

Abstract
A growing number of studies show that different types of ion channels localize in caveolae and are regulated by the level of membrane cholesterol. Furthermore, it has been proposed that cholesterol-induced regulation of ion channels might be attributed to partitioning into caveolae and association with caveolin-1 (Cav-1). We tested, therefore, whether Cav-1 regulates the function of inwardly rectifying potassium channels Kir2.1 that play major roles in the regulation of membrane potentials of numerous mammalian cells. Our earlier studies demonstrated that Kir2.1 channels are cholesterol sensitive. In this study, we show that Kir2.1 channels co-immunoprecipitate with Cav-1 and that co-expression of Kir2.1 channels with Cav-1 in HEK293 cells results in suppression of Kir2 current indicating that Cav-1 is a negative regulator of Kir2 function. These observations are confirmed by comparing Kir currents in bone marrow-derived macrophages isolated from Cav-1−/− and wild-type animals. We also show, however, that Kir2 channels maintain their sensitivity to cholesterol in HEK293 cells that have very low levels of endogenous Cav-1 and in bone marrow-derived macrophages isolated from Cav-1−/− knockout mice. Thus, these studies indicate that Cav-1 and/or intact caveolae are not required for cholesterol sensitivity of Kir channels. Moreover, a single point mutation of Kir2.1, L222I that abrogates the sensitivity of the channels to cholesterol also abolishes their sensitivity to Cav-1 suggesting that the two modulators regulate Kir2 channels via a common mechanism.
Key points
Inwardly rectifying potassium channels (Kir) play key roles in regulating membrane excitability and K+ homeostasis in multiple cell types. Our earlier studies showed that Kir2 channels, one of the major subfamilies of Kir, are suppressed by membrane cholesterol and that cholesterol stabilizes these channels in a closed ‘silent’ state.
This paper addresses a fundamental question of how Kir2 channels are regulated by caveolins, the major structural proteins of caveolae, and the relationship between the sensitivity of the channels to caveolin and to cholesterol.
In this study, we present direct evidence that caveolin-1 is a negative regulator of Kir2 function and that cholesterol and caveolin-1 regulate the channels by a common mechanism.
This study also challenges a general notion that cholesterol depletion alters ion channel function by disrupting caveolae, demonstrating that neither caveolin-1 nor intact caveolae are required for cholesterol sensitivity of Kir2 channels.
Furthermore, we present first insights into the structural determinants of the cross-talk between the sensitivity of Kir2 channels to caveolin and to cholesterol.
Introduction
Inwardly rectifying potassium channels (Kir) are ubiquitously expressed in multiple tissues and play major roles in regulating membrane excitability and K+ homeostasis. Kir2 channels are a major subfamily of the inward rectifiers responsible for maintaining membrane potential in cardiomyocytes, smooth muscle cells and neurons, as well as in non-excitable cells, such as endothelial cells and macrophages (Bichet et al. 2003; Kubo et al. 2005; Hibino et al. 2010). In previous studies, we demonstrated that Kir2 channels are suppressed by the elevation of membrane cholesterol in endothelial cells in vitro and in vivo (Romanenko et al. 2002; Fang et al. 2006), as well as when expressed in different heterologous expression systems (Romanenko et al. 2004; Rosenhouse-Dantsker et al. 2010). Moreover, cholesterol-induced suppression of Kir channels in aortic endothelial cells dampens the sensitivity of the channels to shear stress, an effect that correlates with hypercholesterolaemia-induced loss of flow-induced vasodilatation (Fang et al. 2006) suggesting that it plays an important role in the regulation of vascular tone.
In terms of the mechanism, our studies suggest that cholesterol inhibits Kir channels by direct specific interaction with the channel protein, which results in stabilization of the channels in the closed state. First, we showed that cholesterol-induced inhibition of Kir channels is stereospecific (Romanenko et al. 2002). We also showed that while increasing membrane cholesterol significantly decreases whole cell Kir currents, it had no effect on cell surface channel expression, or on single channel properties of the channels (unitary conductance, or open probability), which led us to suggest that interaction with cholesterol stabilizes the channels in a ‘silent state’ that has minimal or no activity (Romanenko et al. 2004). Moreover, we and others showed that cholesterol can inhibit purified Kir channels incorporated into liposomes in the absence of any intermediates, including caveolin (Singh et al. 2009; D'Avanzo et al. 2011) and that cholesterol–Kir binding is required for the inhibitory effect (Singh et al. 2011). Identification of putative cholesterol binding sites in Kir2 channels further supports the notion that cholesterol interacts with Kir channels directly (Rosenhouse-Dantsker et al. 2013).
In contrast, several studies have suggested that cholesterol sensitivity of ion channels could be attributed to their interaction with caveolin. More specifically, as it is known that cholesterol depletion results in disruption of caveolae/lipid rafts, it has been proposed that cholesterol depletion modulates ion channel function by dissociation of caveolins from the channel proteins. This hypothesis is supported by recent studies showing that caveolins are negative regulators of several types of K+ channels, including large conductance Ca2+-sensitive K+ channels (Wang et al. 2005; Riddle et al. 2011), ATP-sensitive K+ channels (Garg et al. 2009; Davies et al. 2010) and renal outer medullary K+ channels (Lin et al. 2011). Moreover, earlier studies also showed that Kir2 channels partition into cholesterol-enriched membrane microdomains and that cholesterol depletion results in partial translocation of the channels from cholesterol-rich to cholesterol-poor membrane fractions whereas cholesterol enrichment has the opposite effect (Tikku et al. 2007). Indeed, the fact that cholesterol can inhibit purified Kir channels directly does not exclude the possibility that in mammalian cells it might also exert its effect at least in part by regulating the association of the channels with caveolins. In this study, we address this issue by testing whether caveolin-1 (Cav-1) regulates Kir2 channels and whether it is required for the sensitivity of the channels to cholesterol. Our results show that Cav-1 is a negative regulator of Kir2 channels but is not required to confer cholesterol sensitivity to the channels. Furthermore, these studies demonstrate that while cholesterol regulation of Kir2 channels cannot be attributed to the association of the channels with Cav-1, they do indicate that cholesterol and Cav-1 regulate Kir2 channels by a similar mechanism.
Methods
Cells and transfection
HEK293 cells were grown as previously described in minimum essential medium (MEM) containing GlutaMAX, 10% fetal bovine serum, 1% MEM non-essential amino acids, penicillin (50 U ml−1) and streptomycin (50 U ml−1) in a 5% CO2 humidified atmosphere at 37°C. All media and reagents were from Invitrogen (CA, USA). Cells were transiently cotransfected with: (i) Kir2.1 and GFP; (ii) Kir2.1 and Cav-1-YFP (Cav-1 tagged with YFP); (iii) Kir2.1-L222I with GFP; or (iv) Kir2.1-L222I with Cav-1-YFP using Lipofectamine 2000 Transfection Reagent (Invitrogen) according to the manufacturer's protocol. Earlier studies from our group showed that Cav-1-YFP constructs support the normal formation of caveolae (Chen et al. 2012; Bakhshi et al. 2013) indicating that the chimeric protein is fully functional and that it is able to traffic to the correct membrane domains. Experiments were conducted 2–3 days after transfection. pcDNA3-Kir2.1-haemagglutinin (pcDNA3-Kir2.1-HA) was a gift from Dr. Carol Vandenberg (University of California, Santa Barbara). cDNAs for Cav-1-YFP, Kir2.1 and GFP were prepared using a plasmid midi-kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions.
Isolation of bone marrow-derived macrophages
Freshly isolated bone marrow-derived macrophages (BMDMs) are commonly used to investigate macrophage function (e.g. Vicente et al. 2003; Joo et al. 2009). BMDMs were obtained from Cav-1−/− knockout (KO) mice generated on the background of B6/129SJ2 with B6 wild-type (WT) animals used as controls. Mice aged 3–6 weeks were used for BMDM isolation according to the method of Celada et al. (1984) with minor modifications. Briefly, after humane killing (30% CO2 inhalation for 10 min followed by cervical dislocation), bone marrow was flushed from the femurs, washed, and resuspended in DMEM with 10% endotoxin-free fetal bovine serum and 10% (vol/vol) L929 cell-conditioned medium as a biological source of macrophage colony-stimulating factor. The medium was replenished on day 4 and non-adherent cells were removed. The cells were used for experiments on days 7–9. The procedure was approved by the Office of Animal Care and Institutional Biosafety Committees of the University of Illinois at Chicago.
Cholesterol modulation
HEK293 cells transfected with Kir2.1 or Cav-1 were depleted or enriched with cholesterol by incubating with methyl-β-cyclodextrin (MβCD) alone or following saturation with cholesterol as described previously (Tikku et al. 2007). MβCD (5 mm) solution in DMEM without serum mixed with saturated cholesterol was sonicated and shaken overnight at 37°C. HEK293 cells were incubated with the MβCD solution with or without cholesterol for 1 h to enhance or reduce the cellular cholesterol level. Amplex Red cholesterol assay kit (Molecular Probes, Oregon, USA) was used to measure cellular cholesterol content in HEK293 cells according to manufacturer specifications.
Electrophysiology studies
Whole cell recordings
Kir2.1 currents in HEK293 cells were recorded using a standard whole cell patch clamp configuration. Pipettes were pulled (SG10 glass, 1.20 mm ID, 1.60 mm; Richland Glass, Richland, NJ, USA) to give a final resistance of 2–6 MΩ. Currents were recorded using an EPC9 amplifier (HEKA Electronik, Lambrecht, Germany) and accompanying acquisition and analysis software (Pulse & PulseFit; HEKA Electronik). The external solution contained (in mm): NaCl 150, KCl 6, MgCl2 1.0, CaCl2 1.5, Hepes 10 and EGTA 1.0 at pH 7.3 (pH adjusted with NaOH). The pipette solution contained (mm) KCl 145, MgCl2 1.0, Hepes 10, EGTA 1.0, ATP 4 at pH 7.3 (pH adjusted with KOH). Currents were elicited with 500 ms linear voltage ramps from –100 to +60 mV at an interpulse interval of 3 s. The holding potential was −60 mV. Pipette and whole cell capacitances were automatically compensated. Only the cells exhibiting fluorescence were used for recordings. All the recordings were carried out at room temperature (22–25°C). The traces were accepted for analysis if membrane resistance was at least 500 MΩ with most of the recordings being >1 GΩ and access resistance was <10 MΩ. Single channel recordings were performed in the cell-attached configuration using an Axopatch 200B amplifier. Records were acquired using QuBIO software and filtered online at 2 kHz. Both the extracellular solution and pipette solution contained (in mm): KCl 156, Hepes 10, MgCl2 1. CaCl2 1.5, EGTA 1.0 and pH 7.3 (pH adjusted with KOH). Once the cell-attached configuration was obtained, voltage steps were made to increasingly negative membrane potentials in increments of 20 mV and held at those potentials for several minutes for acquisition. All the recordings were carried out at room temperature (22–25°C). Analysis was performed in QuB software post-acquisition. The slope conductance was obtained from the slope of the line of best fit passing through the plot of current amplitudes at −40 mV, −60 mV and −80 mV (for Kir2.1-GFP and that for Kir2.1-Cav-1-YFP). Open probability (NPo) was calculated from the sum of the occupancies of discernible open levels obtained from idealizing the current record. More specifically, the product, iNPo, where i refers to the unitary current is the mean current or equivalent to the relative area of the open state in the amplitude histogram. Briefly, we idealized the current recordings to determine the number of the channels in the patch based on a maximum likelihood fit to the raw data (Nicolai & Sachs, 2014). Even though multiple channels were observed in most patches (n = 3–8), the QUB software (Milescu et al. 2005) estimates the single channel kinetics and the number of open channels.
Co-immunoprecipitation and Western blotting
HEK293 cells were rinsed with ice-cold phosphate-buffered saline (PBS; pH 7.4) and homogenized in ice-cold lysis buffer (25 mm Tris–HCl, 250 mm NaCl, 10 mm EDTA, pH 7.6) containing 1% Triton X-100 and 2 mm phenylmethylsulphonyl fluoride with protease inhibitor tablets (Roche, Basel, Switzerland). The lysates were precleared by incubation with r-protein-G agarose (Invitrogen; 1 h, 4°C) before they were incubated with primary antibody for 2 h at 4°C. Cell homogenates were immunoprecipitated with anti-Cav-1 antibody. Antigen–antibody complexes were captured with r-protein-G agarose (4°C, 2 h). Agarose beads were washed four times with lysis buffer before removal of bound proteins by boiling in SDS sample buffer. Samples were resolved by SDS–PAGE (10% acrylamide gel) and transferred on to a nitrocellulose membrane. The transferred blots were blocked and then incubated for 1 h with primary antibody. After washing, the blots were incubated with peroxidase-conjugated secondary antibodies for 45 min and developed using the ECL detection system (Amersham, Amersham, UK).
Flow cytometry
Cells were fixed with 4% paraformaldehyde solution, incubated with 1% bovine serum albumin (BSA) PBS solution for 2 h to block non-specific binding, and then incubated with primary antibodies (1:600 of anti-HA mouse monoclonal antibody; Covance Mouse mono HA.11 that recognizes HA-Kir constructs) in PBS solution containing 1% BSA overnight at 4°C. For visualization, cells were incubated in Alexa Fluor 555-conjugated secondary antibody solution (Invitrogen Molecular Probes, CA, USA; antimouse IgG, 1:200 dilution in PBS containing 1% BSA, 1 h), washed and imaged using a Zeiss Axiovert 200M microscope (Zeiss, Oberkochen, Germany). Flow cytometry analysis was carried out by FACS analyser (Becton Dickinson Fortessa, NJ, USA), employing 561 nm laser excitation. The fluorescence Alexa Fluor 555 dye was measured via 582/15 nm filter. To eliminate signals due to cellular fragments, only those events with forward and side light scattering comparable to single whole cells were analysed. All fluorescence signals were logarithmically displayed. Ten thousand cells were run for each sample and data were collected in the list mode. The analysis of flow cytometry data was performed using FlowJo flow cytometry application software (Oregon, USA). The autofluorescence signal was subtracted from the fluorescence of all cells. Results were expressed as the geometric mean (Gm) of cells expressing Kir with the HA tag.
Statistical analysis
Group data are presented as means ± s.e.m. The comparison between two groups was performed using an unpaired t test. When more than two groups were compared or when the interaction was of interest, one-way or two-way ANOVA analysis respectively was used with log transformation to the outcome as the normality assumption was violated. Subsequent analysis then was conducted if a significant result was found in ANOVA. By doing this, the issue of multiple comparisons can be ignored due to this protected test approach (Johnson, 1998) Two-tailed P < 0.05 was considered statistically significant.
Results
Caveolin-1 is a negative regulator of Kir2.1 function
Physical interaction between Kir2.1 and caveolin-1
Previous studies have shown that Kir2 channels, specifically Kir2.1 and Kir2.3, partition into caveolin-enriched membrane microdomains (Tikku et al. 2007) suggesting that these channels may physically interact with caveolin. To test whether this is the case, Kir2.1 and Cav-1 were expressed in HEK293 cells (a cell line that expresses nominal levels of both proteins) and the physical interaction between transfected proteins was tested by co-immunoprecipitation. Kir2.1 channels contained an HA-tag, inserted into the outer loop of the channel, which was shown previously not to interfere with its function (Epshtein et al. 2009). The co-immunoprecipitation was performed either by precipitating the complex with anti-Cav-1 antibody and then probed with anti-HA antibody to identify Kir2.1-HA channels (Fig. 1Aa) or by precipitating with anti-HA antibody and probing for Cav-1 (Fig. 1Ab). Strong bands for both Kir2.1 and Cav-1 were detected using either Cav-1 or HA as precipitating agents indicating that Kir2.1 channels physically interact with Cav-1. Non-specific IgG antibody was used as a negative control.
Figure 1. Cav-1 is a negative regulator of Kir2.1 function.
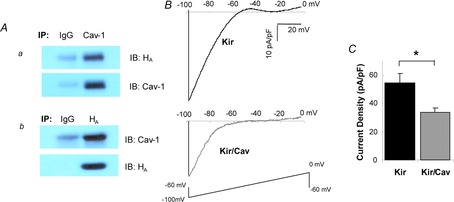
A, co-immunoprecipitation of Cav-1 with Kir2.1 channels. For immunoprecipitation, the same amount of IgG and Cav-1 or HA antibodies were added to the same amounts of lysates from cells transfected with Kir2.1 and Cav-1. The samples were then probed with HA and Cav-1 antibodies after running gels as indicated. B, representative current traces for HEK293 cells expressing Kir2.1/GFP or Kir2.1/Cav-1-YFP. C, average peak current densities at −97 mV for HEK293 cells transfected with Kir2.1/GFP or Kir2.1/Cav-1-YFP (n = 36–38, *P < 0.05). Cav-1, caveolin-1; HA, haemagglutinin.
Co-expression with caveolin-1 decreases Kir2.1 whole cell current
To determine the functional impact of Cav-1 on Kir2.1 channels, Kir2.1 was co-expressed with Cav-1 in HEK293 cells and the currents were recorded in the whole cell mode. Cav-1 was tagged with YFP to identify cells that are successfully transfected. In control cells, Kir2.1 was co-expressed with GFP alone. Currents were recorded only from cells expressing either GFP or Cav-1-YFP and, as expected, most of the fluorescent cells showed typical Kir currents (see traces in Fig. 1B). Co-expression of Kir2.1 with Cav-1 resulted in a significant decrease in Kir2.1 current density, apparent both from the typical traces recorded from cells expressing Kir2.1 alone vs. cells that co-expressed Kir2.1 and Cav-1 (Fig. 1B) and from the average Kir2.1 current densities in the two experimental cell populations (Fig. 1C). To exclude a possibility that overexpression of Cav-1 alters the level of cellular cholesterol in HEK293 cells, we measured cholesterol levels in HEK293 cells with and without overexpression of Cav-1 and found no effect on cholesterol level (cellular cholesterol levels were 9.04 + 2.8 μg mg−1 protein and 9.10 + 2.9 μg mg−1 protein in control cells and in cells overexpressing Cav-1 respectively).
Co-expression with caveolin-1 has no effect on the surface expression of Kir2.1 channels
A decrease in whole cell current is typically accounted for either by lower expression of the channels on the plasma membrane or by a decrease in either unitary conductance or open probability of the channels. To address the first possibility, HA-Kir2.1 channels were co-expressed with Cav-1-YFP in HEK293 cells, immuno-stained with anti-HA antibodies and the level of Kir2.1 expression was quantified by flow cytometry. As described above, in control cells Kir2.1 were co-expressed with GFP. Notably, in these experiments channels were HA-tagged on their extracellular domain making them accessible to antibodies without membrane permeabilization, which allowed us to identify the channel population inserted into the plasma membrane. Our observations show that co-expression of Kir2.1 with Cav-1 does not have any effect on surface (plasma membrane) expression of Kir2.1 (Fig. 2).
Figure 2. Caveolin has no effect on the surface expression for Kir2.1.
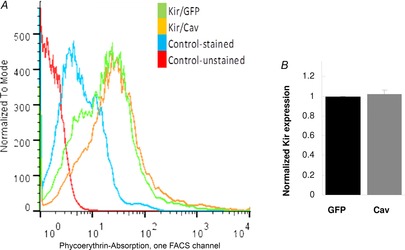
A, representative overlay of flow cytometry histograms of HEK293 cells expressing Kir2.1 with GFP or with Cav-1-YFP. The cells were stained with primary HA antibody and Alexa Fluor 555 secondary antibody. B, expression of Kir2.1 co-expressed with Cav-1 normalized to Kir2.1 co-expressed with GFP from flow cytometry data (n = 3). Cav, caveolin; HA, haemagglutinin.
Co-expression with caveolin-1 has no effect on the single channel properties of Kir2.1
To address the second possibility, we performed single channel recordings of Kir2.1 expressed in HEK293 cells that were either transfected with Kir2.1 co-transfected with Cav-1-YFP or GFP. In both cases, we observed single channel events with a unitary conductance of 23–24 pS, typical for Kir2.1 channels, as reported in previous studies (Kubo et al. 1993, 2005; Fang et al. 2005) and, as expected, no significant channel activity was observed in HEK293 cells that did not express Kir2.1. Most importantly, our data show that there is no difference either in the unitary conductance or in the open probability of Kir2.1 channels in the presence and in the absence of Cav-1 (Fig. 3). These observations are also strikingly similar to our earlier studies showing that while cholesterol decreases whole cell current of Kir2.1, it has no effect on the surface expression of the channels or on their single channel properties (Romanenko et al. 2004).
Figure 3. Caveolin has no effect on Kir2.1 single channel current amplitude and cumulative open probability.
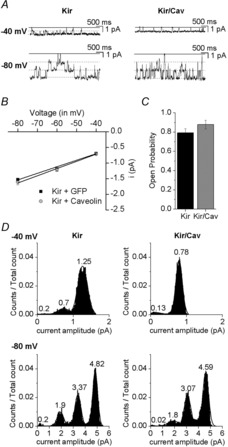
A, representative current traces from −40 mV and −80 mV from HEK293T cells co-transfected with either Kir2.1+GFP (Kir) or Kir2.1+Cav-YFP (Kir/Cav) in a cell-attached configuration. Multiple channels were observed in most membrane patches in both conditions. In any given patch more channels were routinely observed at a more negative membrane voltage (−80 mV) compared to a less negative membrane voltage (−40 mV). The closed level is indicated by the continuous line and the open levels are indicated by dotted lines. B, average unitary conductance of Kir2.1 channels was calculated from the slopes of the linear fits between –40 mV and −80 mV. In the control condition (absence of caveolin) the conductance of Kir was 21 pS and in the presence of caveolin the conductance of Kir was 23 pS. Therefore unitary conductance of Kir2.1 channels was the same relatively unaltered by overexpression of caveolin. C, bar graph summarizing the mean open probabilities are shown. In the case of cells transfected with Kir2.1+GFP (Kir) the mean open probability 0.794 ± 0.0414 and with Kir2.1+Cav-YFP (Kir/Cav) mean open probability was 0.878 ± 0.044. D, representative all-point histograms of channel records at −40 mV and −80 mV are shown for cells transfected with either Kir2.1+GFP (Kir) or Kir2.1+Cav-YFP (Kir/Cav). Cav, caveolin.
Caveolin-1 is not required for cholesterol sensitivity of Kir2 channels in HEK293 cells
As our observations demonstrate that Cav-1 has an inhibitory effect on Kir2.1 function, the next question was whether Cav-1 plays a role in cholesterol-induced suppression of the channels. To address this question, we tested whether Kir2.1 channels are sensitive to the level of cellular cholesterol in HEK293 cells, which as was pointed out above, express only nominal levels of Cav-1. The level of cellular cholesterol was modulated by exposing the cells to MβCD or MβCD-cholesterol resulting in approximately 40–50% cholesterol depletion or enrichment, respectively ( Fig 4, inset). We show here that Kir2.1 current that results from expressing the channels in HEK293 cells is suppressed by enriching the cells with cholesterol and enhanced by cholesterol depletion demonstrating that Cav-1 is not necessary to confer cholesterol sensitivity to Kir2.1 (Fig. 4A and B). Interestingly, however, overexpression of Cav-1 shifted the sensitivity of Kir2.1 to cholesterol resulting in the loss of the cholesterol enrichment effect and an increase in the cholesterol depletion effect on the Kir current (Fig. 4C and D). In terms of cholesterol content of the cells, however, overexpression of Cav-1 had no effect on the degree of cholesterol enrichment or on the degree of cholesterol depletion (Fig. 4, inset). These observations indicate that while Cav-1 is not required for cholesterol sensitivity of the channels, they suggest that there is a crosstalk between the two agents in regulating the channels.
Figure 4. Caveolin is not required but shifts cholesterol sensitivity of Kir2 channels in HEK293 cells.
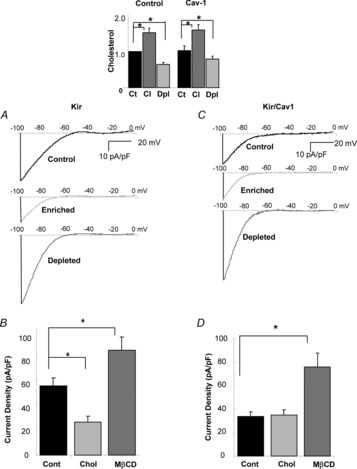
Inset: Cellular cholesterol levels in HEK293 cells co-expressing Kir2.1 with GFP or Cav-1-YFP in control (Ct), MβCD (Dpl) and MβCD/cholesterol (Cl) treated conditions. The numbers were normalized using Kir2.1/GFP in control cells as a reference (n = 3, *P < 0.05). A, representative current traces recorded from HEK293 cells expressing Kir2.1 with GFP as a marker for control cells or cells exposed to 5 mm MβCD or MβCD/cholesterol for 1 h to deplete the cells of cholesterol or to enrich cells with cholesterol, respectively. B, average peak current densities at −97 mV for control, MβCD and MβCD/cholesterol treated HEK293 cells expressing Kir2.1 (n = 14–36, *P < 0.05). C, representative current traces for HEK293 cells co-expressing Kir2.1 and Cav-1-YFP in control, MβCD and MβCD/cholesterol treatment. D, average peak current densities at −97 mV for the same cells (n = 14–44, *P < 0.05). Ct, control; Cl, MβCD/cholesterol; Dpl, MβCD; MβCD, methyl-β-cyclodextrin.
Abrogation of cholesterol sensitivity is associated with the loss of caveolin-1 sensitivity of Kir2.1
Our earlier studies identified several mutations of Kir2.1 that either diminish or abrogate cholesterol sensitivity of the channels (Epshtein et al. 2009; Rosenhouse-Dantsker et al. 2010, 2011). These mutants, therefore, provide a unique tool to test whether the sensitivities of the channels to cholesterol and to Cav-1 share a similar mechanism. In this study, we chose one of the ‘cholesterol-insensitive’ mutants where leucine in position 222 is substituted with isoleucine (Kir2.1-L222I), a mutation that abrogates the sensitivity of Kir2.1 to cholesterol in CHO cells and in Xenopus oocytes (Epshtein et al. 2009; Rosenhouse-Dantsker et al. 2010). As expected, Kir2.1-L222I currents in HEK293 cells were also not affected by cholesterol enrichment or by cholesterol depletion (inset). Furthermore, we show here that while Kir2.1-L222I is similar to Kir2.1-WT in that it physically interacts with Cav-1, as demonstrated by co-immunoprecipitation (Fig. 5A) Cav-1 has no effect on Kir2.1-L222I current indicating that this mutation abrogates the sensitivity of the channels to Cav-1 (Fig. 5B and C).
Figure 5. Cav-1 has no effect on cholesterol insensitive Kir mutant L222I.
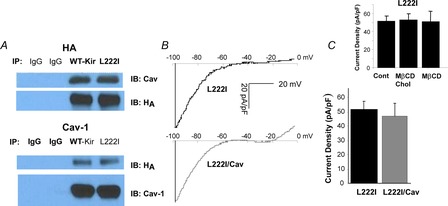
Inset: Average peak current densities at −97 mV for HEK293 cells expressing Kir2.1-L222I under different cholesterol conditions [control, cholesterol-enriched (MβCD-cholesterol) and cholesterol-depleted (MβCD)]. A, co-immunoprecipitation of caveolin-1 with Kir2.1-L222I channels. B, representative current traces for HEK293 cells expressing L222I with and without Cav-1. C, average peak current densities at −97 mV for HEK293 cells expressing Kir2.1 with and without Cav-1 (n = 20–33). Cav-1, caveolin-1; Cont, control; MβCD, methyl-β-cyclodextrin.
Genetic deletion of caveolin-1 increases Kir activity in primary macrophages
To test whether Cav-1 also acts as a negative regulator of Kir2 channels in vivo, we compared Kir currents in BMDMs isolated from Cav-1 KO and WT mice. In addition, while HEK293 cells express a low level of Cav-1, cells isolated from Cav-1 KO mice are completely devoid of this protein. We chose to focus on BMDMs in this part of the study because macrophages are well-known to express Kir2 channels (Kubo et al. 1993; Vicente et al. 2003; Thomas et al. 2011). Moreover, we have recently shown that macrophage Kir current is completely inhibited by a dominant-negative subunit of Kir2.1 (dnKir2.1) indicating that Kir2.1 is the dominant Kir channel in macrophages (Thomas et al. 2011). In the current study, we demonstrate that genetic deletion of Cav-1 results in a significant increase (∼30%) in the Kir current density indicating that, similar to our co-expression study in HEK293 cells, endogenous Cav-1 also acts as a negative regulator of Kir2 channels (Fig. 6). The levels of cellular cholesterol in BMDMs isolated from Cav-1 KO and from WT mice were very similar (25 ± 4 μg mg−1 protein vs. 25 ± 3 μg mg−1 protein respectively). Furthermore, consistent with the observations described above, macrophage Kir current in Cav-1 KO BMDMs is significantly enhanced by cholesterol depletion indicating that expression of Cav-1 is not required to confer cholesterol sensitivity to Kir channels in primary macrophages (Fig. 6). However, similar to that observed in HEK293 cells, the effect of cholesterol depletion on Kir currents was lower in BMDMs that do not express Cav-1 (the ratios between Kir currents in cholesterol depleted vs. control cells were 1.9 ± 0.16 in WT cells and 1.45 ± 0.11 in Cav-1 KO cells, P < 0.05).
Figure 6. Genetic deletion of Cav-1 results in an increase in Kir current but does not abrogate Kir cholesterol sensitivity in primary macrophages.
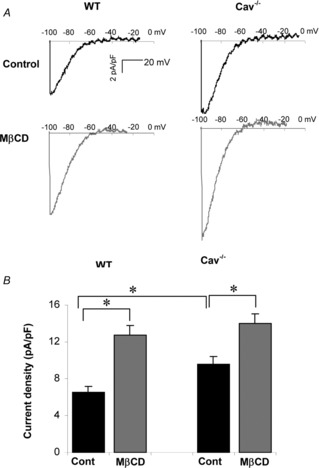
A, representative current traces of K+ currents in WT and Cav-1 KO BMDMs under control conditions and in cells depleted of cholesterol. B, average peak current densities at −97 mV of Kir currents under control and cholesterol depleted conditions (n = 26–30, *P < 0.05).
Discussion
Interaction between Kir2.1 and caveolin-1
Previous studies have shown that Cav-1 physically interacts with the subunits of the ATP-sensitive Kir channels (Kir6.1) (Sampson et al. 2004) and with kidney Kir channels (renal outer medullary K+ channels) (Lin et al. 2011). In addition, we have shown previously that Kir2.1 and Kir2.3 channels partition into caveolin-enriched membrane fractions (Tikku et al. 2007) suggesting that they may also interact with Cav-1. In this study we demonstrate that indeed Kir2.1 physically interacts with Cav-1 and that the loss of Cav-1 expression results in an increase in Kir2.1 current both in the heterologous expression system and in native cells.
Caveolin-1 binding motifs in Kir2.1
We also examined whether Kir2.1 has any of the caveolin binding motifs (CBMs) that have been identified in previous studies. Specifically, consensus binding motifs with the composite forms of ϕXϕXXXXϕ, ϕXXXXϕXXϕ and ϕXϕXXXXϕXXϕ, where ϕ is the aromatic amino acid Trp, Phe or Tyr, have been associated with caveolin binding (Couet et al. 1997). In this study, we identified the ϕXϕXXXXϕ CBM in two separate segments of Kir2.1. One segment was located at the interface between the outer transmembrane helix and the N-terminus of the channel (residues 81–88), thus residing close to the inner surface of the membrane, and the second segment was in the outer transmembrane helix close to the extracellular domain of the channel (residues 96–103). As it is known that Cav-1 resides in the inner leaflet of the membrane with its intramembrane domain spanning the single lipid monolayer of the inner leaflet (Hoop et al. 2012), it is probable that it would bind to the first of the segments described above that is close to the intracellular part of the membrane and not to the second segment that is close to the extracellular side. Figure 7A depicts the location of the CBM at the interface with the cytosolic domain in a surface presentation of the crystallized structure of Kir2.2. Alignment of all Kir channels shows that this CBM is present in the majority of channels, including the four Kir2 channels (Kir2.1–2.4) and most of G-protein coupled Kirs (GIRK or Kir3 channels) (see Table 1).
Figure 7. Kir2 channels have a caveolin binding motif at the interface between the TM and cytosolic domains.
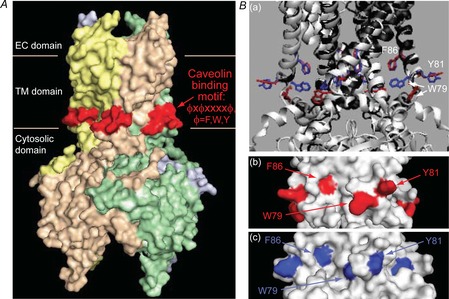
A, location of the caveolin ϕXϕXXXXϕ binding motif (red) at the interface between the outer transmembrane helix and the N-terminus of the channels (residues 81–88 in Kir2.1) in a surface presentation of the crystal structure of Kir2.2. Ba, comparison of the locations of the aromatic residues of the caveolin binding motif in the closed conformation of Kir2.2 (PDB ID 3jyc; the ribbon representation of the channel is coloured black and the aromatic residues in a stick representation are red) and in the conformation obtained in the presence of PIP2 that activates the channel (PDB ID 3spi; the ribbon representation of the channel is coloured white and the aromatic residues in a stick representation are blue). The structures were generated and aligned using the Visual Molecular Dynamics package. b–c surface representations of the region in Kir2.2 that includes the caveolin binding motif showing the locations of the aromatic residues depicted in Fig. 6Ba, in b the closed conformation (red) and Bc the conformation obtained in the presence of PIP2 (blue). Fig. 6A, and 6Bb–c, were generated using Pymol. PIP2, phosphatidylinositol 4,5-bisphosphate.
Table 1.
Sequence alignment of Kir channels to a inner helix segment of a putative caveolin binding motif of Kir2.1
 |
Shown are the residues that are located at the equivalent position to residues 81–88 in Kir2.1 in the inner transmembrane helix at the interface with the cytosolic domain. The residues critical for caveolin binding are highlighted in black.
Recently, the role of CBMs in caveolin signalling has become the subject of debate due to new structural information and bioinformatics analysis (Byrne et al. 2012; Collins et al. 2012). Specifically, it has been pointed out that if a CBM is to function as a caveolin interaction motif, two physical requirements must be met: First, the putative CBM should be exposed in the folded protein structure and accessible to caveolin. Second, the functional CBM must lie in a flexible (or disordered) region of the interacting protein whose conformational space would be restricted upon caveolin interaction. While it is not clear whether all the CBMs that have been identified in other proteins meet these requirements (Byrne et al. 2012; Collins et al. 2012) the CBM-based putative caveolin binding site located at the inner surface of the membrane of Kir2.2 does. First, this CBM is accessible in the closed conformation of the channel, and second, the region in which it is located undergoes structural changes during gating and therefore, caveolin is expected to restrict it to the closed conformation upon binding.
Implications for Kir gating
Earlier studies showed that the region identified above as a putative Cav-1 binding site is critical for channel gating as evident by comparing the closed and the open conformations of the channels obtained from the crystal structures (Tao et al. 2009; Hansen et al. 2011). In particular, the first aromatic residue in the CBM (W79) is displaced during channel gating (see Fig. 7Ba) and it becomes partially obscured when the channel opens (see Fig. 7Bb and Bc). The implication of this structural change is that Cav-1 should preferentially bind the channel in the closed state because the ability of Cav-1 to interact with the channel in the open state is expected to be reduced by the displacement of one of the residues of the binding motif. We propose, therefore, that preferential binding of Cav-1 to the closed state of the channels may result in stabilization of the channels in their closed state, which would contribute to the inhibitory effect of Cav-1 on the channel function. Furthermore, our previous analysis (Rosenhouse-Dantsker et al. 2011) demonstrated that the leucine residue at position 222 shown here to be critical for the Cav-1-sensitivity of Kir2.1, exhibits correlated motion with glutamate 303 located in the G-loop that has been proposed to act as the cytosolic gate of Kir2.1 (Bichet et al. 2003; Pegan et al. 2005; Nishida et al. 2007). This correlation supports the notion that Cav-1 regulates Kir2.1 channels by interfering with the gating mechanism. As we proposed earlier for cholesterol-induced suppression of Kir2.1, we propose here that Cav-1 also regulates the channels by shifting them into a ‘silent mode’. Comparative analysis of cholesterol and Cav-1 effects is described below.
Cross-talk between caveolin-1 and cholesterol in regulation of Kir channels
Cholesterol sensitivity of Kir2.1 cannot be attributed to the association with caveolin
Numerous studies demonstrated that a variety of ion channels partition into cholesterol and caveolin-rich membrane microdomains leading to the hypothesis that partitioning into these domains is the basis of cholesterol sensitivity of ion channels (reviewed by Levitan et al. 2010). Furthermore, cholesterol and caveolin were shown to act in parallel to suppress the activity of channels further supporting the idea that changes in cellular cholesterol may regulate channel activity by controlling their association with caveolins. The actual relationship between caveolins and cholesterol in the molecular regulation of ion channel function, however, remained poorly understood. It was also not clear whether partitioning into lipid rafts is a prerequisite for cholesterol sensitivity of the channels. In the present study, we show that while Cav-1 is a negative regulator of Kir2.1 channels, neither Cav-1 per se nor the integrity of caveolae are required for the sensitivity of Kir2.1 to cholesterol. Indeed, Cav-1 was shown to be the dominant form of caveolin in macrophages and while caveolin-2 is also expressed to some degree (Kiss et al. 2000), genetic deletion of Cav-1 results in destabilization and loss of caveolin-2 and the loss of caveolae (Drab et al. 2001). It is also known that while Cav-1 and -2 are co-expressed in most cell types, expression of the third form of caveolin, Cav-3, is muscle-specific (Williams & Lisanti, 2004). Therefore, our observations that Kir2.1 channels are sensitive to changes in membrane cholesterol in macrophages isolated from Cav-1−/− mice indicate that partitioning into caveolae is not a prerequisite for cholesterol sensitivity of Kir2.1 channels.
Cholesterol and caveolin-1 regulate Kir2.1 channels by a common mechanism
Our earlier studies have shown that an increase in membrane cholesterol results in a decrease in Kir2.1 whole cell currents without affecting surface expression unitary conductance, or open probability of the channels (Romanenko et al. 2004). These observations led us to hypothesize that cholesterol stabilizes the channels in a closed state creating a population of ‘silent channels’ that, while expressed on the plasma membrane, exist in a long-lived closed state that is too long to be picked up in a standard single channel recording, resulting in ‘silent channels’ being invisible as was discussed in previous studies (Romanenko et al. 2004; Jackson, 2006). Here we show exactly the same phenomenon in cells that overexpress Cav-1. Furthermore, we find that the same mutation, Kir2.1-L222I that renders the channels insensitive to cholesterol, also renders them insensitive to Cav-1. These observations suggest that cholesterol and Cav-1 regulate Kir2.1 channels by a similar mechanism. We do not propose, however, that cholesterol and Cav-1 bind to the channels at the same sites. In contrast, our observations suggest that the binding regions for the two modulators are clearly distinct. Specifically, as described above, a Cav-1 binding motif was identified at the protein–lipid interface in the transmembrane domain of Kir2.1. Putative cholesterol binding sites, on the other hand, which were identified in our recent studies, are non-annular sites in between the transmembrane helices (Rosenhouse-Dantsker et al. 2013). It is interesting to note that a Cav-1 binding motif was found in the vicinity of one of the putative cholesterol binding regions that resides on the interface between the transmembrane domain and the cytosolic domains of the channel.
We propose that there are two possible mechanisms to account for the cross-talk between cholesterol and Cav-1 in regulating Kir channels: One possibility is that Cav-1 does not regulate the channels by itself but increases the sensitivity of the channels to cholesterol. This hypothesis is based on observations that an increase in Cav-1 expression suppresses Kir2.1 channels under normal cholesterol conditions but not in cholesterol-enriched cells and that cholesterol has no further effect on Kir when co-expressed with Cav-1. It is also attractive to suggest that as Cav-1 is a cholesterol-binding protein it could enhance the interaction between cholesterol and the channels. Alternatively, however, it is also possible that Cav-1 and cholesterol act separately but their effects converge in regulating Kir2.1 gating through a L222I-sensitive mechanism. Currently, we cannot fully discriminate between these possibilities.
Acknowledgments
We are very grateful to Mr Sang Joon Ahn, Mr Alan Ropsky and Dr Yi-Fan Chen for their help with ANOVA statistical analysis. We also thank Mr Gregory Kowalsky for his help in formatting the figures.
Glossary
- BMDM
bone marrow-derived macrophage
- Cav-1
caveolin-1
- CBM
caveolin binding motif
- Kir
inwardly rectifying potassium channel
- MβCD
methyl-β-cyclodextrin
- WT
wild-type
Additional information
Competing interests
The authors have no conflict of interest to disclose.
Author contributions
H.H. designed and performed experiments, analysed and interpreted the data and wrote the manuscript. A.R.-D. performed and interpreted analysis and wrote the manuscript. R.G., Y.E. and Z.C. performed and analysed experiments. F.S. and R.D.M. advised in performing the experiments and interpreted data. I.L. designed, analysed and interpreted the data and wrote the manuscript. All experiments were performed at the University of Illinois at Chicago with the exception of the single channel experiments that were performed at the University at Buffalo, SUNY. All authors have read and approved the final version of the manuscript.
Funding
The work was supported by National Institutes of Health grants HL073965 and HL083298 (I.L.), HL071626 and HL060678 (R.D.M.), HL054887 (F.S.), DOD grant W81XWH-11-2-0125 (F.S.) and a Scientist Development Grant (11SDG5190025) from the American Heart Association (A.R.-D.). Statistical analysis was supported by the CCTS grant UL1TR000050.
References
- Bakhshi FR, Mao M, Shajahan AN, Piegeler T, Chen Z, Chernaya O, Sharma T, Elliott WM, Szulcek R, Bogaard HJ, Comhair S, Erzurum S, van Nieuw Amerongen GP, Bonini MG, Minshall RD. Nitrosation-dependent caveolin 1 phosphorylation, ubiquitination, and degradation and its association with idiopathic pulmonary arterial hypertension. Pulm Circ. 2013;3:816–830. doi: 10.1086/674753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichet D, Haass FA, Jan LY. Merging functional studies with structures of inward-rectifier K+ channels. Nat Rev Neurosci. 2003;4:957–967. doi: 10.1038/nrn1244. [DOI] [PubMed] [Google Scholar]
- Byrne DP, Dart C, Rigden DJ. Evaluating caveolin interactions: Do proteins interact with the caveolin scaffolding domain through a widespread aromatic residue-rich motif. PLoS One. 2012;7:e44879. doi: 10.1371/journal.pone.0044879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celada A, Gray PW, Rinderknecht E, Schreiber RD. Evidence for a gamma-interferon receptor that regulates macrophage tumoricidal activity. J Exp Med. 1984;160:55–74. doi: 10.1084/jem.160.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Bakhshi FR, Shajahan AN, Sharma T, Mao M, Trane A, Bernatchez P, van Nieuw Amerongen GP, Bonini MG, Skidgel RA, Malik AB, Minshall RD. Nitric oxide dependent Src activation and resultant caveolin-1 phosphorylation promote eNOS/caveolin-1 binding and eNOS inhibition. Mol Biol Cell. 2012;23:1388–1398. doi: 10.1091/mbc.E11-09-0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BM, Davis MJ, Hancock JF, Parton RG. Structure-based reassessment of the caveolin signaling model: Do caveolae regulate signaling through caveolin-protein interactions. Dev Cell. 2012;23:11–20. doi: 10.1016/j.devcel.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Identification of peptide and protein ligands for the caveolin-scaffolding domain. J Biol Chem. 1997;272:6525–6533. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- D'Avanzo N, Hyrc K, Enkvetchakul D, Covey DF, Nichols CG. Enantioselective protein-sterol interactions mediate regulation of both prokaryotic and eukaryotic inward rectifier K+ channels by cholesterol. PLoS One. 2011;6:e19393. doi: 10.1371/journal.pone.0019393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies LM, Purves GI, Barrett-Jolley R, Dart C. Interaction with caveolin-1 modulates vascular ATP-sensitive potassium (KATP) channel activity. J Physiol. 2010;588:3255–3266. doi: 10.1113/jphysiol.2010.194779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- Epshtein Y, Chopra AP, Rosenhouse-Dantsker A, Kowalsky GB, Logothetis DE, Levitan I. Identification of a C-terminus domain critical for the sensitivity of Kir2.1 channels to cholesterol. Proc Natl Acad Sci U S A. 2009;106:8055–8060. doi: 10.1073/pnas.0809847106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Schram G, Romanenko VG, Shi C, Conti L, Vandenberg CA, Davies PF, Nattel S, Levitan I. Functional expression of Kir2.x in human aortic endothelial cells: the dominant role of Kir2.2. Am J Physiol Cell Physiol. 2005;289:C1134–C1144. doi: 10.1152/ajpcell.00077.2005. [DOI] [PubMed] [Google Scholar]
- Fang Y, Mohler ER, III, Hsieh E, Osman H, Hashemi SM, Davies PF, Rothblat GH, Wilensky RL, Levitan I. Hypercholesterolemia suppresses inwardly rectifying K+ channels in aortic endothelium in vitro and in vivo. Circ Res. 2006;98:1064–1071. doi: 10.1161/01.RES.0000218776.87842.43. [DOI] [PubMed] [Google Scholar]
- Garg V, Sun W, Hu K. Caveolin-3 negatively regulates recombinant cardiac KATP channels. Biochem Biophys Res Commun. 2009;385:472. doi: 10.1016/j.bbrc.2009.05.100. [DOI] [PubMed] [Google Scholar]
- Hansen SB, Tao X, MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature. 2011;477:495. doi: 10.1038/nature10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- Hoop CL, Sivanandam VN, Kodali R, Srnec MN, van der Wel PCA. Structural characterization of the caveolin scaffolding domain in association with cholesterol-rich membranes. Biochemistry. 2012;51:90. doi: 10.1021/bi201356v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson WF. Silent inward rectifier K+ channels in hypercholesterolemia. Circ Res. 2006;98:982–984. doi: 10.1161/01.RES.0000222140.93190.7f. [DOI] [PubMed] [Google Scholar]
- Johnson DE. Applied Multivariate Statistics. Belmont, CA: Duxbury Press; 1998. [Google Scholar]
- Joo M, Kwon M, Cho Y-J, Hu N, Pedchenko TV, Sadikot RT, Blackwell TS, Christman JW. Lipopolysaccharide-dependent interaction between PU.1 and cJun determines production of lipocalin-type prostaglandin D synthase and prostaglandin D2 in macrophages. Am J Physiol Lung Cell Mol Physiol. 2009;296:L771–L779. doi: 10.1152/ajplung.90320.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss A, Túri A, Müllner N, Tímár J. Caveolin isoforms in resident and elicited rat peritoneal macrophages. Eur J Cell Biol. 2000;79:343–349. doi: 10.1078/S0171-9335(04)70038-2. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 1993;362:127–132. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Adelman JP, Clapham DE, Jan LY, Karschin A, Kurachi Y, Lazdunski M, Nichols CG, Seino S, Vandenberg CA. International Union of Pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol Rev. 2005;57:509–526. doi: 10.1124/pr.57.4.11. [DOI] [PubMed] [Google Scholar]
- Levitan I, Fang Y, Rosenhouse-Dantsker A. Cholesterol and ion channels. In: Harris JR, Romanenko V, editors. Cholesterol Binding and Cholesterol Transport Proteins. Springer Science; 2010. pp. 509–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D-H, Yue P, Pan C, Sun P, Wang W-H. MicroRNA 802 Stimulates ROMK channels by suppressing caveolin-1. J Am Soc Nephrol. 2011;22:1087–1098. doi: 10.1681/ASN.2010090927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milescu LS, Akk G, Sachs F. Maximum likelihood estimation of ion channel kinetics from macroscopic currents. Biophys J. 2005;88:2494–2515. doi: 10.1529/biophysj.104.053256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai C, Sachs F. Fitting random data to state models with QuB software. Biophysical Reviews. 2014 In Press. [Google Scholar]
- Nishida M, Cadene M, Chait BT, MacKinnon R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 2007;26:4005–4015. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegan S, Arrabit C, Zhou W, Kwiatkowski W, Collins A, Slesinger PA, Choe S. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci. 2005;8:279–287. doi: 10.1038/nn1411. [DOI] [PubMed] [Google Scholar]
- Riddle MA, Hughes JM, Walker BR. Role of caveolin-1 in endothelial BKCa channel regulation of vasoreactivity. Am J Physiol Cell Physiol. 2011;301:C1404–C1414. doi: 10.1152/ajpcell.00013.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanenko VG, Rothblat GH, Levitan I. Modulation of endothelial inward rectifier K+ current by optical isomers of cholesterol. Biophys J. 2002;83:3211–3222. doi: 10.1016/S0006-3495(02)75323-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanenko VG, Fang Y, Byfield F, Travis AJ, Vandenberg CA, Rothblat GH, Levitan I. Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys J. 2004;87:3850–3861. doi: 10.1529/biophysj.104.043273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenhouse-Dantsker A, Leal-Pinto E, Logothetis DE, Levitan I. Comparative analysis of cholesterol sensitivity of Kir channels: Role of the CD loop. Channels. 2010;4:63–66. doi: 10.4161/chan.4.1.10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenhouse-Dantsker A, Logothetis DE, Levitan I. Cholesterol sensitivity of KIR2.1 is controlled by a belt of residues around the cytosolic pore. Biophys J. 2011;100:381. doi: 10.1016/j.bpj.2010.11.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenhouse-Dantsker A, Noskov S, Durdagi S, Logothetis DE, Levitan I. Identification of novel cholesterol-binding regions in Kir2 channels. J Biol Chem. 2013;288:31154–31164. doi: 10.1074/jbc.M113.496117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson LJ, Hayabuchi Y, Standen NB, Dart C. Caveolae localize protein kinase A signalling to arterial ATP-sensitive potassium channels. Circ Res. 2004;95:1012–1018. doi: 10.1161/01.RES.0000148634.47095.ab. [DOI] [PubMed] [Google Scholar]
- Singh DK, Rosenhouse-Dantsker A, Nichols CG, Enkvetchakul D, Levitan I. Direct regulation of prokaryotic Kir channel by cholesterol. J Biol Chem. 2009;284:30727–30736. doi: 10.1074/jbc.M109.011221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Shentu T-P, Enkvetchakul D, Levitan I. Cholesterol regulates prokaryotic Kir channel by direct binding to channel protein. Biochim Biophys Acta. 2011;1808:2527. doi: 10.1016/j.bbamem.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Avalos JL, Chen J, MacKinnon R. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 Å resolution. Science. 2009;326:1668–1674. doi: 10.1126/science.1180310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J, Epshtein Y, Chopra A, Ordog B, Ghassemi M, Christman JW, Nattel S, Cook JL, Levitan I. Anthrax lethal factor activates K+ channels to induce IL-1b secretion in macrophages. J Immunol. 2011;186:5236–5243. doi: 10.4049/jimmunol.1001078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikku S, Epshtein Y, Collins H, Travis AJ, Rothblat GH, Levitan I. Relationship between Kir2.1/Kir2.3 activity and their distribution between cholesterol-rich and cholesterol-poor membrane domains. Am J Physiol Cell Physiol. 2007;293:C440–C450. doi: 10.1152/ajpcell.00492.2006. [DOI] [PubMed] [Google Scholar]
- Vicente R, Escalada A, Coma M, Fuster G, Sanchez-Tillo E, Lopez-Iglesias C, Soler C, Solsona C, Celada A, Felipe A. Differential voltage-dependent K+ channel responses during proliferation and activation in macrophages. J Biol Chem. 2003;278:46307–46320. doi: 10.1074/jbc.M304388200. [DOI] [PubMed] [Google Scholar]
- Wang X-L, Ye D, Peterson TE, Cao S, Shah VH, Katusic ZS, Sieck GC, Lee H-C. Caveolae targeting and regulation of large conductance Ca2+-activated K+ channels in vascular endothelial cells. J Biol Chem. 2005;280:11656–11664. doi: 10.1074/jbc.M410987200. [DOI] [PubMed] [Google Scholar]
- Williams TM, Lisanti MP. The caveolin genes: from cell biology to medicine. Ann Med. 2004;36:584–595. doi: 10.1080/07853890410018899. [DOI] [PubMed] [Google Scholar]