

Abstract
The glomerular filtration barrier comprises a fenestrated capillary endothelium, glomerular basement membrane and podocyte slit diaphragm. Over the past decade we have come to realise that permselectivity depends on size and not necessarily charge, that the molecular sieve depends on the podocyte contractile apparatus and is highly dynamic, and that protein uptake by proximal tubular epithelial cells stimulates signalling and the production of transcription factors and inflammatory mediators. Alport syndrome is the second commonest monogenic cause of renal failure after autosomal dominant polycystic kidney disease. Eighty per cent of patients have X-linked disease caused by mutations in the COL4A5 gene. Most of these result in the replacement of the collagen IV α3α4α5 network with the α1α1α2 heterotrimer. Affected membranes also have ectopic laminin and increased matrix metalloproteinase levels, which makes them more susceptible to proteolysis. Mechanical stress, due to the less elastic membrane and hypertension, interferes with integrin-mediated podocyte–GBM adhesion. Proteinuria occurs when urinary levels exceed tubular reabsorption rates, and initiates tubulointerstitial fibrosis. The glomerular mesangial cells produce increased TGFβ and CTGF which also contribute to glomerulosclerosis. Currently there is no specific therapy for Alport syndrome. However treatment with angiotensin converting enzyme (ACE) inhibitors delays renal failure progression by reducing intraglomerular hypertension, proteinuria, and fibrosis. Our greater understanding of the mechanisms underlying the GBM changes and their consequences in Alport syndrome have provided us with further novel therapeutic targets.
 |
Judy Savige trained as a physician specialising in nephrology at the University of Melbourne and the Royal Melbourne Hospital. She completed her PhD in immunology at the Hammersmith Hospital laboratory and returned to a post-doctoral position at the Royal Melbourne Hospital and the Howard Florey Institute. Subsequent positions include Professor of Medicine at the University of Melbourne and Foundation Professor, Chairman of the Division of Medicine, and Director of Clinical Training at Northern Health. She has recently returned to the Royal Melbourne Hospital as a Professorial Fellow. Professor Savige's research interests have been inherited renal disease, especially Alport syndrome and Thin basement membrane nephropathy, the demonstration of ocular abnormalities in inherited renal disease and mutation databases. Her laboratory demonstrated the genes affected in thin basement membrane nephropathy and that this represented the carrier state for autosomal recessive Alport syndrome. Her laboratory co-curates the mutation databases for Alport syndrome with the Guy's Genetics laboratory, and for Dense deposit disease.
Overview of glomerular filtration barrier
The nephron is the basic structural and functional unit of the kidney, and comprises the glomerulus, proximal convoluted tubule, loop of Henle, distal convoluted tubule and collecting duct. The glomerulus consists of capillary loops from which water and low molecular weight molecules (glucose, amino acids, creatinine, urea) move across the filtration barrier into Bowman's space, and then into the proximal tubular lumen. Normally humans produce 180 L of filtrate daily from plasma with 10 kg protein, but less than 1 g of protein is found in the urine (Hausmann et al. 2010). Proteinuria is a risk factor for progressive renal failure in all forms of kidney disease, and tubulointerstitial inflammation, fibrosis and tubular atrophy correlate directly with the amount of proteinuria.
The filtration barrier consists of a fenestrated capillary endothelium; the glomerular basement membrane (GBM); and the slit diaphragm between podocyte foot processes (Fig. 1). The endothelial fenestrations and glycocalyx allow direct access of plasma proteins to the GBM (Kriz, 2008), the GBM results from the fusion of the basement membranes produced by the endothelial cells and podocytes (Abrahamson et al. 2009), and the slit diaphragm is formed by the lateral interaction of proteins from adjacent podocyte foot processes. Podocytes enclose the outer surface of the glomerular capillaries with narrow ‘foot processes’ that interdigitate with those of neighbouring cells. Adjacent foot processes are linked to each other by the slit diaphragms. Podocytes also interact basally with the GBM through integrin–cell matrix interactions that signal to the cytoskeleton and help regulate the GBM architecture, and porosity (Suh & Miner, 2013; Fig. 2).
Figure 1. Factors in the pathogenesis of glomerular disease in Alport syndrome.

A, increased biomechanical strain from hypertension, abnormal GBM, altered podocyte cytoskeleton and cell adhesion. B, lamellated or thinned GBM with different collagen IV and laminin molecules, and increased MMP proteolysis. C, podocyte α2β1 integrin and DDR1 detect abnormal collagen IV. D, altered podocyte adhesion and actin/cytoskeleton. E, nuclear peroxisome proliferator-activated receptor γ (PPARγ) downregulation of SD slit diaphragm gene transcription and podocyte apoptosis (based on Noone & Licht, 2013).
Figure 2. Pathogenesis of tubulointerstitial disease and disease progression in Alport syndrome.
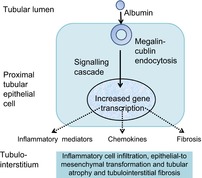
The figure is based on Noone & Licht, 2013.
The glomerular filtration barrier prevents the passage of molecules based on their size, that is, their molecular weight and radius (Ohlson et al. 2000). The nature and location of the size barrier, and the relative contributions of the slit diaphragm, GBM and endothelium are not known. Whether the glomerular filtration barrier also selects for charge is controversial (Harvey & Miner, 2008).
Fenestrated endothelium
The endothelial fenestrae may contribute to protein sieving (Jeansson & Haraldsson, 2006), not through size since the fenestrae are too large, but possibly based on the negatively charged cell surface glycocalyx.
GBM
The GBM comprises mainly collagen IV, laminin, nidogen and the heparan sulphate proteoglycan, agrin. Collagen IV is a heterotrimer of α1–α6 chains. The individual chains all have non-collagenous termini and an intermediate triple helix with a Gly-Xaa-Yaa sequence (Hudson et al. 2003). Unlike fibrillar collagen, the collagen IV termini are retained and interact through their amino and carboxy ends via disulphide and sulfilimine bonds, and lysyl-oxidase-mediated cross-links (Hudson, 2004; Vanacore et al. 2009) to form a ‘chicken wire-like’ structure. Collagen IV is found in α1α1α2, α3α4α5 or α5α5α6 networks. The α1α1α2 network occurs in the GBM at birth but is replaced, in infancy, by the α3α4α5 heterotrimer in the glomerulus, cochlea and eye. Collagen IV is highly metabolically active with many binding partners (Parkin et al. 2011).
Laminin is a heterotrimer of α, β and γ chains, with variants of each of these chains producing at least 15 different isoforms (Miner & Yurchenco, 2004). Each molecule comprises a cruciform structure in which the N-termini of each of the three chains form a short arm and the carboxy termini combine to form a single long arm. The laminin trimers are named for their α, β and γ chains in this order, and four isoforms (laminin-111, -511 -521 and small amounts of laminin-332) are found in the kidney. Laminin-511 predominates in the GBM at birth but switches to laminin-521 in infancy when the collagen IV networks change. Laminin is linked to neighbouring podocytes by integrins and dystroglycan. The laminin–integrin interaction physically links the GBM to the podocyte actin cytoskeleton (through integrin and actin-associated proteins such as talin and α- actinin; Faul et al. 2007), and is reinforced by CD151.
Nidogen acts as a bridge between collagen IV and laminin, and probably stabilises the GBM. Agrin links laminin and the GBM to adjacent cells (Bezakova & Ruegg, 2003), but mice lacking agrin have no significant structural or functional defects (Suh & Miner, 2013).
Podocyte slit diaphragm
Podocyte foot process effacement and slit diaphragm abnormalities occur in nearly all cases of proteinuria. The significance of foot process effacement in the development of proteinuria is not clear.
The slit diaphragm consists of multiple layers of nephrin strands that connect adjacent podocyte foot processes (Kestila et al. 1998). This meshwork has channels that can accommodate albumin molecules (Wartiovaara et al. 2004). However, the slit diaphragm is located distally in the glomerular filtration barrier and is unlikely to be significant even if its pores are smaller than albumin (Jarad & Miner, 2009). Furthermore the amount of proteinuria does not correlate directly with slit diaphragm damage (Jarad & Miner, 2009).
Proteinuria
Tubular protein reabsorption
Albumin is the most abundant molecule in the glomerular filtrate and is reabsorbed by the proximal tubular epithelial cells. Excessive albumin loss in the filtrate and reabsorption by the proximal tubular cells results in abnormal signalling and a proinflammatory environment.
Proximal tubular epithelial cells have specific binding sites for albumin which is absorbed via receptor-mediated endocytosis. Megalin and cubulin form a complex that mediates albumin uptake. Megalin is a member of the lipoprotein receptor (LDL-R) family with a large extracellular domain and many complement-type, growth factor-repeats and an epidermal growth factor-repeat. The cytoplasmic tail has sequence motifs for phosphorylation probably for signalling. Cubulin, on the other hand, is almost entirely extracellular with only a small membrane anchoring domain.
For many ligands, megalin is processed via regulated intramembrane proteolysis (RIP). Phosphokinase C-dependent matrix metalloproteinase cleaves the receptor, a γ-secretase cleaves the remaining transmembrane–cytosolic receptor fragment, and the released fragment translocates to the nucleus where it upregulates gene transcription (Li et al. 2008). The cleaved megalin extracellular domain with its epidermal growth factor (EGF)-like structure binds to EGF-R and activates signalling.
Thus, excess urinary albumin results in a complex signalling cascade and upregulation of various chemotactic (MCP1, RANTES; Ninichuk et al. 2005), inflammatory (tumour necrosis factor α, TNFα) and profibrotic (transforming growth factor-β, TGFβ, and connective tissue growth factor, CTGF) mediators (Table 1; Baines & Brunskill, 2011). Albuminuria also appears to have a role in tubular cell apoptosis through inhibiting the nuclear factor κB (NFκB)-dependent survival pathways (Takase et al. 2008).
Table 1.
Disease pathogenesis in Alport syndrome and targets of treatment
| Mechanism | Strategy | Status as treatment and efficacy |
|---|---|---|
| Genetic mutation resulting in loss of collagen IV α3α4α5 heterotrimer from the GBM | Correction of gene defect at genetic level | Not possible currently |
| Nonsense mutations – inhibition of nonsense-mediated decay | No evidence yet | |
| Nonsense mutations – read through of mutation | Some evidence in other diseases such as Duchenne muscular dystrophy (Finkel et al. 2013) | |
| Stem cell therapy | Not possible currently in humans but appears to work in mice even with a damaged GBM | |
| Gene replacement therapy | Adenoviral vectors with LacZ expression cassette (Heikkila et al. 1996; Parpala-Sparman et al. 1999) and renal perfusion. Results in increased gene expression | |
| Bone marrow transplantation | Some expression of collagen IV α3α4α5 and improved renal function in mice (Prodromidi et al. 2006; Sugimoto et al. 2006); improved renal function and life span but only marginally (LeBleu et al. 2009) | |
| Missense mutations – Chaperone therapy | Appears to work partially in cell lines (Pieri et al. 2014) | |
| Mechanical strain | Renin–angiotensin–aldosterone blockade: Anti-hypertensive (reduces mechanical strain), antiproteinuric, antifibrotic | Efficacious in humans, delays onset of end-stage renal failure by up to 10 years (Gross et al. 2003) |
| Aldosterone antagonist: antihypertensive and antiproteinuric | Reduces proteinuria and TGFβ1 (Giani et al. 2013) | |
| Other antihypertensives: reduce mechanical strain and renal failure progression | Less effective than ACE inhibition | |
| Matrix metalloproteinase degradation of GBM | Inhibition of MMP-2, -3 and -9 | Before proteinuria, delays onset of proteinuria and prolongs survival in mice (Zeisberg et al. 2006) |
| Inhibition of MMP12 through CCR2 | Prevents GBM damage in mice (Rao et al. 2006) | |
| Inhibition of MMP-12 through genetic ablation of USAG1 | Inhibits disease progression and doubles lifespan in mice (Tanaka et al. 2010) | |
| Integrin α1 chain knockout in podocytes and mesangial cells | Further protection against GBM destruction (Sayers et al. 1999) | |
| DDR1/2 inhibition | (Kruegel et al. 2013) | |
| Inflammation | Chemokine receptor antagonists, reduce inflammation | May not be specific |
| Irradiation, reduces neutrophil infiltration | Small improvement in renal function in mice (Katayama et al. 2008) | |
| Complement inhibitors | Inhibit proximal tubular epithelial cell production of inflammatory and profibrotic cytokines (Tang et al. 2009) | |
| Fibrosis | TGFβ soluble receptor inhibitor | Minor improvement in normal mice; more pronounced when combined with α1 blockade (Sayers et al. 1999) |
| Reduced matrix accumulation, and expression of TGFβ1 and CTGF, through HMG-Co-A reductase inhibitors | Improves renal function and extends life span (Koepke et al. 2007) | |
| BMP7 antagonists potentially prevent tubular atrophy | No evidence yet in vivo |
There are also other proximal tubular cell albumin receptors including the neonatal Fc receptor and CD36 (Susztak et al. 2005; Roopenian & Akilesh, 2007)
Protein filtration
The last decade has seen two novel hypotheses proposed to explain the mechanisms underlying proteinuria (Smithies, 2003; Comper et al. 2008). The ‘gel permeation/diffusion hypothesis’ envisages protein diffusion through GBM behaving like a modified gel (Smithies, 2003). Increased protein concentration in the glomerular filtrate results from altered gel or GBM, from a reduced surface area for filtration with foot process effacement, or from reduced endothelial fenestration. These increase hydraulic resistance but diffusion of plasma proteins remains constant, that is, the same amount of protein is diluted in a smaller volume. The increased protein concentration in the filtrate then overwhelms tubular resorption. The more conventional explanation is that electrical forces determine glomerular permselectivity and there is evidence in non-mammalian systems for this (Hausmann et al. 2010).
The other novel mechanism describes a controversial route for tubular protein absorption. The conventional view is that small amounts of albumin are absorbed through the proximal tubule, that the rate of albumin take-up corresponds with fluid, and that most of the absorbed albumin is degraded and transported through the cells (Park & Maack, 1984). There is preliminary evidence that suggests more albumin leaks through the glomerular filtration barrier, and that much of this is returned to the circulation intact rather than being degraded (Comper et al. 2008).
Alport syndrome
Increasingly diseases are considered to affect the glomerular filtration barrier rather than any of its individual components. Alport syndrome represents one such condition.
Alport syndrome is an inherited form of progressive kidney failure with hearing loss and ocular abnormalities (Gubler et al. 1981; Savige et al. 2013). It affects one in 10,000 individuals, and 80% of families have X-linked inheritance while most of the others have autosomal recessive disease. X-linked disease is caused by mutations in the COL4A5 gene (Barker et al. 1990), and autosomal recessive disease by two mutations in either the COL4A3 or COL4A4 gene. These genes code for the collagen IV α3–α5 chains. Collagen IV represents the major component of basement membranes in the glomerulus, cochlea, lens capsule and retina, which explains the clinical features. Heterozygotes for COL4A3 or COL4A4 mutations are carriers of autosomal recessive Alport syndrome, and have thin basement membrane nephropathy. This occurs in 1% of the population and is characterised by persistent isolated haematuria without proteinuria, hypertension or renal impairment. Hearing and ocular examination are normal.
Clinical features
Renal failure is the most significant risk of Alport syndrome, and males with X-linked inheritance develop end-stage disease before the age of 30 years (‘early onset’, usually with extrarenal features) or after 30 (‘late onset’, often with only hearing loss), and require dialysis or transplantation. Clinical features are generally milder in females with X-linked disease because of X chromosome inactivation, and about 15% develop renal failure by the age of 60 years (less than the published figure which was derived from hospital-based patients; Jais et al. 2003), and, rarely, the lenticonus or central retinopathy.
The GBM in Alport syndrome is lamellated, and there is secondary glomerulosclerosis and tubulointerstitial fibrosis. The rate of deterioration of renal function correlates better with the amount of tubulointerstitial fibrosis than with glomerulosclerosis (Hood et al. 2002).
Haematuria occurs through breaks in the GBM (Collar et al. 2001). The origin of the proteinuria is more complex. The observation that proteinuria occurs relatively late in Alport syndrome suggests that the abnormal collagen IV is not primarily responsible for any increased permeability (Abrahamson et al. 2007; Suh & Miner, 2013). In Alport syndrome, proteinuria occurs with the changes to the filtration barrier and, later, with compensatory hyperfiltration damage and glomerulosclerosis. This contrasts with Pierson disease where mutations in the laminin β2 gene produce proteinuria from birth (Pierson et al. 1963).
Hypertension is common in Alport syndrome too and poorly controlled hypertension increases the rate of renal failure progression.
All patients with Alport syndrome have a high tone sensorineural hearing loss that is helped with hearing aids. This is obvious from childhood in males with X-linked disease but plateaus in middle age. Hearing loss is also common in females with X-linked disease. The stria vascularis membrane in the cochlea is lamellated with an identical appearance to the GBM. The pathogenesis of the hearing defect is not well understood but the membrane appearance resembles that of the GBM and mechanical forces are probably important in both.
The ocular abnormalities do not affect vision significantly but are useful pointers to the diagnosis of Alport syndrome and its severity (Colville & Savige, 1997). The lenticonus or conical anterior protrusion of the lens eventually requires lens replacement and does not recur post-operatively. The central perimacular fleck retinopathy progresses until middle age but does not require treatment. Ultrastructurally the lens capsule is thinned and fractured, and the retinal membranes are thinned too.
What accounts for the differences in clinical features and outcome in Alport syndrome and thin basement membrane nephropathy? Individuals with thin basement membrane nephropathy have a heterozygous mutation in a collagen IV chain gene, and affected membranes are thinned rather than lamellated. The thinned membranes have reduced amounts of the collagen IV α3α4α5 network but still sufficient that there is minimal replacement with the α1α1α2 network, and probably no consequent increase in proteolysis, or mechanical stress. Thus both proteinuria and hypertension are uncommon in thin basement membrane nephropathy. Where proteinuria and renal impairment occur in Thin basement membrane nephropathy, it is likely that there is an additional mutation in the podocin gene (NPHS2; Savige et al. 2003; Tonna et al. 2008), a superimposed glomerular abnormality such as IgA glomerulonephritis, or the thinning is due to misdiagnosed X-linked Alport syndrome.
Diagnosis
The diagnosis of Alport syndrome is made with demonstration of a lamellated GBM on ultrastructural examination of the renal biopsy. In addition, there may be abnormalities in the collagen IV composition on immunohistochemical examination (Kashtan et al. 1989). In X-linked disease, the collagen IV α5 chain, as well as the α3 and α4 chains are often absent from the GBM, and also from the epidermal basement membrane; and in autosomal recessive disease, the α3α4α5 network is absent from the GBM but the α5 chain persists in the α5α5α6 network in the epidermis.
The recent Expert guidelines on the diagnosis and management of Alport syndrome and thin basement membrane nephropathy have indicated that genetic testing should be used to confirm the diagnosis of Alport syndrome (Savige et al. 2013). The advantages of genetic testing are that it makes the diagnosis with certainty, identifies the mode of inheritance, and, for X-linked Alport syndrome, the nature of the underlying mutation suggests the likelihood of early onset renal failure and the need for urgent treatment.
Genetic mutations
More than 1100 unique variants have been described in the COL4A5 gene in X-linked Alport syndrome (https://grenada.lumc.nl/LOVD2/COL4A/home.php?select_db=COL4A5). Large deletions, rearrangements, small deletions and insertions, nonsense mutations, and carboxy terminus missense mutations usually produce early onset renal failure (Jais et al. 2000). Missense mutations at the amino terminus result in the later, adult onset disease, and splice mutations are associated with intermediate severity.
Nonsense mutations or changes that result in a downstream nonsense mutation account for 40% of all mutations in X-linked Alport syndrome, and missense mutations account for a further 40% (Hertz et al. 2012). Little is known about how these mutations cause disease, but in other collagen diseases, nonsense mutations result in nonsense-mediated decay, destruction of the collagen IV α5 mRNA and loss of the corresponding α5 chain from affected tissues (Bateman et al. 2009). Missense mutationis in other similar diseases result in the accumulation of misfolded collagen IV chains in the endoplasmic reticulum (ER) and their failure to be exported into the extracellular matrix (Pieri et al. 2014).
Thus, most mutations in X-linked Alport syndrome, at least in males, result in the loss of the collagen IV α5 chain from affected tissues. The lack of any one of the collagen IV α3, α4 or α5 chains results in the loss of the other chains which form the heterotrimer, and prevents its formation. There is a compensatory increase in the collagen IV α1α1α2 network, which is usually a minor component on the subendothelial GBM surface. The resulting GBM is thinned but functions nearly normally for years until it becomes split, thickened and vacuolated.
Pathogenesis
Understanding the pathogenesis of Alport syndrome is the first step in developing effective treatments (Table 1) (Noone & Licht, 2013).
Disease initiation
Three interdependent mechanisms are responsible for the initiation of the typical Alport GBM abnormalities. The replacement of the collagen network with the α1α1α2 heterotrimer is most important. The replacement network is more susceptible to biomechanical stress and to proteolysis (Kalluri et al. 1997). The collagen α1 and α2 chains have fewer cysteines than the α3 and α4 chains, and hence less cross-linking within and between heterotrimers (Zhou & Reeders, 1996; Gunwar et al. 1998). These chains also have more proteolytic cleavage sites (Parkin et al. 2011). Alport glomeruli also have increased levels of matrix metalloproteinases, MMP2, MMP-3, MMP-9 and especially MMP-12 (Zeisberg et al. 2006; Rao et al. 2006), probably secondary to hypertension-induced biomechanical strain (Meehan et al. 2009).
Secondly, the replacement of the normal collagen IV network with the α1α1α2 heterotrimer results in the ectopic overproduction of laminins α1 and α5 and a disrupted architecture. Laminin-111 is increased in the regions of focally thickened GBM which are more permeable to protein (Abrahamson et al. 2007). Ectopic laminin deposition occurs through both podocycte signalling and mesangial cell activation. It further exacerbates the tendency to biomechanical strain and increases proteinuria.
Thirdly, the collagen IV α1α1α2 network is less structurally sound and more susceptible to the mechanical strain caused by the intraglomerular hypertension and ultrafiltration. Biomechanical strain is present early since Alport glomeruli from preproteinuric mice are more deformable (Wyss et al. 2011). The normal glomerulus already operates under high hydrostatic pressure, but the pressures are even higher in Alport glomeruli because of the abnormal GBM, fewer cross-links and altered deformability. Pressures are also high within the cochlea, and together with the greater abundance of collagen IV α3α4α5 in the kidney and ear, may explain why these tissues, but not the lung and small bowel, where they also occur, are affected.
Podocytes
The podocyte detects altered collagen IV through its α2β1 integrin receptor. Integrins act as mechanosensors linking the extracellular matrix with the podocyte actin cytoskeleton, and a defective signal upregulates podocyte extracellular matrix deposition (Borza et al. 2012).
The podocyte also detects altered collagen IV through the discoidin domain receptor 1 (DDR1), a transmembrane tyrosine kinase receptor at the lateral margin of the podocyte foot processes between the GBM and slit diaphragm (Vogel et al. 1997), independent of integrin binding (Vogel et al. 2000). The podocyte upregulates levels of the cytokines and growth factors needed to repair the GBM, but these also increase inflammation and fibrosis, and hence disease progression (Gross et al. 2004, 2010).
The mechanical stresses on the adhesive interfaces between the podocytes and GBM, which are largely mediated through laminin-521–integrin α3β1 interactions, further promote the accumulation of the abnormal laminins. Cell adhesion, cytoskeletal dynamics and cell signalling machinery are affected too (Cummins et al. 2007).
Mesangial cells, ectopic laminin deposition and cytokines
The mesangium is also important. Biomechanical stress on the glomerular capillary tuft initiates integrin α1β1-mediated activation of Rac1/CDC42 and mesangial process invasion of the capillary loops (Zallocchi et al. 2013). These contribute to the deposition of laminin-211 which further exacerbates the mesangial invasion. The mesangial cytokines (TGFβ1 and CTGF) and MMPs also contribute to the altered structural and functional properties of the Alport GBM with irregular thickening, splitting, and increased permeability. These events all affect podocyte cell health.
Proximal tubular epithelial cells, proteinuria, interstitial inflammation and fibrosis
Many cells within the kidney including podocytes, mesangial cells and proximal tubular cells produce chemokines (Sayyed et al. 2011), that attract leucocytes and macrophages to damaged tissue. Activated macrophages result in increased TNFα (Ryu et al. 2011), which is both pro-inflammatory and pro-apoptotic.
Matrix metalloproteinases are important in breaking down tubular basement membrane and allowing cells access to the tubulointerstititum (Cheng & Lovett, 2003).TGFβ1 produced by podocytes activates fibroblasts, myofibroblast formation and epithelial-to-mesenchymal transition (EMT). The role of EMT is controversial but, while usually involved in repair, here results in fibrosis, with glomerulosclerosis and interstitial damage (Sayers et al. 1999). Complement activation appears to be important in EMT through TGFβ1, since C3a receptor-deficient mice have less proteinuria, glomerulosclerosis and tubulointerstitial damage (Tang et al. 2009).
Disease progression
Mechanical strain, hypertension and proteinuria all contribute to disease progression in Alport syndrome (Meehan et al. 2009). Mechanical strain activates the rennin–angiotensin system (Durvasula et al. 2004) and induces SPARC (Secreted Protein Acidic and Rich in Cysteine; Durvasula & Shankland, 2005), both of which contribute to glomerulosclerosis. Hypertension accelerates proteinuria and induces matrix metalloproteinase production. TGFβ has a major role in disease progression and interstitial fibrosis. As the disease progresses and nephron mass is lost, glomerular hypertension further increases, exacerbating the biomechanical strain and the downstream disease mechanisms affected by it.
Treatment
Currently there is no specific treatment for Alport syndrome, and all males with X-linked disease and individuals with autosomal recessive disease eventually develop end-stage renal failure. However, it has become obvious that treatment with rennin–angiotensin blockade particularly before the onset of microalbuminuria slows down the rate of progression to end-stage disease (Gross et al. 2003, 2012). Renin–angiotensin blockade has many potential actions in the treatment of Alport syndrome: blockade is antihypertensive, antiproteinuric and antifibrotic. A reduction in blood pressure puts less stress on the defective GBM, on the adjacent podocyte, and on the whole glomerulus (Wyss et al. 2011). Renin–angiotensin blockade is safe but only partially effective. Dialysis and renal transplantation in end-stage disease are effective and relatively safe, and potential new treatments must have greater efficacy and fewer risks than these approaches.
Is it possible to repair already established GBM defects? Recent experiments in a mouse model have confirmed that abnormal GBM can be repaired, for example, by the secretion of the wild-type collagen IV α3α4α5 heterotrimer, and that even imperfect repair delays the onset of proteinuria and end-stage renal failure (Lin et al. 2013). The authors concluded that their results ‘validated the pursuit of therapeutic approaches aimed at normalising the GBM to prolong kidney function’ (Lin et al. 2013).
What strategies might these treatments target? Potential strategies are described in Table 1. It is becoming apparent that it will be important to know the underlying genetic mutation before starting treatment in Alport syndrome. Mutations that result in early onset disease warrant more aggressive treatment. The response to treatment is likely to be different even in individuals with different mutations for reasons we do not understand. It is unlikely that any of these treatments will have a uniform benefit in all patients. The benefit of any treatment must always be weighed up against the risks. It will be important to start treatment early, even before the onset of microalbuminuria, as we have seen with ACE inhibitor treatment. None of these treatments is likely to be completely effective and combinations are likely.
Conclusions
There are thus three approaches for the treatment of Alport syndrome based on our understanding of its pathogenesis: firstly, to correct the genetic mutation, for example, by replacing mutant podocytes with cells that produce the normal collagen IV α3α4α5 network; secondly, to prevent the development of the abnormal GBM and secondary glomerulosclerosis; and thirdly, to attenuate the compensatory glomerular and tubulointerstitial inflammation (Lin et al. 2013).
Glossary
- ACE
angiotensin converting enzyme
- CTGF
connective tissue growth factor
- DDR1
discoidin domain receptor 1
- EGF
epidermal growth factor
- EMT
epithelial-to-mesenchymal transition
- ER
endoplasmic reticulum
- GBM
glomerular basement membrane
- MMP
matrix metalloproteinase
- TGFβ
transforming growth factor-β
- TNFα
tumour necrosis factor α
Additional information
Competing interests
None declared.
Funding
Funding was provided by grants from the Alport Syndrome Foundation, Macquarie, Pedersen family, Kidney Foundation of Canada, and by the Alport Foundation of Australia.
References
- Abrahamson DR, Hudson BG, Stroganova L, Borza DB, St John PL. Cellular origins of type IV collagen networks in developing glomeruli. J Am Soc Nephrol. 2009;20:1471–1479. doi: 10.1681/ASN.2008101086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahamson DR, Isom K, Roach E, Stroganova L, Zelenchuk A, Miner JH, St John PL. Laminin compensation in collagen alpha3(IV) knockout (Alport) glomeruli contributes to permeability defects. J Am Soc Nephrol. 2007;18:2465–2472. doi: 10.1681/ASN.2007030328. [DOI] [PubMed] [Google Scholar]
- Baines RJ, Brunskill NJ. Tubular toxicity of proteinuria. Nat Rev Nephrol. 2011;7:177–180. doi: 10.1038/nrneph.2010.174. [DOI] [PubMed] [Google Scholar]
- Barker DF, Hostikka SL, Zhou J, Chow LT, Oliphant AR, Gerken SC, Gregory MC, Skolnick MH, Atkin CL, Tryggvason K. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science. 1990;248:1224–1227. doi: 10.1126/science.2349482. [DOI] [PubMed] [Google Scholar]
- Bateman JF, Boot-Handford RP, Lamande SR. Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat Rev Genet. 2009;10:173–183. doi: 10.1038/nrg2520. [DOI] [PubMed] [Google Scholar]
- Bezakova G, Ruegg MA. New insights into the roles of agrin. Nat Rev Mol Cell Biol. 2003;4:295–308. doi: 10.1038/nrm1074. [DOI] [PubMed] [Google Scholar]
- Borza CM, Su Y, Chen X, Yu L, Mont S, Chetyrkin S, Voziyan P, Hudson BG, Billings PC, Jo H, Bennett JS, Degrado WF, et al. Inhibition of integrin α2β1 ameliorates glomerular injury. J Am Soc Nephrol. 2012;23:1027–1038. doi: 10.1681/ASN.2011040367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Lovett DH. Gelatinase A (MMP-2) is necessary and sufficient for renal tubular cell epithelial–mesenchymal transformation. Am J Pathol. 2003;162:1937–1949. doi: 10.1016/S0002-9440(10)64327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collar JE, Ladva S, Cairns TD, Cattell V. Red cell traverse through thin glomerular basement membranes. Kidney Int. 2001;59:2069–2072. doi: 10.1046/j.1523-1755.2001.00721.x. [DOI] [PubMed] [Google Scholar]
- Colville DJ, Savige J. Alport syndrome. A review of the ocular manifestations. Ophthalmic Genet. 1997;18:161–173. doi: 10.3109/13816819709041431. [DOI] [PubMed] [Google Scholar]
- Comper WD, Haraldsson B, Deen WM. Resolved: normal glomeruli filter nephrotic levels of albumin. J Am Soc Nephrol. 2008;19:427–432. doi: 10.1681/ASN.2007090997. [DOI] [PubMed] [Google Scholar]
- Cummins PM, von Offenberg Sweeney N, Killeen MT, Birney YA, Redmond EM, Cahill PA. Cyclic strain-mediated matrix metalloproteinase regulation within the vascular endothelium: a force to be reckoned with. Am J Physiol Heart Circ Physiol. 2007;292:H28–H42. doi: 10.1152/ajpheart.00304.2006. [DOI] [PubMed] [Google Scholar]
- Durvasula RV, Petermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, Pichler R, Griffin S, Couser WG, Shankland SJ. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004;65:30–39. doi: 10.1111/j.1523-1755.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- Durvasula RV, Shankland SJ. Mechanical strain increases SPARC levels in podocytes: implications for glomerulosclerosis. Am J Physiol Renal Physiol. 2005;289:F577–F584. doi: 10.1152/ajprenal.00393.2004. [DOI] [PubMed] [Google Scholar]
- Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–437. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Finkel RS, Flanigan KM, Wong B, Bonnemann C, Sampson J, Sweeney HL, Reha A, Northcutt VJ, Elfring G, Barth J, Peltz SW. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS One. 2013;8:e81302. doi: 10.1371/journal.pone.0081302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giani M, Mastrangelo A, Villa R, Turolo S, Marra G, Tirelli AS, Hopfer H, Edefonti A. Alport syndrome: the effects of spironolactone on proteinuria and urinary TGF-β1. Pediatr Nephrol. 2013;28:1837–1842. doi: 10.1007/s00467-013-2490-z. [DOI] [PubMed] [Google Scholar]
- Gross O, Beirowski B, Harvey SJ, McFadden C, Chen D, Tam S, Thorner PS, Smyth N, Addicks K, Bloch W, Ninomiya Y, Sado Y, Weber M, Vogel WF. DDR1-deficient mice show localized subepithelial GBM thickening with focal loss of slit diaphragms and proteinuria. Kidney Int. 2004;66:102–111. doi: 10.1111/j.1523-1755.2004.00712.x. [DOI] [PubMed] [Google Scholar]
- Gross O, Beirowski B, Koepke ML, Kuck J, Reiner M, Addicks K, Smyth N, Schulze-Lohoff E, Weber M. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003;63:438–446. doi: 10.1046/j.1523-1755.2003.00779.x. [DOI] [PubMed] [Google Scholar]
- Gross O, Girgert R, Beirowski B, Kretzler M, Kang HG, Kruegel J, Miosge N, Busse AC, Segerer S, Vogel WF, Muller GA, Weber M. Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease. Matrix Biol. 2010;29:346–356. doi: 10.1016/j.matbio.2010.03.002. [DOI] [PubMed] [Google Scholar]
- Gross O, Licht C, Anders HJ, Hoppe B, Beck B, Tonshoff B, Hocker B, Wygoda S, Ehrich JH, Pape L, Konrad M, Rascher W, et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012;81:494–501. doi: 10.1038/ki.2011.407. [DOI] [PubMed] [Google Scholar]
- Gubler M, Levy M, Broyer M, Naizot C, Gonzales G, Perrin D, Habib R. Alport's syndrome. A report of 58 cases and a review of the literature. Am J Med. 1981;70:493–505. doi: 10.1016/0002-9343(81)90571-4. [DOI] [PubMed] [Google Scholar]
- Gunwar S, Ballester F, Noelken ME, Sado Y, Ninomiya Y, Hudson BG. Glomerular basement membrane. Identification of a novel disulfide-cross-linked network of α3, α4, and α5 chains of type IV collagen and its implications for the pathogenesis of Alport syndrome. J Biol Chem. 1998;273:8767–8775. doi: 10.1074/jbc.273.15.8767. [DOI] [PubMed] [Google Scholar]
- Harvey SJ, Miner JH. Revisiting the glomerular charge barrier in the molecular era. Curr Opin Nephrol Hypertens. 2008;17:393–398. doi: 10.1097/MNH.0b013e32830464de. [DOI] [PubMed] [Google Scholar]
- Hausmann R, Kuppe C, Egger H, Schweda F, Knecht V, Elger M, Menzel S, Somers D, Braun G, Fuss A, Uhlig S, Kriz W, et al. Electrical forces determine glomerular permeability. J Am Soc Nephrol. 2010;21:2053–2058. doi: 10.1681/ASN.2010030303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkila P, Parpala T, Lukkarinen O, Weber M, Tryggvason K. Adenovirus-mediated gene transfer into kidney glomeruli using an ex vivo and in vivo kidney perfusion system – first steps towards gene therapy of Alport syndrome. Gene Ther. 1996;3:21–27. [PubMed] [Google Scholar]
- Hertz JM, Thomassen M, Storey H, Flinter F. Clinical utility gene card for: Alport syndrome. Eur J Hum Genet. 2012;20 doi: 10.1038/ejhg.2011.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood JC, Dowling J, Bertram JF, Young RJ, Huxtable C, Robinson W, Savige J. Correlation of histopathological features and renal impairment in autosomal dominant Alport syndrome in Bull terriers. Nephrol Dial Transplant. 2002;17:1897–1908. doi: 10.1093/ndt/17.11.1897. [DOI] [PubMed] [Google Scholar]
- Hudson BG. The molecular basis of Goodpasture and Alport syndromes: beacons for the discovery of the collagen IV family. J Am Soc Nephrol. 2004;15:2514–2527. doi: 10.1097/01.ASN.0000141462.00630.76. [DOI] [PubMed] [Google Scholar]
- Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med. 2003;348:2543–2556. doi: 10.1056/NEJMra022296. [DOI] [PubMed] [Google Scholar]
- Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO, Flinter F, Pirson Y, Dahan K, et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study. J Am Soc Nephrol. 2003;14:2603–2610. doi: 10.1097/01.asn.0000090034.71205.74. [DOI] [PubMed] [Google Scholar]
- Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO, Flinter F, Pirson Y, Verellen C, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. 2000;11:649–657. doi: 10.1681/ASN.V114649. [DOI] [PubMed] [Google Scholar]
- Jarad G, Miner JH. Update on the glomerular filtration barrier. Curr Opin Nephrol Hypertens. 2009;18:226–232. doi: 10.1097/mnh.0b013e3283296044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeansson M, Haraldsson B. Morphological and functional evidence for an important role of the endothelial cell glycocalyx in the glomerular barrier. Am J Physiol Renal Physiol. 2006;290:F111–F116. doi: 10.1152/ajprenal.00173.2005. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Shield CF, Todd P, Hudson BG, Neilson EG. Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J Clin Invest. 1997;99:2470–2478. doi: 10.1172/JCI119431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashtan CE, Atkin CL, Gregory MC, Michael AF. Identification of variant Alport phenotypes using an Alport-specific antibody probe. Kidney Int. 1989;36:669–674. doi: 10.1038/ki.1989.244. [DOI] [PubMed] [Google Scholar]
- Katayama K, Kawano M, Naito I, Ishikawa H, Sado Y, Asakawa N, Murata T, Oosugi K, Kiyohara M, Ishikawa E, Ito M, Nomura S. Irradiation prolongs survival of Alport mice. J Am Soc Nephrol. 2008;19:1692–1700. doi: 10.1681/ASN.2007070829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, et al. Positionally cloned gene for a novel glomerular protein – nephrin – is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- Koepke ML, Weber M, Schulze-Lohoff E, Beirowski B, Segerer S, Gross O. Nephroprotective effect of the HMG-CoA-reductase inhibitor cerivastatin in a mouse model of progressive renal fibrosis in Alport syndrome. Nephrol Dial Transplant. 2007;22:1062–1069. doi: 10.1093/ndt/gfl810. [DOI] [PubMed] [Google Scholar]
- Kriz W. Fenestrated glomerular capillaries are unique. J Am Soc Nephrol. 2008;19:1439–1440. doi: 10.1681/ASN.2008060583. [DOI] [PubMed] [Google Scholar]
- Kruegel J, Rubel D, Gross O. Alport syndrome – insights from basic and clinical research. Nat Rev Nephrol. 2013;9:170–178. doi: 10.1038/nrneph.2012.259. [DOI] [PubMed] [Google Scholar]
- LeBleu V, Sugimoto H, Mundel TM, Gerami-Naini B, Finan E, Miller CA, Gattone VH, 2nd, Lu L, Shield CF, 3rd, Folkman J, Kalluri R. Stem cell therapies benefit Alport syndrome. J Am Soc Nephrol. 2009;20:2359–2370. doi: 10.1681/ASN.2009010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Cong R, Biemesderfer D. The COOH terminus of megalin regulates gene expression in opossum kidney proximal tubule cells. Am J Physiol Cell Physiol. 2008;295:C529–C537. doi: 10.1152/ajpcell.00037.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Suh JH, Go G, Miner JH. Feasibility of repairing glomerular basement membrane defects in Alport syndrome. J Am Soc Nephrol. 2013 doi: 10.1681/ASN.2013070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehan DT, Delimont D, Cheung L, Zallocchi M, Sansom SC, Holzclaw JD, Rao V, Cosgrove D. Biomechanical strain causes maladaptive gene regulation, contributing to Alport glomerular disease. Kidney Int. 2009;76:968–976. doi: 10.1038/ki.2009.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol. 2004;20:255–284. doi: 10.1146/annurev.cellbio.20.010403.094555. [DOI] [PubMed] [Google Scholar]
- Ninichuk V, Gross O, Reichel C, Khandoga A, Pawar RD, Ciubar R, Segerer S, Belemezova E, Radomska E, Luckow B, Perez de Lema G, Murphy PM, et al. Delayed chemokine receptor 1 blockade prolongs survival in collagen 4A3-deficient mice with Alport disease. J Am Soc Nephrol. 2005;16:977–985. doi: 10.1681/ASN.2004100871. [DOI] [PubMed] [Google Scholar]
- Noone D, Licht C. An update on the pathomechanisms and future therapies of Alport syndrome. Pediatr Nephrol. 2013;28:1025–1036. doi: 10.1007/s00467-012-2272-z. [DOI] [PubMed] [Google Scholar]
- Ohlson M, Sorensson J, Haraldsson B. Glomerular size and charge selectivity in the rat as revealed by FITC-ficoll and albumin. Am J Physiol Renal Physiol. 2000;279:F84–F91. doi: 10.1152/ajprenal.2000.279.1.F84. [DOI] [PubMed] [Google Scholar]
- Park CH, Maack T. Albumin absorption and catabolism by isolated perfused proximal convoluted tubules of the rabbit. J Clin Invest. 1984;73:767–777. doi: 10.1172/JCI111270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin JD, San Antonio JD, Pedchenko V, Hudson B, Jensen ST, Savige J. Mapping structural landmarks, ligand binding sites, and missense mutations to the collagen IV heterotrimers predicts major functional domains, novel interactions, and variation in phenotypes in inherited diseases affecting basement membranes. Hum Mutat. 2011;32:127–143. doi: 10.1002/humu.21401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpala-Sparman T, Lukkarinen O, Heikkila P, Tryggvason K. A novel surgical organ perfusion method for effective ex vivo and in vivo gene transfer into renal glomerular cells. Urol Res. 1999;27:97–102. doi: 10.1007/s002400050094. [DOI] [PubMed] [Google Scholar]
- Pieri M, Stefanou C, Zaravinos A, Erguler K, Stylianou K, Lapathitis G, Karaiskos C, Savva I, Paraskeva R, Dweep H, Sticht C, Anastasiadou N, et al. Evidence for Activation of the Unfolded Protein Response in collagen IV nephropathies. J Am Soc Nephrol. 2014;25:260–275. doi: 10.1681/ASN.2012121217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson M, Cordier J, Hervouuet F, Rauber G. An unusual congenital and familial congenital malformative combination involving the eye and kidney. J Genet Hum. 1963;12:184–213. [PubMed] [Google Scholar]
- Prodromidi EI, Poulsom R, Jeffery R, Roufosse CA, Pollard PJ, Pusey CD, Cook HT. Bone marrow-derived cells contribute to podocyte regeneration and amelioration of renal disease in a mouse model of Alport syndrome. Stem Cells. 2006;24:2448–2455. doi: 10.1634/stemcells.2006-0201. [DOI] [PubMed] [Google Scholar]
- Rao VH, Meehan DT, Delimont D, Nakajima M, Wada T, Gratton MA, Cosgrove D. Role for macrophage metalloelastase in glomerular basement membrane damage associated with Alport syndrome. Am J Pathol. 2006;169:32–46. doi: 10.2353/ajpath.2006.050896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- Ryu M, Kulkarni OP, Radomska E, Miosge N, Gross O, Anders HJ. Bacterial CpG-DNA accelerates Alport glomerulosclerosis by inducing an M1 macrophage phenotype and tumor necrosis factor-α-mediated podocyte loss. Kidney Int. 2011;79:189–198. doi: 10.1038/ki.2010.373. [DOI] [PubMed] [Google Scholar]
- Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter F. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol. 2013;24:364–375. doi: 10.1681/ASN.2012020148. [DOI] [PubMed] [Google Scholar]
- Savige J, Rana K, Tonna S, Buzza M, Dagher H, Wang YY. Thin basement membrane nephropathy. Kidney Int. 2003;64:1169–1178. doi: 10.1046/j.1523-1755.2003.00234.x. [DOI] [PubMed] [Google Scholar]
- Sayers R, Kalluri R, Rodgers KD, Shield CF, Meehan DT, Cosgrove D. Role for transforming growth factor-β1 in Alport renal disease progression. Kidney Int. 1999;56:1662–1673. doi: 10.1046/j.1523-1755.1999.00744.x. [DOI] [PubMed] [Google Scholar]
- Sayyed SG, Ryu M, Kulkarni OP, Schmid H, Lichtnekert J, Gruner S, Green L, Mattei P, Hartmann G, Anders HJ. An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int. 2011;80:68–78. doi: 10.1038/ki.2011.102. [DOI] [PubMed] [Google Scholar]
- Smithies O. Why the kidney glomerulus does not clog: a gel permeation/diffusion hypothesis of renal function. Proc Natl Acad Sci U S A. 2003;100:4108–4113. doi: 10.1073/pnas.0730776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto H, Mundel TM, Sund M, Xie L, Cosgrove D, Kalluri R. Bone-marrow-derived stem cells repair basement membrane collagen defects and reverse genetic kidney disease. Proc Natl Acad Sci U S A. 2006;103:7321–7326. doi: 10.1073/pnas.0601436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh JH, Miner JH. The glomerular basement membrane as a barrier to albumin. Nat Rev Nephrol. 2013;9:470–477. doi: 10.1038/nrneph.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susztak K, Ciccone E, McCue P, Sharma K, Bottinger EP. Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. 2005;2:e45. doi: 10.1371/journal.pmed.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takase O, Minto AW, Puri TS, Cunningham PN, Jacob A, Hayashi M, Quigg RJ. Inhibition of NF-κB-dependent Bcl-xL expression by clusterin promotes albumin-induced tubular cell apoptosis. Kidney Int. 2008;73:567–577. doi: 10.1038/sj.ki.5002563. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Asada M, Higashi AY, Nakamura J, Oguchi A, Tomita M, Yamada S, Asada N, Takase M, Okuda T, Kawachi H, Economides AN, et al. Loss of the BMP antagonist USAG-1 ameliorates disease in a mouse model of the progressive hereditary kidney disease Alport syndrome. J Clin Invest. 2010;120:768–777. doi: 10.1172/JCI39569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Lu B, Hatch E, Sacks SH, Sheerin NS. C3a mediates epithelial-to-mesenchymal transition in proteinuric nephropathy. J Am Soc Nephrol. 2009;20:593–603. doi: 10.1681/ASN.2008040434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonna S, Wang YY, Wilson D, Rigby L, Tabone T, Cotton R, Savige J. The R229Q mutation in NPHS2 may predispose to proteinuria in thin-basement-membrane nephropathy. Pediatr Nephrol. 2008;23:2201–2207. doi: 10.1007/s00467-008-0934-7. [DOI] [PubMed] [Google Scholar]
- Vanacore R, Ham AJ, Voehler M, Sanders CR, Conrads TP, Veenstra TD, Sharpless KB, Dawson PE, Hudson BG. A sulfilimine bond identified in collagen IV. Science. 2009;325:1230–1234. doi: 10.1126/science.1176811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel W, Brakebusch C, Fassler R, Alves F, Ruggiero F, Pawson T. Discoidin domain receptor 1 is activated independently of β1 integrin. J Biol Chem. 2000;275:5779–5784. doi: 10.1074/jbc.275.8.5779. [DOI] [PubMed] [Google Scholar]
- Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997;1:13–23. doi: 10.1016/s1097-2765(00)80003-9. [DOI] [PubMed] [Google Scholar]
- Wartiovaara J, Ofverstedt LG, Khoshnoodi J, Zhang J, Makela E, Sandin S, Ruotsalainen V, Cheng RH, Jalanko H, Skoglund U, Tryggvason K. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J Clin Invest. 2004;114:1475–1483. doi: 10.1172/JCI22562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss HM, Henderson JM, Byfield FJ, Bruggeman LA, Ding Y, Huang C, Suh JH, Franke T, Mele E, Pollak MR, et al. Biophysical properties of normal and diseased renal glomeruli. Am J Physiol Cell Physiol. 2011;300:C397–C405. doi: 10.1152/ajpcell.00438.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallocchi M, Johnson BM, Meehan DT, Delimont D, Cosgrove D. α1β1 integrin/Rac1-dependent mesangial invasion of glomerular capillaries in Alport syndrome. Am J Pathol. 2013;183:1269–1280. doi: 10.1016/j.ajpath.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M, Khurana M, Rao VH, Cosgrove D, Rougier JP, Werner MC, Shield CF, 3rd, Werb Z, Kalluri R. Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PLoS Med. 2006;3:e100. doi: 10.1371/journal.pmed.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Reeders ST. The alpha chains of type IV collagen. Contrib Nephrol. 1996;117:80–104. doi: 10.1159/000424808. [DOI] [PubMed] [Google Scholar]