

Abstract
Histone deacetylase (HDAC) enzymes have been demonstrated as critical components in maintaining chromatin homeostasis, CNS development, and normal brain function. Evidence in mouse models links HDAC expression to learning, memory, and mood-related behaviors; small molecule HDAC inhibitor tool compounds have been used to demonstrate the importance of specific HDAC subtypes in modulating CNS-disease-related behaviors in rodents. So far, no direct evidence exists to understand the quantitative changes in HDAC target engagement that are necessary to alter biochemistry and behavior in a living animal. Understanding the relationship between target engagement and in vivo effect is essential in refining new ways to alleviate disease. We describe here, using positron emission tomography (PET) imaging of rat brain, the in vivo target engagement of a subset of class I/IIb HDAC enzymes implicated in CNS-disease (HDAC subtypes 1, 2, 3, and 6). We found marked differences in the brain penetrance of tool compounds from the hydroxamate and benzamide HDAC inhibitor classes and resolved a novel, highly brain penetrant benzamide, CN147, chronic treatment with which resulted in an antidepressant-like effect in a rat behavioral test. Our work highlights a new translational path for understanding the molecular and behavioral consequences of HDAC target engagement.
Keywords: Neuroimaging epigenetics, preclinical, translational, rodent, radiotracer
Histone deacetylase (HDAC) enzymes, particularly isoforms from class I (HDAC 1, 2, 3, and 8), class IIa (HDAC 4, 5, 7, and 9), and class IIb (HDAC 6 and 10), have been demonstrated as critical components in maintaining chromatin homeostasis, CNS development, and normal brain function. Altered HDAC expression has been linked to CNS diseases including Alzheimer’s disease, bipolar disorder, schizophrenia, and major depressive disorder via post-mortem human brain research.1−4 Modulating HDAC expression in mice has been shown to impact learning, memory, and mood-related behaviors.5−7 Further, small molecule HDAC inhibitor tool compounds have been used to demonstrate the importance of specific HDAC subtypes in modulating CNS-disease-related behaviors in rodents.5,8,9
However, only indirect evidence, obtained via invasive, destructive methods, exists to demonstrate the engagement of HDAC targets in brain. Inhibitor-induced changes in acetylation of histone proteins and altered gene expression from dissected brain tissue are related to HDAC function but do not provide information about the quantitative changes in HDAC target engagement necessary to alter biochemistry or behavior in a living animal.
We describe here, using positron emission tomography (PET) imaging of rat brain, the in vivo target engagement of a subset of class I/IIb HDAC enzymes implicated in regulating CNS-disease-related behaviors. Using a [11C]labeled probe that permits direct visualization of HDAC subtypes 1, 2, 3, and 6,10 we demonstrate evidence of the mechanism of HDAC inhibitors in brain with known and novel HDAC inhibitors from the hydroxamate and ortho-aminoanilide chemical classes.
Understanding the relationship between target engagement and in vivo effect is essential in refining new ways to alleviate disease. This concept is established for dopamine D2-receptor drug studies, where binding measured by the D2/D3 probe raclopride (among many others) has facilitated dose-finding for human trials and provided insight into the phamacodynamic origins of clinical dopaminergic drug effects.11−13 Here, our work highlights a new translational path for understanding the molecular and behavioral consequences of HDAC target engagement.
Results
The target engagement of HDAC subtypes 1, 2, 3, and 6 in living rodent brain has not yet been evaluated, impeding the interpretation of treatment effect relationships for myriad HDAC inhibitors in preclinical research. Therefore, we used [11C]martinostat, a radiotracer that we have recently described10 with robust potency and selectivity for a subset of HDACs that make it an excellent in vivo PET probe (Figure 1 A and synthesis Scheme S1, Supporting Information). We first measured “baseline” tracer uptake and averaged binding in whole brain in vehicle-treated rats (n = 9). Dynamic uptake is represented in the averaged time–activity curve (Figure 2B), with raw data scaled to 100% whole brain uptake at time = 600 s post-[11C]martinostat administration. Time–activity curve profiles were consistent across all scans, with percent radiotracer bound declining from a scaled mean of 100% (t = 600 s) to 95% ± 2.2% (t = 3600 s, mean ± SEM). The average variance for this time range was 12.4%.
Figure 1.

[11C]Martinostat and HDAC inhibitor tool compounds. (A) [11C]-labeled martinostat is a versatile HDAC imaging probe characterized by (i) potent induction of histone acetylation in cells (EC50 = 100 nM for histone H3 lysine 9 acetylation and histone H4 lysine 12 acetylation compared with >1900 for SAHA) and (ii) robust selectivity for HDAC subtypes 1, 2, 3, and 6 (IC50 = 0.3–4.0 nM) in a recombinant human enzyme assay. (B) Hydroxamate HDAC inhibitor tool compounds. (C) ortho-Aminoanilide tool compounds and the short-chain fatty acid, valproic acid.
Figure 2.

Uptake of [11C]martinostat in rat brain. (A) Timeline schematic of blocking dose administration and PET/CT imaging. All animals were stabilized on anesthesia at least 20 min prior to [11C]martinostat administration. (B) Dynamic tracer uptake in whole brain was evaluated using ROI analysis on dynamic imaging data sets from n = 9 “baseline” rats blocked with vehicle (10% DMSO, 10% Tween 80, 80% saline) 5 min prior to tracer administration. Data are expressed as percent uptake in whole brain relative to uptake at time = 600 s (mean ± standard deviation). (C) Self-blocking of [11C]martinostat tracer binding (change in % of whole brain [11C] uptake) is demonstrated via pretreatment (5 min, iv) with a dose range (0.001–1 mg/kg, n = 1/group or 2.0 mg/kg, n = 3/group) of unlabeled martinostat and compared with baseline controls, described in panel B. (D) Quantification of [11C] activity in whole brain at time 3600 s measured via trend in accumulated radioactivity from time 10 min to time 60 min for baseline (white bar, 0 mg/kg) and each blocking condition (grayscale). (E) Spearman correlation with directional t test identified significant dose–response relationships for relative [11C] blockade in whole brain in rat (gray triangles, r = 0.89; p = 0.006) and whole-brain volume of distribution (VT) from analogous PET experiments in nonhuman primate (NHP; white squares, r = 0.90; p = 0.042); * indicates blocking doses scaled for NHP equivalent dose (mg/kg). (F) Pretreatment with unlabled martinostat (1 mg/kg) 5 min, 4 h, or 24 h prior to tracer administration demonstrated time-dependent effects of target engagement, calculated as difference from baseline (100% – % blocked binding).
Specific target binding was demonstrated with self-blocking experiments. Radioprobe bound in whole brain was decreased following intravenous (iv) pretreatment with a dose range of unlabeled ([12C])martinostat, 5 min prior to radiotracer administration (Figure 2C). Blocked binding of the radiotracer by the nonradioactive martinostat was quantified by measuring the change in the radioactivity accumulated from time 600 s to time 3600 s, and demonstrated tracer binding was reduced dose-dependently from 86.7% to 36.5% of baseline levels by pretreatment with martinostat at 0.001–2 mg/kg. (Figure 2D). We measured the blocked binding with replicate experiments at the 2 mg/kg pretreatment dose of martinostat and resolved good reproducibility from three independent experiments (36.5% ± 1.75%, mean ± SEM, Figure 2D), with an average variance over the time range of 10.7%, consistent with the baseline group. Formal comparison by Student’s t test revealed a significant difference in radiotracer binding between rats pretreated with 2 mg/kg martinostat and baseline controls (Figure 2D, *p = 2.0 × 10–8). Given the similarity in variance at low (baseline) and high (2 mg/kg self-blocking) measures of HDAC target engagement, we performed power analysis calculations (two-sample z test) using a representative standard deviation of 6.2% (Supportive Information, Table S1). These results showed that for blocked binding measurements ≤85% of baseline, statistical significance could be achieved with ≤3 replicates, a feasible group size for preclinical imaging throughput. Moreover, we calculated that blocked binding measurements ≤92.5% of baseline could resolve statistically significant differences using a higher, but achievable 15 animals/group. Therefore, we hereafter considered blocked binding experiments demonstrating ≤92.5% of baseline tracer uptake in brain as positive initial evidence for measurable target engagement.
Quantification of absolute radiotracer uptake via total distribution volume (VT) analysis requires normalization to full arterial input function from blood sampling and was not feasible in rats. However, VT quantification was achieved at baseline and in a range of self-blocking doses in analogous experiments conducted in nonhuman primates (NHPs) (described in detail in a forthcoming manuscript). We found that the dose-dependent decreases observed in NHP VT were significantly correlated (Figure 2E, white squares, r = 0.90; p = 0.042). We also observed a significant correlation by the same test using scaled whole brain uptake levels of total [11C] activity in rat (Figure 2E, gray triangles, r = 0.89; p = 0.006). This comparison gave confidence that in rat, in the absence of a blood input function, relative HDAC target binding could be resolved using the time–activity curve slope, given the unique tracer kinetics of [11C]martinostat, which show a relatively nondynamic time–activity curve within the scan duration.
Using this method, we then varied the pretreatment time for nonradioactive martinostat (1 mg/kg) from 5 min to 4 h and 24 h before radiotracer administration. Target binding was blocked most (40% reduction) after a short, 5 min, blocking dose pretreatment (Figure 2F) with a lesser blockade observed 4 h after pretreatment and no appreciable change observed 24 h after nonradioactive martinostat pretreatment (n = 1 rat/blocking condition; 5 min time point same data as in Figure 2C,D.) These data demonstrate a relative peak of binding of the nonradioactive martinostat at the 5 min pretreatment time point and are consistent with the established, fast-binding kinetics of hydroxamate HDAC inhibitors like martinostat.
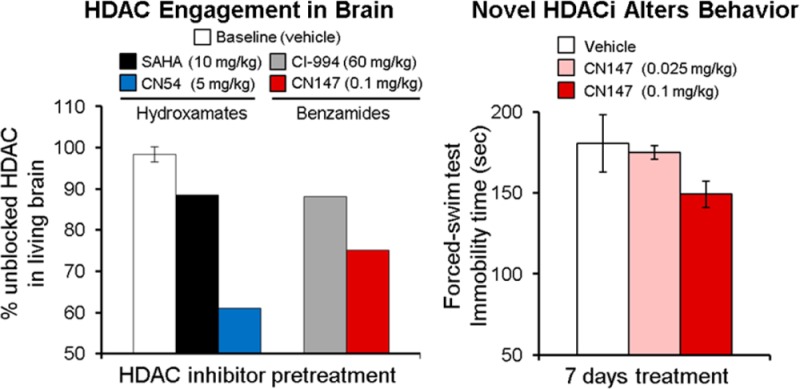
The hydroxamic acid class of HDAC inhibitors has been reported to modulate behavioral response in CNS-disease-related paradigms in rodents; however the extent of target engagement in the brain has not yet been demonstrated. Therefore, we next applied the metric of [11C]martinostat target binding in whole rat brain to investigate the relative HDAC1–3 and 6 engagement of structurally distinct hydroxamate HDAC inhibitors (structures shown in Figure 1B). Anesthetized rats were pretreated with known and novel hydroxamate HDAC inhibitors as indicated (iv pretreatement, 3–10 min prior to tracer administration). Quantification of dynamic imaging data revealed that the hydroxamates SAHA and givinostat both resulted in only subtle blockade of radiotracer binding in whole brain (Figure 3A), consistent with recent reports demonstrating poor brain penetrance of the prototypical hydroxamic acid HDAC inhibitor, SAHA.14,15 Poor brain penetrance was overcome in the development of martinostat by the addition of an adamantyl group, a chemical moiety frequently used developing in CNS-penetrant compounds.10 Therefore, we tested PK5 and PK6, two hydroxamate HDAC inhibitor compounds containing heterocyclic capping groups often found in CNS drugs.16 We found that pretreatment resulted in a modest ∼20% decrease in whole brain target binding of [11C]martinostat (n = 2 replicates/condition, mean ± SEM). Further, we quantified a 40% reduction in whole brain target binding of [11C]martinostat following pretreatment with CN54, a heterocycle-capped hydroxamate HDAC inhibitor identified from the patent literature base17 that showed a robust 4.5:1 ratio of brain/plasma partitioning after iv treatment. Taken together, these data support that [11C]martinostat binding can quantify target engagement of structurally distinct, brain-penetrant hydroxamate HDAC inhibitors in living rat brain.
Figure 3.

HDAC inhibitor pretreatment blocks [11C]martinostat uptake in brain. (A) Pretreatment with hydroxamic acids (iv) resulted in a 10–14% blockade of tracer uptake in whole brain for the known hydroxamic acids, SAHA and givinostat (n = 1/group), with greater, 18–22%, blockade for the two novel compounds, PK5 and PK6 (n = 2/group; mean ± SEM). Imaging experiments revealed robust 40% blockade of whole brain [11C] levels by the hydroxamic acid, CN54 (n = 1/group). (B) Pretreatment with the protypical orthoaminoanilide benzamide, CI-994, via ip injection revealed modest blockade (9–14%) with limited impact of dose (15–60 mg/kg) acute pretreatment blocking time (2.5–4 h) or duration of treatment (7 days treatment with final treatment 4 h prior to tracer administration). (C) Pretreatment (ip) with the HDAC subtype-selective benzamides Cpd60 and RGFP966 also resulted in limited, 7–10%, blockade of [11C]martinostat binding in whole brain. A 500 mg/kg ip dose of sodium valproate, administered 30 min prior to tracer administration blocked uptake by 16%. (D) Brain penetrance of novel benzamides was evaluated via blocked tracer binding for the tool compound CN147. CN147 demonstrated robust 23% tracer blockade at 5 mg/kg (ip, 2.5 h pretreatment). At 1 mg/kg (ip), maximal blocking effects were observed 4 h after pretreatment and were maintained at a lower dose of 0.1 mg/kg. Consistent with the slow-binding kinetics of benzamide HDAC inhibitors, no appreciable blockade was observed following a short, 5 min, pretreatment time with CN147, 5 mg/kg, ip (data not shown).
HDAC inhibitor tool compounds from the ortho-aminoanilide (OAA) class (Figure 1C) have been demonstrated to selectively inhibit a subset of class I HDAC isoforms and as a result have emerged as a way to evaluate selective inhibition of specific HDAC isoforms in the CNS.5,8,9,18,19 We investigated the impact of benzamide HDAC inhibitor treatment on [11C]martinostat target binding in whole brain. Given the slow kinetics described for members of the OAA class of HDAC inhibitors,9,18 we administered compounds via intraperitoneal (ip) injection 2.5–7.5 h prior to tracer administration. Treatment with the prototypical benzamide-based OAA HDAC inhibitor, CI-994, a selective inhibitor for HDAC subtypes 1, 2, and 3 recently shown to enhance memory reconsolidation in mice,5 revealed that radiotracer binding was reduced by <14% (Figure 3B). The reduction in target binding was not altered by CI-994 dose (15, 30, or 60 mg/kg) or by protracted treatment (Figure 3B, striped bar; 15 mg/kg, 7 days of daily ip treatment with an additional treatment on the day of PET imaging, 4 h before tracer administration).
Radiotracer target binding in whole brain was also blocked, at 7–10%, by Cpd60 and RGFP966, two selective OAA HDAC inhibitors previously shown to modulate behavior in rodents.8,9 These two compounds differ in their reported selectivity for recombinant HDACs with Cpd60 demonstrated as a selective nanomolar inhibitor of HDAC 1 and 29 and RGFP966 reported as selective for HDAC3.8 We observed a more robust 16% reduction in basal HDAC target engagement following pretreatment with 500 mg/kg of sodium valproate, a short-chain fatty acid containing anticonvulsant and mood stabilizer with selective class I HDAC inhibitory properties (Figure 3C).20,21 These data are consistent with prior findings indicating limitations for brain penetrance of OAA as a compound class.9,22,23 They also support the notion that valproic acid may function as an HDAC inhibitor in vivo within the dose ranges that result in behavioral effects.24−26
To test whether benzamide brain penetrance could be improved using a strategy of adamantane incorporation, we synthesized the novel compound CN147 (MW 429.3, Figure 1C and synthesis in Scheme S2, Supporting Information). In HDAC biochemical assays with recombinant human enzymes, this compound demonstrated HDAC selectivity (HDAC 1, 2, 3) and potency (IC50 150–600 nM) similar to CI-994, a close structural homologue. We then evaluated target engagement in brain by [11C]martinostat PET imaging. We identified a 25% blockade of tracer uptake that varied by dose (5, 1, or 0.1 m/kg) and pretreatment time (ip 2.5, 4, or 7.5 h prior to tracer administration; Figure 3D, solid grayscale bars).
Given the robust HDAC engagement indicated for CN147 by our PET data, we wanted to confirm presence in brain using an independent method. Therefore, we obtained pharmacokinetic data for CN147 (1 mg/kg) in rat brain tissue and in blood using established (invasive and destructive) LC-MS/MS methodology (Figure 4A).27 The brain pharmacokinetic profile for CN147 revealed a Cmax of 315 nM, a half-life of 6.8 h, and that the compound was present in the brain 8–24 h after ip or iv administration, a result similar to that reported for other benzamides.9,28 The brain to plasma exposure profile for CN147 was in marked contrast to that obtained for CI-994 (brain/plasma ratio 1:10), as measured by the same method, following a 10 mg/kg ip treatment (Figure 4B). The robust 20:1 brain partitioning of CN147 compared with CI-994 was further illustrated by coplotting brain/plasma ratios for (Figure 4C).
Figure 4.
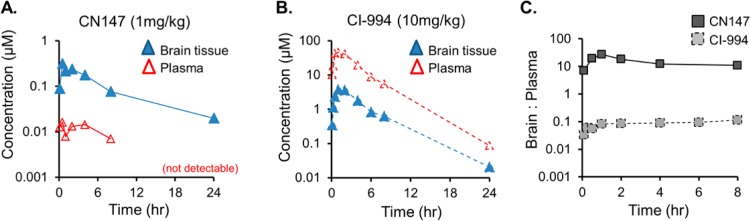
Pharmacokinetics in rat brain and plasma. Acute treatment following single systemic treatment with (A) CN147 (1 mg/kg) or (B) CI-994 (10 mg/kg) was used to evaluate the concentration of each compound by LC-MS/MS in plasma (red) or brain tissue (blue) 5 min to 24 h after administration using n = 3 rats per compound per time point. (C) Brain/plasma ratios for CN147 and CI-994 demonstrate dramatic differences in CNS vs systemic exposure.
A growing number of reports have described the beneficial impact of OAA-based HDAC inhibitors in CNS-relevant rodent behavioral tests.5,8,9 On the basis of the promising brain penetration data and evidence for HDAC target engagement from our PET imaging, we prioritized CN147 for evaluating the impact of HDAC inhibition on behavior. As a first step to provide evidence of behavioral changes induced by CN147, we chose the modified forced swim test because another OAA, Cpd60 was recently demonstrated to be ameliorative in the mouse FST after 7–10 days systemic treatment at 45 mg/kg.9 Given that the brain/plasma exposure of Cpd60 was 0.5 (1:20) in mice after a single 45 mg/kg ip dose, we rationalized (equilibrating for species differences in body surface area)29 that a dose of CN147 in rat of 0.025–1 mg/kg would result in brain exposure close to that achieved by Cpd60 in mice.
We observed an antidepressant-like effect after 7 days ip treatment with 0.1 mg/kg CN147 treatment compared with vehicle-treated controls (two-tailed Student’s t test p = 0.16, n = 4/treatment group; Figure 5A). Rats treated with 0.1 mg/kg CN147 showed a 17% reduction in FST immobility time compared with controls, albeit not significant by Student’s t test (p = 0.17). Notably, comparison of the reduced FST immobility times (0.1 mg/kg) with rats treated at a dose of 0.025 mg/kg (n = 4/group) revealed a significant difference (p = 0.035). Further, regression analysis by Spearman correlation indicated immobility time and CN147 dose (0, 0.025, or 0.1 mg/kg) were significantly correlated (r = −0.6072; two-tailed p = 0.0196), supporting the observed antidepressant-like effects by chronic 0.1 mg/kg CN147 treatment (Figure 5A). We confirmed our ability to detect reduced FST immobility in an independent cohort of rats using an established subchronic administration of the antidepressant imipramine, which significantly reduced immobility times (104 ± 3.2 s, mean ± SEM) compared with rats injected with vehicle (185.7 ± 19.8 s; n = 3 rats/treatment group; Student’s t test p = 0.014).
Figure 5.

Initial behavioral impact of the novel brain penetrant HDAC inhibitor, CN147. (A) Chronic (7 days) treatment with CN147 at 0.1 mg/kg resulted in a 17% reduction in forced-swim test immobility compared with vehicle-treated controls as evaluated by a trained scorer blinded to treatment groups. Treatment with CN147 at a lower dose (0.025 mg/kg) had no appreciable effect on FST immobility score compared with controls, and analysis by Spearman correlation revealed that CN147 dose and FST immobility times were significantly correlated (Spearman r = −0.0671, two-tailed p = 0.0192). (B) Locomotor effects of CN147 treatment were evaluated after 7 days treatment with 0.5 mg/kg or 2.0 mg/kg compared with vehicle-treated controls; distance traveled (cm) was lower in CN147-treated rats than in control animals and was not considered a confound to interpretation of decreased immobility observed in the forced-swim test. No treatment-group differences were observed in rat weight gain (data not shown).
Locomotor activity in a novel open field was evaluated in the same vehicle/CN147-treated rats, following a subsequent one-week treatment paradigm with vehicle or a 20-fold increased dose of CN147 (treatment group assignments were increased in dose, but not scrambled). Results from a 10 min session showed that rats treated with CN147 had a lower distance traveled in a novel open field compared with vehicle-treated controls. The locomotor activity that we observed in vehicle-treated rats during the 10 min session was consistent with previously reported independent results for the same strain.30 This result clarifies that CN147 treatment did not induce locomotor stimulatory effects and, thus, did not confound the observed antidepressant-like effect in the FST (Figure 5B). Furthermore, it suggests that CN147, and by inference class I HDACs, may impact neurocircuits involved in both mood and anxiety-related behavior.31
Discussion
We demonstrate here the immediate preclinical utility of investigating the brain uptake and in vivo target engagement of known and novel small molecule HDAC inhibitors. Increasing reports indicate the therapeutic benefit of HDAC inhibitor compounds in preclinical models, including behavioral testing of wild-type rodents,9,32,33 in ameliorating behavioral deficits in a genetic5,6 or etiological (stress) model of brain disease.34 Until now, no tool has been available to profile the density or engagement of HDAC targets in such models. This, in part, has impeded translational studies of novel HDAC inhibitor drugs in humans with CNS dysfunction, despite the fact that HDAC density changes have been reported in post-mortem human brain.1−4 We begin here to understand the relationship between HDAC density and occupancy and disease and treatment related behaviors in a living animal.
The expression of HDACs in the brain is robust and widespread,35,36 and the optimal reference region to evaluate nonspecific binding of [11C]martinostat has not yet been resolved due to the recent development of this tool. We have shown that relative measures of binding can be made in the absence of a reference region or blood sampling, providing a readily applicable HDAC imaging tool for rodents.
Identifying lead HDAC inhibitor molecules in preclinical and translational research can be catalyzed using PET imaging with [11C]martinostat, which demonstrated a high proportion of specific binding, with more than 60% of probe binding in brain prevented in homologous ([12C]martinostat) and heterologous (CN54) in vivo blocking experiments.
Our imaging results highlight a novel, highly brain penetrant HDAC inhibitor, CN147, which demonstrated an antidepressant-like effect in the forced swim test after chronic treatment. OAA compounds with similar features have been previously shown to result in beneficial changes in preclinical behavioral responses relevant to mood and memory disorders, including Cpd60, CI-994, and RGFP966.5,8,9 Investigating the impact of CN147 in additional behavioral paradigms and disease models will further clarify the role of HDAC targets in the brain. Additionally, it will be important to investigate the behavioral impact of other novel HDAC inhibitors to clarify whether a low level of HDAC target occupancy in CNS is sufficient to induce meaningful molecular and behavioral changes, which is relevant for current efforts to determine whether HDACs can be targeted for beneficial therapeutic effects in a range of CNS disorders.37−40
One potential explanation for a mechanism by which low engagement of HDAC targets could differentially result in such profound changes at the level of behavior is inhibitor binding selectivity at the level of HDAC multiprotein complexes. HDAC subtypes, particularly HDAC 1, 2, and 3, are members of distinct protein complexes. Efficient engagement of HDAC inhibitors to HDAC targets in vivo likely depends in part on other components of HDAC protein complexes and, potentially, on activity-dependent conformational changes.41 Therefore, it is fully possible that in vivo, OAA HDAC inhibitors only bind to class I HDAC targets in a subset of protein complexes. Further work using isolation of endogenous HDAC complexes from specific tissues of interest like the brain along with detailed analysis of selectivity and potency in combination with the use of autoradiography methods36 will likely provide an additional layer of clarification and complexity to the impact of HDAC inhibitors on transcription, neural signaling, and brain output. In doing so, this strategy holds promise to facilitate improvements in HDAC radiotracer development and the application of neuroimaging to novel therapeutic development efforts.
Acknowledgments
This research was carried out at the Athinoula A. Martinos Center for Biomedical Imaging at the Massachusetts General Hospital, using resources provided by the Center for Functional Neuroimaging Technologies, P41EB015896, a P41 Regional Resource supported by the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health. This work also involved the use of instrumentation supported by the NIH Shared Instrumentation Grant Program or High-End Instrumentation Grant Program, specifically, Grant Numbers S10RR017208, S10RR026666, S10RR022976, S10RR019933, and S10RR029495. We thank members of the Hooker and Haggarty laboratories for helpful discussions. We especially thank the radiopharmacy team and imaging support staff who enabled all imaging experiments.
Glossary
Abbreviations
- HDAC
histone deacetylase
- PET
positron emission tomography
- OAA
ortho-aminioanilide
- iv
intravenous
- ip
intraperitoneal
- SEM
standard error of the mean
- NHP
nonhuman primate
- VT
total volume of distribution
- FST
forced swim test
Supporting Information Available
Materials and methods, chemical synthesis schemes and characterization for martinostat and CN147 and statistical power analyses are included. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Imaging experiments were contributed by F.A.S., C.W., G.C.V., H.Y.W., R.N., W.R.T., S.J.H., and J.M.H. Chemistry and radiochemistry were contributed by C.W., R.N., A.K., E.B.H., S.J.H., and J.M.H. Biochemistry and in vitro assays were contributed to by Y.L.Z., J.G., S.A.R., F.A.S., E.B.H., and S.J.H. Behavioral experiments were contributed by F.A.S., G.C.V., S.J.H., and J.M.H. F.A.S. drafted the manuscript with contributions from C.W., G.C.V., H.Y.W., J.G., W.R.T., S.J.H. and J.M.H.
Research was supported by the National Institute of Drug Abuse (NIDA) of the National Institutes of Health under Grant Number R01DA030321 (J.M.H.; S.J.H) with additional support under Grant R01DA028301 (S.J.H.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Graff J.; Rei D.; Guan J. S.; Wang W. Y.; Seo J.; Hennig K. M.; Nieland T. J.; Fass D. M.; Kao P. F.; Kahn M.; Su S. C.; Samiei A.; Joseph N.; Haggarty S. J.; Delalle I.; Tsai L. H. (2012) An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington H. E. 3rd; Maze I.; LaPlant Q. C.; Vialou V. F.; Ohnishi Y. N.; Berton O.; Fass D. M.; Renthal W.; Rush A. J. 3rd; Wu E. Y.; Ghose S.; Krishnan V.; Russo S. J.; Tamminga C.; Haggarty S. J.; Nestler E. J. (2009) Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 29, 11451–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes F. M.; Lim B.; Matzilevich D.; Walsh J. P.; Subburaju S.; Minns M. (2007) Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc. Natl. Acad. Sci. U. S. A. 104, 10164–10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R. P.; Grayson D. R.; Gavin D. P. (2008) Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: analysis of the National Brain Databank microarray collection. Schizophr. Res. 98, 111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J.; Joseph N. F.; Horn M. E.; Samiei A.; Meng J.; Seo J.; Rei D.; Bero A. W.; Phan T. X.; Wagner F.; Holson E.; Xu J.; Sun J.; Neve R. L.; Mach R. H.; Haggarty S. J.; Tsai L. H. (2014) Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell 156, 261–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J. S.; Haggarty S. J.; Giacometti E.; Dannenberg J. H.; Joseph N.; Gao J.; Nieland T. J.; Zhou Y.; Wang X.; Mazitschek R.; Bradner J. E.; DePinho R. A.; Jaenisch R.; Tsai L. H. (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakovcevski M.; Bharadwaj R.; Straubhaar J.; Gao G.; Gavin D. P.; Jakovcevski I.; Mitchell A. C.; Akbarian S. (2013) Prefrontal cortical dysfunction after overexpression of histone deacetylase 1. Biol. Psychiatry 74, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M.; McQuown S. C.; Rogge G. A.; Astarabadi M.; Jacques V.; Carreiro S.; Rusche J. R.; Wood M. A. (2013) HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. U. S. A. 110, 2647–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder F. A.; Lewis M. C.; Fass D. M.; Wagner F. F.; Zhang Y. L.; Hennig K. M.; Gale J.; Zhao W. N.; Reis S.; Barker D. D.; Berry-Scott E.; Kim S. W.; Clore E. L.; Hooker J. M.; Holson E. B.; Haggarty S. J.; Petryshen T. L. (2013) A selective HDAC 1/2 inhibitor modulates chromatin and gene expression in brain and alters mouse behavior in two mood-related tests. PLoS One 8, e71323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Schroeder F. A.; Wey H. Y.; Borra R.; Wagner F. F.; Reis S. A.; Kim S. W.; Holson E. B.; Haggarty S. J.; Hooker J. M. (2014) In vivo imaging of histone deacetylases (HDACs) in the central nervous system and major peripheral organs. J. Med. Chem. 10.1021/jm500872p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P. (2014) Clozapine, a fast-off-D2 antipsychotic. ACS Chem. Neurosci. 5, 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P.; Tallerico T. (1999) Rapid release of antipsychotic drugs from dopamine D2 receptors: An explanation for low receptor occupancy and early clinical relapse upon withdrawal of clozapine or quetiapine. Am. J. Psychiatry 156, 876–884. [DOI] [PubMed] [Google Scholar]
- Volkow N. D.; Tomasi D.; Wang G. J.; Logan J.; Alexoff D. L.; Jayne M.; Fowler J. S.; Wong C.; Yin P.; Du C. (2014) Stimulant-induced dopamine increases are markedly blunted in active cocaine abusers. Mol. Psychiatry 10.1038/mp.2014.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson J. E.; La H.; Plise E.; Chen Y. H.; Ding X.; Hanania T.; Sabath E. V.; Alexandrov V.; Brunner D.; Leahy E.; Steiner P.; Liu L.; Scearce-Levie K.; Zhou Q. (2013) SAHA enhances synaptic function and plasticity in vitro but has limited brain availability in vivo and does not impact cognition. PLoS One 8, e69964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Eessalu T. E.; Barth V. N.; Mitch C. H.; Wagner F. F.; Hong Y.; Neelamegam R.; Schroeder F. A.; Holson E. B.; Haggarty S. J.; Hooker J. M. (2013) Design, synthesis, and evaluation of hydroxamic acid-based molecular probes for in vivo imaging of histone deacetylase (HDAC) in brain. Am. J. Nucl. Med. Mol. Imaging 4, 29–38. [PMC free article] [PubMed] [Google Scholar]
- Binaschi M.; Boldetti A.; Gianni M.; Maggi C. A.; Gensini M.; Bigioni M.; Parlani M.; Giolitti A.; Fratelli M.; Valli C.; Terao M.; Garattini E. (2010) Antiproliferative and differentiating activities of a novel series of histone deacetylase inhibitors. ACS Med. Chem. Lett. 1, 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers K., Patzke H. (2009) Methods for treating cognitive disorders using inhibitors of histone deacetylase, International patent WO2009137462 A2.
- Chou C. J.; Herman D.; Gottesfeld J. M. (2008) Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J. Biol. Chem. 283, 35402–35409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot J. L.; Hoffman D. M.; Witter D. J.; Stanton M. G.; Harrington P.; Hamblett C.; Siliphaivanh P.; Wilson K.; Hubbs J.; Heidebrecht R.; Kral A. M.; Ozerova N.; Fleming J. C.; Wang H.; Szewczak A. A.; Middleton R. E.; Hughes B.; Cruz J. C.; Haines B. B.; Chenard M.; Kenific C. M.; Harsch A.; Secrist J. P.; Miller T. A. (2014) Delayed and Prolonged Histone Hyperacetylation with a Selective HDAC1/HDAC2 Inhibitor. ACS Med. Chem. Lett. 5, 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fass D. M.; Shah R.; Ghosh B.; Hennig K.; Norton S.; Zhao W. N.; Reis S. A.; Klein P. S.; Mazitschek R.; Maglathlin R. L.; Lewis T. A.; Haggarty S. J. (2011) Short-Chain HDAC Inhibitors Differentially Affect Vertebrate Development and Neuronal Chromatin. ACS Med. Chem. Lett. 2(1), 39–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel C. J.; Zhang F.; Huang E. Y.; Guenther M. G.; Lazar M. A.; Klein P. S. (2001) Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 276, 36734–36741. [DOI] [PubMed] [Google Scholar]
- Hooker J. M.; Kim S. W.; Alexoff D.; Xu Y.; Shea C.; Reid A.; Volkow N.; Fowler J. S. (2010) Histone deacetylase inhibitor, MS-275, exhibits poor brain penetration: PK studies of [C]MS-275 using Positron Emission Tomography. ACS Chem. Neurosci. 1, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y. J.; Kang Y.; Muench L.; Reid A.; Caesar S.; Jean L.; Wagner F.; Holson E.; Haggarty S. J.; Weiss P.; King P.; Carter P.; Volkow N. D.; Fowler J. S.; Hooker J. M.; Kim S. W. (2014) Image-guided synthesis reveals potent blood-brain barrier permeable histone deacetylase inhibitors. ACS Chem. Neurosci. 5, 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong E.; Chen Y.; Gavin D. P.; Grayson D. R.; Guidotti A. (2010) Valproate induces DNA demethylation in nuclear extracts from adult mouse brain. Epigenetics 5, 730–735. [DOI] [PubMed] [Google Scholar]
- Ralph-Williams R. J.; Paulus M. P.; Zhuang X.; Hen R.; Geyer M. A. (2003) Valproate attenuates hyperactive and perseverative behaviors in mutant mice with a dysregulated dopamine system. Biol. Psychiatry 53, 352–359. [DOI] [PubMed] [Google Scholar]
- Shephard R. A.; Stevenson D.; Jenkinson S. (1985) Effects of valproate on hyponeophagia in rats: Competitive antagonism with picrotoxin and non-competitive antagonism with RO 15-1788. Psychopharmacology (Berlin, Ger.) 86, 313–317. [DOI] [PubMed] [Google Scholar]
- Fass D. M.; Reis S. A.; Ghosh B.; Hennig K. M.; Joseph N. F.; Zhao W. N.; Nieland T. J.; Guan J. S.; Kuhnle C. E.; Tang W.; Barker D. D.; Mazitschek R.; Schreiber S. L.; Tsai L. H.; Haggarty S. J. (2013) Crebinostat: A novel cognitive enhancer that inhibits histone deacetylase activity and modulates chromatin-mediated neuroplasticity. Neuropharmacology 64, 81–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder F. A.; Chonde D. B.; Riley M. M.; Moseley C. K.; Granda M. L.; Wilson C. M.; Wagner F. F.; Zhang Y. L.; Gale J.; Holson E. B.; Haggarty S. J.; Hooker J. M. (2013) FDG-PET imaging reveals local brain glucose utilization is altered by class I histone deacetylase inhibitors. Neurosci. Lett. 550, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan-Shaw S.; Nihal M.; Ahmad N. (2008) Dose translation from animal to human studies revisited. FASEB J. 22, 659–661. [DOI] [PubMed] [Google Scholar]
- Mechan A. O.; Moran P. M.; Elliott M.; Young A. J.; Joseph M. H.; Green R. (2002) A comparison between Dark Agouti and Sprague-Dawley rats in their behaviour on the elevated plus-maze, open-field apparatus and activity meters, and their response to diazepam. Psychopharmacology (Berlin, Ger.) 159, 188–195. [DOI] [PubMed] [Google Scholar]
- Fass D. M.; Schroeder F. A.; Perlis R. H.; Haggarty S. J. (2014) Epigenetic mechanisms in mood disorders: Targeting neuroplasticity. Neuroscience 264, 112–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy P. J.; Feng J.; Robison A. J.; Maze I.; Badimon A.; Mouzon E.; Chaudhury D.; Damez-Werno D. M.; Haggarty S. J.; Han M. H.; Bassel-Duby R.; Olson E. N.; Nestler E. J. (2013) Class I HDAC inhibition blocks cocaine-induced plasticity by targeted changes in histone methylation. Nat. Neurosci. 16, 434–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W. Y.; Kim S.; Kim J. H. (2008) Chronic microinjection of valproic acid into the nucleus accumbens attenuates amphetamine-induced locomotor activity. Neurosci. Lett. 432, 54–57. [DOI] [PubMed] [Google Scholar]
- Espallergues J.; Teegarden S. L.; Veerakumar A.; Boulden J.; Challis C.; Jochems J.; Chan M.; Petersen T.; Deneris E.; Matthias P.; Hahn C. G.; Lucki I.; Beck S. G.; Berton O. (2012) HDAC6 regulates glucocorticoid receptor signaling in serotonin pathways with critical impact on stress resilience. J. Neurosci. 32, 4400–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broide R. S.; Redwine J. M.; Aftahi N.; Young W.; Bloom F. E.; Winrow C. J. (2007) Distribution of histone deacetylases 1–11 in the rat brain. J. Mol. Neurosci. 31, 47–58. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang Y. L.; Hennig K.; Gale J. P.; Hong Y.; Cha A.; Riley M.; Wagner F.; Haggarty S. J.; Holson E.; Hooker J. (2013) Class I HDAC imaging using [ (3)H]CI-994 autoradiography. Epigenetics 8, 756–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A.; Sananbenesi F.; Mungenast A.; Tsai L. H. (2010) Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol. Sci. 31, 605–617. [DOI] [PubMed] [Google Scholar]
- Graff J.; Tsai L. H. (2013) The potential of HDAC inhibitors as cognitive enhancers. Annu. Rev. Pharmacol. Toxicol. 53, 311–330. [DOI] [PubMed] [Google Scholar]
- Haggarty S. J.; Tsai L. H. (2011) Probing the role of HDACs and mechanisms of chromatin-mediated neuroplasticity. Neurobiol. Learn. Mem. 96, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiwer M.; Lewis M. C.; Wagner F. F.; Holson E. B. (2013) Therapeutic potential of isoform selective HDAC inhibitors for the treatment of schizophrenia. Future Med. Chem. 5, 1491–1508. [DOI] [PubMed] [Google Scholar]
- Becher I.; Dittmann A.; Savitski M. M.; Hopf C.; Drewes G.; Bantscheff M. (2014) Chemoproteomics Reveals Time-Dependent Binding of Histone Deacetylase Inhibitors to Endogenous Repressor Complexes. ACS Chem. Biol. 9, 1736–1746. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.