

Abstract
Methylation of the fifth carbon of cytosine was the first epigenetic modification to be discovered in DNA. Recently, three new DNA modifications have come to light: hydroxymethylcytosine, formylcytosine, and carboxylcytosine, all generated by oxidation of methylcytosine by Ten Eleven Translocation (TET) enzymes. These modifications can initiate full DNA demethylation, but they are also likely to participate, like methylcytosine, in epigenetic signalling per se. A scenario is emerging in which coordinated regulation at multiple levels governs the participation of TETs in a wide range of physiological functions, sometimes via a mechanism unrelated to their enzymatic activity. Although still under construction, a sophisticated picture is rapidly forming where, according to the function to be performed, TETs ensure epigenetic marking to create specific landscapes, and whose improper build-up can lead to diseases such as cancer and neurodegenerative disorders.
Keywords: DNA modifications, epigenetics, human diseases, hydroxymethylation, TET proteins
Introduction
DNA methylation was observed more than a century ago by W. G. Ruppel, who discovered in Mycobacterium tuberculosis, the causative agent of tuberculosis, a non-canonical nucleotide that he believed to be a methylated pyrimidine (Ruppel, 1898). Since this initial discovery, the modified base has been identified as 5-methylcytosine (5mC), and a plethora of papers have shown it to play many roles in both normal physiology and disease. This epigenetic mark is made de novo by the DNA methyltransferases DNMT3A and DNMT3B, and is propagated during DNA replication by the maintenance methyltransferase DNMT1 (Denis et al, 2011). In diseases such as certain cancers and neuronal disorders, 5mC patterns are often altered. For example, neoplasia is frequently characterized by both global hypomethylation, rendering the genome unstable, and local hypermethylation and repression of tumour suppressor genes needed to fight cancer (You & Jones, 2012). In 2009, two seminal papers reported the existence of another modified form of cytosine: 5-hydroxymethylcytosine (5hmC). This modified base is generated by the Ten Eleven Translocation (TET) enzymes through oxidation of 5mC (Kriaucionis & Heintz, 2009; Tahiliani et al, 2009). The TET enzyme family comprises three TETs (TET1, TET2, TET3), which all derive from a common ancestor that was triplicated in a jawed vertebrate (Pastor et al, 2013). They use Fe2+ and α-ketoglutarate for catalysis, and all three TETs can also iteratively oxidize 5-hydroxymethylcytosine to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (Tahiliani et al, 2009; Ito et al, 2011; Blaschke et al, 2013; Minor et al, 2013; Yin et al, 2013). In tissues, the abundance of these DNA modifications tends to vary in the order 5mC > 5hmC > 5fC > 5caC, where both 5fC and 5caC abundance are respectively found at nearly one and two orders of magnitude lower than 5hmC. In two recent studies, TET-mediated catalysis was further investigated by analysing the crystal structures of human TET2 and an amoebal homologue of mammalian TET1 (NgTET1 from Naegleria gruberi). It seems that the negatively charged DNA binds the basic amino acids located at the surface of the TET protein and that the enzyme flips the methylcytosine out of the DNA double helix into its double-stranded β-helix catalytic pocket where it is stabilized by hydrogen bonds. DNMTs also show such a base-flipping mechanism. Importantly, the cys-rich DNA binding domain located in the carboxyterminal portion of human TET2 also plays a role in stabilizing the double-stranded β-helix domain (Hu et al, 2013; Hashimoto et al, 2014). Through their catalytic activity, the TETs act as initiators of DNA demethylation, either via passive dilution of 5hmC with DNA replication as seen in maturating primordial germ cells (PGCs) and fertilized zygotes, or through the action of DNA repair enzymes (the base excision repair (BER) machinery), which enzymatically remove the modified base and replace it with a cytosine (Guo et al, 2011; He et al, 2011; Maiti & Drohat, 2011; Zhang et al, 2012). This last process can occur through direct removal of 5fC and 5caC by the TDG glycosylase, followed by DNA repair. Alternatively, an initial deamination of 5hmC to 5-hydroxymethyluridine (5-hmU) by AID/APOBEC enzymes followed by BER also leads to complete DNA demethylation (Guo et al, 2011). Intriguingly, decarboxylation of 5caC has been observed in mouse embryonic stem cell (mESC) extracts, but no putative decarboxylase has yet been discovered (Schiesser et al, 2012). Lastly, Chen et al (2012) reported in 2012 that DNMT3A and DNMT3B are also DNA 5-hydroxymethylcytosine dehydroxymethylases, and that incubation of these methyltransferases with 5hmC-containing PCR products leads to partial dehydroxymethylation, this activity being increased in an oxidizing environment.
In summary, many routes of TET-triggered DNA demethylation (replication dependent, BER, deamination followed by BER, decarboxylation or dehydroxymethylation) are opened by a first oxidation of 5mC to 5hmC. Yet we should bear in mind that gene-rich regions in some tissues, such as those of the nervous system, display significant “stable” enrichment in hydroxymethylcytosine (Kriaucionis & Heintz, 2009), suggesting that TET-mediated 5hmC formation not only primes demethylation but also creates a unique epigenetic landscape in these cells.
Previous reviews discussing DNA modifications created by TET proteins have notably dealt with general aspects, their genomic distribution, and their roles in embryonic development (e.g. (Delatte & Fuks, 2013; Pastor et al, 2013; Wu & Zhang, 2014). The present review covers more recent discoveries in the exciting world of TETs. It first offers a glimpse at new TETs modes of action that are now surfacing thanks to studies providing novel insights into TET regulation, TET partners, and readers of DNA modifications. The focus then shifts to the roles played by TETs in key physiological processes and to the involvement of TET malfunction in disease, with special emphasis on non-cancer pathologies.
Novel insights into the mechanisms of TET-mediated DNA modifications
While our picture of how TET enzymes contribute to the dynamic methylation/demethylation cycle is still blurred, researchers have begun to lift the veil on how TET enzymatic activity is regulated, how TETs are targeted to precise DNA sequences, and how the 5mC oxidation signal is further read by “interpreters”. Below, we describe significant recent advances in this area.
Modes of direct regulation of TET enzymes
Direct regulation of TET enzymatic activity should be a rather straightforward means of modulating DNA modification levels (Fig 1). For 5mC oxidation, the TET proteins use Fe(II) and O2 as cofactors along with α-ketoglutarate, a metabolite of the Krebs cycle, which is converted to succinate (Tahiliani et al, 2009). In several cancers, including 80–90% of adult grade II/III gliomas (Losman & Kaelin, 2013), the isocitrate dehydrogenases 1 and 2 (IDH1 and IDH2) are mutated and the oncometabolite 2-hydroxyglutarate (2-HG) is produced. As 2-HG can compete directly with α-ketoglutarate, it unsurprisingly inhibits various dioxygenases, including the TET enzymes (Xu et al, 2011a). As the TET and IDH proteins act in concert, acute myeloid leukaemias with IDH mutations display both a global and a specific hypermethylation signature, possibly due to decreased TET activity (Figueroa et al, 2010).
Figure 1. Regulation of TETs/methylcytosine oxidation.
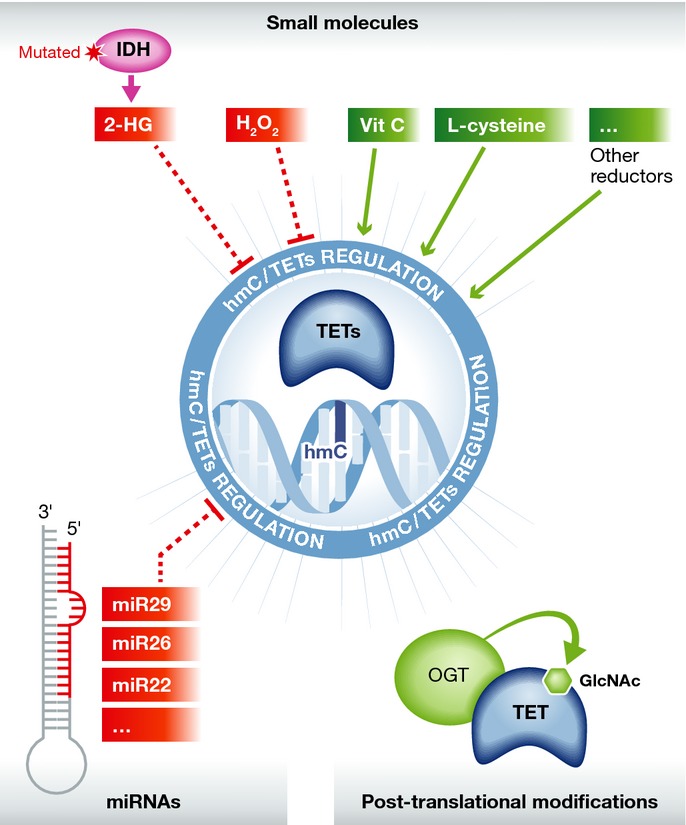
TET proteins can be regulated at multiple levels, all having a potential impact on global or local methylcytosine oxidation. Small molecules such as 2-hydroxyglutarate (2-HG) or hydrogen peroxide (H2O2) inhibit the catalytic activity of TETs while others, such as vitamin C, enhance their activity. Similarly, small microRNAs (miRs) can also affect TET-mediated 5hmC formation by direct downregulation of TET expression. Finally, TETs are connected to and regulated by chromatin related proteins. For example, TET bind the OGT GlcNAc transferase, which can glycosylate and possibly stabilize TETs.
Novel insights into the direct regulation of TET activity were obtained in 2013. Several groups independently reported that ascorbic acid (vitamin C) can significantly enhance TET activity in tubo, in cultured cells, and in vivo (Blaschke et al, 2013; Minor et al, 2013; Yin et al, 2013). Vitamin C deficiency has long been known to result in scurvy, a human disease caused by impaired collagen synthesis. The characteristic symptoms of scurvy are due to decreased prolyl 4-hydroxylase activity and incomplete hydroxylation of collagen fibres (Chojkier et al, 1983; Peterkofsky, 1991). In mouse embryonic fibroblasts (MEFs), Minor et al (2013) observed a 50% increase of the 5hmC level after 1 h of treatment with 10 μM ascorbate, and up to a five-fold increase after longer exposure times. Yin et al further demonstrated both in cultured cells and with purified TET domains that vitamin C causes rapid formation of 5hmC, 5fC, and 5caC, probably by reducing Fe(III) back to Fe(II) after enzymatic catalysis. In keeping with the ability of the 5hmC-5fC-5caC pathway to induce demethylation, mESCs treated with vitamin C display a significant concomitant demethylation. These results were confirmed in vivo in mice deficient in ascorbic acid synthesis (Yin et al, 2013), and genome-wide mapping of 5hmC after vitamin C treatment of mESCs evidenced a global increase in hydroxymethylation and a decrease in 5mC (Blaschke et al, 2013; Yin et al, 2013). Interestingly, Blaschke et al found vitamin C to both induce DNA demethylation and increase expression of germline genes, whereas some regions such as imprinted loci and intracisternal A particle (IAP) retroelements proved refractory to DNA demethylation. Furthermore, although various chemical reducing agents (GSH, vitamin B1, vitamin E) appeared not to affect 5-methylcytosine oxidation patterns, L-cysteine was shown to increase 5hmC by nearly 20% and 5fC by 50% (Blaschke et al, 2013; Yin et al, 2013). Similarly, hydroquinone, a potent reducing agent used for skin depigmentation, significantly increases hydroxymethylcytosine (Coulter et al, 2013). It remains to be seen whether oxidizing agents negatively impact TET activity, perhaps by oxidizing the Fe(II) catalytic centre. Unpublished data from our lab point to inactivation of the TET enzymes by hydrogen peroxide treatment, and we have observed a significant decrease in 5hmC in both cells treated with various oxidants and GPxI/II-knockout mice (GPx enzymes being the major natural antioxidants in the cells). As oxidative stress is involved in various diseases such as cancers and neuronal disorders, it will be important to assess whether alteration of global and local 5hmC patterns might explain the manifestations of these life-threatening diseases (B. Delatte & F. Fuks, unpublished data).
Besides regulation by small molecules, recent reports indicate that TET proteins can be post-transcriptionally regulated by microRNAs (miRNAs or miRs) (Cheng et al, 2013; Fu et al, 2013; Morita et al, 2013; Song et al, 2013a,b). For example, Morita et al (2013) have shown that miR-29-family of microRNAs can repress TET1 and thymine-DNA glycosylase (TDG) without greatly altering DNA methylation, possibly because they can also downregulate DNMT3A and DNMT3B. Zhang et al (2013a) have found miR-29 to inhibit TET1-TET2 and TET3 as well as TDG, this being accompanied by a small but significant decrease in hydroxymethylcytosine. MiR-26 can also repress all three TETs and TDG, emphasizing a quite complex regulation of DNA demethylation enzymes by small RNAs (Fu et al, 2013). Interestingly, transgenic mice overexpressing miR-26 display a decreased level of 5hmC and an increased proliferation and differentiation of pancreatic cells in vivo. This suggests that TETs might play a role in pancreatic differentiation and also in pancreas-related diseases such as cancer and diabetes (Fu et al, 2013). As miR-26 and many other miRs are deregulated in human malignancies and can function either as oncogenes or as tumour-suppressor genes (Fu et al, 2013), and as a decreased hydroxymethylcytosine level is suggested to be a hallmark of cancer (see below; cf. also Delatte & Fuks, 2013), TET proteins might be directly downregulated by miR networks during tumorigenesis. It is noteworthy in this regard that overexpression of miR-22 in the haematopoietic or mammary gland compartment inhibits TET-mediated hydroxymethylation and promotes leukaemogenesis or breast cancer, respectively. In fact, myelodysplastic patients displaying a decreased level of TET2 show poor survival and an inverse correlation between the TET2 and miR-22 levels. A similar anti-correlation has been observed in breast cancer patients exhibiting reduced TET1/TET2 expression (Song et al, 2013a,b). TET2 being a target of miR-29 and miR-26 has been recently confirmed by Chen et al, who studied the effects of overexpressing nearly 500 miR constructs in H293T cells. They observed no deregulation by miR-22, possibly for technical reasons such as the length of the 3′-untranslated regions used in their experiments. Remarkably, they identified another ten miR families that regulate TET2, notably miR-101 and miR-125, which induce abnormal haematopoiesis and a malignant phenotype in xenograft experiments, directly via TET2 downregulation (Cheng et al, 2013). These reports highlight the possible role of miRNAs in mediating the post-transcriptional regulation of TETs during pathogenesis.
Another mode of TET regulation to be considered is post-translational modifications (PTM). Covalent PTMs (such as acetylation, methylation, phosphorylation, and glycosylation) are well known to provide a vast indexing potential and to expand the range of functions of histones and non-histone proteins. Little has been published on the possible regulation of TETs by post-translational modifications. We do know that all three TETs can be modified by the O-linked N-acetylglucosamine (O-GlcNAc) transferase (OGT) (Shi et al, 2013; Vella et al, 2013; Ito et al, 2014). This glycosyltransferase adds O-GlcNAc moieties on various cytosolic and nuclear proteins (Hu et al, 2010). Protein O-glycosylation is known to antagonize phosphorylation, by direct competition for site-occupancy at a single hydroxyl group or via steric hindrance at proximal sites in the same polypeptide (Wang et al, 2008). As glycosylation of threonine 535 of TET1 enhances the protein's stability (Shi et al, 2013), it is tempting to propose that phosphorylation of this residue in the absence of GlcNAc might flag the protein for ubiquitin-mediated degradation and thereby cause a significant decrease in 5mC oxidation marks (Shi et al, 2013). One might expect TETs, like many other epigenetic enzymes (Denis et al, 2011), to be decorated by various PTMs affecting their enzymatic activity.
In summary, the body of available evidence suggests that small molecules, microRNAs, and PTMs may regulate hydroxymethylcytosine levels by directly modulating TET expression and/or enzymatic activity. Future studies may bring to light additional modes of regulation, notably involving direct amino acid modifications.
An expanding TET interaction network
Since the original discovery of TETs in 2009, another approach to elucidating TET functions has been to identify proteins with which these enzymes interact. The search for TET interactants has brought to light a plethora of transcription-related factors: transcription factors/nuclear receptors (CXXC4, NANOG, PPARγ, PU.1, EBF1, PRDM14) and chromatin-associated proteins involved in transcriptional activation (OGT and SET1/COMPASS complex) or repression (SIN3A/HDACs, NURD). Below we describe in more detail what we know about these TET partners and how they might help TETs to perform their functions (Fig 2).
Figure 2. The expanding TET interaction network.

Several proteins and catalytically active complexes have been shown to interact with TET enzymes, some having a role in cell reprogramming or differentiation, TET degradation, or in shaping the chromatin landscape by favouring an open or closed chromatin state. Some factors also seem to impact TET function via an indirect mechanism. This is the case for PARP1 and EZH2 (marked with an asterisk). EBF1 is double-marked to emphasize the fact that, although it can play a role in differentiation, the “EBF1-TET” context has been identified in a chondrosarcoma cell line.
The strongest TET partner identified so far is OGT, which we and others have recently reported (Chen et al, 2012; Deplus et al, 2013; Shi et al, 2013; Vella et al, 2013; Ito et al, 2014). Recent evidence of an epigenetic function for OGT has emerged with the discovery that it can glycosylate histone H2B on serine 112, causing its ubiquitinylation and the subsequent trimethylation of histone H3 at lysine 4 (H3K4me3). This modification is known to “decorate” active genes and to be mainly deposited by the SET1/COMPASS complex (Fujiki et al, 2011, 2013). Depending on TETs and OGT abundance and on the cell context, TET1, TET2, and TET3 all seem able to interact physically with OGT. The published data now enable us to propose a “hydroxymethylation independent” model of gene activation, in which TETs associate with OGT on promoters (mainly at CpG rich islands) and enhance its glycosyltransferase activity. This would in turn favour glycosylation of serine 112 of H2B (H2BS112GlcNAc) and ubiquitinylation of lysine 120 (H2BK120ub). In parallel, TET-promoted OGT activity would stabilize by glycosylation the SET1/COMPASS complex, this resulting in SET1/COMPASS binding to H2BK120ub, trimethylation of H3K4, and transcriptional activation (Chen et al, 2013; Deplus et al, 2013; Vella et al, 2013). Efforts should be made to unravel the physiological role(s) of this strong TET-OGT connection. Studies on neuronal cells might prove particularly fruitful, as both OGT and the TETs appear most abundant in these cells and are essential to neuronal development (Okuyama & Marshall, 2003; Kriaucionis & Heintz, 2009).
Genome-wide studies have shown that TET1 is associated not only with active “H3K4me3 only” promoters but also with bivalent poised “H3K4me3/H3K27me3” promoters and repressed “H3K27me3 only” promoters. In mESCs, furthermore, TET1 knockdown leads to activation of a number of genes, and this suggests that TET1 may have a transcriptional repressor role (Wu et al, 2011). In 2011, Wu et al found TET1 depletion to decrease binding of the H3K27me3 responsible enzyme EZH2 to co-bound genes. While TET1 has been reported to interact with EZH2 (Cartron et al, 2013), other investigators have failed to observe such an association (Williams et al, 2011a; Wu et al, 2011; our unpublished work). It thus seems likely that the TET1-EZH2 link is an indirect one, in which TET1 would “prepare the ground” for EZH2 binding by “shaping” the promoter's DNA modification state. In 2011, the Helin's laboratory described the SIN3A corepressor complex as a binding partner of TET1 (Williams et al, 2011b). SIN3A is known to associate with histone deacetylases HDAC1 and HDAC2, enzymes that maintain histones in a deacetylated form, thereby repressing transcription. Williams et al (2011b) further showed that TET1 clusters around transcription start sites with SIN3A, recruits SIN3A to a subset of promoters and induces transcriptional silencing of the co-bound genes. HDAC 1 and 2 are parts of other corepressor complexes such as NuRD or coREST, and NuRD has also been found to interact with TET1 in mESCs (Yildirim et al, 2011; Kelly & Cowley, 2013; Shi et al, 2013). As described below, the MBD3 NurD component can bind 5-hmC via its MBD domain and the genome-wide profile of MBD3 is similar to that of TET1 (Yildirim et al, 2011). These combined results thus constitute evidence of a link between TET1, 5hmC, and histone deacetylase systems in transcriptional gene repression. TETs are likely to be found to cooperate with yet other chromatin complexes - both activating and repressive - in controlling gene expression.
TET1 and TET3 can bind DNA via their N-terminal CXXC and C-terminal cys-rich domains, but TET2 has lost its CXXC domain during evolution, which is now encoded by a separate gene called CXXC4 or IDAX, a reported inhibitor of the Wnt/β-catenin pathway. Interestingly, Anjana Rao's laboratory has discovered that CXXC4 can associate with TET2 and recruit it to promoters. More surprisingly, binding of CXXC4 to DNA stimulates caspase-dependent TET2 degradation. This is a unique case of epigenetic marking through recruitment of a protein to DNA and its subsequent degradation (Ko et al, 2013). Other transcription factors can recruit TET enzymes to their genomic targets. This happens during cell reprogramming to pluripotency: physical interaction between the NANOG and TET1/TET2 proteins has been shown to facilitate reprogramming of neural stem cells in a manner that depends on methylcytosine hydroxylase activity (Costa et al, 2013). TET recruitment has also been observed during differentiation. One example concerns the differentiation of pre-adipocytes to adipocytes, triggered by the nuclear receptor PPARγ. During adipogenesis and in a ligand-dependent manner, PPARγ switches binding partners from a co-repressor to a co-activator complex on the PPAR-response elements (PPREs) of numerous genes expressed specifically in adipocytes. PPARγ poly(ADP-ribosyl)ation (PARylation), a post-translational protein modification mediated by PARPs, is known to regulate PPARγ transcriptional activity, and this may involve DNA demethylation (Fujiki et al, 2013). In a recent study, Fujiki et al showed that during differentiation, TET1 interacts with PPARγ after PARylation of the nuclear receptor. They further revealed that TET1 and TET2 can bind to PAR polymers and propose a model in which active DNA demethylation of key adipocyte-specific genes occurs via direct binding of TET1 and TET2 to the modified PPARγ receptor (Fujiki et al, 2013). During osteoclast differentiation, likewise, the transcription factor PU.1 interacts with TET2 and recruits it to genes that end up hypomethylated (de la Rica et al, 2013). Interestingly, PU.1 is required to generate most myeloid lineages (including macrophages, neutrophils, and dendritic cells) and for B-cell differentiation; it can be suppressed or mutated in leukaemia (Shereen & Rodney, 2011). It can also interact with the spliceosome machinery and participate in alternative splice site selection (Guillouf et al, 2006). These observations suggest an unanticipated involvement of the TET2-PU.1 association in cancer and in essential processes such as mRNA metabolism. On the basis of a search for consensus binding sites surrounding hypermethylated positions in a meta-analysis of IDH-mutant cancers, Guilhamon et al identified EBF1 as an interacting partner of TET2 in a human chondrosarcoma cell line. Further experiments showed that EBF1 and TET2 co-occupy certain genomic loci (Guilhamon et al, 2013). It is known that EBF1 is required for B lymphopoiesis and a significant fraction of genes, co-bound by EBF1 and the transcription factor E2-alpha (E2A), correlates with histones modifications of activated chromatin but also with poised or repressed regions in pro-B cells (Hagman et al, 2012). Knowing the interaction with EBF1 and that TET2 has a critical function in the haematopoietic compartment, it will be of great interest to assess its role in B-lymphocytes signaling and differentiation (Delatte & Fuks, 2013).
TET1 and TET2 also interact with the transcriptional regulator PRDM14, whose overexpression in mESCs leads to increased hydroxymethylation, demethylation, and subsequent activation of a set of pluripotency, germline, and imprinted genes (Okashita et al, 2013). As mentioned above, PGCs undergo global demethylation during their maturation, as a result of global conversion of methylcytosine to hydroxymethylcytosine and replication-dependent dilution. During this global wave of methylation resetting, imprinted loci also become demethylated, and TET1 has been found to play a role in erasing paternal and maternal genomic imprints, this being particularly evident in a TET1 knockout mouse which also display a defect in meiosis of developed gametes and some developmental arrest between E16.5 and E18.5. As PRDM14 is highly expressed in PGCs and interacts with TET1, it is tempting to speculate that these two proteins might participate together in imprinting erasure during PGC reprogramming (Okashita et al, 2013; Yamaguchi et al, 2013).
All in all, the functional diversity of the TET interaction network (Fig 2) suggests that the TET proteins are more versatile than initially believed and participate in other processes besides demethylation of CpG dinucleotides. ChIP-seq experiments on histone marks in relevant tissues will help to unravel the physiological roles of the TETs beyond differentiation and reprogramming. Also, metabolic and signalling pathways (such as those involved in OGT metabolism or Wnt/β-catenin) are likely to modify the TET-related epigenome. An important future task could thus be to compare maps of OGT or CXXC4 with maps of TET2 and 5hmC, for example, under various conditions affecting these pathways.
Readers of oxidized methylcytosines
In mammals, 5hmC levels vary according to the tissue and cell type. The brain and spinal cord show the highest levels, and organs such as the testes, spleen, and thymus display the lowest. An intermediate level is recorded in embryonic stem cells were approximately 0.1% of all bases in these cells are hydroxymethylcytosines (Globisch et al, 2010; Ito et al, 2010; Szwagierczak et al, 2010). The abundance of this oxidized methylcytosine, particularly in the brain, has led the scientific community to seek proteins that can bind to 5hmC and interpret/translate the encoded information. Several studies have shown that methyl-binding proteins (MBDs) can to some extend bind to 5hmC DNA, most often within a hemi-hydroxymethylated template and with lower affinity than to fully methylated DNA. This is the case of MeCP2, MBD1, MBD2, and MBD4 (Jin et al, 2010; Mellen et al, 2012; Otani et al, 2013; Hashimoto et al, 2014). MBD3 has been known for some time not to bind 5mC on its own, but in 2011, Yildirim and coworkers reported that the repressive NurD complex can bind both 5hmC-containing and non-modified DNA through its MBD3 subunit (Yildirim et al, 2011). The jury is still out as to whether MBD3 is a true hmC reader, as it has not been detected by others (Spruijt et al, 2013). The fact that MeCP2 can recognize 5hmC in vitro will force the community to re-evaluate the MethylCap assay, which employs the MeCP2 MBD domain to interrogate 5mC in genomic DNA. This applies particularly to tissues such as brain tissue, where almost half of the methylcytosine is in fact hydroxymethylcytosine (Brinkman et al, 2010). Further in silico and in vitro studies have examined whether the DNMT1 partner UHRF1 might also bind to hemi- or fully hydroxymethylated DNA via its SRA domain. It appears that UHRF1 does bind 5hmC-containing DNA, but the function of this binding is not yet clear. The protein is known to flip the base out of the DNA double helix. This might enhance further oxidation of 5hmC to 5fC and 5caC by making the hydroxymethylated base more accessible to the TET enzymes (Frauer et al, 2011). Accordingly, H293T cells overexpressing the UHRF1 homologue UHRF2, which also binds hydroxymethylcytosine, display oxidation of 5mC to 5hmC, with a remarkable increase in 5fC and 5caC (Spruijt et al, 2013).
The above-mentioned studies mostly describe in vitro approaches where recombinant proteins are incubated with modified DNA fragments and then used in electrophoretic mobility shift assays. Two groups have reported identifying readers of 5hmC, 5fC, and 5caC by quantitative mass spectrometry (Iurlaro et al, 2013; Spruijt et al, 2013). Interestingly, the first group found some readers to be specific to mESCs, neural progenitor cells (NPCs), or adult mouse brain tissue, while the second revealed proteins whose binding to 5hmC or 5fC depends on the CpG composition. This indicates (i) that hydroxymethylcytosine probably behaves differently in different cell types and (ii) that some presumptive readers bind oxidized methylcytosines in a sequence-specific manner. The screens also identified several specific 5fC and 5caC readers, among them TDG, supporting the view that this protein can act as a DNA demethylase (Iurlaro et al, 2013; Spruijt et al, 2013).
In summary, specific proteins are recruited to 5hmC, 5fC, or 5caC sites, and some of them are likely to regulate gene expression or to drive active DNA demethylation. More in-depth characterization by ChIP-seq and the use of knockout mice should help to show whether the proteins identified so far are true “readers” that can interpret 5hmC, 5fC, or 5caC modifications and translate them into an activating or repressing signal, or merely “binders” with some affinity for modified cytosines.
Emerging roles of TETs in normal physiology and disease
TETs/hmC in cell reprogramming and differentiation
The TET proteins participate in two waves of global DNA demethylation that occur during reprogramming of primordial germ cells as well as of paternal pronucleus in the zygote. Such reprogrammings are needed to ensure resetting of the epigenomes for pluripotency (Gu et al, 2011; Inoue et al, 2011; Iqbal et al, 2011; Wossidlo et al, 2011; Hackett et al, 2012; Nakamura et al, 2012). When PGCs migrate to the future gonads, high levels of TET1 and TET2 allows conversion of 5mC into 5hmC and this, not only globally but also more locally (e.g. at genomic imprinted loci). This is followed by subsequent passive “replication-coupled” dilution of 5hmC during several round of cell divisions with a progressive loss of hydroxymethylation (Hackett et al, 2012). A second wave of global DNA demethylation appears when the spermatozoon fertilizes the oocyte. In this case, TET3 hydroxylates the paternal DNA while PGC7 (also called DPPA3/STELLA) maintains normal methylation levels on the maternal genome as well as on imprinted genes by binding to the repressive H3K9me2 histone mark and by repelling TET3 (Szabo & Pfeifer, 2012). Worth mentioning, it has been observed that in the paternal pronucleus, 5hmC is further oxidized into 5fC and 5caC and that those modifications are also passively diluted through cell divisions (Gu et al, 2011; Inoue & Zhang, 2011; Inoue et al, 2011; Iqbal et al, 2011; Wossidlo et al, 2011; Nakamura et al, 2012).
The initial discovery of TET enzymes in ESCs has led many groups to assess their importance in pluripotency maintenance and cell differentiation. Although some reports suggest a role of TET1 in maintenance of pluripotency, especially via direct binding and demethylation of the NANOG promoter, other papers do not support this view (Ito et al, 2010; Ficz et al, 2011; Koh et al, 2011; Williams et al, 2011a; Wu et al, 2011; Xu et al, 2011b; Doege et al, 2012; Freudenberg et al, 2012). Furthermore, in vivo results obtained with TET1 knockout and TET1/TET2 double-knockout mice (under conditions where TET3 is unlikely to compensate significantly for the knockout) suggest that ESCs stemness can be maintained without these methyldioxygenases (Dawlaty et al, 2011, 2013). In fact, although a fraction of the double KO mice displayed midgestation abnormalities and perinatal lethality, viable and fertile mice were also obtained. Those however display a reduced size and weight as compared to their wt counterparts and the females also show smaller ovaries and reduced fertility. This indicates an overtly normal development with variable phenotypes probably due to variable penetrance of epigenetic defects. This double knockout is in sharp contrast with the TET3 knockout embryos from Xu's group that show apparent lethality either around embryonic day 11.5 or after birth, highlighting the crucial role of TET3 in reprogramming the paternal pronucleus after fertilization (Gu et al, 2011). While the jury is still out on whether TETs play a role in maintaining pluripotency in ESCs, another phenomenon involving TETs is coming to light: reprogramming of developmentally committed cells such as MEFs to produce induced pluripotent stem cells (iPSCs). iPSCs, phenotypically comparable to ESCs, can be produced in vitro upon overexpression of the Yamanaka “OSKM” factors (OCT4, SOX2, KLF4 and C-MYC). During this process profound epigenetic reprogramming takes place, with loss of methylation on the promoters of several pluripotency genes (Takahashi & Yamanaka, 2006). Several studies have stressed the importance of TETs in iPSC reprogramming. In the first of these studies, TET2 and PARP1 were identified as key factors in this process: both are upregulated in MEFs upon OSKM treatment, and both bind to ESRRB and NANOG to shape the chromatin landscape essential for correct gene expression and iPSC reprogramming (Doege et al, 2012). As mentioned earlier, another report revealed that NANOG can interact with TET1 and TET2 and facilitate reprogramming of neural stem cells in a TET-activity-dependent manner (Costa et al, 2013). Remarkably, Gao et al (2013) were able to induce iPSC formation in experiments where they replaced OSKM with “TSKM” (TET1, SOX2, KLF4, and C-MYC), probably because OCT4 is a direct binding target of TET1. These results highlight the importance of TET1 and TET2 in the demethylation and activation of key pluripotency genes such as ESRRB, NANOG, and OCT4 during reprogramming of differentiated cells, and thus suggest an important role for methyldioxygenases in regenerative medicine studies.
Recent reports pinpoint a participation of TETs in terminal differentiation as well. In 2013, Hahn et al found the 5hmC pattern to be very dynamic during NPC differentiation to neurons. In gene bodies they observed a global increase in 5hmC without any subsequent change in DNA methylation. They further showed that TET2 and TET3 are required for correct NPC differentiation and that their loss leads to abnormal accumulation of cell clusters along the radial axis in the intermediate and ventricular zones (Hahn et al, 2013). A similar increase in hydroxymethylcytosine is seen during olfactory neuron differentiation, and TET3 overexpression disrupts both the olfactory receptor expression pattern and the targeting of axons to the olfactory bulb (Colquitt et al, 2013). These two studies strongly suggest that the hydroxymethylation landscape is important for the correct formation of brain tissues, as already reported by Shi's group, which found TET3 to be required for normal development of the eye and brain in Xenopus laevis (Xu et al, 2012). Three recent papers have shed further light on the involvement of TET enzymes in brain development (Kaas et al, 2013; RudenKo et al, 2013; Zhang et al, 2013b). Zhang et al found that TET1 is needed for NPC proliferation and that in vivo depletion of TET1 affects neuron production in the hippocampus. Further characterization of the mice in Morris water maze experiments revealed that their short-term memory was reduced (Zhang et al, 2013b). Rudenko et al (2013) reached different conclusions: in fear conditioning and Morris water maze tests, they found a normal neuronal density in the hippocampus of TET1-knockout mice, with no short-term memory impairment but with a significant decrease in memory extinction (which affects long-term memory), possibly due to alteration of the long-term depression pathway. As these last results have been partially reproduced in mice overexpressing wild-type or mutant TET1 catalytic domains in the hippocampus, TET1 appears to act as an “inhibitor” of long-term memory, independently of its catalytic function (Kaas et al, 2013). While a consensus is hard to reach at this stage, it would seem that TETs affect both short- and long-term memory as well as NPC proliferation and differentiation. The discrepancies between Zhang's and Rudenko's results may be due to the use of different knockouts: Zhang et al targeted exons 11–13 of the TET1 protein, whereas Rudenko et al used knockout mice with a deleted exon 4 (previously described by Jaenisch and colleagues Rudenko et al, 2013; Zhang et al, 2013b). Further studies, including systematic conditional hippocampal knockout of TET1, TET2, and TET3 (and also double and triple knockouts), should help to assess these divergences and to distinguish the exact roles of the three TETs in neurogenesis.
Also beyond the neuronal context, reports show that the hydroxymethylcytosine pattern is highly dynamic during terminal cell differentiation, as described for preadipocytes, germ cells, and differentiating monocytes (Fujiki et al, 2013; Gan et al, 2013; Klug et al, 2013; de la Rica et al, 2013). During adipogenesis, the genome displays a global increase in 5hmC and local hydroxymethylation on genes that are later expressed in mature adipocytes (Fujiki et al, 2013). A similar initial global increase in 5hmC, followed by a global decrease, and the presence of differentially hydroxymethylated regions (dhMRs) on certain coding and non-coding genes are also observed during spermatogenesis (Gan et al, 2013). Finally, Klug et al (2013) and de la Rica et al (2013) have provided evidence that TETs, especially TET2, are essential to the proper differentiation of monocytes to osteoclasts: during this process, DNA demethylation preceded by 5hmC deposition is observed.
All in all, it thus seems that while 5hmC may decrease when mESCs lose their pluripotency, a global increase in this epigenetic mark can occur during terminal differentiation, sometimes followed by a subsequent decrease. This highlights the dynamicity of the hydroxymethylcytosine pattern and suggests a crucial role for the TET enzymes in cell differentiation and organ (e.g. brain) formation as well as a key role in cell reprogramming. With proteins so essential and so versatile, it comes as no surprise that their loss, or an alteration of the 5hmC pattern, can lead to diseases such as cancer or neurological disorders. Roles of altered TET functioning and hydroxymethylation in disease are discussed below.
TETs/modified cytosines in cancer and non-cancer diseases
TETs are on the one hand recognized as tumour suppressor and a hallmark of many cancers seems to be a significant decrease in 5hmC. This may be partly due to the well-known global hypomethylation that takes place during cell transformation, but in breast cancers, melanomas, and leukaemias without MLL rearrangements, reports have additionally revealed either a mutation in the TET2 gene (although the level of one TET does not always correlate with the global level of hydroxymethylcytosine) or substantial downregulation of all three TETs, for example by micro-RNAs (Tefferi et al, 2009; Haffner et al, 2011; Nestor et al, 2011; Hsu et al, 2012; Lian et al, 2012; Yang et al, 2012; Huang et al, 2013; Song et al, 2013a,b). In 2012, Hsu et al (2012) reported TET1 to be downregulated in prostate and breast cancer tissues, and found its loss to facilitate cell invasion in xenograft mouse models, at least via deregulation of “inhibitor of metalloproteinase” proteins (TIMPs). In human melanomas, which show a marked genome-wide decrease in hydroxymethylcytosine and more subtle changes in methylation patterns, the most decreased TET is TET2. When TET2 is overexpressed in human melanoma cells xenografted onto immunodeficient NSG mice, tumour growth is suppressed. This suggests a crucial involvement of TET2 and 5hmC in melanoma development (Lian et al, 2012). Furthermore, TET2 is often mutated in myeloid malignancies,. In 4–13% of myeloproliferative neoplasms and 20–25% of myelodysplastic syndromes, somatic deletions and TET2-inactivating mutations are found, but do not correlate clearly with prognosis (Tefferi et al, 2009; Bejar et al, 2011). TET2 mutations are also found in 7–23% of AMLs, where they correlate with poor prognosis. A major role of TET2 in leukaemia has been confirmed in TET2-knockout mice, where loss of the enzyme appears to increase the haematopoietic stem cell compartment and to skew cell differentiation towards the myeloid compartment, causing symptoms resembling those associated with TET2 mutations (Li et al, 2011; Moran-Crusio et al, 2011; Quivoron et al, 2011).
In fascinating contrast to the above, there are contexts where TETs appear to have an oncogenic action. They owe the designation “Ten-Eleven translocation enzyme” to a rare translocation of the histone methyl transferase gene MLL and of the TET1 coding sequence in acute myeloid leukaemia (AML), yielding a 5′ MLL-TET1 3′ chimera (Lorsbach et al, 2003). The role of this translocation in leukaemia is unclear, and it seems that oncogenic transformation driven by the rearranged MLL is observed with different translocation partners (de Boer et al, 2013). Yet Huang et al revealed in 2013 that TET1 plays an essential oncogenic role in MLL-rearranged leukaemia. It is aberrantly overexpressed in MLL-rearranged AML, while the TET2 and TET3 expression levels remain unchanged. ChIP-qPCR and expression experiments have further shown that the TET1 promoter is a direct target of MLL-fusion proteins and that the increase in TET1 leads to a global genomic increase of hydroxymethylcytosine. Furthermore, MLL fusions and TET1 both co-regulate genes such as HOXA9 or MEIS1 that inhibit apoptosis and enhance cell proliferation, causing transformation and leukaemogenesis (Huang et al, 2013).
In summary, the hydroxymethylcytosine level is often deregulated in cancer, but not always in the same manner: globally there may be an increase, as seen in MLL-rearranged leukaemia, or a marked decrease, as observed in many other cancer types. Recently, some elegant techniques have been created to precisely map 5hmC at single-nucleotide resolution (Booth et al, 2012; Yu et al, 2012). This could make this epigenetic modification an interesting marker for use in diagnosing cancers, especially blood cancers, for which cell samples can easily be obtained.
Growing attention is also focusing on the involvement of TET dysregulation in non-cancer diseases. There is increasing evidence that epigenetic dysfunction and resulting changes in gene expression may contribute at least partly to the aetiology of various pathologies, especially neurological disorders (see Table1) (Jakovcevski & Akbarian, 2012). As mentioned above, the mammalian brain is very rich in hydroxymethylcytosine. In Rett syndrome, mutations in MeCP2 impair the binding of this protein to 5hmC, suggesting that the altered deposition of this mark by TET methyldioxygenases may also play a role in neurological diseases (Kriaucionis & Heintz, 2009; Mellen et al, 2012). Accordingly, several reports have revealed altered TET expression and an abnormal 5hmC pattern in the brains of patients suffering from psychosis, Huntington's disease, Friedreich's ataxia, and fragile X syndrome (Campuzano et al, 1996; Tassone et al, 2000; Saveliev et al, 2003; Sadri-Vakili & Cha, 2006; Dong et al, 2012; Guidotti et al, 2012; Al-Mahdawi et al, 2013; Auta et al, 2013; Chouliaras et al, 2013; Villar-Menendez et al, 2013; Wang et al, 2013; Yao et al, 2013; Coppieters et al, 2014). Psychosis is a broad category encompassing symptoms of schizophrenia and bipolar disorder. Psychotic patients have been found to display downregulated expression of several vulnerability genes, including GAD67, reelin, NR2A, and GAT1, in parallel with hypermethylation of the corresponding promoter regions (Dong et al, 2012). Recently, two groups additionally reported hyper-hydroxymethylation of the GAD67 promoter in the inferior parietal lobe, both in a psychotic cohort and in mice born with psychosis-like disorders (Dong et al, 2012; Matrisciano et al, 2012). To evaluate the possible role of 5hmC in GAD67 repression, it would be interesting to see if variation of the 5hmC level in GAD67 might correlate with TET binding to one or more corepressors such as SIN3A, and NurD. In addition, Dong et al (2012) revealed local variations in hydroxymethylcytosine content, a global increase in both TET1 and 5hmC in schizophrenic and bipolar patients, and persons suffering from depression displayed a similar, albeit insignificant trend. A TET1 increase has likewise been observed in psychotic patients with a history of alcohol abuse and interestingly, in lymphocytes of schizophrenic patients. The latter finding suggests that it might be possible to identify peripheral methylation/hydroxymethylation biomarkers for use in diagnosis/prognosis when a biopsy is not an option (Guidotti et al, 2012; Auta et al, 2013).
Table 1.
This table summarizes the involvement of TETs and methylcytosine oxidation in neuronal diseases
| TETs/hmC connection in neuronal diseases | ||
|---|---|---|
| Pathologies | Observations | References |
| Rett syndrome | Decrease of MeCP2 binding on 5hmC | Kriaucionis & Heintz (2009) Mellen et al (2012) |
| Psychosis | GAD67 promoter hyper-hydroxymethylation | Dong et al (2012) Matrisciano et al (2012) |
| Global increase of 5hmC Increase of TET1 expression |
Dong et al (2012) Guidotti et al (2012) Auta et al (2013) |
|
| Huntington's disease | Decrease of 5hmC on ADORA2A promoter | Villar-Menendez et al (2013) |
| Global decrease of 5hmC Decrease of TET1 expression |
Wang et al (2013) | |
| Friedreich's ataxia | Global increase of 5hmC | Al-Mahdawi et al (2013) |
| Fragile X syndrome | Global decrease of 5hmC on gene bodies and promoters Subtle increase of 5hmC on enhancers and repetitive elements |
Yao et al (2013) |
| Alzheimer's disease | Global increase of 5hmC in hippocampus Global decrease of 5hmC in temporal tissues |
Coppieters et al (2014) Chouliaras et al (2013) |
Huntington's disease (HD) is a genetic disorder characterized by expansion of glutamine residues in the protein huntingtin, due to accumulation of the CAG triplet in the first exon of the corresponding gene. The symptoms include difficulties in motor coordination and also psychiatric disturbances, cognitive disorders, and weight loss (Sadri-Vakili & Cha, 2006). In 2013, Villar-Menendez et al (2013) showed that the promoter of the ADORA2A gene, encoding the G-protein coupled receptor A2AR known to be downregulated in HD and involved in the disease, displays a decreased 5hmC level in a mouse model of HD and in the putamens of HD patients. In contrast to the global increase in hydroxymethylcytosine found in psychosis, dot-blot experiments and analysis of 5hmC patterns by deep sequencing in an HD mouse model revealed a rather global decrease in 5hmC, accompanied by a significant decrease in TET1 expression. Analysis of the differentially hydroxymethylated regions further uncovered an alteration of canonical pathways involved in neuronal development and differentiation pathways. Hence, the loss of hydroxymethylcytosine may impair neuronal function in HD brains and appears as a novel epigenetic marker of the disease (Wang et al, 2013).
Friedreich's ataxia (FRDA) is another neurodegenerative disease due to expansion of a triplet (GAA). This expansion, here in the first intron of the FXN gene, leads to heterochromatin formation and gene silencing. Upstream from the repeats lay CpGs that appear hypermethylated in patients suffering from the disease. In the studies concerned, however, the techniques used to map 5mC could not distinguish this mark from 5hmC (Campuzano et al, 1996; Saveliev et al, 2003; Al-Mahdawi et al, 2013). In 2013, Al-Mahdawi et al (2013) addressed this issue, showing that nearly all the methylcytosine is in fact hydroxymethylcytosine in both FRDA and normal human cerebellar tissues, and that 5hmC is more abundant in the former than in the latter. They further hypothesized that the observed 5hmC increase might enhance production of FAST-1 antisense RNA, which could in turn mediate heterochromatin formation and FXN downregulation, causing the typical symptoms of the disease. Lastly, the fragile X genetic syndrome, triggered by FMR1 gene repression, is a widespread inherited cause of autism and mental retardation in boys (Tassone et al, 2000; Yao et al, 2013). While the disorder is known to be associated with hypermethylation of the FMR1 promoter and subsequent transcriptional gene silencing, possible global fragile-X-associated changes in DNA methylation/hydroxymethylation were not addressed until recently, when Yao et al reported a global decrease in 5hmC in gene bodies and promoters and a more subtle increase in cerebellum-specific enhancers and some repetitive elements. The same report also presents dhMR analyses highlighting changes affecting functional pathways in neuronal development. As these results were obtained from a knockin mouse model, it will be important to assess the global level of hydroxymethylcytosine in fragile X syndrome patients (Yao et al, 2013).
In each of the above-mentioned neuronal diseases, the global level of hydroxymethylation thus seems to be altered, either upward or downward according to the disease. Also, some disorders may involve several tissues in the brain, and both the global and specific 5hmC patterns may depend on the analysed tissue. This is exemplified by two recent studies showing that in Alzheimer's disease, the most common form of dementia in humans, hippocampal regions display a significant increase in 5hmC, while the middle frontal and temporal gyri show a significant decrease (Chouliaras et al, 2013; Coppieters et al, 2014).
In summary, the TET methyldioxygenase machinery seems altered in various diseases, notably cancers and neudegenerative disorders but also probably other important diseases such as oxidative-stress related disorders (see modes of direct regulation of TET enzymes). This strongly suggests that 5hmC patterns are important in many pathologies. As DNA can be recovered easily from tissues and is stable over time, 5hmC profiles might be used to find clinically relevant epigenetic biomarkers. Yet questions remain to be answered. It will notably be important to identify clearly the pathways affecting 5hmC profile changes and those affected by them. We also need to distinguish whether variations of the global level of this mark are causes or consequences of the diseases concerned. These important questions will surely keep a lot of laboratories excited and busy for the next few years.
Concluding remarks
Since their discovery in 2009, the TET enzymes have sparked tremendous interest in the epigenetic community. Key studies have highlighted the importance of active or passive TET-primed demethylation in both cell reprogramming and cell differentiation, and the functional diversity of TET partner proteins suggests a more multifaceted action than anticipated. TETs associate with diverse chromatin-related machineries involved in transcriptional activation or repression, and the marks they create may act as epigenetic signals per se, i. e. as more than just intermediates in DNA demethylation. TETs also seem to play roles that do not depend on their catalytic activity.
Challenges abound in the world of TETs: untangling the intricate web of TET interactions with the chromatin environment, answering burning questions such as: is the frequently observed positive correlation of transcription with hydroxymethylcytosine in gene bodies the cause or the consequence of gene expression? What proteins act as true 5hmC, 5fC, and 5caC “readers” and what are their precise biological roles?
It comes as no surprise that perturbed TET functioning leads to disease, given the importance of TETs in so many key cellular processes. In many cancers a global loss of hydroxymethylcytosine is observed, but it remains uncertain whether altered hydroxymethylation patterns, like altered DNA methylation patterns, can be viewed as true hallmarks of certain cancers. As various cancers display altered TET-gene expression or TET mutations, and as TETs play a role in generating fC and caC, the possible link between cancers and changes in fC and/or caC patterns is certainly worth investigating. This applies also to neuronal disorders, as several show a perturbed 5hmC distribution. There remain crucial gaps in our understanding of how TET dysfunctioning/dysregulation contributes to disease aetiology, and filling them is likely to be rewarding. Once we do understand the link between TET functions and particular diseases, we can hope to develop therapeutic tools. Just as known DNMT-modulating drugs (e.g. selective non-nucleoside inhibitors) are commonly used to treat myelodysplastic syndromes (Sekeres & Cutler, 2013), TET modulators (inhibitors or activators) may prove clinically useful. Small molecules and TET-microRNAs antagonists could be promising candidates. In fact, the therapeutic use of microRNAs antagonists might be just around the corner: a phase II study is in progress on Miravirsen (Santaris Pharma), a miR-122 antisense Locked Nucleic Acid that significantly reduces the level of viral RNA in hepatitis-C-virus-infected patients (Janssen et al, 2013). Alternatively, TET enzymes might be targeted to specific genomic loci to restore the normal epigenetic landscape. Joung's group has already exploited this idea, successfully using TALE-TET fusion proteins to target the TET1 catalytic domain to known methylated genes (Maeder et al, 2013).
No wonder the world of TET is in ferment! Although still under construction, a picture is emerging in which TETs both contribute to building “DNA modification block” patterns through creation and erasure of epigenetic marks and participate with diverse partners in a more intricate scheme. As results accumulate with their share of surprises, the simplistic view presented in Fig 3 is bound to become much more elaborate. The challenge ahead will be to fully relate the many levels in which TETs participate to the patterns they must properly create.
Figure 3. The evolving “TETris playground”.
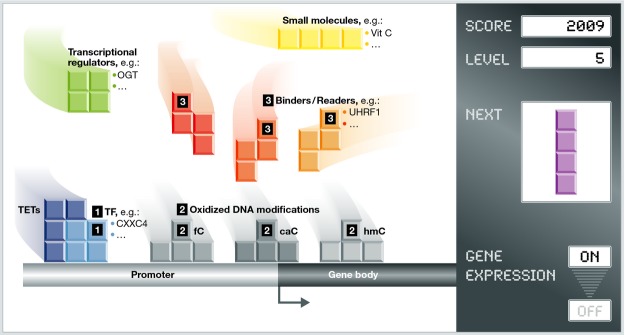
A picture is emerging in which TETs build up specific “DNA modification blocks” (hmC, fC, caC) through their coordinated regulation at multiple levels. The following sequence of events can therefore be suggested: (i) Transcription factors will result in (ii) 5hmC, 5fC, and 5caC formation with 5hmC being more abundant in gene bodies. (iii) These modifications can be further bound by various “binders” or “readers” that will either participate in DNA demethylation or translate the epigenetic signal to transcriptional activation or repression. Other active players are transcriptional regulators and small molecules such as vitamin C that directly affect TET activity and hence the deposition of 5hmC, 5fC, and 5caC. These DNA modification blocks, TETs, and TET regulators can assemble in a gene- and/or a cell-state-dependent manner creating precise landscapes which will ultimately affect gene expression.
Glossary
- 2-HG
2-Hydroxyglutarate
- BER
Base Excision Repair
- NPC
Neural Progenitor Cell
- OSKM
Oct4 Sox2 Klf4 c-Myc
- PGC
Primordial Germ Cell
- PPRE
PPAR-Response Elements
- TET
Ten Eleven Translocation
- TIMPS
Tissue Inhibitors of Metalloproteinases
Author contributions
BD and FF wrote the manuscript, RD designed the figures.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Al-Mahdawi S, Sandi C, Mouro Pinto R, Pook MA. Friedreich ataxia patient tissues exhibit increased 5-hydroxymethylcytosine modification and decreased CTCF binding at the FXN locus. PLoS ONE. 2013;8:e74956. doi: 10.1371/journal.pone.0074956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auta J, Smith RC, Dong E, Tueting P, Sershen H, Boules S, Lajtha A, Davis J, Guidotti A. DNA-methylation gene network dysregulation in peripheral blood lymphocytes of schizophrenia patients. Schizophr Res. 2013;150:312–318. doi: 10.1016/j.schres.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, Ebert BL. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–2506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke K, Ebata KT, Karimi MM, Zepeda-Martinez JA, Goyal P, Mahapatra S, Tam A, Laird DJ, Hirst M, Rao A, Lorincz MC, Ramalho-Santos M. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500:222–226. doi: 10.1038/nature12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer J, Walf-Vorderwulbecke V, Williams O. In focus: MLL-rearranged leukemia. Leukemia. 2013;27:1224–1228. doi: 10.1038/leu.2013.78. [DOI] [PubMed] [Google Scholar]
- Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, Balasubramanian S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–937. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- Brinkman AB, Simmer F, Ma K, Kaan A, Zhu J, Stunnenberg HG. Whole-genome DNA methylation profiling using MethylCap-seq. Methods. 2010;52:232–236. doi: 10.1016/j.ymeth.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- Cartron PF, Nadaradjane A, Lepape F, Lalier L, Gardie B, Vallette FM. Identification of TET1 partners that control its DNA-demethylating function. Genes Cancer. 2013;4:235–241. doi: 10.1177/1947601913489020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Wang KY, Shen CK. The mammalian de novo DNA methyltransferases DNMT3A and DNMT3B are also DNA 5-hydroxymethylcytosine dehydroxymethylases. J Biol Chem. 2012;287:33116–33121. doi: 10.1074/jbc.C112.406975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561–564. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Guo S, Chen S, Mastriano SJ, Liu C, D'Alessio AC, Hysolli E, Guo Y, Yao H, Megyola CM, Li D, Liu J, Pan W, Roden CA, Zhou XL, Heydari K, Chen J, Park IH, Ding Y, Zhang Y, et al. An extensive network of TET2-targeting microRNAs regulates malignant hematopoiesis. Cell Rep. 2013;5:471–481. doi: 10.1016/j.celrep.2013.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojkier M, Spanheimer R, Peterkofsky B. Specifically decreased collagen biosynthesis in scurvy dissociated from an effect on proline hydroxylation and correlated with body weight loss. In vitro studies in guinea pig calvarial bones. J Clin Invest. 1983;72:826–835. doi: 10.1172/JCI111053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouliaras L, Mastroeni D, Delvaux E, Grover A, Kenis G, Hof PR, Steinbusch HW, Coleman PD, Rutten BP, van den Hove DL. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer's disease patients. Neurobiol Aging. 2013;34:2091–2099. doi: 10.1016/j.neurobiolaging.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquitt BM, Allen WE, Barnea G, Lomvardas S. Alteration of genic 5-hydroxymethylcytosine patterning in olfactory neurons correlates with changes in gene expression and cell identity. Proc Natl Acad Sci USA. 2013;110:14682–14687. doi: 10.1073/pnas.1302759110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppieters N, Dieriks BV, Lill C, Faull RL, Curtis MA, Dragunow M. Global changes in DNA methylation and hydroxymethylation in Alzheimer's disease human brain. Neurobiol Aging. 2014;35:1334–1344. doi: 10.1016/j.neurobiolaging.2013.11.031. [DOI] [PubMed] [Google Scholar]
- Costa Y, Ding J, Theunissen TW, Faiola F, Hore TA, Shliaha PV, Fidalgo M, Saunders A, Lawrence M, Dietmann S, Das S, Levasseur DN, Li Z, Xu M, Reik W, Silva JC, Wang J. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature. 2013;495:370–374. doi: 10.1038/nature11925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter JB, O'Driscoll CM, Bressler JP. Hydroquinone increases 5-hydroxymethylcytosine formation through ten eleven translocation 1 (TET1) 5-methylcytosine dioxygenase. J Biol Chem. 2013;288:28792–28800. doi: 10.1074/jbc.M113.491365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Ganz K, Powell BE, Hu YC, Markoulaki S, Cheng AW, Gao Q, Kim J, Choi SW, Page DC, Jaenisch R. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell. 2011;9:166–175. doi: 10.1016/j.stem.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Breiling A, Le T, Raddatz G, Barrasa MI, Cheng AW, Gao Q, Powell BE, Li Z, Xu M, Faull KF, Lyko F, Jaenisch R. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell. 2013;24:310–323. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delatte B, Fuks F. TET proteins: on the frenetic hunt for new cytosine modifications. Brief Funct Genomics. 2013;12:191–204. doi: 10.1093/bfgp/elt010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis H, Ndlovu MN, Fuks F. Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep. 2011;12:647–656. doi: 10.1038/embor.2011.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E, Shih AH, Levine RL, Bernard O, Mercher T, Solary E, Urh M, Daniels DL, Fuks F. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013;32:645–655. doi: 10.1038/emboj.2012.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doege CA, Inoue K, Yamashita T, Rhee DB, Travis S, Fujita R, Guarnieri P, Bhagat G, Vanti WB, Shih A, Levine RL, Nik S, Chen EI, Abeliovich A. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature. 2012;488:652–655. doi: 10.1038/nature11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong E, Gavin DP, Chen Y, Davis J. Upregulation of TET1 and downregulation of APOBEC3A and APOBEC3C in the parietal cortex of psychotic patients. Transl Psychiatry. 2012;2:e159. doi: 10.1038/tp.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauer C, Hoffmann T, Bultmann S, Casa V, Cardoso MC, Antes I, Leonhardt H. Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS ONE. 2011;6:e21306. doi: 10.1371/journal.pone.0021306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenberg JM, Ghosh S, Lackford BL, Yellaboina S, Zheng X, Li R, Cuddapah S, Wade PA, Hu G, Jothi R. Acute depletion of Tet1-dependent 5-hydroxymethylcytosine levels impairs LIF/Stat3 signaling and results in loss of embryonic stem cell identity. Nucleic Acids Res. 2012;40:3364–3377. doi: 10.1093/nar/gkr1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Jin L, Wang X, Luo A, Hu J, Zheng X, Tsark WM, Riggs AD, Ku HT, Huang W. MicroRNA-26a targets ten eleven translocation enzymes and is regulated during pancreatic cell differentiation. Proc Natl Acad Sci USA. 2013;110:17892–17897. doi: 10.1073/pnas.1317397110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S, Imai Y, Kim J, He HH, Igarashi K, Kanno J, Ohtake F, Kitagawa H, Roeder RG, Brown M, Kato S. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature. 2011;480:557–560. doi: 10.1038/nature10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki K, Shinoda A, Kano F, Sato R, Shirahige K, Murata M. PPARgamma-induced PARylation promotes local DNA demethylation by production of 5-hydroxymethylcytosine. Nat Commun. 2013;4:2262. doi: 10.1038/ncomms3262. [DOI] [PubMed] [Google Scholar]
- Gan H, Wen L, Liao S, Lin X, Ma T, Liu J, Song CX, Wang M, He C, Han C, Tang F. Dynamics of 5-hydroxymethylcytosine during mouse spermatogenesis. Nat Commun. 2013;4:1995. doi: 10.1038/ncomms2995. [DOI] [PubMed] [Google Scholar]
- Gao Y, Chen J, Li K, Wu T, Huang B, Liu W, Kou X, Zhang Y, Huang H, Jiang Y, Yao C, Liu X, Lu Z, Xu Z, Kang L, Wang H, Cai T, Gao S. Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell. 2013;12:453–469. doi: 10.1016/j.stem.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Globisch D, Munzel M, Muller M, Michalakis S, Wagner M, Koch S, Bruckl T, Biel M, Carell T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi L, He X, Jin SG, Iqbal K, Shi YG, Deng Z, Szabo PE, Pfeifer GP, Li J, Xu GL. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–610. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- Guidotti A, Dong E, Gavin DP, Veldic M, Zhao W, Bhaumik DK, Pandey SC, Grayson DR. DNA methylation/demethylation network expression in psychotic patients with a history of alcohol abuse. Alcohol Clin Exp Res. 2012;37:417–424. doi: 10.1111/j.1530-0277.2012.01947.x. [DOI] [PubMed] [Google Scholar]
- Guilhamon P, Eskandarpour M, Halai D, Wilson GA, Feber A, Teschendorff AE, Gomez V, Hergovich A, Tirabosco R, Fernanda Amary M, Baumhoer D, Jundt G, Ross MT, Flanagan AM, Beck S. Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nat Commun. 2013;4:2166. doi: 10.1038/ncomms3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillouf C, Gallais I, Moreau-Gachelin F. Spi-1/PU.1 oncoprotein affects splicing decisions in a promoter binding-dependent manner. J Biol Chem. 2006;281:19145–19155. doi: 10.1074/jbc.M512049200. [DOI] [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, Down TA, Surani MA. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2012;339:448–452. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner MC, Chaux A, Meeker AK, Esopi DM, Gerber J, Pellakuru LG, Toubaji A, Argani P, Iacobuzio-Donahue C, Nelson WG, Netto GJ, De Marzo AM, Yegnasubramanian S. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget. 2011;2:627–637. doi: 10.18632/oncotarget.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagman J, Ramirez J, Lukin K. B lymphocyte lineage specification, commitment and epigenetic control of transcription by early B cell factor 1. Curr Top Microbiol Immunol. 2011;356:17–38. doi: 10.1007/82_2011_139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MA, Qiu R, Wu X, Li AX, Zhang H, Wang J, Jui J, Jin SG, Jiang Y, Pfeifer GP, Lu Q. Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell Rep. 2013;3:291–300. doi: 10.1016/j.celrep.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Pais JE, Zhang X, Saleh L, Fu ZQ, Dai N, Correa IR, Zheng Y, Cheng X. Structure of a Naegleria Tet-like dioxygenase in complex with 5-methylcytosine DNA. Nature. 2014;506:391–395. doi: 10.1038/nature12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CH, Peng KL, Kang ML, Chen YR, Yang YC, Tsai CH, Chu CS, Jeng YM, Chen YT, Lin FM, Huang HD, Lu YY, Teng YC, Lin ST, Lin RK, Tang FM, Lee SB, Hsu HM, Yu JC, Hsiao PW, et al. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2012;2:568–579. doi: 10.1016/j.celrep.2012.08.030. [DOI] [PubMed] [Google Scholar]
- Hu P, Shimoji S, Hart GW. Site-specific interplay between O-GlcNAcylation and phosphorylation in cellular regulation. FEBS Lett. 2010;584:2526–2538. doi: 10.1016/j.febslet.2010.04.044. [DOI] [PubMed] [Google Scholar]
- Hu L, Li Z, Cheng J, Rao Q, Gong W, Liu M, Shi YG, Zhu J, Wang P, Xu Y. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell. 2013;155:1545–1555. doi: 10.1016/j.cell.2013.11.020. [DOI] [PubMed] [Google Scholar]
- Huang H, Jiang X, Li Z, Li Y, Song CX, He C, Sun M, Chen P, Gurbuxani S, Wang J, Hong GM, Elkahloun AG, Arnovitz S, Szulwach K, Lin L, Street C, Wunderlich M, Dawlaty M, Neilly MB, Jaenisch R, et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc Natl Acad Sci USA. 2013;110:11994–11999. doi: 10.1073/pnas.1310656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Zhang Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science. 2011;334:194. doi: 10.1126/science.1212483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Shen L, Dai Q, He C, Zhang Y. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 2011;21:1670–1676. doi: 10.1038/cr.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Jin SG, Pfeifer GP, Szabo PE. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc Natl Acad Sci USA. 2011;108:3642–3647. doi: 10.1073/pnas.1014033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito R, Katsura S, Shimada H, Tsuchiya H, Hada M, Okumura T, Sugawara A, Yokoyama A. TET3-OGT interaction increases the stability and the presence of OGT in chromatin. Genes Cells. 2014;19:52–65. doi: 10.1111/gtc.12107. [DOI] [PubMed] [Google Scholar]
- Iurlaro M, Ficz G, Oxley D, Raiber EA, Bachman M, Booth MJ, Andrews S, Balasubramanian S, Reik W. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 2013;14:R119. doi: 10.1186/gb-2013-14-10-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurological disease. Nat Med. 2012;18:1194–1204. doi: 10.1038/nm.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen HL, Kauppinen S, Hodges MR. HCV infection and miravirsen. N Engl J Med. 2013;369:878. doi: 10.1056/NEJMc1307787. [DOI] [PubMed] [Google Scholar]
- Jin SG, Kadam S, Pfeifer GP. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 2010;38:e125. doi: 10.1093/nar/gkq223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas GA, Zhong C, Eason DE, Ross DL, Vachhani RV, Ming GL, King JR, Song H, Sweatt JD. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron. 2013;79:1086–1093. doi: 10.1016/j.neuron.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RD, Cowley SM. The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem Soc Trans. 2013;41:741–749. doi: 10.1042/BST20130010. [DOI] [PubMed] [Google Scholar]
- Klug M, Schmidhofer S, Gebhard C, Andreesen R, Rehli M. 5-Hydroxymethylcytosine is an essential intermediate of active DNA demethylation processes in primary human monocytes. Genome Biol. 2013;14:R46. doi: 10.1186/gb-2013-14-5-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, An J, Bandukwala HS, Chavez L, Aijo T, Pastor WA, Segal MF, Li H, Koh KP, Lahdesmaki H, Hogan PG, Aravind L, Rao A. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature. 2013;497:122–126. doi: 10.1038/nature12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, Lahesmaa R, Orkin SH, Rodig SJ, Daley GQ, Rao A. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, Yang FC, Xu M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118:4509–4518. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K, Xu W, Tan L, Hu Y, Zhan Q, Lee CW, Hu D, Lian BQ, Kleffel S, Yang Y, Neiswender J, Khorasani AJ, Fang R, Lezcano C, Duncan LM, et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150:1135–1146. doi: 10.1016/j.cell.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23) Leukemia. 2003;17:637–641. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, Ho QH, Sander JD, Reyon D, Bernstein BE, Costello JF, Wilkinson MF, Joung JK. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. 2013;31:1137–1142. doi: 10.1038/nbt.2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrisciano F, Tueting P, Dalal I, Kadriu B, Grayson DR, Davis JM, Nicoletti F, Guidotti A. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology. 2012;68:184–194. doi: 10.1016/j.neuropharm.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor EA, Court BL, Young JI, Wang G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem. 2013;288:13669–13674. doi: 10.1074/jbc.C113.464800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, Perna F, Pandey S, Madzo J, Song C, Dai Q, He C, Ibrahim S, Beran M, Zavadil J, Nimer SD, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita S, Horii T, Kimura M, Ochiya T, Tajima S, Hatada I. miR-29 represses the activities of DNA methyltransferases and DNA demethylases. Int J Mol Sci. 2013;14:14647–14658. doi: 10.3390/ijms140714647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Liu YJ, Nakashima H, Umehara H, Inoue K, Matoba S, Tachibana M, Ogura A, Shinkai Y, Nakano T. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature. 2012;486:415–419. doi: 10.1038/nature11093. [DOI] [PubMed] [Google Scholar]
- Nestor CE, Ottaviano R, Reddington J, Sproul D, Reinhardt D, Dunican D, Katz E, Dixon JM, Harrison DJ, Meehan RR. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2011;22:467–477. doi: 10.1101/gr.126417.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okashita N, Kumaki Y, Ebi K, Nishi M, Okamoto Y, Nakayama M, Hashimoto S, Nakamura T, Sugasawa K, Kojima N, Takada T, Okano M, Seki Y. PRDM14 promotes active DNA demethylation through the Ten-eleven translocation (TET)-mediated base excision repair pathway in embryonic stem cells. Development. 2013;141:269–280. doi: 10.1242/dev.099622. [DOI] [PubMed] [Google Scholar]
- Okuyama R, Marshall S. UDP-N-acetylglucosaminyl transferase (OGT) in brain tissue: temperature sensitivity and subcellular distribution of cytosolic and nuclear enzyme. J Neurochem. 2003;86:1271–1280. doi: 10.1046/j.1471-4159.2003.01939.x. [DOI] [PubMed] [Google Scholar]
- Otani J, Arita K, Kato T, Kinoshita M, Kimura H, Suetake I, Tajima S, Ariyoshi M, Shirakawa M. Structural basis of the versatile DNA recognition ability of the methyl-CpG binding domain of methyl-CpG binding domain protein 4. J Biol Chem. 2013;288:6351–6362. doi: 10.1074/jbc.M112.431098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14:341–356. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterkofsky B. Ascorbate requirement for hydroxylation and secretion of procollagen: relationship to inhibition of collagen synthesis in scurvy. Am J Clin Nutr. 1991;54(Suppl. 6):1135S–1140S. doi: 10.1093/ajcn/54.6.1135s. [DOI] [PubMed] [Google Scholar]
- Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, Godley L, Opolon P, Tilly H, Solary E, Duffourd Y, Dessen P, Merle-Beral H, Nguyen-Khac F, Fontenay M, Vainchenker W, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- de la Rica L, Rodriguez-Ubreva J, Garcia M, Islam AB, Urquiza JM, Hernando H, Christensen J, Helin K, Gomez-Vaquero C, Ballestar E. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 2013;14:R99. doi: 10.1186/gb-2013-14-9-r99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudenko A, Dawlaty MM, Seo J, Cheng AW, Meng J, Le T, Faull KF, Jaenisch R, Tsai LH. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron. 2013;79:1109–1122. doi: 10.1016/j.neuron.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppel GW. Zur chemie der tuberkelbacillen. Z Physiol Chem. 1898;26:218–232. [Google Scholar]
- Sadri-Vakili G, Cha JH. Mechanisms of disease: histone modifications in Huntington's disease. Nat Clin Pract Neurol. 2006;2:330–338. doi: 10.1038/ncpneuro0199. [DOI] [PubMed] [Google Scholar]
- Saveliev A, Everett C, Sharpe T, Webster Z, Festenstein R. DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature. 2003;422:909–913. doi: 10.1038/nature01596. [DOI] [PubMed] [Google Scholar]
- Schiesser S, Hackner B, Pfaffeneder T, Muller M, Hagemeier C, Truss M, Carell T. Mechanism and stem-cell activity of 5-carboxycytosine decarboxylation determined by isotope tracing. Angew Chem Int Ed Engl. 2012;51:6516–6520. doi: 10.1002/anie.201202583. [DOI] [PubMed] [Google Scholar]
- Sekeres MA, Cutler C. How we treat higher-risk myelodysplastic syndromes. Blood. 2013;123:829–836. doi: 10.1182/blood-2013-08-496935. [DOI] [PubMed] [Google Scholar]
- Shereen AT, Rodney PD. The transcription factor PU.1 is a critical regulator of cellular communication in the immune system. Arch Immunol Ther Exp. 2011;59:431–440. doi: 10.1007/s00005-011-0147-9. [DOI] [PubMed] [Google Scholar]
- Shi FT, Kim H, Lu W, He Q, Liu D, Goodell MA, Wan M, Songyang Z. Ten-eleven translocation 1 (Tet1) is regulated by O-linked N-acetylglucosamine transferase (Ogt) for target gene repression in mouse embryonic stem cells. J Biol Chem. 2013;288:20776–20784. doi: 10.1074/jbc.M113.460386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song SJ, Ito K, Ala U, Kats L, Webster K, Sun SM, Jongen-Lavrencic M, Manova-Todorova K, Teruya-Feldstein J, Avigan DE, Delwel R, Pandolfi PP. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell. 2013a;13:87–101. doi: 10.1016/j.stem.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song SJ, Poliseno L, Song MS, Ala U, Webster K, Ng C, Beringer G, Brikbak NJ, Yuan X, Cantley LC, Richardson AL, Pandolfi PP. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell. 2013b;154:311–324. doi: 10.1016/j.cell.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, Munzel M, Wagner M, Muller M, Khan F, Eberl HC, Mensinga A, Brinkman AB, Lephikov K, Muller U, Walter J, Boelens R, van Ingen H, Leonhardt H, Carell T, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Szabo PE, Pfeifer GP. H3K9me2 attracts PGC7 in the zygote to prevent Tet3-mediated oxidation of 5-methylcytosine. J Mol Cell Biol. 2012;4:427–429. doi: 10.1093/jmcb/mjs038. [DOI] [PubMed] [Google Scholar]
- Szwagierczak A, Bultmann S, Schmidt CS, Spada F, Leonhardt H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010;38:e181. doi: 10.1093/nar/gkq684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Chamberlain WD, Hagerman PJ. Transcription of the FMR1 gene in individuals with fragile X syndrome. Am J Med Genet. 2000;97:195–203. doi: 10.1002/1096-8628(200023)97:3<195::AID-AJMG1037>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM, Hanson CA, Pardanani A, Gilliland DG, Levine RL. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009;23:1343–1345. doi: 10.1038/leu.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella P, Scelfo A, Jammula S, Chiacchiera F, Williams K, Cuomo A, Roberto A, Christensen J, Bonaldi T, Helin K, Pasini D. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol Cell. 2013;49:645–656. doi: 10.1016/j.molcel.2012.12.019. [DOI] [PubMed] [Google Scholar]
- Villar-Menendez I, Blanch M, Tyebji S, Pereira-Veiga T, Albasanz JL, Martin M, Ferrer I, Perez-Navarro E, Barrachina M. Increased 5-methylcytosine and decreased 5-hydroxymethylcytosine levels are associated with reduced striatal A2AR levels in Huntington's disease. Neuromolecular Med. 2013;15:295–309. doi: 10.1007/s12017-013-8219-0. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gucek M, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc Natl Acad Sci USA. 2008;105:13793–13798. doi: 10.1073/pnas.0806216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Yang Y, Lin X, Wang JQ, Wu YS, Xie W, Wang D, Zhu S, Liao YQ, Sun Q, Yang YG, Luo HR, Guo C, Han C, Tang TS. Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington's disease. Hum Mol Genet. 2013;22:3641–3653. doi: 10.1093/hmg/ddt214. [DOI] [PubMed] [Google Scholar]
- Williams K, Christensen J, Helin K. DNA methylation: TET proteins-guardians of CpG islands? EMBO Rep. 2011a;13:28–35. doi: 10.1038/embor.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J, Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011b;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, Arand J, Nakano T, Reik W, Walter J. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun. 2011;2:241. doi: 10.1038/ncomms1240. [DOI] [PubMed] [Google Scholar]
- Wu H, D'Alessio AC, Ito S, Xia K, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–393. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156:45–68. doi: 10.1016/j.cell.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011a;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wu F, Tan L, Kong L, Xiong L, Deng J, Barbera AJ, Zheng L, Zhang H, Huang S, Min J, Nicholson T, Chen T, Xu G, Shi Y, Zhang K, Shi YG. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell. 2011b;42:451–464. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Xu C, Kato A, Tempel W, Abreu JG, Bian C, Hu Y, Hu D, Zhao B, Cerovina T, Diao J, Wu F, He HH, Cui Q, Clark E, Ma C, Barbara A, Veenstra GJ, Xu G, Kaiser UB, et al. Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell. 2012;151:1200–1213. doi: 10.1016/j.cell.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Shen L, Liu Y, Sendler D, Zhang Y. Role of Tet1 in erasure of genomic imprinting. Nature. 2013;504:460–464. doi: 10.1038/nature12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu J, Xu ZD, Zhu HG, Ling ZQ, Ye D, Guan KL, Xiong Y. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene. 2012;32:663–669. doi: 10.1038/onc.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao B, Lin L, Street RC, Zalewski ZA, Galloway JN, Wu H, Nelson DL, Jin P. Genome-wide alteration of 5-hydroxymethylcytosine in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum Mol Genet. 2013;23:1095–1107. doi: 10.1093/hmg/ddt504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim O, Li R, Hung JH, Chen PB, Dong X, Ee LS, Weng Z, Rando OJ, Fazzio TG. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. 2011;147:1498–1510. doi: 10.1016/j.cell.2011.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin R, Mao SQ, Zhao B, Chong Z, Yang Y, Zhao C, Zhang D, Huang H, Gao J, Li Z, Jiao Y, Li C, Liu S, Wu D, Gu W, Yang YG, Xu GL, Wang H. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc. 2013;135:10396–10403. doi: 10.1021/ja4028346. [DOI] [PubMed] [Google Scholar]
- You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, Min JH, Jin P, Ren B, He C. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Lu X, Lu J, Liang H, Dai Q, Xu GL, Luo C, Jiang H, He C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol. 2012;8:328–330. doi: 10.1038/nchembio.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Huang B, Xu X, Sessa WC. Ten-eleven translocation (Tet) and thymine DNA glycosylase (TDG), components of the demethylation pathway, are direct targets of miRNA-29a. Biochem Biophys Res Commun. 2013a;437:368–373. doi: 10.1016/j.bbrc.2013.06.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RR, Cui QY, Murai K, Lim YC, Smith ZD, Jin S, Ye P, Rosa L, Lee YK, Wu HP, Liu W, Xu ZM, Yang L, Ding YQ, Tang F, Meissner A, Ding C, Shi Y, Xu GL. Tet1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell. 2013b;13:237–245. doi: 10.1016/j.stem.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]