

Abstract
Cilia and flagella are formed and maintained by intraflagellar transport (IFT) and play important roles in sensing and moving across species. At the distal tip of the cilia/flagella, IFT complexes turn around to switch from anterograde to retrograde transport; however, the underlying regulatory mechanism is unclear. Here, we identified ICK localization at the tip of cilia as a regulator of ciliary transport. In ICK-deficient mice, we found ciliary defects in neuronal progenitor cells with Hedgehog signal defects. ICK-deficient cells formed cilia with mislocalized Hedgehog signaling components. Loss of ICK caused the accumulation of IFT-A, IFT-B, and BBSome components at the ciliary tips. In contrast, overexpression of ICK induced the strong accumulation of IFT-B, but not IFT-A or BBSome components at ciliary tips. In addition, ICK directly phosphorylated Kif3a, while inhibition of this Kif3a phosphorylation affected ciliary formation. Our results suggest that ICK is a Kif3a kinase and essential for proper ciliogenesis in development by regulating ciliary transport at the tip of cilia.
Keywords: ciliary transport, ciliogenesis, kinase, kinesin
Introduction
Cilia are microtubule-based organelles that extend from the surface of various types of cells in vertebrates (Fliegauf et al, 2007; Gerdes et al, 2009; Nigg & Raff, 2009). Upon cell differentiation, some types of cells change their ciliary function by altering their protein components and morphology. For example, neuronal progenitor cells possess short cilia that contain components of Sonic Hedgehog (Shh) signaling such as Smo and Gli. Cilia are known to be important for Shh signal transduction (Huangfu & Anderson, 2005). In the absence of Shh pathway stimulation, Smo rarely localizes in the cilia. In this condition, low levels of Gli transcription factors localize to the tip of cilia. However, Shh pathway stimulation induces the accumulation of Smo and Gli in the cilia and then triggers the transcription of target genes (Corbit et al, 2005; Haycraft et al, 2005; Kim et al, 2009). In the developing central nervous system (CNS), Shh signaling in the cilia plays an essential role for neuronal progenitor cell proliferation and differentiation (Chizhikov et al, 2007; Breunig et al, 2008; Han et al, 2008; Spassky et al, 2008; Willaredt et al, 2008; Besse et al, 2011). In contrast, mature neurons in the adult brain develop long primary cilia, on which several types of G-protein-coupled receptors (GPCRs) for neurotransmitters such as melanin-concentrating hormone and somatostatin are localized (Bishop et al, 2007; Berbari et al, 2008; Arellano et al, 2012). Although cilia are morphologically and functionally diverse among different cells as observed in CNS development, the regulatory mechanisms underlying such cell type-specific ciliogenesis are almost unknown.
Cilia are formed and maintained by the evolutionarily conserved process called intraflagellar transport (IFT), the bidirectional protein trafficking of IFT particles along ciliary microtubules. The IFT particles are composed of two subcomplexes called IFT-A and IFT-B. Anterograde transport of these particles is driven by kinesin-2, whereas retrograde transport is mediated by cytoplasmic dynein-2 (Rosenbaum & Witman, 2002). Anterograde transport is switched to retrograde transport at the ciliary tip, where axonemal tubulins are turned over (Ishikawa & Marshall, 2011). In Chlamydomonas and C. elegans, IFT-A and IFT-B dissociate after reaching the ciliary/flagellar tip and then IFT-B reassociates with IFT-A before retrograde transport (Pedersen et al, 2006; Wei et al, 2012). Although the coordinated traffic switching at ciliary tips has been suggested as required for normal ciliary formation and maintenance (Pedersen et al, 2006; Wei et al, 2012), the regulatory mechanisms underlying transport switching at ciliary tips are poorly understood.
Chlamydomonas LF4, Leishmania LmxMPK9, C. elegans Dyf-5, and mouse Mak belong to the evolutionarily conserved MAP kinase subfamily, which negatively regulates ciliary length (Asleson & Lefebvre, 1998; Berman et al, 2003; Bengs et al, 2005; Burghoorn et al, 2007; Omori et al, 2010). In contrast to cell type-specific expression of Mak, another murine orthologue of Chlamydomonas LF4, intestinal cell kinase (ICK), shows ubiquitous expression including in the developing CNS (Togawa et al, 2000). It was reported that ICK is a substrate of cell cycle-related kinase (Ccrk) (Fu et al, 2006). A mutant of Dyf-18, the C. elegans orthologue of Ccrk, occasionally forms long-curved cilia (Phirke et al, 2011). Broad-minded (Bromi), which interacts with Ccrk, is required for the formation of proper structure in cilia (Ko et al, 2010). There was a single case report of a novel neonatal lethal recessive disorder with multiple anomalies involving the endocrine, cerebral, and skeletal systems (endocrine-cerebro-osteodysplasia, ECO) with a homozygous missense mutation in human ICK, suggesting a key role for ICK in the development of multiple organ system (Lahiry et al, 2009). However, the exact biological functions of ICK have not yet been elucidated.
In the current study, we generated and analyzed retina- or brain-specific ICK-deficient mutant mice as well as conventional ICK-deficient mutant mice. These mice showed phenotypes defective in Hedgehog (Hh) signal transduction in multiple organs. Unexpectedly, we found that ICK, which localizes at the tips of cilia, is required for ciliogenesis in neural progenitor cells, but not in mature neurons. Disruption of ICK leads to the accumulation of both IFT-A and IFT-B particles. ICK-deficient MEFs have shortened and stumpy cilia with an accumulation of Shh signaling molecules. Furthermore, overexpression of ICK promoted the localization of IFT-B, but not IFT-A, components at ciliary tips. We also found that ICK directly phosphorylates Kif3a, a subunit of kinesin-2, and that Kif3a phosphorylation is required for normal cilia formation in vivo. These results suggest that ICK is a Kif3a kinase and required for ciliogenesis by regulating ciliary transport at ciliary tips.
Results
ICK-deficient mice show neonatal lethality with skeletal, lung, and brain abnormalities
We first examined the expression pattern of ICK in the developing CNS. We observed that ICK mRNA is expressed in the embryonic day 10.5 (E10.5) neural tube and E15.5 brain including the cerebral cortex (Supplementary Fig S1A and B). ICK mRNA was detected in ganglion cells and weakly in progenitor cells at E17.5 in the retina (Supplementary Fig S1C). We did not detect ICK mRNA at postnatal day 3 (P3) or P21 in the retina (Supplementary Fig S1D and E). In the brain, the expression of ICK reached its peak at P2 and gradually decreased at later stages (Supplementary Fig S1F).
We generated ICK-floxed mice by flanking ICK exon 3 with two loxP sites (Supplementary Fig S1G and H). We first mated ICK-floxed mice with CAG-Cre mice, which express Cre recombinase in female germ cells (Sakai & Miyazaki, 1997), and generated conventional ICK-deficient (ICK−/−) mice. Absence of ICK mRNA and protein expression in the ICK−/− mice was confirmed (Supplementary Fig S2A and B). Ccrk mRNA expression was not upregulated in ICK−/− MEFs (Supplementary Fig S2C). We did not detect Mak mRNA expression in either ICK+/+ or ICK−/− MEFs. ICK+/− mice were viable and fertile and developed without an obvious phenotypic abnormality. In contrast, ICK−/− mice died around birth probably because of respiratory failure. ICK−/− mice exhibited preaxial polydactyly in both fore and hind limbs (Fig 1A–C). All four limbs were severely shortened in the ICK−/− mice at E18.5 (Fig 1D and E). We found that the lungs of ICK−/− embryos at E17.5 show the normal arrangement of four right lobes and one left lobe; however, the lobes were markedly smaller than those of wild-type embryos (Supplementary Fig S2D and E). In contrast to the lung abnormality, other organs, including the liver, kidney, and adrenal gland, were normally developed at E17.5. We observed round-shaped olfactory bulbs (Supplementary Fig S2F, arrowheads) and enlarged cerebral cortexes in the ICK−/− embryos at E17.5 (Supplementary Fig S2G and H). ICK−/− embryos showed hydrocephalus (Fig 1F and G). The expression of Gli1, a downstream gene of the Shh signaling cascade, decreased in the ICK−/− brain (Supplementary Fig S2I).
Figure 1. Loss of ICK causes defects in development and ciliogenesis.
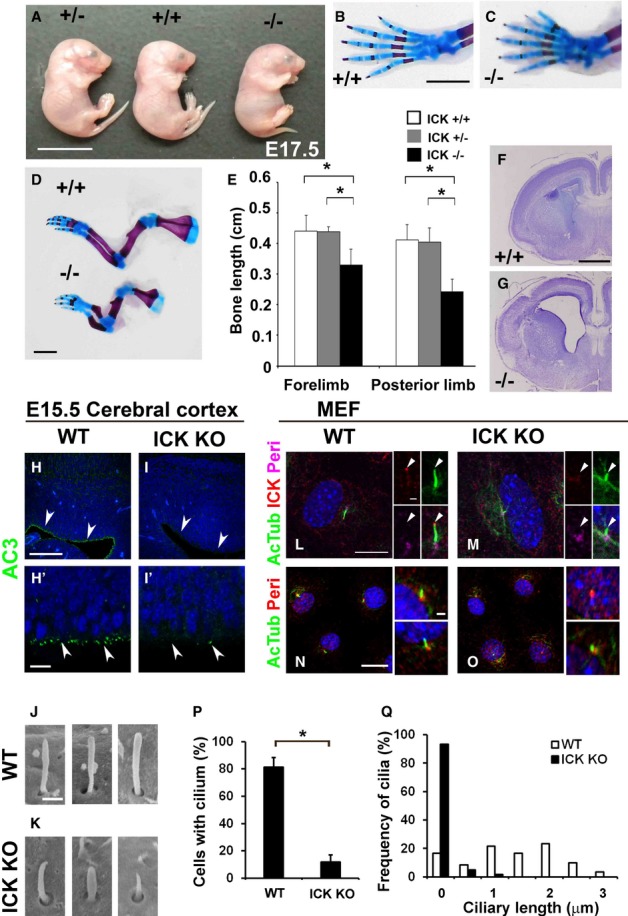
A Image of ICK+/+ (center), ICK+/− (left), and ICK−/− (right) embryos at E17.5.
B–E Skeletal defects in ICK−/− limbs and digits. (B–D) Alizarin red and alcian blue staining of forelimbs from ICK+/+ and ICK−/− mice at E18.5. (B, C) Forelimbs exhibited preaxial polydactyly in ICK−/− embryos. (D, E) The distal long bone length of both forelimb and posterior limb was shorter in ICK−/− mice compared with that in ICK+/+ mice.
F, G Nissl-stained coronal sections from ICK+/+ (F) and ICK−/− (G) mice at E17.5. ICK−/− mice showed hydrocephalus (G).
H–I′ The cilia in the cerebral cortex of ICK+/+ (H, H′) and ICK−/− (I, I′) mice at E15.5 were stained with an anti-adenylate cyclase 3 (AC3) antibody (green). Ciliary numbers on the neuroepithelial cells (arrowheads) in the cerebral cortex are reduced in ICK−/− mice.
J, K Scanning electron microscopic analysis of ICK+/+ (J) and ICK−/− (K) neural tube cilia at E10.5. Cilia are shorter in the ICK−/− neural tube.
L, M ICK is localized at cilia tips. ICK+/+ (L) and ICK−/− (M) MEFs were immunostained with antibodies against ICK (red), acetylated α-tubulin (a marker for the ciliary axoneme, green), and pericentrin (a marker for centrosomes, magenta). Arrowheads indicate ciliary tips.
N–Q Ciliary defects in ICK−/− MEFs. ICK+/+ (N) and ICK−/− (O) MEFs were immunostained with antibodies against pericentrin (red) and acetylated α-tubulin (green). The numbers (P) and length (Q) of the cilia stained with an antibody against acetylated α-tubulin were measured. The cilia in ICK−/− MEFs are markedly fewer and shorter.
Data information: Nuclei were stained with DAPI (blue). Scale bars, 10 mm (A), 2 mm (B–D), 1 mm (F, G), 100 μm (H, I), 20 μm (left panels in N, O), 10 μm (H′, I′, left panels in L, M), 2 μm (right panels in N, O), 1 μm (center and right panels in L, M), and 500 nm (J, K). Error bars show the SD. *P < 0.03.
ICK is required for proper ciliogenesis of neural progenitor cells and embryonic fibroblasts
ICK−/− embryos displayed phenotypes such as defects in cilia formation and/or Hh signaling; therefore, we analyzed ciliary formation in ICK−/− mice. Since we found morphological malformations of the brain in ICK−/− mice at E17.5, we focused on ICK function at an earlier stage in the CNS. At E15.5, neuronal progenitor cells still proliferate in the ventricular zone of the cerebral cortex (Dehay & Kennedy, 2007). We tested whether loss of ICK affects ciliary formation of neural progenitors in the E15.5 cerebral cortex. We observed that neural progenitor cells extend the cilia into ventricles in the wild-type brain (Fig 1H and H′); however, we found few cilia in the ICK−/− brain (Fig 1I and I′). In contrast, epithelial cilia in the nasal pit were normal in the ICK−/− mice (Supplementary Fig S2J–K′). To further investigate the effect of loss of ICK on ciliary formation, we observed cilia in the kidney, skin, and intestine at E15.5 (Supplementary Fig S2L–Q). Cilia in the nephric duct exhibited no obvious change between ICK+/+ and ICK−/− embryos (Supplementary Fig S2L and M). We found fewer cilia in the epidermis and dermis of the skin of ICK−/− embryos (Supplementary Fig S2N and O). In addition, there were markedly fewer cilia in the intestinal muscular layers and submucosa of ICK−/− mice (Supplementary Fig S2P and Q). These results suggest that ICK is essential for ciliogenesis in a tissue-specific manner.
Loss of genes required for ciliogenesis often causes defects in dorsal-ventral neural tube patterning, which is tightly regulated by the Shh morphogen (Dessaud et al, 2008). We examined the expression patterns of HNF3B, Shh, Pax6, Pax7, HB9, Nkx2.2, Nkx6.1, and Islet1/2 at E10.5; however, we did not find any significant differences in their expression patterns between ICK−/− and wild-type neural tubes (Supplementary Fig S3A–P). To investigate the ciliary integrity of neural progenitors, we examined cilia in the neural tube by immunohistochemistry. We found that cilia numbers and lengths decrease in the ICK−/− neural tube (Supplementary Fig S3Q–T). To analyze the ultrastructure of neural tube cilia, we performed scanning electron microscopic analysis. We found that ciliary length decreases in the ICK−/− neural tube (Fig 1J and K). Mutations in Dync2h1, which encodes the heavy chain of the cytoplasmic dynein-2 motor, or Bromi cause morphological changes in the cilia (Ko et al, 2010; Ocbina et al, 2011). Unlike Dync2h1 or Bromi mutants, cilia in the ICK−/− neural tube did not show a swollen morphology. Although we observed ciliary defects in the ICK−/− neural tube, it may not be severe enough to disrupt neural tube patterning.
To analyze ICK function in ciliogenesis, we examined the cilia in mouse embryonic fibroblasts (MEFs) from ICK−/− embryos at E13.5. We generated an anti-ICK antibody and analyzed subcellular localization of ICK in MEFs. We found that ICK is localized at the tips of the cilia in wild-type MEFs (Fig 1L). We confirmed the loss of ICK in ICK−/− MEF cilia (Fig 1M). We observed background signals in the cytoplasm of both ICK+/+ and ICK−/− MEFs (Fig 1L and M). We found that ciliated cells are fewer and the ciliary length is shorter in ICK−/− MEFs (Fig 1N–Q; Supplementary Fig S3U–Z). This result suggests that ICK is essential for ciliary elongation, in contrast to the function of an ICK paralog, Mak, which is required for the negative regulation of ciliary length (Omori et al, 2010).
ICK is required for the ciliogenesis of retinal progenitor cells, but not for photoreceptor cells
To investigate the in vivo function of ICK in the developing retina, we mated the ICK-floxed mouse line with the Dkk3-Cre transgenic mouse line, in which Cre-mediated recombination begins in almost all retinal progenitors at around E10.5 (Sato et al, 2007). We generated ICKflox/flox; Dkk3-Cre+ (ICK Dkk3 CKO) mice and found that they were viable and fertile. We confirmed almost complete loss of the ICK mRNA in the ICK Dkk3 CKO retina (Supplementary Fig S4A). ICK protein expression was lost in the ICK Dkk3 CKO retina (Supplementary Fig S4B).
We found that retinal thickness was reduced in ICK Dkk3 CKO mice compared to control mice at P0 (Supplementary Fig S4C–G). The numbers of proliferating cells decreased in the ICK Dkk3 CKO retina (Supplementary Fig S4C–F, H, and I). We found that the numbers of cilia markedly decreased in the P0 ICK Dkk3 CKO retinal progenitor cells (Fig 2A–C). It is known that cilia are essential for Hh signal transduction and that Shh signaling controls cell proliferation of retinal progenitor cells (Corbit et al, 2005; Huangfu & Anderson, 2005; Wang et al, 2005; Sakagami et al, 2009). Decreased cell proliferation of retinal progenitor cells in the ICK Dkk3 CKO retina is most likely due to the lack of cilia and Shh signaling in retinal progenitor cells.
Figure 2. Loss of ICK affects retinal and cerebellar development.
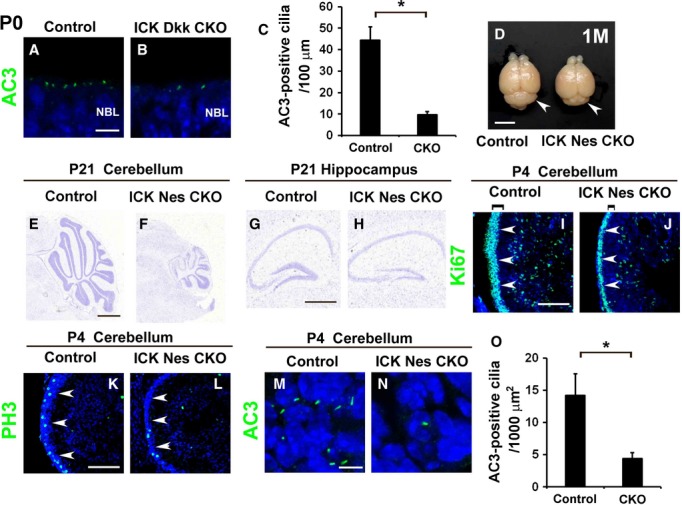
A–C The cilia in retinal neuroepithelial cells from the P0 control and ICK Dkk3 CKO mice were immunostained with an antibody against AC3 [green in (A) and (B)]. The numbers of AC3-positive cilia in P0 control and ICK Dkk3 CKO retinas were counted (C). Ciliary numbers were reduced in the ICK Dkk3 CKO retina.
D Dorsal view of the dissected brain from control and ICK Nes CKO mice at the age of 1 month. The cerebellum (arrowheads) is smaller in ICK Nes CKO mice (right) compared to that in control mice (left).
E–H Nissl staining of cerebellar (E, F) and hippocampal (G, H) sections from P21 control (E, G) and ICK Nes CKO (F, H) mice. The lobes of the cerebellum are markedly smaller in ICK Nes CKO mice (F) compared to those in control mice (E). The hippocampal DG is smaller in ICK Nes CKO mice (H) compared to that in control mice (G).
I–L Immunohistochemical analysis of the ICK Nes CKO cerebellum with cell proliferation markers. Sagittal cerebellar sections were immunostained with antibodies against Ki67 (green in (I) and (J)) and PH3 (green in (K) and (L)). The external granule cell layer (EGL, shown with arrowheads) is thinner in ICK Nes CKO mice.
M–O The cilia in EGL cells of the control (M) and ICK Nes CKO (N) mice were stained with an antibody against AC3. The cilia in the EGL are fewer in the ICK Nes CKO cerebellum. The number of AC3-positive cilia in the control and ICK Nes CKO EGL was counted (O).
Data information: Nuclei were stained with DAPI (blue). Scale bars, 5 mm (D), 1 mm (E, F), 500 μm (G, H), 100 μm (I–L), and 5 μm (A, B, M, N). Error bars show the SD. *P < 0.03. NBL, neuroblastic layer.
To test whether the loss of ICK affects photoreceptor cilia formation and maintenance, we immunostained retinal sections from 1-month-old ICK Dkk3 CKO mice with ciliary markers. We observed no obvious difference in the number and length of the ciliary axoneme and connecting cilia between control and ICK Dkk3 CKO retinas (Supplementary Fig S4J–O). Ciliary localization of Mak was unaltered between control and ICK Dkk3 CKO photoreceptors (Supplementary Fig S4J–K″). These results suggest that ICK plays an essential role in ciliogenesis in retinal progenitors but is dispensable for ciliary assembly and maintenance in mature photoreceptor cells.
Conditional deletion of ICK in the brain causes cerebellar developmental defects
Then, to investigate ICK function in the brain after birth, we generated mice with conditional deletion of ICK in the brain. We crossed ICK-floxed mice with transgenic mice expressing Cre recombinase under the control of the Nestin enhancer in neurons and glia broadly in the brain at E11.5 (Isaka et al, 1999), to generate ICKflox/flox; Nestin-Cre+ (ICK Nes CKO) mice. We confirmed that ICK mRNA expression level is markedly reduced in the cerebellum, hippocampus, cerebral cortex, and whole brain in ICK Nes CKO mice compared to that of the control mice (Supplementary Fig S5A–D). ICK protein expression was lost in the ICK Nes CKO brain (Supplementary Fig S5E). Ccrk mRNA expression was not upregulated in the ICK Nes CKO brain (Supplementary Fig S5F). We did not detect Mak mRNA expression in either control or ICK Nes CKO brains. At P4, ICK Nes CKO and control mice were indistinguishable; however, at the age of 1 month, ICK Nes CKO mice exhibit growth retardation, ataxia, and tremor (Supplementary Fig S5G).
We first analyzed the ICK Nes CKO brain at 1 month of age. We found that the cerebella of ICK Nes CKO mice are obviously smaller than those of the control mice (Fig 2D). We found that the cerebellar lobes of ICK Nes CKO mice were smaller compared to those of the control mice (Fig 2E and F). Similarly, the hippocampal dentate gyrus (DG) was smaller in the ICK Nes CKO mice (Fig 2G and H). In contrast to the ICK−/− mice, no expanded ventricle was observed in the ICK Nes CKO brain at P21 (Supplementary Fig S5H and I).
In the cerebellum, cells in the external granule cell layer (EGL) proliferate to produce the pool of granule cell precursors (GCPs) during the first 2 weeks after birth. GCPs migrate internally past the Purkinje cells to form the inner granule cell layer (IGL) (Sotelo, 2004). To investigate the development of the ICK Nes CKO cerebellum more precisely, we compared the control cerebellum with the ICK Nes CKO cerebellum at P4. We found defective cerebellar foliation in the ICK Nes CKO mice (Supplementary Fig S5J and K). At this stage, the EGL of the ICK Nes CKO cerebellum is thinner compared to that of control cerebellum (Fig 2I–L). Proliferating cell numbers were markedly reduced in the ICK Nes CKO cerebellum (Fig 2I–L; Supplementary Fig S5L and M). Ciliary numbers were significantly smaller in ICK Nes CKO mice compared to that of control mice (Fig 2M–O). To test whether Shh signaling is affected in the ICK Nes CKO cerebellum, we analyzed the expression of Gli1, a downstream gene of the Shh signaling cascade. Gli1 expression significantly decreased in the ICK Nes CKO cerebellum at P4 (Supplementary Fig S5N). These results suggest that the loss of ICK causes ciliary loss in EGL cells and abnormal Shh signaling, resulting in reduction of cell proliferation in the EGL in the ICK Nes CKO cerebellum.
ICK is required for postnatal DG neurogenesis
In the DG, granule neuron precursors proliferate and differentiate into granule neurons at postnatal stages (Altman & Bayer, 1990; Li & Pleasure, 2005). The small size of the DG suggests a possible defect in the postnatal generation of granule neurons in the ICK Nes CKO mice. We found that numbers of proliferating cells decreased in ICK Nes CKO mice (Supplementary Fig S5O–T). This observation suggests that the production of new neurons is defective at postnatal stages in the ICK Nes CKO DG.
To observe the cilia in granule neuron precursors, we immunostained Ki67 and AC3 in the DG. We found that the number of Ki67-positive cells with AC3-positive cilia was severely reduced in the ICK Nes CKO DG (Fig 3A–C). Previously, formation of the primary cilia and Shh signaling were shown to be required for postnatal DG neurogenesis (Breunig et al, 2008; Han et al, 2008). We found that the Gli1 expression level was significantly reduced in the ICK Nes CKO hippocampus (Supplementary Fig S5U). These results suggest that the loss of ICK in the hippocampus causes ciliary loss in DG neural progenitor cells and affects neuronal production through the Shh signal cascade. We did not observe any obvious differences in ependymal cell ciliary formation between the P4 control and ICK Nes CKO brain (Supplementary Fig S5V and W).
Figure 3. ICK is required for ciliogenesis in the developing hippocampal DG.
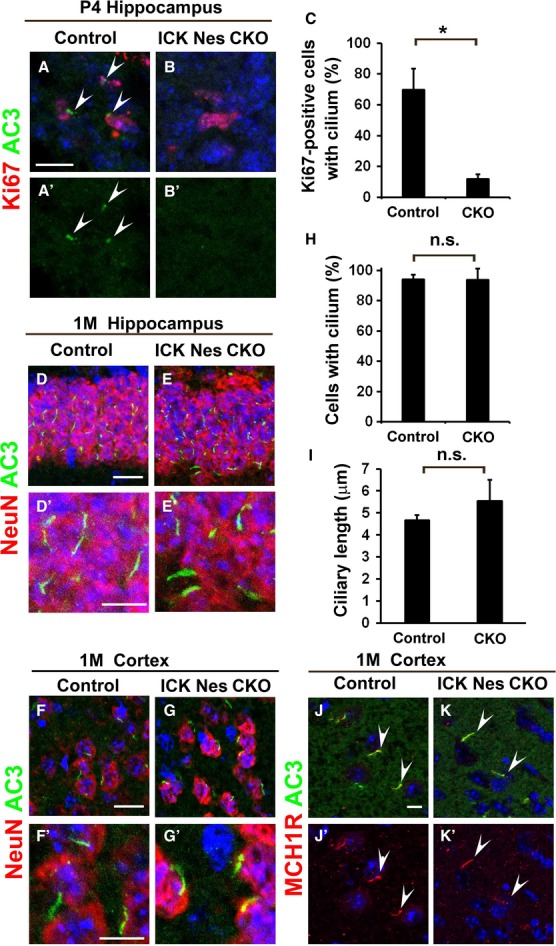
A–B′ Sagittal hippocampal sections from P4 control and ICK Nes CKO mice were immunostained with antibodies against Ki67 (red) and AC3 (green). Numbers of Ki67-positive cells with the cilia were decreased in the ICK Nes CKO DG. Arrowheads indicate cilia on Ki67-positive cells.
C The percentage of Ki67-positive cells with AC3-positive cilia was quantified.
D–G′ Immunohistochemical analysis of the hippocampal DG (D–E′) and cerebral cortex (F–G′) from 1-month-old control (D, D′, F, F′) and ICK Nes CKO (E, E′, G, G′) mice. Coronal sections were stained with antibodies against NeuN (a neuronal marker, red) and AC3 (green). No obvious difference in ciliary number or ciliary length was observed between control and ICK Nes CKO mice.
H, I The numbers (H) and length (I) of the cilia stained with an anti-AC3 antibody were measured. There was no significant difference between the control and ICK Nes CKO cerebral cortex.
J–K′ Ciliary localization of GPCR in the ICK Nes CKO cerebral cortex. Sections from cerebral cortex were stained with ciliary GPCR, MCH1R (red). The cilia were stained with an anti-AC3 antibody (green). MCH1R (arrowheads) was localized in the cilia properly both in control and in ICK Nes CKO mice.
Data information: Nuclei were stained with DAPI (blue). Scale bars, 20 μm (D, E, F, G) and 10 μm (A–B′, D′, E′, F′, G′, J–K′). Error bars show the SD. *P < 0.03. n.s., not significant.
ICK-deficient mature neurons develop cilia
Mature neurons extend cilia in various parts of the brain including the hippocampus and cerebral cortex. Several types of GPCRs including somatostatin receptor 3 (SSTR3) and melanin-concentrating hormone receptor 1 (MCH1R) were found to localize to the neuronal cilia (Handel et al, 1999; Berbari et al, 2008; Marley & von Zastrow, 2010). We investigated the integrity of the neuronal cilia in ICK Nes CKO mice. Unexpectedly, we found that the cilia are normal in the ICK Nes CKO DG and cerebral cortex at 1 month of age (Fig 3D–I).
We examined ciliary localization of GPCRs in mature neurons. We found that MCH1R and SSTR3 are localized properly in the neuronal cilia of the ICK Nes CKO cerebral cortex (Fig 3J–K′; Supplementary Fig S5X–Y′). In contrast to the essential role of ICK for neuronal progenitor ciliogenesis, loss of ICK does not appear to significantly affect ciliary formation and GPCR localization in mature neurons.
ICK is required for proper ciliary localization of Shh pathway components
Shh pathway components including Smo and Gli localize to the cilia, and ciliary localization of these components depends on stimulation of Shh signaling (Corbit et al, 2005; Haycraft et al, 2005). To investigate whether ICK is involved in the regulatory mechanism of ciliary transport of Shh signaling components, we observed Shh signal-dependent ciliary localization of Smo, Gli2, and Gli3 using the ICK−/− MEF (Fig 4A–J; Supplementary Fig S6A–I). In the absence of Shh pathway stimulation, Smo rarely localized in the wild-type MEF cilia; however, Shh pathway stimulation with treatment of a Smo-binding Shh pathway agonist (SAG) induced Smo accumulation in the cilia (Fig 4A, B and I). In contrast, we often observed accumulation of the Smo signal in the ICK−/− MEF cilia even without Shh pathway stimulation (Fig 4C, D, and I). To confirm the abnormal ciliary accumulation of Smo in the ICK−/− MEFs, we co-immunostained Smo with γ-tubulin in the ICK mutant MEFs. We observed that Smo signals in the vicinity of the centriole significantly increased in ICK−/− MEFs compared to those in wild-type MEFs in the absence of Shh pathway activation (Supplementary Fig S6A–D). Unexpectedly, we found the Smo signal displayed on cilia-like structure in the vicinity of centriole in more than 80% ICK−/− MEFs. We found that about 10% of ICK−/− MEFs formed acetylated α-tubulin-positive cilia (Fig 1N–P). Since acetylated α-tubulin is a marker for the ciliary axoneme and Smo is a membrane protein, these data suggest that ICK−/− MEF cilia have ciliary membrane structures without acetylated α-tubulin-positive microtubules. Although AC3 and Arl13b are ciliary membrane proteins, we observed that about 10% of ICK−/− MEFs form AC3-positive cilia and that about 30% of ICK−/− MEFs have Arl13b-positive cilia (Supplementary Fig S3U–Z). The transport system for AC3 and Arl13b in cilia may be different from that for Smo.
Figure 4. Signal-dependent ciliary transport of Shh components is defective in ICK−/− cells.
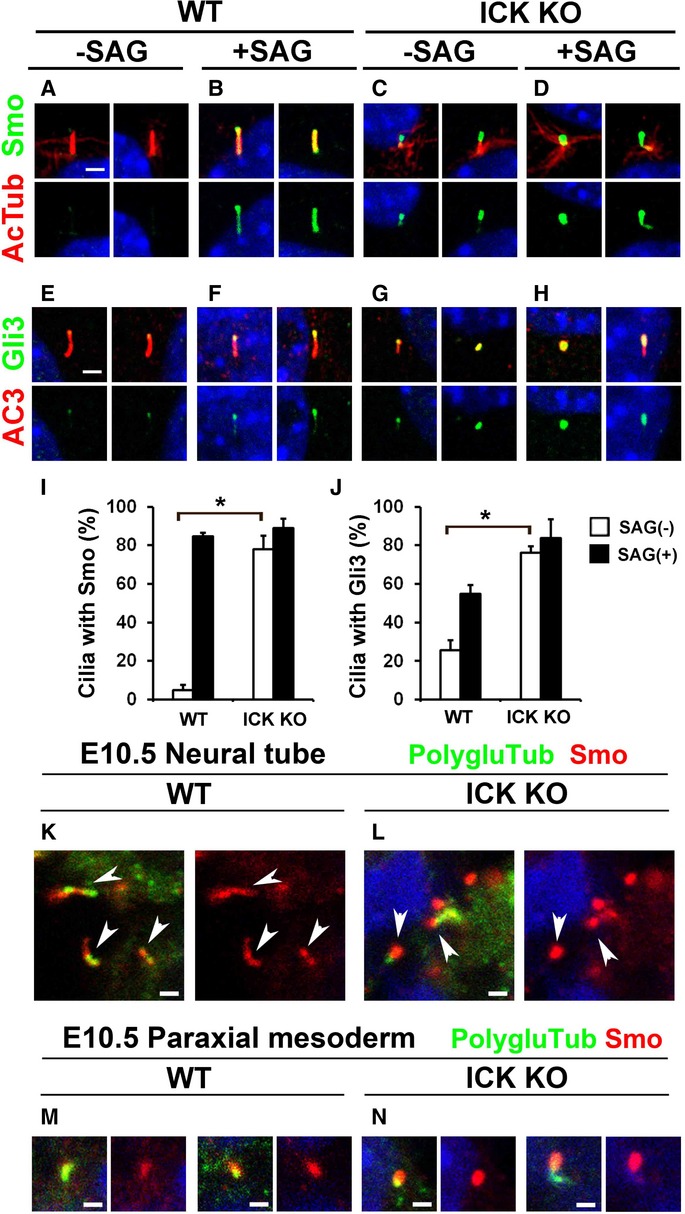
A–H ICK+/+ and ICK−/− MEFs were treated or not treated with Smo agonist (SAG) and immunostained with antibodies against Smo (green in A–D) and Gli3 (green in E–H). The cilia were immunostained with antibodies against acetylated α-tubulin (red in A–D) or AC3 (red in E–H). Smo and Gli3 are aberrantly present in the ICK−/− MEF cilia without stimulation with SAG.
I, J Quantification of the cilia with Smo and Gli3 signals. The percentages of the cilia with Smo (I) and Gli3 (J) signals in ICK+/+ and ICK−/− MEFs with or without treatment of SAG were quantified.
K, L ICK+/+ (K) and ICK−/− (L) neural tube cilia were immunostained with antibodies against Smo (red) and polyglutamylated tubulin (green). Arrowheads indicate Smo-positive cilia. Smo signals increased in ICK−/− neural tube cilia at E10.5.
M, N Cilia in paraxial mesoderm regions of E10.5 ICK+/+ (M) and ICK−/− (N) embryos were immunostained with antibodies against Smo (red) and polyglutamylated tubulin (green). Smo signals in the cilia increased in ICK−/− mice.
Data information: Nuclei were stained with DAPI (blue). Scale bars, 2 μm (A–H) and 1 μm (K–N). Error bars show the SD. *P < 0.03.
In wild-type MEFs, low levels of Gli2 and Gli3 were localized at ciliary tips in the absence of Shh pathway stimulation (Fig 4E and J; Supplementary Fig S6E and I). However, after stimulation with SAG, Gli2 and Gli3 accumulated at ciliary tips (Fig 4F and J; Supplementary Fig S6F and I). In contrast, Gli2 and Gli3 were enriched at cilia tips in the ICK−/− MEFs either with or without Shh pathway stimulation (Fig 4G, H, and J; Supplementary Fig S6G, H, and I). Smo signals in the cilia increased in the E10.5 ICK−/− neural tube and paraxial mesoderm (Fig 4K–N). Gli1 expression was significantly downregulated in ICK−/− MEFs compared to that in wild-type MEFs in the presence of Shh pathway stimulation (Supplementary Fig S6J). These results suggest that ICK regulates the ciliary localization of Shh pathway components.
A kinase-dead mutant and an ECO-associated mutant of ICK have a reduced ability to rescue ciliary formation in ICK−/− MEFs
To investigate the regulatory mechanisms for ciliary formation by ICK, we performed rescue experiments using ICK−/− MEFs. We prepared constructs expressing a FLAG-tagged full-length wild-type ICK (ICK-WT) or a kinase-dead mutant ICK (ICK-KD) (Fig 5A). The ICK-KD construct was generated by substitution of a lysine residue (K33) with an arginine residue (Xia et al, 2002; Fu et al, 2005). We transfected these constructs into ICK−/− MEFs and observed the acetylated α-tubulin-positive cilia (Fig 5B–E). While ICK-WT rescued ciliary formation in ICK−/− MEFs (Fig 5C and E), ICK-KD failed to rescue the cilia in those cells (Fig 5D and E). ICK-WT rescued ciliary length in ICK−/− MEFs (Fig 5F). This result shows that the kinase activity of ICK is essential for its function in cilia formation. We observed that ICK-WT localizes at the ciliary tips (Fig 5C). We found that ICK-KD also localizes in the cilia (Fig 5D), suggesting that kinase activity is not required for ciliary localization of ICK.
Figure 5. A kinase-dead mutant and an ECO-associated mutant of ICK show a decreased ability to rescue shortened cilia phenotype in ICK−/− MEFs.
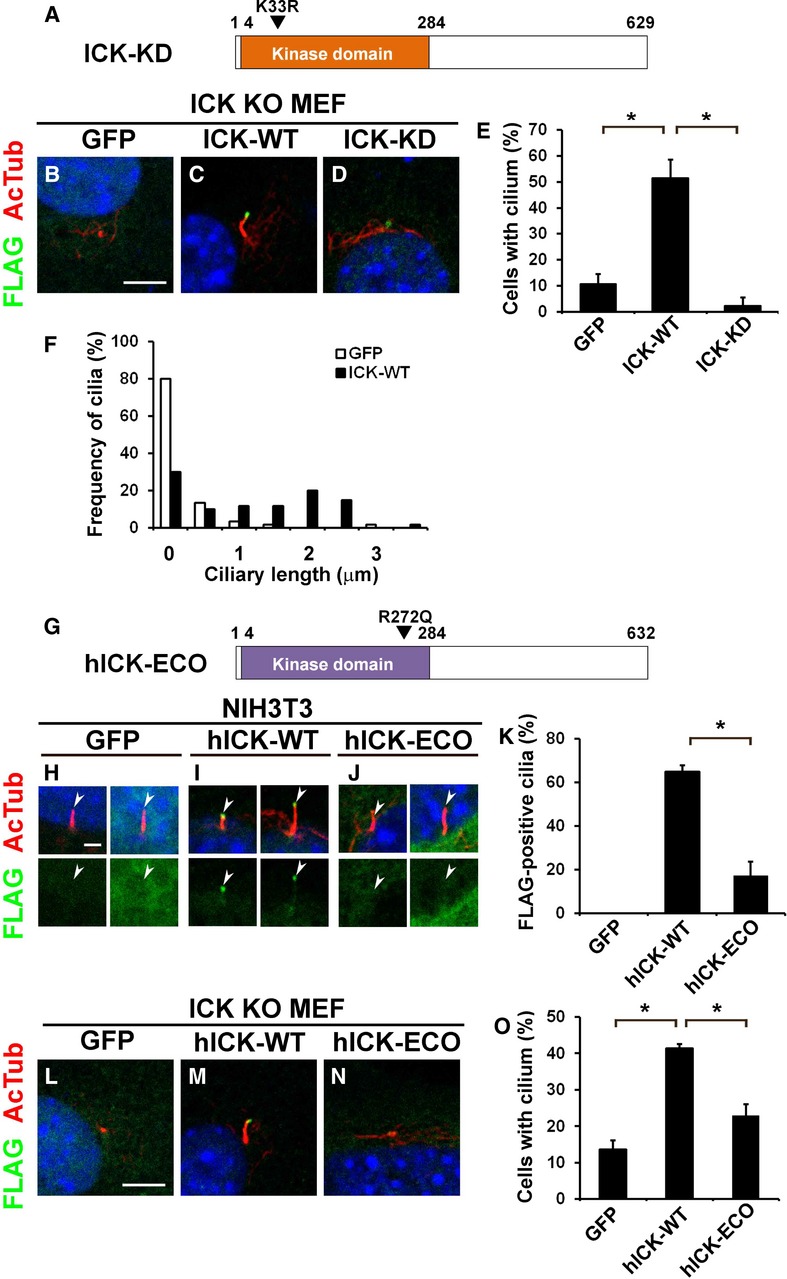
A–F Kinase activity of ICK is required for ciliary formation but not for localization in the cilia. (A) Schematic representation of a kinase-dead ICK. (B–D) Constructs expressing FLAG-tagged GFP (B), wild-type ICK (ICK-WT; C), or kinase-dead ICK K33R (ICK-KD; D) were transfected into ICK−/− MEFs. Localization of FLAG-tagged proteins was observed using anti-FLAG (green) and anti-acetylated α-tubulin (red) antibodies. (E) The number of cilia stained with an anti-acetylated α-tubulin antibody was counted. (F) The length of cilia stained with anti-acetylated α-tubulin antibody was measured.
G–O The ECO-associated mutant of human ICK has a decreased ability to induce ciliary formation. (G) Schematic representation of the ECO-associated mutant of human ICK. (H–J) FLAG-tagged constructs expressing GFP (H), wild-type human ICK (hICK-WT; I), or ECO-associated mutant R272Q of human ICK (hICK-ECO; J) were transfected into NIH3T3 cells. Localization of FLAG-tagged proteins was observed using anti-FLAG (green) and anti-acetylated α-tubulin (red) antibodies. (K) The percentage of the cilia with the FLAG signal was quantified. (L–N) The FLAG-tagged GFP (L)-, hICK-WT (M)-, or hICK-ECO (N)-expressing plasmid was transfected into ICK−/− MEFs. (O) The number of cilia stained with anti-acetylated α-tubulin antibody was counted.
Data information: Nuclei were stained with DAPI (blue). Scale bars, 5 μm (B–D, L–N) and 2 μm (H–J). Error bars show the SD. *P < 0.03. Arrowheads indicate ciliary tips.
It was reported that a missense mutation in human ICK is associated with a developmental disorder, ECO (Lahiry et al, 2009). This mutation results in an amino acid substitution from arginine to glutamine at residue 272 of human ICK (R272Q). To investigate whether this mutation affects ICK function, we generated constructs expressing a FLAG-tagged full-length wild-type human ICK (hICK-WT) and FLAG-tagged ECO-associated mutant R272Q of human ICK (hICK-ECO) (Fig 5G). We transfected these constructs into NIH3T3 cells and observed the subcellular localization of the FLAG-tagged proteins (Fig 5H–K). We found that hICK-WT localizes to the ciliary tips in 65% cells (Fig 5I and K). In contrast, the human mutant ICK had a significantly decreased efficiency in localizing to the ciliary tips compared to that of WT hICK (17%, Fig 5J and K). We next examined the ability of the WT hICK and the human ECO mutant to rescue the formation of acetylated α-tubulin-positive cilia in ICK−/− MEFs (Fig 5L–O). We found that hICK-WT effectively rescues ciliary formation in ICK−/− MEFs, whereas hICK-ECO inefficiently rescues cilia in those cells (Fig 5M–O). These results suggest that the R272Q mutation in human ICK causes loss of proper ICK localization in the cilia, resulting in a functional defect of ICK in the cilia.
ICK regulates the localization of IFT components at ciliary tips
IFT is required for proper ciliary localization of Shh signaling components. To examine whether the loss of ICK affects ciliary transport, we immunostained IFT88 (an IFT-B component) and IFT144 (an IFT-A component) in the cilia of ICK−/− MEFs. We observed that the level of IFT88 and IFT144 localized at ciliary tips markedly increased in ICK−/− MEFs (Fig 6A–E). We next transfected the FLAG-tagged IFT57 (an IFT-B component)-, IFT140 (an IFT-A component)-, or BBS8 (a BBSome component)-expressing plasmids into ICK−/− MEFs, and observed the subcellular localization of the FLAG-tagged proteins. These FLAG-tagged proteins were concentrated at ciliary tips in ICK−/− MEFs (Fig 6F and G; Supplementary Fig S6K–N). These results show that loss of ICK affects IFT machinery. Unlike Ift122sopb mutant mice, which show defects in retrograde IFT (Qin et al, 2011), we did not observe an accumulation of IFT88 at the tips of cilia in the ICK−/− neural tube at E10.5 (Supplementary Fig S6O–P′). To observe whether overexpression of ICK affects ciliary localization of IFT components, we transfected FLAG-tagged Kif3a, IFT88, IFT57, IFT140, or BBS8 expression constructs with or without the ICK-expressing construct (Fig 6H–V). We found that ICK signals at ciliary tips increase in ICK-overexpressing cells (Supplementary Fig S7A and B). Overexpression of ICK induced slight ciliary elongation (Supplementary Fig S7C–E). IFT88 was distributed along the ciliary axoneme without the overexpression of ICK (Fig 6L). In this condition, Kif3a, IFT57, and BBS8 rarely localized to the cilia, and IFT140 only slightly localized at the base of the cilia (Fig 6J, N, R, S, and U). When ICK was overexpressed, Kif3a, IFT88 and IFT57, but not IFT140 or BBS8, more clearly accumulated at ciliary tips (Fig 6K, M, O, R, T, and V). The level of IFT140 localized at ciliary bases increased in ICK-overexpressing cells (Fig 6T). In these cells, IFT88 signals occasionally showed a ring-like structure at the tip of the cilia (Fig 6P and Q). These results show that ICK regulates IFT machinery in the cilia.
Figure 6. Loss or overexpression of ICK affects ciliary localization of IFT components.
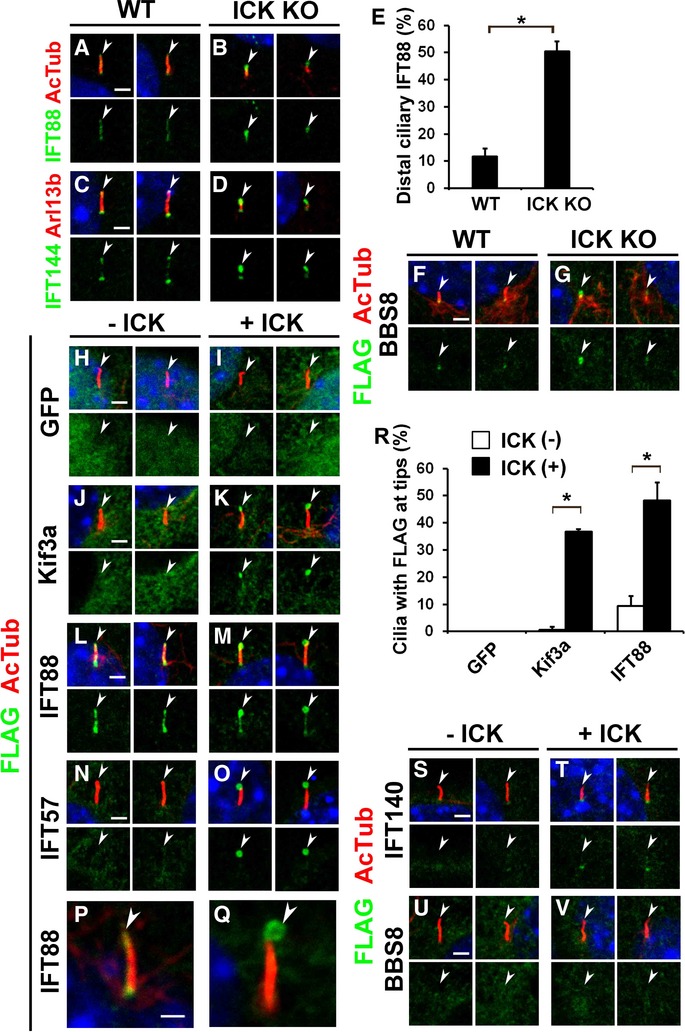
A–G IFT components concentrated at cilia tips in ICK−/− MEFs. (A–D) ICK+/+ and ICK−/− MEFs were stained with anti-IFT88 (A, B) and anti-IFT144 (C, D) antibodies. (E) The number of cilia with a concentration of IFT88 at ciliary tips was measured. (F, G) FLAG-tagged BBS8-expressing plasmid was transfected into ICK+/+ and ICK−/− MEFs. Cells were stained with anti-acetylated α-tubulin (red) and anti-FLAG (green) antibodies.
H–Q FLAG-tagged constructs expressing GFP (H, I), Kif3a (J, K), IFT88 (L, M, P, Q), or IFT57 (N, O) were transfected with or without an ICK expression plasmid into NIH3T3 cells. Localization of FLAG-tagged proteins was observed using anti-FLAG (green) and anti-acetylated α-tubulin (red) antibodies.
R The number of cilia with the FLAG-tagged protein predominantly localized at ciliary tips was counted.
S–V FLAG-tagged constructs expressing IFT140 (S, T) or BBS8 (U, V) were transfected with or without an ICK expression plasmid into NIH3T3 cells. Localization of FLAG-tagged proteins was observed using anti-FLAG (green) and anti-acetylated α-tubulin (red) antibodies.
Data information: Nuclei were stained with DAPI (blue). Scale bars, 2 μm (A–D, F–O, S–V) and 1 μm (P, Q). Error bars show the SD. *P < 0.03. Arrowheads indicate ciliary tips.
Kif3a is phosphorylated by ICK and Kif3a phosphorylation is required for ciliary formation
To identify a phosphorylation target for ICK, we searched for consensus amino acid sequence R-P-X-S/T-P/A/T/S for phosphorylation by ICK in proteins associated with ciliary transport, including components of IFT-A, IFT-B, BBSome, kinesin, and dynein motors. We found that Kif3a contains a highly evolutionarily conserved consensus sequence at position 671–675 in its C-terminal region (Fig 7A and B) (Fu et al, 2006). We performed an in vitro kinase assay using purified GST-ICK and found that Kif3a-C-WT is markedly phosphorylated by ICK, whereas no obvious phosphorylation of GST alone was detected (Fig 7C). We next generated a construct harboring a Thr-to-Ala mutation at residue 674. We found that the phosphorylation level of Kif3a-C-T674A is markedly weaker than that of Kif3a-C-WT (Fig 7C). These results show that ICK directly phosphorylates Kif3a, predominantly at residue 674. To explore whether Kif3a is actually phosphorylated in cells, we made an antibody against phosphorylated Kif3a Thr-674 (p-Kif3a) (Supplementary Fig S8A–C′). We found that p-Kif3a localizes to the cilia in ICK+/+ MEFs and that p-Kif3a signals were often enriched at ciliary bases and tips (Fig 7D). In ICK−/− MEFs, the percentage of cilia with p-Kif3a at the ciliary tips markedly decreased (Fig 7E and F). Interestingly, a majority of shortened cilia lost p-Kif3a signals at the tips of cilia (left and middle panels in Fig 7E), whereas cilia of normal length often had p-Kif3a signals at ciliary tips in ICK−/− MEFs (right panels in Fig 7E). This result suggests that Kif3a is phosphorylated by ICK at ciliary tips and that phosphorylation of Kif3a at residue 674 is linked to ciliary formation.
Figure 7. The C-terminal portion of Kif3a is phosphorylated by ICK and Kif3a phosphorylation is essential for proper ciliary formation.
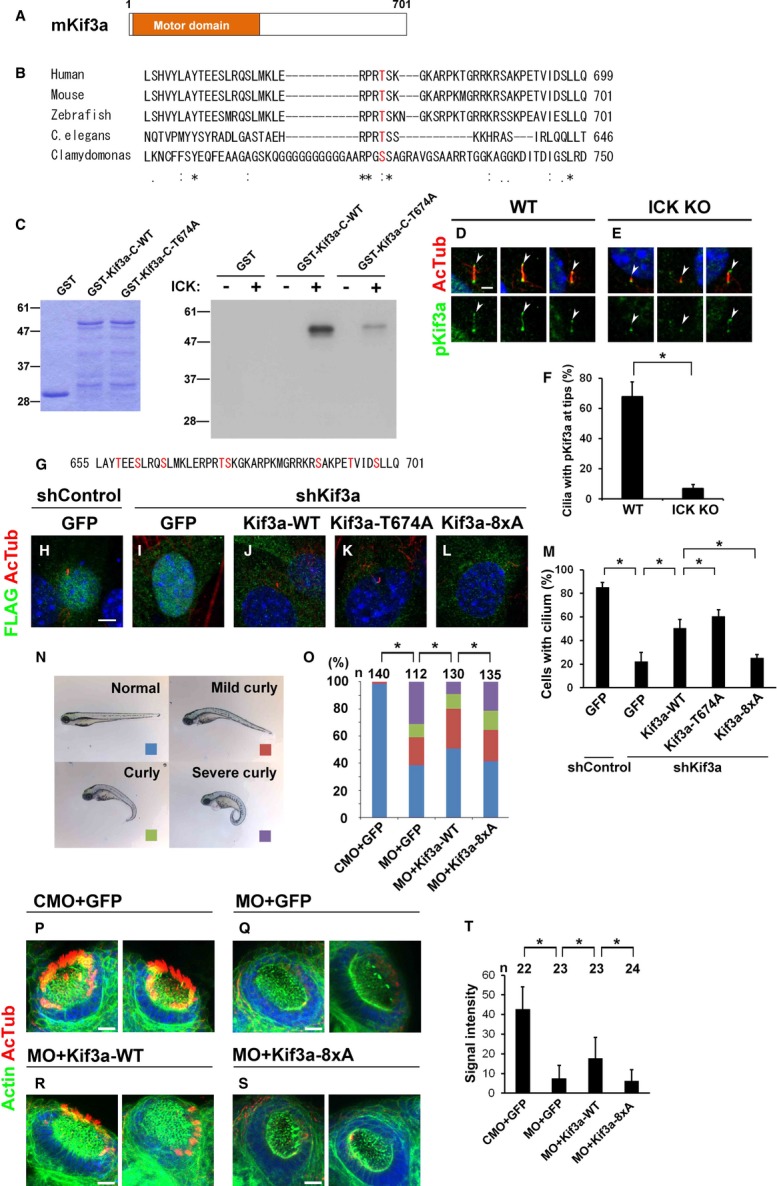
A Schematic representation of mouse full-length Kif3a.
B Amino acid sequence alignment of human KIF3A, mouse Kif3a, zebrafish Kif3a, Caenorhabditis elegans Klp-20, and Chlamydomonas FLA10 proteins. The predicted amino acid sequences of these proteins were aligned by the ClustalW program (http://clustalw.ddbj.nig.ac.jp/). Asterisks, identical amino acids; colons and periods, similar amino acids. Predicted ICK phosphorylation sites are shown in red.
C ICK phosphorylates Kif3a in vitro. GST-tagged C-terminal fragments (residues 455–701) of wild-type Kif3a (GST-Kif3a-C-WT) or T674A Kif3a (GST-Kif3a-C-T674A) were purified from bacterial extracts and stained with Coomassie Brilliant Blue (left panel). GST-Kif3a deletion proteins were applied to the in vitro kinase assay using purified GST-ICK (right panel).
D, E Ciliary localization of p-Kif3a in ICK+/+ (D) and ICK−/− (E) MEFs. Cells were immunostained with antibodies against p-Kif3a (green) and acetylated α-tubulin (red). The proportion of cilia with p-Kif3a signals at ciliary tips decreased in ICK−/− MEFs. Arrowheads indicate ciliary tips.
F The percentage of cilia with p-Kif3a signals at the ciliary tips was quantified.
G The C-terminal amino acid sequence of mouse Kif3a containing eight serine or threonine residues (red).
H–M FLAG-tagged constructs expressing GFP or shRNA-resistant Kif3a (WT, T674A, or 8xA) were transfected with control shRNA or shKif3a-1 into NIH3T3 cells. Cells were immunostained with anti-FLAG (green) and anti-acetylated α-tubulin (red) antibodies (H–L). The number of cilia stained with an anti-acetylated α-tubulin antibody was counted (M).
N Injection with Kif3a antisense morpholino (Kif3a MO) into zebrafish embryos causes curly tail, a phenotype characteristic of ciliary defects.
O Quantification of curly tail phenotype larvae injected with wild-type and mutant Kif3a mRNA and Kif3a MO. Curly tail phenotype is partially rescued by injection with Kif3a-WT mRNA. The numbers of larvae showing curly tail increased with injection with Kif3a-8xA mRNA compared to injection with Kif3a-WT mRNA.
P–T Cilia in nasal pit at 3 dpf larvae were observed by staining of acetylated α-tubulin (red). Representative images of nasal pit from each group were shown (P–S). Actin was stained by phalloidin (green). Kif3a-WT mRNA injection partially rescued the loss of cilia caused by injection of Kif3a MO, whereas Kif3a-8xA mRNA injection failed to rescue that. Signal intensity of acetylated α-tubulin staining of cilia in nasal pit was measured (T).
Data information: Nuclei were stained with DAPI (blue). Scale bars, 20 μm (P–S), 5 μm (H–L), and 2 μm (D, E). Error bars show the SD. *P < 0.03. CMO, control morpholino.
To investigate the role of phosphorylation of Kif3a in ciliary formation, we performed rescue experiments using NIH3T3 cells (Fig 7G–M). We constructed a short hairpin RNA (shRNA) to knockdown Kif3a (Supplementary Fig S8D) and found that shKif3a inhibits ciliary formation (Fig 7I and M). Expression of shRNA-resistant Kif3a-WT rescued shKif3a-mediated inhibition of ciliation (Fig 7J and M). Unexpectedly, shRNA-resistant Kif3a-T674A had a slightly increased ability to rescue shKif3a-induced inhibition of ciliary formation compared to that of Kif3a-WT (Fig 7K and M). Since Kif3a-C-T674A was still phosphorylated by ICK (Fig 7C), we thought that phosphorylation of other serine or threonine residues in the C-terminal region of Kif3a may also be important for Kif3a function in ciliary formation. To further analyze the role of phosphorylation of the Kif3a C-terminal region in ciliary formation, we constructed a plasmid encoding the Kif3a mutant in which 8 Ser or Thr residues clustered in the C-terminal region, including residue 674, are replaced with Ala (Kif3a-8xA) (Fig 7G). Expression of shRNA-resistant Kif3a-8xA failed to rescue shKif3a-mediated inhibition of ciliary formation (Fig 7L and M). These results suggest that phosphorylation of the C-terminal region of Kif3a affects ciliary formation in cultured cells.
To investigate the role of phosphorylation of Kif3a in ciliary formation in vivo, we performed rescue experiments using zebrafish embryos. First, Kif3a antisense morpholino (Kif3a MO), which was designed to inhibit translation of endogenous Kif3a, was injected into zebrafish embryos. At 3 dpf, approximately 60% zebrafish larvae showed a curly body axis, a typical phenotype observed in mutants with ciliary defects (Fig 7N) (Tsujikawa & Malicki, 2004; Omori et al, 2008). In this condition, approximately 30% larvae exhibited severe curly tail phenotype (Fig 7O). In contrast, when we co-injected in vitro transcribed wild-type mouse Kif3a (Kif3a-WT) mRNA with Kif3a MO, the number of larvae with the curly tail phenotype significantly decreased (Fig 7O). However, when we co-injected Kif3a MO with Kif3a-8xA mRNA, the number of larvae with the curly tail phenotype significantly increased compared to co-injection with Kif3a-WT mRNA.
We also investigated ciliogenesis in the nasal pits of the larvae injected with the morpholino and mRNA (Fig 7P–T). At 3 dpf, cilia have developed in nasal pit epithelia in control larvae (Fig 7P and T). In contrast, larvae injected with Kif3a MO and GFP lost cilia at this stage (Fig 7Q and T). Loss of cilia by Kif3a MO injection was partially rescued by co-injection with Kif3a-WT mRNA (Fig 7R and T). However, co-injection with Kif3a-8xA mRNA failed to rescue the ciliary defect by knockdown of Kif3a (Fig 7S and T). Taken together, these results suggest that phosphorylation of the C-terminal portion of Kif3a is essential for normal ciliary formation.
Discussion
Previous studies showed that molecular mechanisms underlying the formation of the cilia are common in most cell types. IFT machinery is essential for the formation of all types of cilia including motile and non-motile (primary) cilia (Louvi & Grove, 2011). For example, a defect of Kif3a causes ciliary loss both in neuronal progenitor cells and in mature neurons (Davenport et al, 2007; Spassky et al, 2008). Similarly, ciliary transition zone (TZ) components are also required for the ciliogenesis in many types of cells (Czarnecki & Shah, 2012). Loss of B9d2, a component of the ciliary TZ, causes the defect of the cilia both in neuronal progenitors and in neurons in the hippocampus (Breunig et al, 2008). In contrast to the phenotypes of these previously reported mutants, we observed that ICK deficiency causes defects only in neural progenitor cells and not in differentiated neurons (Supplementary Fig S8E). This observation suggests that ICK belongs to a new category of factors required for ciliogenesis and that a previously unknown mechanism regulates ciliary assembly in progenitor cells. Why does the loss of ICK cause defects in ciliogenesis in neuronal progenitor cells but not in mature neurons? The first possibility is that ICK deficiency can be functionally compensated by other ciliary kinase(s) in mature neurons, but not in progenitor cells. For example, an ICK paralog, Mak, probably compensates for ICK function in the retinal photoreceptor cells at least. ICK and other kinase(s) may function redundantly in mature neurons. The second hypothesis is that ICK is specifically important for repeating the cycle of formation and reabsorption of the cilia in progenitor cells. Progenitor cells repeatedly assemble and reabsorb cilia during the cell cycle progression. In contrast, mature neurons basically form the cilia only once and never resorb them. Since more IFT activity is probably required for repeated assembly and disassembly of cilia, cilia in progenitor cells may be more sensitive to a defect of protein transport at ciliary tips than those in mature neurons.
We observed that ICK−/− mice display hydrocephalus, whereas ICK Nes CKO mice did not. It was recently reported that ciliary defects cause increased apoptosis and impaired proliferation of NG2+PDGFR-α+ neural progenitors, resulting in neonatal hydrocephalus (Carter et al, 2012), suggesting that the hydrocephalus observed in ICK−/− mice is due to ciliary abnormalities in those progenitors. On the other hand, defects of ependymal cilia cause progressive hydrocephalus at postnatal stages (Tissir et al, 2010). It is known that cilia in ependymal cells are formed after birth (Spassky et al, 2005). We did not observe any obvious differences in ciliary formation of ependymal cells between the P4 control and ICK Nes CKO brain. Perinatal hydrocephalus caused by the absence of ICK may be compensated for in postnatal development.
A missense mutation in ICK was reported in a human ECO patient (Lahiry et al, 2009). The ECO-associated mutant R272Q of human ICK fails to localize at the nucleus and showed diminished kinase activity (Lahiry et al, 2009); however, the molecular mechanism underlying ECO has not yet been elucidated. In our study, we observed that ICK−/− mice display neonatal lethality with multiple organ defects including pulmonary hypoplasia, shortened limbs, polydactyly, cerebral cortex malformation, and ventricular hydrocephalus, which is similar to the clinical and pathological features of ECO patients. Thus, our results demonstrate that ICK loss of function causes some of the phenotypes observed in ECO patients. Interestingly, the phenotypes observed in ICK−/− mice have similarities with those mutants with defects in cilia formation and/or Hh signaling. Indeed, we showed that ICK is required for proper cilia formation and Hh signaling activity and that the human ICK R272Q mutant exhibits a diminished efficiency in localizing to ciliary tips and fails to rescue ciliary formation in ICK−/− MEFs. This suggests that ECO is caused by ciliary defects and/or impaired Hh signaling pathway due to ICK dysfunction. ICK−/− mice can be useful as a mouse model of human ECO and helpful for understanding the pathogenesis of the disorder.
Our and other studies have revealed that Mak and its homologues negatively regulate ciliary length (Asleson & Lefebvre, 1998; Berman et al, 2003; Bengs et al, 2005; Burghoorn et al, 2007; Omori et al, 2010). Since deficiencies in Mak and its homologues produce elongation of the cilia, we first assumed that loss of ICK exhibits a similar elongated ciliary phenotype. Unexpectedly, we observed shortened and stumpy cilia in ICK−/− cells, suggesting that the function of ICK is distinct from that of the previously characterized homologues and paralogues of ICK. The functional difference in ciliary length regulation between Mak and ICK may be due to differences of phosphorylation target specificity, although phosphorylation target specificity is likely to be similar between Mak and ICK because of their well-conserved kinase domains. In contrast to our observations using knockout experiments, a recent study showed that cultured cells knocked down by siRNA against ICK exhibit slightly elongated cilia (Yang et al, 2013). This phenotypic difference might be caused by a small amount of ICK remaining in the knockdown cultured cells. We found that shRNA-resistant Kif3a-T674A has a slightly increased ability to rescue shKif3a-induced inhibition of ciliary formation compared to that of Kif3a-WT. Since ciliary numbers decreased in ICK−/− cells, we first expected that Kif3a-T674A would show reduced ability to rescue shKif3a-induced inhibition of ciliary formation compared to that of Kif3a-WT. The reason why the effect of Kif3a-T674A on ciliary formation in knockdown and rescue experiments using cultured cells is the opposite of the results obtained from ICK knockout experiments is unclear; however, the study by Yang et al reported that knockdown of ICK and Mak promotes ciliary formation in cultured cells (Yang et al, 2013). Suppression of the expression of ICK may promote ciliary formation through reducing the level of Kif3a phosphorylation at residue 674 in cultured cells.
How is ICK involved in cilia formation in progenitor cells? Cilia are assembled by IFT, which is divided into anterograde transport powered by kinesin-2 and retrograde transport driven by cytoplasmic dynein-2 (Rosenbaum & Witman, 2002). IFT trains turnaround and switch from anterograde to retrograde at the ciliary tip. IFT particles and their cargos dissociate after reaching the ciliary tip and then reorganize for retrograde transport (Pedersen et al, 2006). In this study, using overexpression and knockout experiments, we showed that ICK localizes at ciliary tips and regulates the ciliary transport machinery (Fig 8). A mutation in Bromi, which encodes an interaction partner of Ccrk, leads to a swollen or bulbous morphology in cilia (Ko et al, 2010), whereas ICK−/− neural tube cilia do not display that morphology. Although ICK is a substrate of Ccrk (Fu et al, 2006), ICK may have a distinct function from that of Bromi. How does ICK get to ciliary tips? If ICK is transported to ciliary tips by anterograde IFT, the activity of ICK may be suppressed by unknown mechanisms while this protein is being transported as a cargo. This is because proteins that should function at a distinct cellular region must be properly trafficked to that region by kinesin and play their roles only after arriving.
Figure 8. A hypothetical model for ICK function in the regulation of IFT turnaround at ciliary tips.
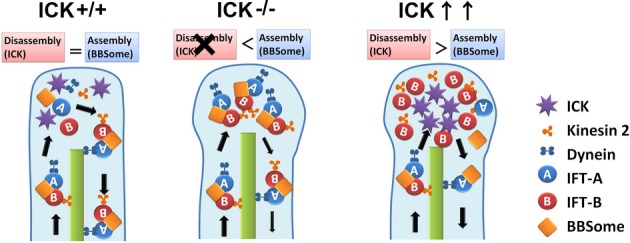
Loss of function of ICK causes the accumulation of components of IFT-A, IFT-B, and BBSome at ciliary tips. Functional defects of cilia observed in ICK-deficient cells were probably due to insufficient turnaround of IFT complexes in cilia. Under ICK overexpression conditions, an excess amount of ICK does not affect ciliogenesis but does cause IFT-B (but not IFT-A) subcomplex accumulation at ciliary tips. Accumulation of IFT-B components occasionally forms ring-like structures at cilia tips. ICK may regulate the disassembly between IFT-A and IFT-B subcomplexes at ciliary tips through phosphorylation of Kif3a C-terminal residues.
It was reported that some factors are implicated in the regulation of IFT at ciliary/flagellar tips in invertebrates. In Chlamydomonas and Tetrahymena, IFT172 is involved in the transport transition between anterograde and retrograde IFT at the tips of cilia and flagella (Pedersen et al, 2005; Tsao & Gorovsky, 2008). The BBSome and DYF-2 (an orthologue of human IFT144) were reported to cooperate to regulate IFT assembly and turnaround at the ciliary tips in C. elegans (Wei et al, 2012). Hypomorphic mutations in bbs-1 and dyf-2 cause the specific accumulation of IFT-B components at ciliary tips, which is similar to the phenotype observed in ICK-overexpressing cells. In contrast, both IFT-A and IFT-B components were concentrated at ciliary tips in ICK−/− MEFs. ICK may regulate disassembly between IFT-A and IFT-B subcomplexes to prepare for retrograde transport. We also observed that BBS8 is concentrated at ciliary tips in ICK−/− MEFs, suggesting that ICK deficiency leads to an excess amount of BBSome at ciliary tips and aberrantly promotes IFT assembly. ICK may control ciliary transport through regulating BBS proteins. To our knowledge, ICK is the only regulatory molecule identified to date that controls transport switching at ciliary tips. How does ICK regulate protein transport at ciliary tips through phosphorylation? We observed that Kif3a is directly phosphorylated by ICK. The phosphorylated form of Kif3a localized to the ciliary tips. The inhibition of phosphorylation of Kif3a affected its function in ciliary formation. Based on these observations, we propose a hypothetical model in which ICK regulates protein transport at the ciliary tips at least partially through phosphorylating Kif3a. Our data on ICK phosphorylation of Kif3a are supported by a previous study showing that loss of function of Dyf-5, the C. elegans orthologue of ICK, affects the function of kinesin-2 motors in cilia (Burghoorn et al, 2007). Since p-Kif3a signals at ciliary tips did not completely disappear in ICK−/− MEFs, ICK may not be the only Kif3a kinase. We observed that Kif3a-8xA more strongly affects ciliary formation than Kif3a-T674A. This observation and the result that Kif3a-C-T674A is still phosphorylated by ICK suggest that ICK regulates Kif3a function through phosphorylation of residue 674 as well as other residues in the Kif3a C-terminal region. However, we cannot exclude the possibility that other kinases contribute to the phosphorylation of that region of Kif3a. We further suppose that there may be other ICK phosphorylation target proteins playing roles in the regulation of ciliary transport. Future studies will advance our understanding of the detailed mechanisms of protein transport at ciliary tips.
Materials and Methods
Generation of ICKflox mice
We subcloned an approximately 12-kb ICK genomic fragment, inserted one loxP site into intron 2 and another loxP site into intron 3, cloned it into a modified pPNT vector to make a targeting construct, and transfected the linearized targeting construct into the TC1 embryonic stem (ES) cell line (Deng et al, 1996). The culture, electroporation, and selection of TC1 were performed as described previously (Muranishi et al, 2011). ES cells that were heterozygous for the targeted gene disruption were microinjected into C57BL/6 blastocysts to obtain chimeric mice.
Generation of ICK knockout (KO) mice and ICK conditional knockout (CKO) mice
We mated the ICKflox mouse line with a CAG-Cre transgenic mouse line, which expresses Cre recombinase under control of the CAG promoter, to obtain a null allele of ICK (Sakai & Miyazaki, 1997). We also mated the ICKflox mouse line with transgenic mice expressing Cre recombinase under control of the Nestin promoter (Nestin-Cre) (Isaka et al, 1999) or the Dkk3 promoter (Dkk3-Cre) (Sato et al, 2007).
Antibodies
We used the following primary antibodies for immunostaining: mouse monoclonal anti-acetylated α-tubulin (Sigma, 6-11B-1, 1:1000), anti-Ki67 (BD Pharmingen, 556003, 1:200), anti-Gli3 (gift from Dr. S. J. Scales, 1:1000) (Wen et al, 2010), anti-γ-tubulin (Sigma, GTU-88, 1:300), anti-FLAG-M2 (Sigma, F1804, 1:1000), anti-NeuN (Millipore, MAB377, 1:500), and anti-polyglutamylated tubulin (Adipogen, GT335, 1:500); mouse polyclonal anti-IFT144 (Abnova, H00057728-B01P, 1:250); rabbit polyclonal anti-AC3 (Santa Cruz, sc-588, 1:300), anti-phospho-histone H3 (Millipore, 06-570, 1:300), anti-Smo (gift from Dr. K. V. Anderson, 1:500) (Ocbina et al, 2011), anti-IFT88 (gift from Dr. G. J. Pazour, 1:1000) (Pazour et al, 2002), anti-pericentrin (Abcam, ab4448, 1:1000), anti-Rpgr (gift from Dr. T. Li, 1:1000) (Hong et al, 2003), anti-FLAG (Sigma, F7425, 1:1000), anti-(pThr674) Kif3a (1:250), anti-IFT88 (Proteintech Group, 13967-1-AP, 1:500), and anti-Arl13b (Proteintech Group, 17711-1-AP, 1:500); goat polyclonal anti-MCH1R (Santa Cruz, C-17, 1:500) and anti-SSTR3 (Santa Cruz, M-18, 1:500); guinea pig polyclonal anti-ICK (1:50), anti-Gli2 (gift from Dr. J. T. Eggenschwiler, 1:1000) (Cho et al, 2008), and anti-Mak (1:1000) (Omori et al, 2010) antibodies. Mouse monoclonal antibodies against Islet1/2, HNF3B, HB9, Nkx2.2, Pax6, Pax7, Shh, and Nkx6.1 were obtained from the Developmental Studies Hybridoma Bank. We used Cy3-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, 1:500), Alexa Fluor 488-conjugated secondary antibodies (Sigma, 1:500), and DyLight 649-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, 1:500).
Plasmid constructs
A full-length cDNA fragment of mouse ICK was amplified by PCR using the RIKEN full-length enriched library clone (GenBank accession no. AK087484) containing a missense mutation (Leu260Pro) as a template, and cloned into the pCAGGSII expression vector (Omori et al, 2010). This missense mutation was corrected by PCR primers. A full-length cDNA fragment of human ICK was amplified by PCR using a human ICK clone obtained from PlasmID (GenBank accession no. BC136420) as a template, and cloned into the pCAGGSII vector. The mouse ICK K33R mutation and human ICK R272Q mutation were introduced by PCR primers. Full-length cDNA fragments of mouse IFT57 and BBS8 were amplified by PCR using the RIKEN full-length enriched library clone (IFT57, GenBank accession no. AK047217; BBS8, GenBank accession no. AK081697) as a template, and cloned into the pCAGGSII expression vector. A full-length cDNA fragment of mouse Kif3a was amplified by PCR using a mouse Kif3a clone obtained from Open Biosystems (GenBank accession no. BC052707) as a template, and cloned into the pCAGGSII vector. Full-length cDNA fragments of mouse IFT88 and IFT140 were amplified by PCR using a mouse P0 retinal cDNA library and cloned into the pCAGGSII expression vector. A cDNA fragment encoding a partial sequence of mouse Kif3a (Kif3a-C, residues 455–701) was amplified by PCR using a mouse Kif3a clone obtained from Open Biosystems (GenBank accession no. BC052707) as a template, and cloned into the pGEX4T-1 vector (GE Healthcare). For Kif3a knockdown, pBAsi-mU6 was used for DNA vector-based shRNA synthesis. Three target sequences, GGGTGAACTTGGAGAAGATGG (shKif3a-1), GGACCTTCTGAAAGCCCAACA (shKif3a-2), and GCCTGAGACCGTAATTGATTC (shKif3a-3), were selected from different positions in the mouse Kif3a open reading frame and subcloned into the pBAsi-mU6 vector. To construct plasmids encoding shKif3a-1-resistant Kif3a, seven silent mutations in the target sequence were introduced by PCR-based mutagenesis.
Statistical analysis
Data are presented as means ± SD. Statistical significance was evaluated using a Mann–Whitney U-test (Fig 7O) or a Student's t-test (other Figures), and P < 0.05 was taken to be statistically significant.
Acknowledgments
We thank Dr. R. Kageyama (Kyoto University) for the Nestin-Cre mouse; Drs. S. J. Scales (Genentech), K. V. Anderson (Sloan-Kettering Institute), G. J. Pazour (University of Massachusetts Medical School), T. Li (National Institutes of Health), and J. T. Eggenschwiler (Princeton University) for reagents; Drs. S. Kondo and M. Watanabe (Osaka University) for help with zebrafish embryos; M. Kadowaki, A. Tani, A. Ishimaru, Y. Saioka, H. Abe, and S. Kennedy for technical assistance. This work was supported by CREST and PRESTO from Japan Science and Technology Agency, a grant for Molecular Brain Science, Grants-in-Aid for Scientific Research on Priority Areas, Grant-in-Aid for Scientific Research (B), Young Scientists (B), Specially Designated Research Promotion and Scientific Research on Innovative Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan, The Takeda Science Foundation, The Uehara Memorial Foundation, Research Foundation for Opto-Science and Technology, The Mitsubishi Foundation, Suzuken Memorial Foundation, and Japan Foundation for Applied Enzymology. A part of this work was supported by ‘Nanotechnology Platform’ (project No. 12024046) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
Author contributions
TC, YO, and TF designed the project. TC, YO, and TF performed the molecular and in situ hybridization experiments. TF generated the floxed mice. TC and YO carried out immunohistochemical analysis. TC performed cell culture experiments. YO carried out rescue experiments using zebrafish embryos. TC and RK performed scanning electron microscopic analysis. TC, YO, and TF wrote the manuscript. TF supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Altman J, Bayer SA. Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J Comp Neurol. 1990;301:365–381. doi: 10.1002/cne.903010304. [DOI] [PubMed] [Google Scholar]
- Arellano JI, Guadiana SM, Breunig JJ, Rakic P, Sarkisian MR. Development and distribution of neuronal cilia in mouse neocortex. J Comp Neurol. 2012;520:848–873. doi: 10.1002/cne.22793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asleson CM, Lefebvre PA. Genetic analysis of flagellar length control in Chlamydomonas reinhardtii: a new long-flagella locus and extragenic suppressor mutations. Genetics. 1998;148:693–702. doi: 10.1093/genetics/148.2.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengs F, Scholz A, Kuhn D, Wiese M. LmxMPK9, a mitogen-activated protein kinase homologue affects flagellar length in Leishmania mexicana. Mol Microbiol. 2005;55:1606–1615. doi: 10.1111/j.1365-2958.2005.04498.x. [DOI] [PubMed] [Google Scholar]
- Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci USA. 2008;105:4242–4246. doi: 10.1073/pnas.0711027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman SA, Wilson NF, Haas NA, Lefebvre PA. A novel MAP kinase regulates flagellar length in Chlamydomonas. Curr Biol. 2003;13:1145–1149. doi: 10.1016/s0960-9822(03)00415-9. [DOI] [PubMed] [Google Scholar]
- Besse L, Neti M, Anselme I, Gerhardt C, Ruther U, Laclef C, Schneider-Maunoury S. Primary cilia control telencephalic patterning and morphogenesis via Gli3 proteolytic processing. Development. 2011;138:2079–2088. doi: 10.1242/dev.059808. [DOI] [PubMed] [Google Scholar]
- Bishop GA, Berbari NF, Lewis J, Mykytyn K. Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain. J Comp Neurol. 2007;505:562–571. doi: 10.1002/cne.21510. [DOI] [PubMed] [Google Scholar]
- Breunig JJ, Sarkisian MR, Arellano JI, Morozov YM, Ayoub AE, Sojitra S, Wang B, Flavell RA, Rakic P, Town T. Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc Natl Acad Sci USA. 2008;105:13127–13132. doi: 10.1073/pnas.0804558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghoorn J, Dekkers MP, Rademakers S, de Jong T, Willemsen R, Jansen G. Mutation of the MAP kinase DYF-5 affects docking and undocking of kinesin-2 motors and reduces their speed in the cilia of Caenorhabditis elegans. Proc Natl Acad Sci USA. 2007;104:7157–7162. doi: 10.1073/pnas.0606974104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CS, Vogel TW, Zhang Q, Seo S, Swiderski RE, Moninger TO, Cassell MD, Thedens DR, Keppler-Noreuil KM, Nopoulos P, Nishimura DY, Searby CC, Bugge K, Sheffield VC. Abnormal development of NG2+ PDGFR-alpha+ neural progenitor cells leads to neonatal hydrocephalus in a ciliopathy mouse model. Nat Med. 2012;18:1797–1804. doi: 10.1038/nm.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizhikov VV, Davenport J, Zhang Q, Shih EK, Cabello OA, Fuchs JL, Yoder BK, Millen KJ. Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J Neurosci. 2007;27:9780–9789. doi: 10.1523/JNEUROSCI.5586-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho A, Ko HW, Eggenschwiler JT. FKBP8 cell-autonomously controls neural tube patterning through a Gli2- and Kif3a-dependent mechanism. Dev Biol. 2008;321:27–39. doi: 10.1016/j.ydbio.2008.05.558. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Czarnecki PG, Shah JV. The ciliary transition zone: from morphology and molecules to medicine. Trends Cell Biol. 2012;22:201–210. doi: 10.1016/j.tcb.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, Yoder BK. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol. 2007;17:1586–1594. doi: 10.1016/j.cub.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay C, Kennedy H. Cell-cycle control and cortical development. Nat Rev Neurosci. 2007;8:438–450. doi: 10.1038/nrn2097. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Dessaud E, McMahon AP, Briscoe J. Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development. 2008;135:2489–2503. doi: 10.1242/dev.009324. [DOI] [PubMed] [Google Scholar]
- Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- Fu Z, Schroeder MJ, Shabanowitz J, Kaldis P, Togawa K, Rustgi AK, Hunt DF, Sturgill TW. Activation of a nuclear Cdc2-related kinase within a mitogen-activated protein kinase-like TDY motif by autophosphorylation and cyclin-dependent protein kinase-activating kinase. Mol Cell Biol. 2005;25:6047–6064. doi: 10.1128/MCB.25.14.6047-6064.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z, Larson KA, Chitta RK, Parker SA, Turk BE, Lawrence MW, Kaldis P, Galaktionov K, Cohn SM, Shabanowitz J, Hunt DF, Sturgill TW. Identification of yin-yang regulators and a phosphorylation consensus for male germ cell-associated kinase (MAK)-related kinase. Mol Cell Biol. 2006;26:8639–8654. doi: 10.1128/MCB.00816-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes JM, Davis EE, Katsanis N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 2009;137:32–45. doi: 10.1016/j.cell.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S, Alvarez-Buylla A. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci. 2008;11:277–284. doi: 10.1038/nn2059. [DOI] [PubMed] [Google Scholar]
- Handel M, Schulz S, Stanarius A, Schreff M, Erdtmann-Vourliotis M, Schmidt H, Wolf G, Hollt V. Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience. 1999;89:909–926. doi: 10.1016/s0306-4522(98)00354-6. [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DH, Pawlyk B, Sokolov M, Strissel KJ, Yang J, Tulloch B, Wright AF, Arshavsky VY, Li T. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest Ophthalmol Vis Sci. 2003;44:2413–2421. doi: 10.1167/iovs.02-1206. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaka F, Ishibashi M, Taki W, Hashimoto N, Nakanishi S, Kageyama R. Ectopic expression of the bHLH gene Math1 disturbs neural development. Eur J Neurosci. 1999;11:2582–2588. doi: 10.1046/j.1460-9568.1999.00699.x. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Marshall WF. Ciliogenesis: building the cell's antenna. Nat Rev Mol Cell Biol. 2011;12:222–234. doi: 10.1038/nrm3085. [DOI] [PubMed] [Google Scholar]
- Kim J, Kato M, Beachy PA. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc Natl Acad Sci USA. 2009;106:21666–21671. doi: 10.1073/pnas.0912180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko HW, Norman RX, Tran J, Fuller KP, Fukuda M, Eggenschwiler JT. Broad-minded links cell cycle-related kinase to cilia assembly and hedgehog signal transduction. Dev Cell. 2010;18:237–247. doi: 10.1016/j.devcel.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiry P, Wang J, Robinson JF, Turowec JP, Litchfield DW, Lanktree MB, Gloor GB, Puffenberger EG, Strauss KA, Martens MB, Ramsay DA, Rupar CA, Siu V, Hegele RA. A multiplex human syndrome implicates a key role for intestinal cell kinase in development of central nervous, skeletal, and endocrine systems. Am J Hum Genet. 2009;84:134–147. doi: 10.1016/j.ajhg.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Pleasure SJ. Morphogenesis of the dentate gyrus: what we are learning from mouse mutants. Dev Neurosci. 2005;27:93–99. doi: 10.1159/000085980. [DOI] [PubMed] [Google Scholar]
- Louvi A, Grove EA. Cilia in the CNS: the quiet organelle claims center stage. Neuron. 2011;69:1046–1060. doi: 10.1016/j.neuron.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marley A, von Zastrow M. DISC1 regulates primary cilia that display specific dopamine receptors. PLoS ONE. 2010;5:e10902. doi: 10.1371/journal.pone.0010902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranishi Y, Terada K, Inoue T, Katoh K, Tsujii T, Sanuki R, Kurokawa D, Aizawa S, Tamaki Y, Furukawa T. An essential role for RAX homeoprotein and NOTCH-HES signaling in Otx2 expression in embryonic retinal photoreceptor cell fate determination. J Neurosci. 2011;31:16792–16807. doi: 10.1523/JNEUROSCI.3109-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139:663–678. doi: 10.1016/j.cell.2009.10.036. [DOI] [PubMed] [Google Scholar]
- Ocbina PJ, Eggenschwiler JT, Moskowitz I, Anderson KV. Complex interactions between genes controlling trafficking in primary cilia. Nat Genet. 2011;43:547–553. doi: 10.1038/ng.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori Y, Zhao C, Saras A, Mukhopadhyay S, Kim W, Furukawa T, Sengupta P, Veraksa A, Malicki J. Elipsa is an early determinant of ciliogenesis that links the IFT particle to membrane-associated small GTPase Rab8. Nat Cell Biol. 2008;10:437–444. doi: 10.1038/ncb1706. [DOI] [PubMed] [Google Scholar]
- Omori Y, Chaya T, Katoh K, Kajimura N, Sato S, Muraoka K, Ueno S, Koyasu T, Kondo M, Furukawa T. Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc Natl Acad Sci USA. 2010;107:22671–22676. doi: 10.1073/pnas.1009437108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Baker SA, Deane JA, Cole DG, Dickert BL, Rosenbaum JL, Witman GB, Besharse JC. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol. 2002;157:103–113. doi: 10.1083/jcb.200107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen LB, Miller MS, Geimer S, Leitch JM, Rosenbaum JL, Cole DG. Chlamydomonas IFT172 is encoded by FLA11, interacts with CrEB1, and regulates IFT at the flagellar tip. Curr Biol. 2005;15:262–266. doi: 10.1016/j.cub.2005.01.037. [DOI] [PubMed] [Google Scholar]
- Pedersen LB, Geimer S, Rosenbaum JL. Dissecting the molecular mechanisms of intraflagellar transport in chlamydomonas. Curr Biol. 2006;16:450–459. doi: 10.1016/j.cub.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Phirke P, Efimenko E, Mohan S, Burghoorn J, Crona F, Bakhoum MW, Trieb M, Schuske K, Jorgensen EM, Piasecki BP, Leroux MR, Swoboda P. Transcriptional profiling of C. elegans DAF-19 uncovers a ciliary base-associated protein and a CDK/CCRK/LF2p-related kinase required for intraflagellar transport. Dev Biol. 2011;357:235–247. doi: 10.1016/j.ydbio.2011.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Lin Y, Norman RX, Ko HW, Eggenschwiler JT. Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proc Natl Acad Sci USA. 2011;108:1456–1461. doi: 10.1073/pnas.1011410108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- Sakagami K, Gan L, Yang XJ. Distinct effects of Hedgehog signaling on neuronal fate specification and cell cycle progression in the embryonic mouse retina. J Neurosci. 2009;29:6932–6944. doi: 10.1523/JNEUROSCI.0289-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Miyazaki J. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem Biophys Res Commun. 1997;237:318–324. doi: 10.1006/bbrc.1997.7111. [DOI] [PubMed] [Google Scholar]
- Sato S, Inoue T, Terada K, Matsuo I, Aizawa S, Tano Y, Fujikado T, Furukawa T. Dkk3-Cre BAC transgenic mouse line: a tool for highly efficient gene deletion in retinal progenitor cells. Genesis. 2007;45:502–507. doi: 10.1002/dvg.20318. [DOI] [PubMed] [Google Scholar]
- Sotelo C. Cellular and genetic regulation of the development of the cerebellar system. Prog Neurobiol. 2004;72:295–339. doi: 10.1016/j.pneurobio.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Spassky N, Merkle FT, Flames N, Tramontin AD, Garcia-Verdugo JM, Alvarez-Buylla A. Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J Neurosci. 2005;25:10–18. doi: 10.1523/JNEUROSCI.1108-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassky N, Han YG, Aguilar A, Strehl L, Besse L, Laclef C, Ros MR, Garcia-Verdugo JM, Alvarez-Buylla A. Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev Biol. 2008;317:246–259. doi: 10.1016/j.ydbio.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Qu Y, Montcouquiol M, Zhou L, Komatsu K, Shi D, Fujimori T, Labeau J, Tyteca D, Courtoy P, Poumay Y, Uemura T, Goffinet AM. Lack of cadherins Celsr2 and Celsr3 impairs ependymal ciliogenesis, leading to fatal hydrocephalus. Nat Neurosci. 2010;13:700–707. doi: 10.1038/nn.2555. [DOI] [PubMed] [Google Scholar]
- Togawa K, Yan YX, Inomoto T, Slaugenhaupt S, Rustgi AK. Intestinal cell kinase (ICK) localizes to the crypt region and requires a dual phosphorylation site found in map kinases. J Cell Physiol. 2000;183:129–139. doi: 10.1002/(SICI)1097-4652(200004)183:1<129::AID-JCP15>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Tsao CC, Gorovsky MA. Different effects of Tetrahymena IFT172 domains on anterograde and retrograde intraflagellar transport. Mol Biol Cell. 2008;19:1450–1461. doi: 10.1091/mbc.E07-05-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujikawa M, Malicki J. Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron. 2004;42:703–716. doi: 10.1016/s0896-6273(04)00268-5. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dakubo GD, Thurig S, Mazerolle CJ, Wallace VA. Retinal ganglion cell-derived sonic hedgehog locally controls proliferation and the timing of RGC development in the embryonic mouse retina. Development. 2005;132:5103–5113. doi: 10.1242/dev.02096. [DOI] [PubMed] [Google Scholar]
- Wei Q, Zhang Y, Li Y, Zhang Q, Ling K, Hu J. The BBSome controls IFT assembly and turnaround in cilia. Nat Cell Biol. 2012;14:950–957. doi: 10.1038/ncb2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Lai CK, Evangelista M, Hongo JA, de Sauvage FJ, Scales SJ. Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol Cell Biol. 2010;30:1910–1922. doi: 10.1128/MCB.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willaredt MA, Hasenpusch-Theil K, Gardner HA, Kitanovic I, Hirschfeld-Warneken VC, Gojak CP, Gorgas K, Bradford CL, Spatz J, Wolfl S, Theil T, Tucker KL. A crucial role for primary cilia in cortical morphogenesis. J Neurosci. 2008;28:12887–12900. doi: 10.1523/JNEUROSCI.2084-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L, Robinson D, Ma AH, Chen HC, Wu F, Qiu Y, Kung HJ. Identification of human male germ cell-associated kinase, a kinase transcriptionally activated by androgen in prostate cancer cells. J Biol Chem. 2002;277:35422–35433. doi: 10.1074/jbc.M203940200. [DOI] [PubMed] [Google Scholar]
- Yang Y, Roine N, Makela TP. CCRK depletion inhibits glioblastoma cell proliferation in a cilium-dependent manner. EMBO Rep. 2013;14:741–747. doi: 10.1038/embor.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.