfiltration, decreased urinary albumin excretion, preserved kidney function, and reduced mesangial expansion, glomerulosclerosis and tubulointerstitial fibrosis (Tuttle, 2008). Regarding clinical studies, a randomized, double-blind, placebo-controlled, multicentre trial showed that in patients with type 2 diabetes and nephropathy, treatment with ruboxistaurin reduced albuminuria and maintained the estimated GFR over 1 year (Tuttle et al. 2005). In addition, a study in patients with type 1 diabetes showed that PKC-β appears to play a role in the maintenance of hyperfiltration, and its inhibition resulted in a decrease in GFR, suggesting that the impact of ruboxistaurin depends on filtration status (Cherney et al. 2009).
Endothelial dysfunction is a key element in the pathogenesis and progression of microvascular complications of diabetes, including DKD. Endothelin-1 (ET-1) is a potent vasoconstrictor with additional proliferative, profibrotic and proinflammatory properties. Diabetes is associated with increased ET-1 and elevated renal expression of the endothelin A receptor. Recent studies based on the use of the endothelin receptor blocker atrasentan have shown a significant reduction of proteinuria and urine neutrophil gelatinase-associated lipocalin (NGAL) (Kohan et al. 2011; Andress et al. 2012), suggesting that the addition of endothelin receptor blockade to currently established therapies with RAAS blockers may result in synergistic renoprotection. However, caution is necessary with these kinds of drugs based on previous studies. Thus, the ASCEND trial (‘Avosentan on time to doubling of serum creatinine, end-stage renal disease or death in patients with type 2 diabetes mellitus and diabetic nephropathy’) (Mann et al. 2010) examined in a prospective, randomized, double-blind fashion the effect of avosentan on time to doubling of serum creatinine, ESRD, or death, in people with type 2 diabetes and overt nephropathy. Unfortunately, the steering committee terminated the trial prematurely because of an excess of cardiovascular events with avosentan, mainly congestive heart failure and fluid overload.
Diabetes is a condition closely associated with low vitamin D levels (Takiishi et al. 2010), while it has been reported that individuals with lower vitamin D are more likely to have diabetes (Mehrotra et al. 2009). In experimental studies, vitamin D has been demonstrated to attenuate renin expression, reduce the expression of inflammatory mediators, promote survival of podocytes by inducing differentiation and preventing apoptosis, and reduce albuminuria and glomerulosclerosis (Panichi et al. 1998; Schwarz et al. 1998; Kuhlmann et al. 2004). Specifically, in animal models of DKD, combination therapy with ARB and vitamin D markedly ameliorated renal injury (Zhang et al. 2008). Regarding clinical data, a recent substudy of the Spanish ‘Progresión de nefropatía diabética (PRONEDI)’ trial has shown that 25(OH) vitamin D deficiency was associated with accelerated progression of DKD in patients over a long-term follow-up (Fernández-Juárez et al. 2013). From a therapeutic perspective, both cholecalciferol and the selective vitamin D receptor activator paricalcitol have been shown to reduce albuminuria in patients with type 2 diabetes (de Zeeuw et al. 2010; Kim, 2011).
There is evidence that inflammation is a cardinal process in the development and progression of DKD, with the participation of diverse inflammatory molecules and pathways in a complex network (Navarro-González & Mora-Fernández, 2008; Rivero et al. 2009). Pentoxifylline (PTF) is a methylxanthine derivate phosphodiesterase inhibitor with anti-inflammatory effects that has been used as a new approach for the treatment of DKD. Experimental studies in animal models of DKD have shown that PTF administration prevented the increased renal expression, synthesis and excretion of the proinflammatory cytokine tumour necrosis factor-α (TNFα), with a decrease in the renal protein content and in glomerular volume, as well as a reduction of renal sodium retention and renal hypertrophy (Gunduz et al. 2004; DiPetrillo & Gesek, 2004; Navarro et al. 2006). Together with these experimental studies, clinical studies have shown that PTF reduces urinary protein excretion in diabetic subjects, both with normal renal function and renal insufficiency (Guerrero-Romero et al. 1995; Navarro et al. 1999, 2003). Furthermore, the addition of PTF to RAAS blockers is associated with a significant additional anti-proteinuric effect (Harmankaya et al. 2003; Navarro et al. 2005).
A better understanding of the role of these new targets in the context of DKD will facilitate the development of novel therapies and strategies that can be translated successfully into clinical applications.
Acknowledgments
The research activity of J.F.N.G. is supported by Programa de Intensificación de la Actividad Investigadora, Instituto de Salud Carlos III, Ministry of Economy and Competitiveness (Convenio ISCIII-Comunidad Autónoma Canarias).
Glossary
- ACE
angiotensin-converting enzyme
- ACEI
ACE inhibitor
- AGEs
advanced glycation end-products
- AI
angiotensin I
- AII
angiotensin II
- ARB
angiotensin receptor blocker
- BP
arterial blood pressure
- DKD
diabetic kidney disease
- ESRD
end-stage renal disease
- GBM
glomerular basement membrane
- GFB
glomerular filtration barrier
- GFR
glomerular filtration rate
- PTF
pentoxifylline
- PKC
protein kinase C
- RAAS
renin–angiotensin–aldosterone system
- UAE
urinary albumin excretion
Additional information
Competing interests
None of the authors have any conflicts of interest.
Funding
Research studies by the authors have been funded by the Spanish Ministry of Health, Instituto de Salud Carlos III (EC07/90021), Sociedad Española de Nefrología and ACINEF. We also acknowledge cofunding by the European Regional Development Funds (ERDF).
References
- Adler AI, Stevens RJ, Manley SE, Bilous RW, Cull CA, Holman RR UKPDS Group. Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64) Kidney Int. 2003;63:225–232. doi: 10.1046/j.1523-1755.2003.00712.x. [DOI] [PubMed] [Google Scholar]
- American Diabetes Association. Standards of medical care in diabetes–2014. Diabetes Care. 2014;37(Suppl. 1):S14–S80. doi: 10.2337/dc14-S014. [DOI] [PubMed] [Google Scholar]
- Andress DL, Coll B, Pritchett Y, Brennan J, Molitich M, Kohan DE. Clinical efficacy of the selective endothelin A receptor antagonist, atrasentan, in patients with diabetes and chronic kidney disease (CKD) Life Sci. 2012;91:739–742. doi: 10.1016/j.lfs.2012.01.011. [DOI] [PubMed] [Google Scholar]
- Basu R, Lee J, Wang Z, Patel VB, Fan D, Das SK, Liu GC, John R, Scholey JW, Oudit GY, Kassiri Z. Loss of TIMP3 selectively exacerbates diabetic nephropathy. Am J Physiol Renal Physiol. 2012;303:F1341–F1352. doi: 10.1152/ajprenal.00349.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett WL, Maruthur NM, Singh S, Segal JB, Wilson LM, Chatterjee R, Marinopoulos SS, Puhan MA, Ranasinghe P, Block L, Nicholson WK, Hutfless S, Bass EB, Bolen S. Comparative effectiveness and safety of medications for type 2 diabetes: an update including new drugs and 2-drug combinations. Ann Intern Med. 2011;154:602–613. doi: 10.7326/0003-4819-154-9-201105030-00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton W, Cattran D, Williams M, Adler S, Appel G, Cartwright K, Foiles PG, Freedman B, Raskin P, Ratner R, Spinowitz B, Whittier F, Wuerth J ACTION I Investigator Group. Am J Nephrol. 2004;24:32–40. doi: 10.1159/000075627. [DOI] [PubMed] [Google Scholar]
- Bomback AS, Kshirsagar AV, Amamoo MA, Klemmer PJ. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: a systematic review. Am J Kidney Dis. 2008;51:199–211. doi: 10.1053/j.ajkd.2007.10.040. [DOI] [PubMed] [Google Scholar]
- Burrows NR, Li Y, Geiss LS. Incidence of treatment for end-stage renal disease among individuals with diabetes in the U.S. continues to decline. Diabetes Care. 2010;33:73–77. doi: 10.2337/dc09-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WW, Zhang L, Yao YS, Su H, Jin YL, Chen Y. Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism and susceptibility to diabetic nephropathy in Chinese type 2 diabetic patients: a meta-analysis. Ren Fail. 2013;35:1038–1043. doi: 10.3109/0886022X.2013.810542. [DOI] [PubMed] [Google Scholar]
- Cherney DZ, Konvalinka A, Zinman B, Diamandis EP, Soosaipillai A, Reich H, Lorraine J, Lai V, Scholey JW, Miller JA. Effects of protein kinase Cβ inhibition on renal hemodynamic function and urinary biomarkers in humans with type 1 diabetes: a pilot study. Diabetes Care. 2009;32:91–93. doi: 10.2337/dc08-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlan MT, Forbes JM, Cooper ME. Role of AGE crosslink breaker, alagebrium, as a renoprotective agent in diabetes. Kidney Int. 2007;72(Suppl. 106):54–60. doi: 10.1038/sj.ki.5002387. [DOI] [PubMed] [Google Scholar]
- Cui W, Du B, Zhou W, Jia Y, Sun G, Sun J, Zhang D, Yuan H, Xu F, Lu X, Luo P, Miao L. Relationship between five GLUT1 gene single nucleotide polymorphisms and diabetic nephropathy: a systematic review and meta-analysis. Mol Biol Rep. 2012;39:8551–8558. doi: 10.1007/s11033-012-1711-z. [DOI] [PubMed] [Google Scholar]
- DCCT. Effect of intensive therapy on the development and progression of diabetic nephropathy in the Diabetes Control and Complications Trial. The Diabetes Control and Complications (DCCT) Research Group. Kidney Int. 1995;47:1703–1720. doi: 10.1038/ki.1995.236. [DOI] [PubMed] [Google Scholar]
- DCCT. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. N Eng J Med. 2000;342:381–389. doi: 10.1056/NEJM200002103420603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellamea BS, Pinto LC, Leitao CB, Santos KG, Canani LH. Endothelial nitric oxide synthase gene polymorphisms and risk of diabetic nephropathy: a systematic review and meta-analysis. BMC Med Genet. 2014;15:9. doi: 10.1186/1471-2350-15-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zeeuw D, Agarwal R, Amdahl M, Audhya P, Coyne D, Garimella T, Parving HH, Pritchett Y, Remuzzi G, Ritz E, Andress D. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet. 2010;376:1543–1551. doi: 10.1016/S0140-6736(10)61032-X. [DOI] [PubMed] [Google Scholar]
- DiPetrillo K, Gesek FA. Pentoxifylline ameliorates renal tumor necrosis factor expression, sodium retention, and renal hypertrophy in diabetic rats. Am J Nephrol. 2004;24:352–359. doi: 10.1159/000079121. [DOI] [PubMed] [Google Scholar]
- Duckworth W, Abraira C, Moritz T, Reda D, Emanuele N, Reaven PD, Zieve FJ, Marks J, Davis SN, Hayward R, Warren SR, Goldman S, McCarren M, Vitek ME, Henderson WG, Huang GD VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360:129–139. doi: 10.1056/NEJMoa0808431. [DOI] [PubMed] [Google Scholar]
- Ewens KG, George RA, Sharma K, Ziyadeh FN, Spielman RS. Assessment of 115 candidate genes for diabetic nephropathy by transmission/disequilibrium test. Diabetes. 2005;54:3305–3318. doi: 10.2337/diabetes.54.11.3305. [DOI] [PubMed] [Google Scholar]
- Fernández-Juárez G, Luño J, Barrio V, García de Vinuesa S, Praga M, Goicoechea M, Lahera V, Casas L, Oliva J on behalf of the PRONEDI Study Group. 25 (OH) vitamin D levels and renal disease progression in patients with type 2 diabetic nephropathy and blockade of the renin-angiotensin system. Clin J Am Soc Nephrol. 2013;8:1870–1876. doi: 10.2215/CJN.00910113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyvbjerg A, Denner L, Schrijvers BF, Tilton RG, Mogensen TH, Paludan SR, Rash R. Long-term renal effects of a neutralizing RAGE antibody in obese type 2 diabetic mice. Diabetes. 2004;53:166–172. doi: 10.2337/diabetes.53.1.166. [DOI] [PubMed] [Google Scholar]
- Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137–188. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- Freedman BI, Wuerth JP, Cartwright K, Bain RP, Dippe S, Hershon K, Mooradian AD, Spinowithz BS. Design and baseline characteristics for the aminoguanidine clinical trial in overt type 2 diabetic nephropathy (ACTION II) Control Clin Trials. 1999;20:493–510. doi: 10.1016/s0197-2456(99)00024-0. [DOI] [PubMed] [Google Scholar]
- Fried LF, Emanuele N, Zhang JH, Brophy M, Conner TA, Duckworth W, Leehey DJ, McCullough PA, O'Connor T, Palevsky PM, Reilly RF, Seliger S, Warren SR, Watnick S, Peduzzi P, Guarino P for the VA NEPHRON-D Investigators. Combined angiotensin inhibition for the treatment of diabetic nephropathy. New Engl J Med. 2013;369:1892–1903. doi: 10.1056/NEJMoa1303154. [DOI] [PubMed] [Google Scholar]
- Gnudi L, Thomas SM, Viberti G. Mechanical forces in diabetic kidney disease: a trigger for impaired glucose metabolism. J Am Soc Nephrol. 2007;18:2226–2232. doi: 10.1681/ASN.2006121362. [DOI] [PubMed] [Google Scholar]
- Grahammer F, Schell C, Huber TB. The podocyte slit diaphragm – from a thin grey line to a complex signalling hub. Nat Rev Nephrol. 2013;9:587–598. doi: 10.1038/nrneph.2013.169. [DOI] [PubMed] [Google Scholar]
- Guerrero-Romero F, Rodríguez-Morán M, Paniagua-Sierra J, García-Bulnes G, Salas-Ramírez M, Amato D. Pentoxifylline reduces proteinuria in insulin-dependent and non-dependent diabetic patients. Clin Nephrol. 1995;43:116–121. [PubMed] [Google Scholar]
- Gunduz Z, Canoz O, Per H, Dusunsel R, Poyrazoglu H, Tez C, Saraymen R. The effects of pentoxifylline on diabetic renal changes in streptozotocin-induced diabetes mellitus. Renal Fail. 2004;26:597–605. doi: 10.1081/jdi-200038329. [DOI] [PubMed] [Google Scholar]
- Halimi JM. The emerging concept of chronic kidney disease without clinical proteinuria in diabetic patients. Diabetes Metab. 2012;38:291–297. doi: 10.1016/j.diabet.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Harmankaya O, Seber S, Yilmaz M. Combination of pentoxifylline with angiotensin converting enzyme inhibitors produces an additional reduction in microalbuminuria in hypertensive type 2 diabetic patients. Ren Fail. 2003;25:465–470. doi: 10.1081/jdi-120021159. [DOI] [PubMed] [Google Scholar]
- Hasslacher C, Ritz E, Wahl P, Michael C. Similar risks of nephropathy in patients with type I or type II diabetes mellitus. Nephrol Dial Transplant. 1989;4:859–863. doi: 10.1093/ndt/4.10.859. [DOI] [PubMed] [Google Scholar]
- Hostetter TH, Troy JL, Brenner BM. The case for intrarenal hypertension in the initiation and progression of diabetic and other glomerulopathies. Am J Med. 1982;72:375–380. doi: 10.1016/0002-9343(82)90490-9. [DOI] [PubMed] [Google Scholar]
- Huber TB, Benzing T. The slit diaphragm: a signaling platform to regulate podocyte function. Curr Opin Nephrol Hypertens. 2005;14:211–216. doi: 10.1097/01.mnh.0000165885.85803.a8. [DOI] [PubMed] [Google Scholar]
- Ismail-Beigi F, Craven T, Banerji MA, Basile J, Calles J, Cohen RM, Cuddihy R, Cushman WC, Genuth S, Grimm RH, Jr, Hamilton BP, Hoogwerf B, Karl D, Katz L, Krikorian A, O'Connor P, Pop-Busui R, Schubart U, Simmons D, Taylor H, Thomas A, Weiss D, Hramiak I ACCORD trial group. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet. 2010;376:419–430. doi: 10.1016/S0140-6736(10)60576-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanwar YS, Sun L, Xie P, Liu F, Chen S. A glimpse of various pathogenic mechanisms of diabetic nephropathy. Ann Rev Pathol. 2011;6:395–423. doi: 10.1146/annurev.pathol.4.110807.092150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassiri Z, Oudit GY, Kandalam V, Awad A, Wang X, Ziou X, Maeda N, Herzenberg AM, Scholey JW. Loss of TIMP3 enhances interstitial nephritis and fibrosis. J Am Soc Nephrol. 2009;20:1223–1235. doi: 10.1681/ASN.2008050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ. Oral cholecalciferol decreases albuminuria and urinary TGF-β1 in patients with type 2 diabetic nephropathy on established renin–angiotensin–aldosterone system inhibition. Kidney Int. 2011;80:851–860. doi: 10.1038/ki.2011.224. [DOI] [PubMed] [Google Scholar]
- Kohan DE, Pritchett Y, Molitch M, Wen S, Garimella T, Audhya P, Andress DL. Addition of atrasentan to renin-angiotensin system blockade reduces albuminuria in diabetic nephropathy. J Am Soc Nephrol. 2011;22:763–772. doi: 10.1681/ASN.2010080869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann A, Haas CS, Gross ML, Reulbach U, Holzinger M, Schwarz U, Ritz E, Amann K. 1,25-Dihydroxyvitamin D3 decreases podocyte loss and podocyte hypertrophy in the subtotally nephrectomized rat. Am J Physiol Renal Physiol. 2004;286:F526–F533. doi: 10.1152/ajprenal.00316.2003. [DOI] [PubMed] [Google Scholar]
- Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008;148:30–48. doi: 10.7326/0003-4819-148-1-200801010-00190. [DOI] [PubMed] [Google Scholar]
- Lewis EJ, Greene T, Spitalewiz S, Blumenthal S, Berl T, Hunsicker LG, Pohl MA, Rohde RD, Raz I, Yerushalmy Y, Yagil Y, Herskovits T, Atkins RC, Reutens AT, Packham DK, Lewis JB Collaborative Study Group. Pyridorin in type 2 diabetic nephropathy. J Am Soc Nephrol. 2012;23:131–136. doi: 10.1681/ASN.2011030272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Huang G, Zhang J, Lin X. Adiponectin gene polymorphisms and susceptibility to diabetic nephropathy: a meta-analysis. Ren Fail. 2014;36:478–487. doi: 10.3109/0886022X.2013.868319. [DOI] [PubMed] [Google Scholar]
- Macía-Heras M, Del Castillo-Rodriguez N, Navarro-González J. The renin-angiotensin-aldosterone system in renal and cardiovascular disease and the effects of its pharmacological blockade. J Diabetes Metab. 2012;3:1–24. [Google Scholar]
- Mann JF, Green D, Jamerson K, Ruilope LM, Kuranoff SJ, Littke T, Viberti G for the ASCEND Study Group. Avosentan for overt diabetic nephropathy. J Am Soc Nephrol. 2010;21:527–535. doi: 10.1681/ASN.2009060593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson PW. The podocyte cytoskeleton in health and in disease. Clin Kidney J. 2012;5:498–501. doi: 10.1093/ckj/sfs153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehdi UF, Adams-Huet B, Raskin P, Vega GL, Toto RD. Addition of angiotensin receptor blockade or mineralocorticoid antagonism to maximal angiotensin-converting enzyme inhibition in diabetic nephropathy. J Am Soc Nephrol. 2009;20:2641–2650. doi: 10.1681/ASN.2009070737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra R, Kermah DA, Salusky IB, Wolf MS, Thadhani RI, Chiu YW, Martins D, Adler SG, Norris KC. Chronic kidney disease, hypovitaminosis D, and mortality in the United States. Kidney Int. 2009;76:977–983. doi: 10.1038/ki.2009.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JH. Organogenesis of the kidney glomerulus: focus on the glomerular basement membrane. Organogenesis. 2011;7:75–82. doi: 10.4161/org.7.2.15275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JH. The glomerular basement membrane. Exp Cell Res. 2012;318:973–978. doi: 10.1016/j.yexcr.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Kidney Foundation. KDOQI clinical practice guideline and clinical practice recommendations for diabetes and chronic kidney disease. Am J Kidney Dis. 2007;49(Suppl. 2):S1–S180. doi: 10.1053/j.ajkd.2006.12.005. [DOI] [PubMed] [Google Scholar]
- National Kidney Foundation. KDOQI clinical practice guideline for diabetes and CKD: 2012 update. Am J Kidney Dis. 2012;60:850–886. doi: 10.1053/j.ajkd.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Navarro JF, Milena FJ, Mora C, León C, García J. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: Effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am J Nephrol. 2006;26:562–570. doi: 10.1159/000098004. [DOI] [PubMed] [Google Scholar]
- Navarro JF, Mora C, Muros M, García J. Additive antiproteinuric effect of pentoxifylline in patients with type 2 diabetes under angiotensin II receptor blockade: a short term, randomised, controlled trial. J Am Soc Nephrol. 2005;16:2119–2126. doi: 10.1681/ASN.2005010001. [DOI] [PubMed] [Google Scholar]
- Navarro JF, Mora C, Muros M, Macía M, García J. Effects of pentoxifylline administration on urinary N-acetyl-β-glucosaminidase excretion in type 2 diabetic patients: a short-term, prospective, randomized study. Am J Kidney Dis. 2003;42:264–270. doi: 10.1016/s0272-6386(03)00651-6. [DOI] [PubMed] [Google Scholar]
- Navarro JF, Mora C, Rivero A, Gallego E, Chahin J, Macía M, Méndez ML, García J. Urinary protein excretion and serum tumor necrosis factor in diabetic patients with advanced renal failure: effects of pentoxifylline administration. Am J Kidney Dis. 1999;33:458–463. doi: 10.1016/s0272-6386(99)70182-4. [DOI] [PubMed] [Google Scholar]
- Navarro-González JF, Mora-Fernández C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol. 2008;19:433–442. doi: 10.1681/ASN.2007091048. [DOI] [PubMed] [Google Scholar]
- Obeidat M, Ballermann BJ. Glomerular endothelium: a porous sieve and formidable barrier. Exp Cell Res. 2012;318:964–972. doi: 10.1016/j.yexcr.2012.02.032. [DOI] [PubMed] [Google Scholar]
- Panichi V, De Pietro S, Andreini B, Bianchi AM, Migliori M, Taccola D, Giovannini L, Tetta C, Palla R. Calcitriol modulates in vivo and in vitro cytokine production: A role for intracellular calcium. Kidney Int. 1998;54:1463–1469. doi: 10.1046/j.1523-1755.1998.00152.x. [DOI] [PubMed] [Google Scholar]
- Parving HH, Brenner BM, McMurray JJV, de Zeeuw D, Haffner SM, Solomon SD, Chatuverdi N, Persson F, Desai AS, Nicolaides M, Richard A, Xiang Z, Brunel P, Pfeffer MA for the ALTITUDE Investigators. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. New Engl J Med. 2012;367:2204–2213. doi: 10.1056/NEJMoa1208799. [DOI] [PubMed] [Google Scholar]
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK AVOID Study Investigators. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008;358:2433–2446. doi: 10.1056/NEJMoa0708379. [DOI] [PubMed] [Google Scholar]
- Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, Marre M, Cooper M, Glasziou P, Grobbee D, Hamet P, Harrap S, Heller S, Liu L, Mancia G, Mogensen CE, Pan C, Poulter N, Rodgers A, Williams B, Bompoint S, de Galan BE, Joshi R, Travert F ADVANCE Collaborative Group. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–2572. doi: 10.1056/NEJMoa0802987. [DOI] [PubMed] [Google Scholar]
- Patterson CC, Gyürüs E, Rosenbauer J, Cinek O, Neu A, Schober E, Parslow RC, Joner G, Svensson J, Castell C, Bingley PJ, Schoenle E, Jarosz-Chobot P, Urbonaité B, Rothe U, Krzisnik C, Ionescu-Tirgoviste C, Weets I, Kocova M, Stipancic G, Samardzic M, de Beaufort CE, Green A, Dahlquist GG, Soltész G. Trends in childhood type 1 diabetes incidence in Europe during 1989–2008: evidence of non-uniformity over time in rates of increase. Diabetologia. 2012;55:2142–2147. doi: 10.1007/s00125-012-2571-8. [DOI] [PubMed] [Google Scholar]
- Pavenstädt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- Remuzzi G, Schieppati A, Ruggenenti P. Nephropathy in patients with type 2 diabetes. New Engl J Med. 2002;346:1145–1151. doi: 10.1056/NEJMcp011773. [DOI] [PubMed] [Google Scholar]
- Ritz E, Rychlik I, Locatelli F, Halimi S. End-stage renal failure in type 2 diabetes: A medical catastrophe of worldwide dimensions. Am J Kidney Dis. 1999;34:795–808. doi: 10.1016/S0272-6386(99)70035-1. [DOI] [PubMed] [Google Scholar]
- Rivero A, Mora C, Muros M, García J, Herrera H, Navarro-González JF. Pathogenic perspectives for the role of inflammation in diabetic nephropathy. Clin Sci. 2009;116:479–492. doi: 10.1042/CS20080394. [DOI] [PubMed] [Google Scholar]
- Satchell SC, Braet F. Glomerular endothelial cell fenestrations: an integral component of the glomerular filtration barrier. Am J Physiol Renal Physiol. 2009;296:F947–F956. doi: 10.1152/ajprenal.90601.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz U, Amann K, Orth SR, Simonaviciene A, Wessels S, Ritz E. Effect of 1,25 (OH)2 vitamin D3 on glomerulosclerosis in subtotally nephrectomized rats. Kidney Int. 1998;53:1696–1705. doi: 10.1046/j.1523-1755.1998.00951.x. [DOI] [PubMed] [Google Scholar]
- Seok SJ, Lee ES, Kim GT, Hyun M, Lee JH, Chen S, Choi R, Kim HM, Lee EY, Chung CH. Blockade of CCL2/CCR2 signalling ameliorates diabetic nephropathy in db/db mice. Nephrol Dial Transplant. 2013;28:1700–1710. doi: 10.1093/ndt/gfs555. [DOI] [PubMed] [Google Scholar]
- Singh A, Satchell SC, Neal CR, McKenzie EA, Tooke JE, Mathieson PW. Glomerular endothelial glycocalyx constitutes a barrier to protein permeability. J Am Soc Nephrol. 2007;18:2885–2893. doi: 10.1681/ASN.2007010119. [DOI] [PubMed] [Google Scholar]
- Soler MJ, Wysocki J, Batlle D. Angiotensin-converting enzyme 2 and the kidney. Exp Physiol. 2008;93:549–556. doi: 10.1113/expphysiol.2007.041350. [DOI] [PubMed] [Google Scholar]
- Stanton RC. Clinical challenges in diagnosis and management of diabetic kidney disease. Am J Kidney Dis. 2014;63(Suppl. 2):S3–S21. doi: 10.1053/j.ajkd.2013.10.050. [DOI] [PubMed] [Google Scholar]
- Takamiya Y, Fukami K, Yamagishi S, Kaida Y, Nakayama Y, Obara N, Iwatani R, Ando R, Koike K, Matsui T, Nishino Y, Ueda S, Cooper ME, Okuda S. Experimental diabetic nephropathy is accelerated in matrix metalloproteinase-2 knockout mice. Nephrol Dial Transplant. 2013;28:55–62. doi: 10.1093/ndt/gfs387. [DOI] [PubMed] [Google Scholar]
- Takiishi T, Gysemans C, Bouillon R, Mathieu C. Vitamin D and diabetes. Endocrinol Metab Clin North Am. 2010;39:419–446. doi: 10.1016/j.ecl.2010.02.013. [DOI] [PubMed] [Google Scholar]
- Tervaert TWC, Mooyaart AL, Amann K, Cohen AH, Cook HT, Drachenberg CB, Ferrario F, Fogo AB, Hass M, de Heer E, Joh K, Noël LH, Radhakrishnan J, Seshan S, Bajema IM, Bruijin JA and on behalf of the Renal Pathology Society. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010;21:556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- Thallas-Bonke V, Lindschau C, Rizkalla B, Bach LA, Boner G, Meier M, Haller H, Cooper ME, Forbes JM. Attenuation of extracellular matrix accumulation in diabetic nephropathy by the advanced glycation end product cross-link breaker ALT-711 via a protein kinase Cα-dependent pathway. Diabetes. 2004;53:2921–2930. doi: 10.2337/diabetes.53.11.2921. [DOI] [PubMed] [Google Scholar]
- Tuttle KR, Bakris GL, Toto RD, McGill JB, Hu K, Anderson PW. The effect of ruboxistaurin on nephropathy in type 2 diabetes. Diabetes Care. 2005;28:2686–2690. doi: 10.2337/diacare.28.11.2686. [DOI] [PubMed] [Google Scholar]
- Tutle KR. Protein kinase Cβ inhibition for diabetic kidney disease. Diabetes Res Clin Pract. 2008;82(Suppl. 1):S70–S74. doi: 10.1016/j.diabres.2008.09.041. [DOI] [PubMed] [Google Scholar]
- UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ. 1998;317:703–713. [PMC free article] [PubMed] [Google Scholar]
- UKPDS. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998a;352:837–853. [PubMed] [Google Scholar]
- UKPDS. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998b;352:854–865. [PubMed] [Google Scholar]
- Wartiovaara J, Ofverstedt LG, Khoshnoodi J, Zhang J, Mäkelä E, Sandin S, Ruotsalainen V, Cheng RH, Jalanko H, Skoglund U, Tryggvason K. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J Clin Invest. 2004;114:2004. doi: 10.1172/JCI22562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng. 2007;9:121–167. doi: 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- Whiting DR, Guariguata L, Weil C, Shaw J. IDF diabetes atlas: global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res Clin Pract. 2011;94:311–321. doi: 10.1016/j.diabres.2011.10.029. [DOI] [PubMed] [Google Scholar]
- Yang S, Zhang J, Feng C, Huang C. MTHFR 677T variant contributes to diabetic nephropathy risk in Caucasian individuals with type 2 diabetes: a meta-analysis. Metabolism. 2013;62:586–594. doi: 10.1016/j.metabol.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Yin YW, Qiao L, Sun QQ, Hu AM, Liu HL, Wang Q, Hou ZZ. Influence of apolipoprotein E gene polymorphism on development of type 2 diabetes mellitus in Chinese Han population: a meta-analysis of 29 studies. Metabolism. 2014;63:532–541. doi: 10.1016/j.metabol.2013.12.008. [DOI] [PubMed] [Google Scholar]
- Yu ZY, Chen LS, Zhang LC, Zhou TB. Meta-analysis of the relationship between ACE I/D gene polymorphism and end-stage renal disease in patients with diabetic nephropathy. Nephrology (Carlton) 2012;17:480–487. doi: 10.1111/j.1440-1797.2012.01592.x. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhang Y, Ning G, Deb DK, Kong J, Li YC. Combination therapy with AT1 blocker and vitamin D analog markedly ameliorates diabetic nephropathy: Blockade of compensatory renin increase. Proc Natl Acad Sci U S A. 2008;105:15896–15901. doi: 10.1073/pnas.0803751105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Zhang L, Li Z, Yang Y, Tang Y, Fu P. Association between TNF-α -308G/A polymorphism and diabetic nephropathy risk: a meta-analysis. Int Urol Nephrol. 2013;45:1653–1659. doi: 10.1007/s11255-013-0490-3. [DOI] [PubMed] [Google Scholar]
- Zheng F, He C, Cai W, Hattori M, Steffes M, Vlassara H. Prevention of diabetic nephropathy in mice by a diet low in glycoxidation products. Diabetes Metab Res Rev. 2002;18:224–237. doi: 10.1002/dmrr.283. [DOI] [PubMed] [Google Scholar]
- Zhou TB, Jian ZP, Qin YH, Drummen GP. Association of transforming growth factor-β1 T869C gene polymorphism with diabetic nephropathy risk. Nephrology (Carlton) 2014;19:107–115. doi: 10.1111/nep.12176. [DOI] [PubMed] [Google Scholar]
- Zhu JM, Wang B, Li J, Chen GM, Fan YG, Feng CC, Pan HF, Ye DQ. D18S880 microsatellite polymorphism of carnosinase gene and diabetic nephropathy: a meta-analysis. Genet Test Mol Biomarkers. 2013;17:289–294. doi: 10.1089/gtmb.2012.0341. [DOI] [PubMed] [Google Scholar]