

Abstract
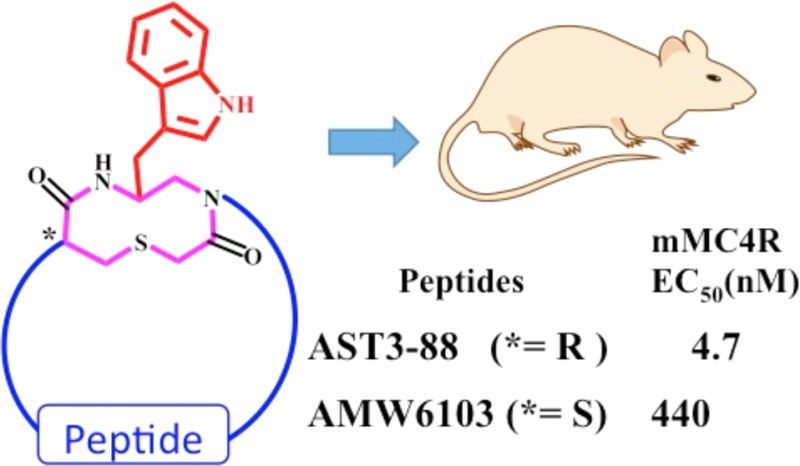
The melanocortin-3 (MC3R) and melanocortin-4 (MC4R) receptors are expressed in the brain and are implicated in the regulation of food intake and energy homeostasis. The endogenous agonist ligands for these receptors (α-, β-, γ-MSH and ACTH) are linear peptides with limited receptor subtype selectivity and metabolic stability, thus minimizing their use as probes to characterize the overlapping pharmacological and physiological functions of the melanocortin receptor subtypes. In the present study, an engineered template, in which the peptide backbone was modified by a heterocyclic reverse turn mimetic at the Trp7 residue, was synthesized using solid phase peptide synthesis and characterized by a β-galactosidase cAMP based reporter gene assay. The functional assay identified a ∼5 nM mouse MC4R agonist (AST3-88) with more than 50-fold selectivity over the mMC3R. Biophysical studies (2D 1H NMR spectroscopy and molecular dynamics) of AST3-88 identified a type VIII β-turn secondary structure spanning the pharmacophore domain stabilized by the intramolecular interactions between the side chains of the His and Trp residues. Enzymatic studies of AST3-88 revealed enhanced stability of AST3-88 over the α-MSH endogenous peptide in rat serum. Upon central administration of AST3-88 into rats, a decreased food intake response was observed. This is the first study to probe the in vivo physiological activity of this engineered peptide-heterocycle template. These findings advance the present knowledge of pharmacophore design for potent, selective, and metabolically stable melanocortin ligands.
Keywords: Melanotropin, obesity, feeding behavior, agouti related protein, AGRP, solid phase synthesis, peptide, small molecule, MC3R, MC4R, GPCR, reverse turn mimetic, serum stability, feeding studies
The melanocortin receptor (MCR) family is a member of the G-protein coupled receptor (GPCR) superfamily that stimulates the adenylate cyclase signal transduction pathway and contains five melanocortin receptor subtypes (MC1–5R).1−6 The melanocortin system includes the endogenous agonists α-, β-, and γ-melanocortin stimulating hormone (MSH) and adrenocorticotropic hormone (ACTH) which are derived from posttranslational modifications of the POMC gene.7,8 Two endogenous antagonists [agouti related protein (AGRP)9 and agouti10] have been discovered to antagonize the centrally expressed MC3R and MC4Rs.11 All the endogenous agonists share a common His-Phe-Arg-Trp (6–9; α-MSH numbering) pharmacophore domain in their primary amino acid sequence.12−14 This tetrapeptide sequence is postulated to be important for melanocortin receptor molecular recognition and ligand-induced receptor activation.15−18 The MC1R, expressed in melanocytes, is involved in the regulation of skin and hair pigmentation.1,2,19 The MC2R is stimulated only by the ACTH agonist and is expressed in the adrenal cortex to regulate steroidogenesis.1 The MC3R is expressed in the gut, placenta, heart, and brain. The MC3R has been reported to be involved in metabolism and energy homeostasis, via mechanism(s) remaining to be characterized.3,20−22 The MC4R is expressed primarily in the brain and regulates feeding behavior, energy homeostasis, and sexual function.5,23−25 The MC5R is expressed in a wide variety of tissues, both centrally and peripherally, and is involved in exocrine gland function in mice.6,26,27 The mouse MC3R and MC4R are expressed in the brain and have 60% similarity in primary amino acid sequence, but possess distinct ligand pharmacological profiles. Central activation of the MC3R and MC4R is postulated to mediate the effects of the melanocortin pathway on energy homeostasis as both the MC3R knockout (KO)21,22 and MC4R KO mice show alterations in energy balance.25 Intracerebroventricular (ICV) administration of the synthetic melanocortin agonist (MTII) decreased food intake, while both the endogenous AGRP antagonist and the synthetic SHU9119 antagonist increased food intake.23,28
Single nucleotide polymorphisms (SNPs) of the hMC4R have been identified in human patients with severe obesity. Farooqi et al. investigated children with severe obesity (under the age of 10) and found that ∼6% of these early onset childhood obesity patients possessed hMC4R single nucleotide polymorphisms (SNPs).29,30 To date, greater than 100 SNPs have been reported in human patients. Seventy of these SNPs have been characterized in vitro (cell culture) in attempts to identify the putative underlying molecular mechanistic defects resulting from the amino acid change(s) in the hMC4R.31−33 In 2011, the MC3R was postulated to be involved in the regulation of food intake by using a mixed pharmacology ligand that possessed partial agonist/antagonist activity at the mMC3R and full agonist activity at the mMC4R.28 Intracerebroventricular administration of this compound in the wild type, MC3RKO, and MC4RKO mice resulted in decreased food intake.28 These, and other genetic and pharmacological studies (involving both animals and humans), support the role of the central melanocortins in the regulation of satiety and energy homeostasis. Selective ligands are needed to understand underlying molecular mechanism(s) by which the MC3R and the MC4R regulate energy balance. The discovery and use of these molecular probes to identify and characterize physiological functions associated with the MCR system are still being sought after.
In continuous efforts to identify potent, selective, and enzymatically stable melanocortin ligands, we have incorporated a bioactive small molecule heterocycle into a peptide in attempts to overcome the inherent problems associated with the endogenous peptides that may limit their consideration for drug development. The strategy applied modifies the peptide backbone resulting in changes in intra- and/or intermolecular interactions postulated to contribute to melanocortin receptor preferred “bioactive” conformation(s) while providing increased protease stability. We have previously reported systematic exploration of the reverse turn mimetic template studied herein with the His6, Phe7, and Arg8 residue domain modifications (α-MSH numbering) that resulted in the identification of a potent agonist ligand at the mMC4R (AMW610).34 However, this compound was only ∼5-fold more selective for the mMC4R versus the mMC3R.
The aromatic indole side chain of the Trp amino acid, a constituent of the melanocortin agonist pharmacophore His6-Phe7-Arg8-Trp9 (α-MSH numbering), has been postulated to play a key role in ligand–receptor interactions.13,14,35,36 Substitution of the Trp residue by an Ala amino acid in various peptide templates was reported to reduce agonist potency at the melanocortin receptor subtypes, particularly at the MC3R.37−39 Therefore, we hypothesized that the modifications of this Trp residue side chain, in the template presented herein, may lead to potent and/or selective analogues at the melanocortin receptors. It has also been postulated that a peptide-heterocyclic scaffold could lead to conformationally constrained ligands that might be more stable for in vitro and in vivo studies as compared to their linear endogenous counterpart(s). The aim of this study was to (i) elucidate the role of the Trp amino acid side chain in the peptide-heterocyclic scaffold in attempts to obtain potent and selective ligands and (ii) study the scope of this novel template on serum stability over the endogenous α-MSH peptide ligand. Peptides, in general, can be promising therapeutic agents by having a number of advantages over small molecules, in terms of specificity and affinity for targets. However, general endogenous peptide low protease stability often traditionally limits their further consideration in drug development. Standard approaches to make peptides less susceptible to serum proteases, for example, using modifications with unnatural amino acids, d-amino acids, β-amino acids, cyclization of peptide chain at the termini, modifications of the peptide backbone, and so forth, have been well established. These approaches often make the peptide structure more constrained and therefore less prone to protease degradation in serum. In the present study, we compared the compound containing a heterocyclic-peptide backbone modification and a disulfide bridge at the N- and C-terminus, which can restrict a peptide’s backbone conformation and modify their stability in serum. In addition, in vivo experiments were performed to assess the physiological effect on food intake of this new chemical probe. Central administration of AST3-88 decreased a cumulative feeding response in rats 48 h post-ICV treatments. Herein, we report the design, functional, and structural characterization of a potent and selective MC4R full agonist and present the studies of proteolytic stability and in vivo properties of this template for the first time. Taken together, this study provides new information on the structural prerequisites for potent, selective, and increased serum stable melanocortin ligands.
Results and Discussion
The development of MC3R and MC4R selective ligands has the therapeutic potential for the treatment of body weight and feeding related disorders. A synergistic role of the MC3R in conjunction with the MC4R for feeding and energy homeostasis are emerging.28,40 Thus, ligands and molecular probes that can discriminate in a subtype specific manner (agonist and/or antagonist), and that can be used to probe the physiological roles of melanocortin receptor subtypes are still needed in the field. Toward this goal, a number of small molecules and peptide ligands have been explored and reported in the literature by a plethora of academic and industrial research laboratories. Our strategy in this study is to combine the use of a bioactive small molecule and incorporate it into a potent cyclic peptide template in attempts to gain further insight into melanocortin receptor molecular recognition, selectivity, and increase protease stability. We report herein the AST3-88 melanocortin agonist that possesses a selective MC4R agonist profile over the MC3R (>50-fold), enhanced serum stability, and results in decreased food intake in vivo.
Synthesis, Structural, and Functional Characterization
The compound AST3-88, and its stereoisomer AMW6103, were synthesized using previously reported method(s) and is summarized in Scheme 1.34,41 To probe the importance of the heterocyclic stereochemistry within the ring moiety, the Cys α-carbons (marked with * in Figure 1) in both AST3-88 (l-configuration) and AMW6103 (d-configuration) were designed and synthesized. The Fmoc-Trp(Boc)-aldehyde was synthesized from the corresponding acid via Fmoc-Trp(Boc)-amide and introduced via a reductive amination method to the resin bound peptide chain. The thioether ring was formed on the resin by mild on-bead cyclization method.34,41 After completion of the synthesis, the ligand was cleavage and the crude peptide was dissolved in a 20% DMSO/water solution and stirred at room temperature to form the disulfide-bridge. The peptides AST3-88 and AMW6103 possessed the correct molecular weights as determined by mass spectrometry. The purity of these peptides (>95%) was assessed by analytical RP-HPLC in two diverse solvent systems (data provided in the experimental section).
Scheme 1.
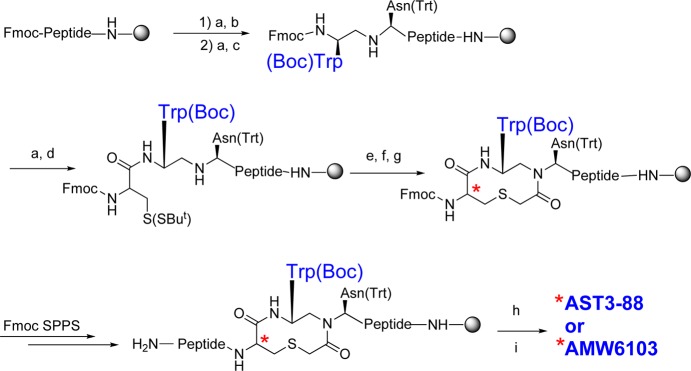
Figure 1.

(A) Illustration of the compounds (i) heterocycle, (ii) AMW610, (iii) AST3-88, and (iv) AMW6103. The stereochemistry of the thioether ring is represented by an asterisk (*). Compounds AST3-88 and AMW610 contain l-Cys, and compound AMW6103 contains a d-Cys at this position. The side chain of His is depicted in green, DPhe in orange, Arg in blue, the heterocycle moiety in pink, and the disulfide bridge is circled in yellow. (B) Amino acid sequences of key endogenous and synthetic melanocortin peptides.
The compounds were tested for agonist functional activity at the mouse MC1R, MC3R, MC4R, and MC5R using a 96-well cAMP based β-galactosidase reporter gene bioassay (Table 1).42 The NDP-MSH (Ac-Ser-Tyr-Ser-Nle-Glu-His-DPhe-Arg-Trp-Gly-Lys-Pro-Val-NH2) peptide43 is one of the standard melanocortin agonists used in the study of melanocortin receptors and was included herein as a reference control and as an internal control for maximal ligand efficacy (100%). The pharmacology of the heterocyclic moiety alone and peptides AMW3-130 and AMW610 (Figure 1) were also included for pharmacological comparisons of the different but related templates. The AMW3-130 molecule was designed as a chimeric cyclic peptide template that incorporated the AGRP based antagonist scaffold that upon substitution of the AGRP Arg-Phe-Phe pharmacophore residues with the agonist His-DPhe-Arg-Trp pharmacophore resulted in the conversion of a weak antagonist into a potent sub-nanometer agonist that was able to functionally rescue polymorphic hMC4R SNPs.33 In 2002, the heterocyclic moiety44 (Figure 1) was reported to possess nanomolar MCR functional agonist activity and initial studies were performed incorporating this moiety into the AMW3-130 peptide template.34,41 The first study identified the AMW610 template (Figure 1) as possessing the most potent nanomolar MCR full agonist functionality of the SAR at the time.34 Biophysical experiments, similar to the ones reported herein, were performed in attempts to correlate SAR and identify solution-phase structural differences between the peptides containing different orientations of the heterocycle.8,34 Thus, for structural and functional comparative purposes, the AMW3-130 and AMW610 ligands have been included herein.
Table 1. Agonist EC50 (nM) Pharmacology of the Modified Peptides at the Mouse Melanocortin Receptorsa.
| mMC1R |
mMC3R |
mMC4R |
mMC5R |
selectivity ratio | |||||
|---|---|---|---|---|---|---|---|---|---|
| peptide | EC50 (nM) | fold change | EC50 (nM) | fold change | EC50 (nM) | fold change | EC50 (nM) | fold change | MC4R vs MC3R |
| NDP-MSH | 0.018 ± 0.003 | 0.14 ± 0.03 | 0.20 ± 0.03 | 0.30 ± 0.05 | 1 | ||||
| AMW3-130b | 0.35 ± 0.17 | 2.00 ± 0.45 | 0.27 ± 0.09 | 2.32 ± 0.57 | 7 | ||||
| heterocycle | 164 ± 22 | 7600 ± 1890 | 650 ± 126 | 335 ± 106 | 12 | ||||
| AMW610c | 33 ± 9 | 1 | 495 ± 215 | 1 | 85 ± 13 | 1 | 72 ± 16 | 1 | 9 |
| AST3-88 | 68.8 ± 17.9 | 2 | 255 ± 24 | (2) | 4.7 ± 0.49 | (18) | 11.9 ± 2.3 | (6) | 54 |
| AMW6103 | 0.81 ± 0.25 | (41) | 5300 ± 4300 | 11 | 440 ± 220 | 5 | 31.0 ± 16.2 | (2) | 12 |
The indicated errors represent the standard error of the mean determined from at least three independent experiments. Parentheses indicate that an increase in potency has resulted. Changes less than 3-fold are considered to be within the inherent experimental assay error.
The AGRP-melanocortin chimeric peptide AMW3-130 has been previously reported in ref (34) as compound 1.
The AMW610 compound has been previously reported in ref (34) as compound 7.
At the mMC1R and mMC3R, AST3-88 resulted in an equipotent compound, within the 3-fold inherent experimental error, as compared to AMW610. At the mMC4R, AST3-88 resulted in a ∼5 nM full agonist that is 18-fold more potent at this receptor than AMW610. At the mMC5R, AST3-88 resulted in 6-fold increased agonist potency as compared to AMW610. The AMW6103 ligand, possessing the D-configuration at the Cys6 residue of the heterocyclic ring (Figure 1), resulted in an agonist with sub nM potency at the mMC1R and equipotent to the cyclic AMW3-130 peptide that lacks the heterocyclic moiety. The AMW6103 ligand also is ∼40-fold more potent at the mMC1R and possessed 11- and 5-fold decreased melanocortin receptor agonist potency at the mMC3R and mMC4Rs, respectively, as compared to the AMW610 template. Compound AMW6103 is a full agonist and equipotent to AMW610 at the mMC5R. The AST3-88 ligand, which has l- configuration at the ring, in general, is more active than its d-counterpart (AMW6103) at the melanocortin receptor isoforms, with the exception of mMC1R, where AMW6103 is 80-fold more potent than the AST3-88. These results are consistent with our earlier study34 where the l-configured analogues generally were more potent than their d-counterparts. Furthermore, the AST3-88 is more than 50-fold selective at the mMC4R than the mMC3R, versus the AMW6103 that only 12-fold selective and possessed decreased agonist potency. The MC4R to MC3R selectivity of the control compounds AMW610, AMW3-130, and heterocyclic ring itself ranged from 7- to 12-fold (Table 1).34,41
Biophysical Structural Studies (NMR and CAMM)
To study the solution-phase structural differences, and how these subtle conformational differences might be associated with changes in the observed pharmacology of these two AST3-88 and AMW6103 analogues, we performed 2D 1H NMR and computer assisted molecular modeling (CAMM) experiments. Both compounds were dissolved in ∼50% (v/v) acetonitrile-d3 and ∼50% H2O for NMR experiments. This solvent system has been previously used by our laboratory on similar peptide template(s) and was used herein so that structures identified from this study could be compared with previous studies under similar experimental conditions.34,41 The criteria used for this particular solvent selection have been previously reported and are included herein: “1) it must completely dissolve all the peptides examined that possessed different biophysical properties, 2) similar solvents have been used previously so that structural comparisons could be performed and discussed, and 3) the amide protons were well resolved in this system versus the other solvents attempted. Other solvents that were attempted but did not result in all the peptides examined being fully dissolved included (i) DMSO-d6, (ii) CDCl3/DMSO, (iii) CD3CN/H2O, (iv) MeOH-d3, (v) MeOH/DMSO, (vi) H2O/D2O.”41 Unfortunately, the AMW6103 compound, in all the different solvent systems we examined and reported previously (indicated above), was unable to result in observable and resolvable peaks necessary for proton assignments, and therefore, this ligand was excluded from further structural studies. The 1H NMR and chemical shift assignments (ppm) for analogue AST3-88 (assigned in the ∼50% (v/v) acetonitrile-d3 and ∼50% H2O solvent system) are provided in the Supporting Information (Table S1). Use of this solvent system and similar experimental conditions provided the opportunity to directly compare the structures of AST3-88 with those of AMW610 and AMW3-130.8,34
In the present study, we compare shifts of specific amino acids and the global patterns of one peptide to another. Figure 2 is a histogram of the difference between experimental and reference random-coil (Δ-RC) shift45 values for the conserved His-DPhe-Arg-Trp-Asn-Ala region of compounds AST3-88 and AMW610 and AMW3-130. The random coil shift values are averages of overall possible conformations that an amino acid can adopt in the random coil. Wishart and Sykes have published a complete set of 1H, 13C, and 15N RC shift values that can be used as references in order to detect regions in the peptide or protein which are not in random coil conformations.45 Given that the compounds have exactly the same amino acids and that they differ only in the position or presence of the heterocycle ring, there are significant differences in random coil shift values between them. The comparison of chemical shifts from one compound to other demonstrates that the chemical shifts of these residues can differ significantly, presumably indicating different conformational structure(s). The random coil shift values of the backbone NH protons in AST3-88 and AMW3-130, especially in the His, DPhe, Arg, and Trp region, have negative random coil shift values between 0.2 and 0.3 ppm. This suggests that the AST3-88 may have a regular backbone conformation with secondary structure in the pharmacophore region similar to AMW3-130, which has been reported to have a reverse turn in the similar region.34 Following resonance assignments, we determined the nuclear Overhauser effect (NOE) values of the residues, providing an estimate of the distance between pairs of protons closer than about 5 Å in space. The NOE patterns can be used as a tool to identify secondary structure in peptides and/or to distinguish structural differences between analogues. Figure 3 summarizes the NOE intensities observed for the AST3-88 along with the comparator compounds AMW610 and AMW3-130 (taken from ref (34)). Several short- and long-range NOE connectivities were observed for AST3-88 in the pharmacophore domain indicating that the region around the His-DPhe-Arg-Trp amino acids possesses a regular secondary structure. To examine the solution based conformational structure(s) of AST3-88 and compare its differences from peptides AMW3-130 and AMW610, restrained molecular dynamics simulation (RMD) experiments were performed. The computational molecular dynamics simulations, based upon NMR experimental values, can be used to provide information on the fluctuations and conformational changes of the peptides examined. This method allows the prediction of the static and dynamic properties of molecules from direct interactions between the molecules. The RMD simulation was initiated by using unambiguous NMR distant constraints on the peptide without the presence of the disulfide bond. Following a 1 ns run, the disulfide bond was formed and the compound was allowed to fully relax by energy minimization before performing a more robust RMD simulation for 10 ns. Following the MD run, 200 equally spaced structures were energy minimized with the NMR based restraints, analyzed, and grouped into conformational families. A superposition of all 200 energy-minimized conformations was not distinguishable, suggesting that the simulations resulted in more than one conformational family. Cluster analysis was performed by comparing the backbone φ and ψ angles of the His-DPhe-Arg-Trp amino acid ligand domain, which resulted in one major family with 40% of all the 200 conformers (Figure 4A). The structures of compounds AMW610 and AMW3-130 were generated by the same method depicted above and included here for comparison. The structure of AST3-88 is represented as the superposition of the conformers of the major family of compound aligned on backbone heavy atoms. The global root-mean-square (RMSD) value of residues in the “His-DPhe-Arg-Trp” region from the average structure is 1.16 ± 0.45 Å. Analysis of the φ–ψ backbone dihedral angles of the average structure (Supporting Information Table S2) resulted in the identification of type VIII β-turn structure spanning DPhe4 and Arg5 residues. Type VIII is reported to be the most common type of β-turn secondary structure observed in peptides and proteins.46 The φ and ψ values of the representative structure from the major family are φ(i + 1) ≈ −69, ψ(i + 1) ≈ −58, φ(i + 2) ≈ −125, and ψ(i + 2) ≈ 41 [idealized values for this type of turn have been reported as φ(i + 1) ≈ −60, ψ(i + 1) ≈ −30, φ(i + 2) ≈ −120, and ψ(i + 2) ≈ 120].46 A putative hydrogen bond was also identified between the backbone carbonyl oxygen of His3 (i residue) and the backbone amide NH of Cys6 (i + 3 residue). Figure 5 represents the φ–ψ distribution of all the residues in the sequence of AST3-88 with the exception of the terminal Tyr residues. The φ–ψ distribution map of AST3-88 resulted in conformers tightly packed in the pharmacophore regions, suggesting a constrained conformation in this region, similar to the case of AMW3-130. Apart from the pharmacophore region, the Phe10 and Cys11 amino acids have very compact conformers, suggesting that the C-terminus of this compound is relatively rigid and involved in regular structure. This observation is consistent with the constraints induced by the disulfide bridge.
Figure 2.

Illustration of the compound deviation from random coil values for (A) AST3-88, (B) AMW610, and (C) AMW3-130.45 The HN and Hα protons are represented by solid bars and open bars, respectively. Vertical axes are in parts per million, and the horizontal axes represent the His-DPhe-Arg-Trp-Asn-Ala region of the peptide.
Figure 3.

Summary of the NOE intensities from 400 ms NOESY data observed for the compound (A) AST3-88, (B) AMW610, and (C) AMW3-130. The height of the bar indicates the strength of the NOE, and these are categorized as strong (1.8–3.0 Å), medium (1.8–3.5 Å), or weak (1.8–5.0 Å).
Figure 4.

Sausage stereoview representations of the major family members of compound (A) AST3-88 (in the H, F, R,W region; RMSD = 1.16 ± 0.45 Å), (B) AMW610, and (C) AMW3-130 (in the H, F, R,W region; RMSD = 0.66 ± 0.3 Å) aligned on the backbone heavy atoms of residues 3–7. The His side chain is indicated in green, the DPhe side chain in yellow, the Arg side chain in blue, and the Trp side chain in orange. A wide gray backbone indicates greater flexibility in that domain. Following restrained molecular dynamics (RMD) simulations for 10 ns, 200 equally spaced structures were energy minimized with the NMR based restraints. The energy minimized structures were grouped into conformational families by comparison of the backbone dihedral angels within the His-Phe-Arg-Trp domain.
Figure 5.

CAMM based φ–ψ angle distribution for ligands (A) AST3-88, (B) AMW610, and (C) AMW3-130 conformational families. The His residue is indicated in green, the DPhe residue in orange, the Arg residue in blue, and the Trp residue in red. All other residues are indicated in purple.
The 2D 1H NMR and CAMM studies were performed to correlate the functional and conformational properties the compounds. In earlier studies, it was postulated that the bioactive conformation of melanocortin ligands such as, MTII and SHU9119 constitute a putative reverse turn in the pharmacophore region.47 In a study of chimeric AGRP-melanocortin template peptides with a lactam bridge, similar to the AMW3-130 template, a reverse turn was found in all the studied peptides.48 The chimeric peptide template used in present study (with a disulfide bridge in place of a lactam bridge) was also found to have Type I′ reverse turn encompassing Arg and Trp residues in pharmacophore domain.34,41 In present study, we found that compound AST3-88, an agonist on all the melanocortin receptor subtypes, possesses a Type VIII reverse turn around DPhe4 (i + 1) and Arg5 (i + 2) regions of the sequence. The reverse turn was stabilized by the hydrogen bond between His3 and Cys6 residues. In the earlier structural findings for the compound AMW610 where the Arg residue resided on the heterocyclic moiety, it was observed that insertion of the heterocyclic ring made the peptide backbone more flexible and resulted in several conformational families with very low populations.34,41 In contrast, positioning of the Trp residue on the heterocyclic ring (AST3-88) in the present study resulted in one major family with 40% of the total studied conformers. The very close proximity of His3 and Trp6 suggests that the structure may be additionally stabilized by aromatic–aromatic and/or cation−π interactions involving side chains of these residues. The His3 and Trp6 residues appeared to be stacked in parallel arrangement. The DPhe4 residue is oriented perpendicular to His3 and Trp6 amino acids. Additionally, the arrangement of aromatic residue side chains of His3, Trp7, and Phe10 are juxtaposed, forming a hydrophobic cluster. The above-mentioned structural features of AST3-88 are in agreement with the reported structure of MTII and linear α-MSH-analogues in terms of having a reverse turn in the pharmacophore region. The AMW3-130 ligand, a potent agonist at the melanocortin receptors, was also identified to possess a type I′ β-turn encompassing Trp7 and Asn8 residues with a constrained backbone conformation. These data support the hypothesis that the modulation in the orientation of Trp residue stabilizes the bioactive conformation via intramolecular interaction and orient the other side chains for efficient receptor interactions. This also suggests that the reverse turn in the pharmacophore region is more favored for the MC4R binding pocket than the MC3R. It has been postulated that the putative hydrophobic pocket of MC3R is different than the MC4R, which explains the difference in ligand–receptor interactions and hence, the difference in EC50 values at these receptor subtypes.
Serum Stability of Compound AST3-88
Modifications in peptide structure by different approaches including cyclization,49,50d-amino acids,51 unnatural amino acids,52 and β-amino acids53 are well recognized strategies for the goal of improving peptide stability in serum and protecting against protease-mediated degradation. Since AST3-88 was prepared by inserting a heterocyclic moiety into the peptide backbone, the stability of AST3-88 in rat serum was compared with two peptides prone for degradation, α-MSH and AST789-13 (Ac-RRWWRF-NH2). It has been reported for α-MSH [13-mer linear peptide with acetylated N-terminus and amidated C-terminus (Ac-SYSMEHFRWGKPV-NH2)] that, during a 4 h incubation, it was rapidly degraded in frog serum and lost half of its activity in rat serum after 1 h of incubation.12,54 The AST789-13 control peptide was reported to possesses a half-life of 1.5 h in human serum.55 In our study, LC-MS analysis of the aliquots collected during 24 h incubations of these three peptides with rat serum revealed differences in their degradation kinetics (Figure 6). The results indicated that AST789-13 was degraded completely within 2 h. The endogenous α-MSH peptide had a half-life of nearly 2.5 h with only ∼10% of the compound remaining in the serum after 6 h. of incubation. In contrast, AST3-88 was far more stable than AST789-13 and α-MSH as ∼40% of AST3-88 remained at 6 h and 12% at 24 h of incubation in rat serum (Figure 6). As the degradation products of AST3-88 were not identified in this study, the degradation pathway of AST3-88 and the mechanism behind the greater stability of AST3-88 over α-MSH and AST789-13 remain undefined. However, it is postulated that the structural properties of AST3-88, including the disulfide bridge joining the N- and C-terminus, inclusion of a d-amino acid in its sequence, and the presence of the heterocyclic modified peptide backbone (Figure 1), contribute to its resistance to both exopeptidase and endopeptidase-mediated degradation.
Figure 6.

Rat serum stability profiles of the peptides AST789-13, α-MSH, and AST3-88. Relative peptide concentrations are based on mass spec ion counts. AST789-13 completely degraded in less than 2 h. Endogenous linear peptide α-MSH rapidly degraded within 6 h with only ∼1% intact peptide remaining at the 24 h point. Compound AST3-88, with the engineered backbone, degraded slowly in rat serum with ∼40% remaining at 6 h, and ∼12% was detected by MS at the 24 h time point.
Effect of ICV Treatment of Compound AST3-88 on Feeding
To determine if compound AST3-88 produces an in vivo physiological response, we administered two doses (2.5 or 5.0 nmol) of compound AST3-88 intracerbroventricularly (ICV) into the third ventricle of cannulated and overnight fasted rats. In earlier studies, administration of MC3R and MC4R agonists inhibited food intake postinjection.23,28 Conversely, ICV administration of MC3R and MC4R antagonists increases food intake.23,28 To test the effects of the MC4R selective compound AST3-88, rats were induced to feed by overnight food deprivation, a validated method to observe modulation in MC4R signaling.23 Animals were injected with either vehicle or compound AST3-88 after completion of overnight fasting with food being returned 90 min after treatment. Figure 7 shows the effect of AST3-88 on food intake up to 48 h after the return of food. The AST3-88 ligand produced a statistically significant decrease in food intake (relative to the vehicle condition) at both doses 3 to 48 h post treatment. Thiele and colleagues have previously reported this type of prolonged activity upon ICV administration of MTII in rats.56 They reported that MTII reduced 48 h food consumption (1.0 nmol dose), and also reduced body weight at 24 and 48 h (0.1 and 1.0 nmol doses, respectively) post treatment. In another example, central administration of agouti-related protein (AGRP), an endogenous antagonist of the MC3R and MC4R, robustly increased food intake in mice with the effect lasting up to 24 h.57,58 It has also been reported that AGRP produced a robust and prolonged food intake response in wild type, MC3RKO, and MC4RKO mice.28 In summary, ICV treatment of compound AST3-88 has a significant effect on inhibition of food intake in rats.
Figure 7.

(A) Effects of AST3-88 ICV treatment on cumulative food intake in rats. Animals were injected with either saline, 2.5 nmol, or 5 nmol of AST3-88 after completion of overnight fasting. Each data point is represents the mean of 4–6 animals ± SEM; *p < 0.05 and ***p < 0.001.
Conclusions
In the current study, it has been demonstrated that an engineered peptide backbone strategy could be utilized to obtain potent, selective, and stable melanocortin receptor ligands. Taken together with previous observations, the results of these studies support the hypothesis that the Trp residue side chain may play an important role in (i) stabilizing the bioactive conformation by intramolecular interactions, (ii) properly orienting the pharmacophore residues to interact with the receptor in the binding pocket, and (iii) specific intermolecular interaction with the receptor residues resulting in differential agonist potencies at different receptors, that can contribute to selectivity. The advantage of this approach also extends to the enhanced metabolic and physiological stability of the engineered peptides. The AST3-88 molecule that possesses nanomolar potency and selectivity for the MC4R versus the MC3R, prolonged biological activity, and resistance to enzymatic proteolysis can serve as a molecular probe to study physiological role(s) of these melanocortin receptors and also further improve the design of selective ligands.
Methods
Chemistry
All chemicals were ACS grade or better, were obtained from commercial suppliers, and were used without further purification. The amino acids Nα 9-fluorenylmethoxycarbonyl (Fmoc)-Cys(Trt), Fmoc-Arg(Pbf), Fmoc-His(Trt), Fmoc-Trp(Boc), Fmoc-d-Phe, Fmoc-Phe, Fmoc-Asn(Trt), Fmoc-Ala, Rink amide p-methylbenzhydrylamine Resin (p-MBHA Resin, 0.47 mequiv/g substitution), and the coupling reagent O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) were purchased from Peptides International (Louisville, KY). The amino acid Fmoc-Tyr (tBu) was purchased from Advanced Chem Tech (Louisville, KY).
Solid Phase Peptide Synthesis Using Microwave Irradiation
The Rink amide MBHA resin was transferred into a 25 mL polypropylene reaction vessel (CEM) and “swelled” in dichloromethane for 1 h. The bottom cap of the vessel was removed after the 1 h swelling process, and the vessel was transferred onto a vacuum filtration manifold. Deprotection of the Fmoc group was achieved in DMF by irradiating the sample at 75 °C, 30 W for 4 min in the CEM Discover SPS instrument. After cooling and washing the resin with DMF, a ninhydrin “Kaiser” test was performed.59 The amino acid coupling step was performed under the same microwave conditions (75 °C, 30 W) for 5 min using HBTU. The coupling of the Cys and His amino acids was performed at lower temperatures (50 °C, 30 W) for 5 min. The heterocycle ring was assembled as previously described.44 The reductive alkylation, acylation of chloroacetic anhydride, and the deprotection of the thio tert-Butyl group steps were performed at the room temperature. The thioether ring closure was performed using N-ethylmorpholine in DMF at elevated temperature (55–60 °C) in an automated synthesizer (Advanced ChemTech 440MOS, Louisville, KY). Bubbling nitrogen gas into the reaction vessel mixed the reagents. The iterative process of the Fmoc deprotection/coupling cycle was performed to elongate the peptide chain. After the final deprotection, the peptide was cleaved from the resin using the cleavage cocktail (91% TFA, 3.0% H2O, 3.0% EDT, and 3.0% TIS) at room temperature for 3 h. Note, as the peptides containing the heterocyclic moiety were synthesized to completion on resin, if racemization occurred during the isolated heterocycle formation as previously reported,44 it was not identified or purified in the major product peak collected by RP-HPLC. After cleavage and side chain deprotection, the solution was concentrated and the peptide was precipitated and washed using cold (4 °C) anhydrous diethyl ether.
The crude linear peptides were dissolved in 20% DMSO in water (1.0 mg/mL) and stirred at room temperature. Progress for the disulfide cyclization was monitored by UV-HPLC, which was generally completed within 24–36 h. The resulting solution was lyophilized to yield the crude cyclic peptide and purified by reversed-phase HPLC (flow rate of 5 mL/min and gradients ranging from 30%–55% acetonitrile/water 0.1% TFA) using a Shimadzu chromatography system with a photodiode array detector and a semipreparative RP-HPLC C18 bonded silica column (Vydac 218TP1010, 1.0 × 25 cm). The purified peptides were >95% pure as determined by RP-HPLC in two diverse solvent systems and had the correct molecular mass. Electrospray ionization mass spectrometry (ESI-MS) was used to record spectra on an ABI 3200Q TRAP instrument. Analytical data for AST3-88: purity > 95%; k′ (MeCN) 4.7, k′ (MeOH) 7.9; exact mass calcd for C77H94N20O15S3, 1634.6; found, 1635.6 (M + 1). Analytical data for AMW6103: purity > 95%; k′ (MeCN) 5.7, k′ (MeOH) 9.8; exact mass calcd for C77H94N20O15S3, 1634.6; found, 1635.7 (M + 1).
Functional Bioassay
The HEK-293 cells stably expressing the mouse melanocortin receptors were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal calf serum and transfected with 4 μg of the CRE/β-galactosidase reporter gene as previously described.42 Briefly, 5000–15 000 post-transfection cells were plated into collagen treated 96-well plates (Nunc) and incubated overnight. Forty-eight hours post-transfection, the cells were stimulated with 100 μL of peptide (10–4–10–12 M) or forskolin (10–4 M) control in assay medium (DMEM containing 0.1 mg/mL BSA and 0.1 mM isobutylmethylxanthine) for 6 h. The assay media was aspirated, and 50 μL of lysis buffer (250 mM Tris-HCl pH = 8.0 and 0.1% Triton X-100) was added. The plates were stored at −80 °C overnight. The plates containing the cell lysates were thawed the following day. Aliquots of 10 μL were taken from each well and transferred to another 96-well plate for relative protein determination. To the cell lysate plates, 40 μL of phosphate-buffered saline with 0.5% BSA was added to each well. Subsequently, 150 μL of substrate buffer (60 mM sodium phosphate, 1 mM MgCl2, 10 mM KCl, 5 mM β-mercaptoethanol, 2 mg/mL ortho-nitrophenyl-β-galactoside [ONPG]) was added to each well and the plates were incubated at 37 °C. The sample absorbance, OD405, was measured using a 96-well plate reader (Molecular Devices). The relative protein was determined by adding 200 μL of 1:5 dilutions Bio Rad G250 protein dye/water to the 10 μL cell lysate sample taken previously, and the OD595 was measured on a 96-well plate reader (Molecular Devices). Data points were normalized both to the relative protein content and nonreceptor dependent forskolin stimulation. Maximal efficacy was compared to that observed for the NDP-MSH control peptide tested simultaneously on each 96-well plate. The agonist EC50 values represent the mean of duplicate wells performed in three or more independent experiments. The EC50 value estimates, and their associated standard errors, were determined by fitting the data to a nonlinear least-squares analysis using the PRISM software program (v4.0, GraphPad Inc.). The results are not corrected for peptide content.
NMR Spectroscopy and Computer-Assisted Molecular Modeling (CAMM)
The ligand NMR samples were prepared (∼1 mM) by dissolving 1.0 mg of the purified peptide in a 750 μL solution containing 400 μL of CD3CN and 350 μL of H2O and adding DSS as an internal standard (0.0 ppm). The NMR data were collected at 34 °C with a Bruker Avance II spectrometer operating at 600 MHz (using a cryoprobe) at the Advanced Magnetic Resonance and Imaging Spectroscopy (AMRIS) facility at the University of Florida. Standard proton TOCSY and NOESY 2D 1H NMR data were collected, processed, and analyzed as described previously.34,41,48,60 The chemical shifts of each of the peptides in this study were assigned using standard TOCSY and NOESY 1H-based strategies.61
Proton–proton distances were calibrated using the well resolved methylene protons of the Cys6 (heterocycle ring) residue based on the relationship r = rref(ηref/η)1/6, where r is the distance between atoms, η is the NOESY cross-peak volume, rref is the known distance, and ηref is the corresponding volume of the NOESY calibration cross-peak. The NOE volumes were categorized as strong (1.8–3.0 Å), medium (1.8–3.5 Å), or weak (1.8–5.0 Å) (Supporting Information). All conformational molecular modeling experiments were performed using the SYBYL v7.0 software from Tripos Inc. (St. Louis, MO) on a Silicon Graphics workstation. Restrained molecular dynamics (RMD) simulations were run in vacuo with a dielectric constant of 4.0, at a temperature of 500 K, and using the Tripos force field and Gastaiger-Hückel partial atomic charges. The peptides were initially built in a fully extended linear conformation. In the first step of modeling, RMD simulations were run for 1 ns. Following the initial 1 ns RMD trajectory, the cysteine residues were oriented next to each other, and disulfide bonds were manually formed and energy minimized without restraints. Finally, all the NMR-based NOE restraints were included, and 10 ns RMD trajectories were collected. Following the RMD simulations, structures from 200 equally spaced points along the dynamics trajectory were energy minimized, analyzed, and grouped into conformational families. Comparing the backbone φ and ψ angles of the His-DPhe-Arg-Trp amino acid ligand domain performed cluster analysis. This process identified representative structures (lowest energy conformers) of the conformational families that were used for further analysis.
Serum Stability Assay
Rat serum (495 μL) was incubated at 37 °C for 15 min, and 5 μL of peptide (10–3 M stock solution; final conc. 10 μM) was added to initiate the assay. The solution (serum + peptide) was mixed intermittently duration the incubation, and 50 μL aliquots were taken at 0, 0.5, 1.5, 3, 6, and 24 h of the incubation. Aliquots were then mixed with 150 μL of cold (4 °C) 66% aqueous acetonitrile and incubated at 4 °C for 10–15 min to precipitate serum proteins. The supernatant was collected after centrifugation at 14 000g for 10 min and then stored at −80 °C immediately.
Liquid Chromatography–Mass Spectrometry (LC-MS) Analysis of Peptide Stability
A 5 μL aliquot of diluted samples was injected into a Waters Acquity ultraperformance liquid chromatography (UPLC) system (Milford, MA) and separated by a gradient of mobile phase ranging from water to 95% aqueous acetonitrile containing 0.1% formic acid over a 10 min run. LC elute was introduced into a Waters SYNAPT QTOF mass spectrometer (QTOF-MS) for accurate mass measurement and ion counting under extended dynamic range mode. Capillary voltage and cone voltage were maintained at 3.2 kV and 30 V, respectively, for positive electrospray ionization (ESI). Source temperature and desolvation temperature were set at 120 and 350 °C, respectively. Nitrogen was used as both cone gas (50 L/h) and desolvation gas (700 L/h). Mass chromatograms and mass spectral data were acquired and processed by MassLynx software (Waters) in centroid format. Peptides were identified by accurate masses and comparison with authentic standards. Signal intensity of interested peptide in spiked serum sample at the beginning of incubation (0 h) was arbitrarily set as 100%.
Feeding Studies in Rats
Animals
Male Sprague–Dawley rats (Charles River) were anesthetized with a mixture of ketamine/xylazine (90/10 mg/kg). In a stereotaxic apparatus with the incisor bar set at −3.3 mm (relative to horizontal zero), cannulas (26 gauge) were implanted with the tip targeted at 2.5 mm posterior and 8.1 mm ventral to bregma. Cannulas were secured to the skull with stainless steel screws and dental cement. All injections were made with a 33 gauge internal cannula that extended 1 mm beyond the tip of the guide cannula. In all cases, the injection volume was 3 μL. The injections were given over a 30 s period, and the internal cannula was left in place an additional 30 s to allow diffusion from the tip. Cannula patency and placement was tested prior to and following data collection. This test consisted of an ICV injection of angiotensin II (30 ng/3 μL). Data from rats that did not drink at least 5 mL of water in the 30 min period after injection in both trials were discarded. The feeding study was approved by the University of Minnesota Institutional Animal Care and Use Committee.
Feeding Regimen
For the data and results presented here, rats were fasted overnight and then given ICV injections of compound 1 (AST3-88) at doses of 0, 2.5, and 5 nmol. The peptide was initially dissolved in a small volume of DMSO, and saline was added to adjust the concentrations. The final concentration of DMSO in the vehicle and both doses was 16.7%. Food was returned 90 min after injections, and food intake (corrected for spillage) was measured (by weight) at 1.5, 3, 24, and 48 h. Rats were weighed prior to injection and 1, 2, and 4 days later. No adverse or toxicity related behaviors were observed in the duration of this study. It should be noted that, prior to this test, these rats had been used in two trials (conducted 7 and 15 days earlier) in which they received ICV injections of AST3-88 (0, 2.5, or 5 nmol) under ad libitum feeding conditions and no statistically significant differences were observed.
Statistics
Data are represented as the average of the mean ± SEM. For statistical analyses, cumulative food intake over time was performed using two-way ANOVA followed by an LSD post hoc test. Statistical significance is considered if p < 0.05.
Glossary
Abbreviations
- ACTH
adrenocorticotropin hormone
- AGRP
agouti-related protein
- ASIP
agouti-signaling protein
- CAMM
computer-assisted molecular modeling
- cAMP
cyclic 5′-adenosine monophosphate
- DCM
dichloromethane
- DMF
N,N-dimethylformamide
- Fmoc
Nα 9-fluorenylmethoxycarbonyl
- GPCR
G protein coupled receptor
- MC1R
melanocortin-1 receptor
- MC2R
melanocortin-2 receptor
- MC3R
melanocortin-3 receptor
- MC4R
melanocortin-4 receptor
- MC5R
melanocortin-5 receptor
- MCR
melanocortin receptor
- MeOH
methanol
- MSH
melanocyte stimulating hormone
- NOE
nuclear Overhauser effect
- POMC
proopiomelanocortin
- ϕ
phi
- ψ
psi
- SAR
structure–activity relationship
- SEM
standard error of the mean
- TFA
trifluoroacetic acid
- TM
transmembrane
- α-MSH
alpha-melanocyte stimulating hormone
- β-MSH
beta-melanocyte stimulating hormone
- γ-MSH
gamma-melanocyte stimulating hormone
- μM
micromolar
- RMD
restrained molecular dynamics
- NDP-MSH (4-Norleucine-7-d-Phenylalanine)
Ac-Ser-Tyr-Ser-Nle-Glu-His-DPhe-Arg-Trp-Gly-Lys-Pro-Val-NH2
- AMW3-130
Tyr-c[Cys-His-DPhe-Arg-Trp-Asn-Ala-Phe-Cys]-Tyr-NH2
- MTII
Ac-Nle-c[Asp-His-DPhe-Arg-Trp-Lys]-NH2
- AST789-13
Ac-RRWWRF-NH2
Supporting Information Available
Proton NMR chemical shift assignments (ppm) for AST3-88 and the backbone dihedral angels of the representative structures in Figure 4. This material is available free of charge via the Internet at http://pubs.acs.org
This work has been supported in part by NIH Grants RO1DK064250 (C.H.-.L), RO1DK091906 (C.H.-L.), and R01DA021280 (A.S.L.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mountjoy K. G.; Robbins L. S.; Mortrud M. T.; Cone R. D. (1992) The cloning of a family of genes that encode the melanocortin receptors. Science 257, 1248–1251. [DOI] [PubMed] [Google Scholar]
- Chhajlani V.; Wikberg J. E. (1992) Molecular cloning and expression of the human melanocyte stimulating hormone receptor cDNA. FEBS Lett. 309, 417–420. [DOI] [PubMed] [Google Scholar]
- Roselli-Rehfuss L.; Mountjoy K. G.; Robbins L. S.; Mortrud M. T.; Low M. J.; Tatro J. B.; Entwistle M. L.; Simerly R. B.; Cone R. D. (1993) Identification of a receptor for gamma melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc. Natl. Acad. Sci. U.S.A. 90, 8856–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountjoy K. G.; Mortrud M. T.; Low M. J.; Simerly R. B.; Cone R. D. (1994) Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol. Endocrinol. 8, 1298–1308. [DOI] [PubMed] [Google Scholar]
- Gantz I.; Miwa H.; Konda Y.; Shimoto Y.; Tashiro T.; Watson S. J.; DelValle J.; Yamada T. (1993) Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J. Biol. Chem. 268, 15174–15179. [PubMed] [Google Scholar]
- Gantz I.; Shimoto Y.; Konda Y.; Miwa H.; Dickinson C.; Yamada T. (1994) Molecular cloning, expression, and characterization of a fifth melanocortin receptor. Biochem. Biophys. Res. Commun. 200, 1214–1220. [DOI] [PubMed] [Google Scholar]
- Eipper B. A.; Mains R. E. (1980) Structure and biosynthesis of pro-adrenocorticotropin/endorphin and related peptides. Endocr. Rev. 1, 1–27. [DOI] [PubMed] [Google Scholar]
- Smith A. I.; Funder J. W. (1988) Proopiomelanocortin processing in the pituitary, central nervous system, and peripheral tissues. Endocr. Rev. 9, 159–179. [DOI] [PubMed] [Google Scholar]
- Ollmann M. M.; Wilson B. D.; Yang Y. K.; Kerns J. A.; Chen Y.; Gantz I.; Barsh G. S. (1997) Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 278, 135–138. [DOI] [PubMed] [Google Scholar]
- Lu D.; Willard D.; Patel I. R.; Kadwell S.; Overton L.; Kost T.; Luther M.; Chen W.; Woychik R. P.; Wilkison W. O.; et al. (1994) Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature 371, 799–802. [DOI] [PubMed] [Google Scholar]
- McNulty J. C.; Jackson P. J.; Thompson D. A.; Chai B.; Gantz I.; Barsh G. S.; Dawson P. E.; Millhauser G. L. (2005) Structures of the Agouti Signaling Protein. J. Mol. Biol. 346, 1059–1070. [DOI] [PubMed] [Google Scholar]
- Castrucci A. M.; Hadley M. E.; Sawyer T. K.; Wilkes B. C.; al-Obeidi F.; Staples D. J.; de Vaux A. E.; Dym O.; Hintz M. F.; Riehm J. P.; et al. (1989) Alpha-melanotropin: the minimal active sequence in the lizard skin bioassay. Gen. Comp. Endrocrinol. 73, 157–163. [DOI] [PubMed] [Google Scholar]
- Hruby V. J.; Wilkes B. C.; Hadley M. E.; Al-Obeidi F.; Sawyer T. K.; Staples D. J.; de Vaux A. E.; Dym O.; Castrucci A. M.; Hintz M. F.; et al. (1987) α-Melanotropin: the minimal active sequence in the frog skin bioassay. J. Med. Chem. 30, 2126–2130. [DOI] [PubMed] [Google Scholar]
- Haskell-Luevano C.; Sawyer T. K.; Hendrata S.; North C.; Panahinia L.; Stum M.; Staples D. J.; Castrucci A. M.; Hadley M. F.; Hruby V. J. (1996) Truncation studies of α-melanotropin peptides identify tripeptide analogues exhibiting prolonged agonist bioactivity. Peptides 17, 995–1002. [DOI] [PubMed] [Google Scholar]
- Holder J. R.; Haskell-Luevano C. (2003) Melanocortin tetrapeptides modified at the N-terminus, His, Phe, Arg, and Trp positions. Ann. N.Y. Acad. Sci. 994, 36–48. [DOI] [PubMed] [Google Scholar]
- Haskell-Luevano C.; Holder J. R.; Monck E. K.; Bauzo R. M. (2001) Characterization of melanocortin NDP-MSH agonist peptide fragments at the mouse central and peripheral melanocortin receptors. J. Med. Chem. 44, 2247–2252. [DOI] [PubMed] [Google Scholar]
- Haskell-Luevano C.; Sawyer T. K.; Hendrata S.; North C.; Panahinia L.; Stum M.; Staples D. J.; Castrucci A. M.; Hadley M. F.; Hruby V. J. (1996) Truncation studies of α-melanotropin peptides identify tripeptide analogues exhibiting prolonged agonist bioactivity. Peptides 17, 995–1002. [DOI] [PubMed] [Google Scholar]
- Hruby V. J. W.; B C.; Cody W. L.; Sawyer T. K.; Hadley M. E. (1984) Melanotropins: Structural, Conformational and Biological Considerations in the Development of Superpotent and Superprolonged Analogs. Pept. Protein Rev. 3, 1–64. [Google Scholar]
- Lerner A. B.; McGuire J. S. (1961) Effect of α- and β-melanocyte stimulating hormones on the skin colour of man. Nature 189, 176–179. [DOI] [PubMed] [Google Scholar]
- Gantz I.; Konda Y.; Tashiro T.; Shimoto Y.; Miwa H.; Munzert G.; Watson S. J.; DelValle J.; Yamada T. (1993) Molecular cloning of a novel melanocortin receptor. J. Biol. Chem. 268, 8246–8250. [PubMed] [Google Scholar]
- Chen A. S.; Marsh D. J.; Trumbauer M. E.; Frazier E. G.; Guan X. M.; Yu H.; Rosenblum C. I.; Vongs A.; Feng Y.; Cao L.; Metzger J. M.; Strack A. M.; Camacho R. E.; Mellin T. N.; Nunes C. N.; Min W.; Fisher J.; Gopal-Truter S.; MacIntyre D. E.; Chen H. Y.; Van der Ploeg L. H. (2000) Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat. Genet. 26, 97–102. [DOI] [PubMed] [Google Scholar]
- Butler A. A.; Kesterson R. A.; Khong K.; Cullen M. J.; Pelleymounter M. A.; Dekoning J.; Baetscher M.; Cone R. D. (2000) A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology 141, 3518–3521. [DOI] [PubMed] [Google Scholar]
- Fan W.; Boston B. A.; Kesterson R. A.; Hruby V. J.; Cone R. D. (1997) Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385, 165–168. [DOI] [PubMed] [Google Scholar]
- Wessells H.; Levine N.; Hadley M. E.; Dorr R.; Hruby V. (2000) Melanocortin Receptor Agonists, Penile Erection, and Sexual Motivation: Human Studies with Melanotan II. Int. J. Impotence Res. 12(Suppl 4), S74–S79. [DOI] [PubMed] [Google Scholar]
- Huszar D.; Lynch C. A.; Fairchild-Huntress V.; Dunmore J. H.; Fang Q.; Berkemeier L. R.; Gu W.; Kesterson R. A.; Boston B. A.; Cone R. D.; Smith F. J.; Campfield L. A.; Burn P.; Lee F. (1997) Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88, 131–141. [DOI] [PubMed] [Google Scholar]
- Chhajlani V.; Muceniece R.; Wikberg J. E. (1993) Molecular cloning of a novel human melanocortin receptor. Biochem. Biophys. Res. Commun. 195, 866–873. [DOI] [PubMed] [Google Scholar]
- Chen W.; Kelly M. A.; Opitz-Araya X.; Thomas R. E.; Low M. J.; Cone R. D. (1997) Exocrine gland dysfunction in MC5-R-deficient mice: evidence for coordinated regulation of exocrine gland function by melanocortin peptides. Cell 91, 789–798. [DOI] [PubMed] [Google Scholar]
- Irani B. G.; Xiang Z.; Yarandi H. N.; Holder J. R.; Moore M. C.; Bauzo R. M.; Proneth B.; Shaw A. M.; Millard W. J.; Chambers J. B.; Benoit S. C.; Clegg D. J.; Haskell-Luevano C. (2011) Implication of the melanocortin-3 receptor in the regulation of food intake. Eur. J. Pharmacol. 660, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi I. S.; Keogh J. M.; Yeo G. S.; Lank E. J.; Cheetham T.; O’Rahilly S. (2003) Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med. 348, 1085–1095. [DOI] [PubMed] [Google Scholar]
- Vaisse C.; Clement K.; Durand E.; Hercberg S.; Guy-Grand B.; Froguel P. (2000) Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J. Clin. Invest. 106, 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z.; Proneth B.; Dirain M. L.; Litherland S. A.; Haskell-Luevano C. (2010) Pharmacological characterization of 30 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists, synthetic agonists, and the endogenous agouti-related protein antagonist. Biochemistry 49, 4583–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z.; Litherland S. A.; Sorensen N. B.; Proneth B.; Wood M. S.; Shaw A. M.; Millard W. J.; Haskell-Luevano C. (2006) Pharmacological characterization of 40 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists and the agouti-related protein (AGRP) antagonist. Biochemistry 45, 7277–7288. [DOI] [PubMed] [Google Scholar]
- Xiang Z.; Pogozheva I. D.; Sorenson N. B.; Wilczynski A. M.; Holder J. R.; Litherland S. A.; Millard W. J.; Mosberg H. I.; Haskell-Luevano C. (2007) Peptide and small molecules rescue the functional activity and agonist potency of dysfunctional human melanocortin-4 receptor polymorphisms. Biochemistry 46, 8273–8287. [DOI] [PubMed] [Google Scholar]
- Singh A.; Wilczynski A.; Holder J. R.; Witek R. M.; Dirain M. L.; Xiang Z.; Edison A. S.; Haskell-Luevano C. (2011) Incorporation of a bioactive reverse-turn heterocycle into a peptide template using solid-phase synthesis to probe melanocortin receptor selectivity and ligand conformations by 2D 1H NMR. J. Med. Chem. 54, 1379–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskell-Luevano C.; Boteju L. W.; Miwa H.; Dickinson C.; Gantz I.; Yamada T.; Hadley M. E.; Hruby V. J. (1995) Topographical modification of melanotropin peptide analogues with β-methyltryptophan isomers at position 9 leads to differential potencies and prolonged biological activities. J. Med. Chem. 38, 4720–4729. [DOI] [PubMed] [Google Scholar]
- Haskell-Luevano C.; Toth K.; Boteju L.; Job C.; Castrucci A. M.; Hadley M. E.; Hruby V. J. (1997) β-Methylation of the Phe7 and Trp9 melanotropin side chain pharmacophores affects ligand-receptor interactions and prolonged biological activity. J. Med. Chem. 40, 2740–2749. [DOI] [PubMed] [Google Scholar]
- Sahm U. G.; Olivier G. W.; Branch S. K.; Moss S. H.; Pouton C. W. (1994) Synthesis and biological evaluation of α-MSH analogues substituted with alanine. Peptides 15, 1297–1302. [DOI] [PubMed] [Google Scholar]
- Bednarek M. A.; Silva M. V.; Arison B.; MacNeil T.; Kalyani R. N.; Huang R. R.; Weinberg D. H. (1999) Structure-function studies on the cyclic peptide MT-II, lactam derivative of α-melanotropin. Peptides 20, 401–409. [DOI] [PubMed] [Google Scholar]
- Holder J. R.; Xiang Z.; Bauzo R. M.; Haskell-Luevano C. (2002) Structure-activity relationships of the melanocortin tetrapeptide Ac-His-D-Phe-Arg-Trp-NH2 at the mouse melanocortin receptors. 4. Modifications at the Trp position. J. Med. Chem. 45, 5736–5744. [DOI] [PubMed] [Google Scholar]
- Lee M.; Kim A.; Conwell I. M.; Hruby V.; Mayorov A.; Cai M.; Wardlaw S. L. (2008) Effects of selective modulation of the central melanocortin-3-receptor on food intake and hypothalamic POMC expression. Peptides 29, 440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.; Dirain M.; Witek R.; Rocca J. R.; Edison A. S.; Haskell-Luevano C. (2013) Structure-activity relationships of peptides incorporating a bioactive reverse-turn heterocycle at the melanocortin receptors: identification of a 5800-fold mouse melanocortin-3 receptor (mMC3R) selective antagonist/partial agonist versus the mouse melanocortin-4 receptor (mMC4R). J. Med. Chem. 56, 2747–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Shields T. S.; Stork P. J.; Cone R. D. (1995) A colorimetric assay for measuring activation of Gs- and Gq-coupled signaling pathways. Anal. Biochem. 226, 349–354. [DOI] [PubMed] [Google Scholar]
- Sawyer T. K.; Sanfillippo P. J.; Hruby V. J.; Engel M. H.; Heward C. B.; Burnett J. B.; Hadley M. E. (1980) 4-Norleucine, 7-D-Phenylalanine-α-Melanocyte-Stimulating Hormone: A Highly Potent α-Melanotropin with Ultra Long Biological Activity. Proc. Natl. Acad. Sci. U.S.A. 77, 5754–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondebjerg J.; Xiang Z.; Bauzo R. M.; Haskell-Luevano C.; Meldal M. (2002) A solid-phase approach to mouse melanocortin receptor agonists derived from a novel thioether cyclized peptidomimetic scaffold. J. Am. Chem. Soc. 124, 11046–11055. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Sykes B. D. (1994) Chemical Shifts as a Tool for Structure Determination. Methods Enzymol. 239, 363–392. [DOI] [PubMed] [Google Scholar]
- Hutchison E. G. T.; J M. (1994) A revised set of potentials for β-turn formation in proteins.. Protein Sci. 3, 2207–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying J.; Kover K. E.; Gu X.; Han G.; Trivedi D. B.; Kavarana M. J.; Hruby V. J. (2003) Solution structures of cyclic melanocortin agonists and antagonists by NMR. Biopolymers 71, 696–716. [DOI] [PubMed] [Google Scholar]
- Wilczynski A.; Wilson K. R.; Scott J. W.; Edison A. S.; Haskell-Luevano C. (2005) Structure-activity relationships of the unique and potent agouti-related protein (AGRP)-melanocortin chimeric Tyr-c[β-Asp-His-DPhe-Arg-Trp-Asn-Ala-Phe-Dpr]-Tyr-NH2 peptide template. J. Med. Chem. 48, 3060–3075. [DOI] [PubMed] [Google Scholar]
- Hess S.; Linde Y.; Ovadia O.; Safrai E.; Shalev D. E.; Swed A.; Halbfinger E.; Lapidot T.; Winkler I.; Gabinet Y.; Faier A.; Yarden D.; Xiang Z.; Portillo F. P.; Haskell-Luevano C.; Gilon C.; Hoffman A. (2008) Backbone cyclic peptidomimetic melanocortin-4 receptor agonist as a novel orally administrated drug lead for treating obesity. J. Med. Chem. 51, 1026–1034. [DOI] [PubMed] [Google Scholar]
- Rozek A.; Powers J. P.; Friedrich C. L.; Hancock R. E. (2003) Structure-based design of an indolicidin peptide analogue with increased protease stability. Biochemistry 42, 14130–14138. [DOI] [PubMed] [Google Scholar]
- Hunter H. N.; Jing W.; Schibli D. J.; Trinh T.; Park I. Y.; Kim S. C.; Vogel H. J. (2005) The interactions of antimicrobial peptides derived from lysozyme with model membrane systems. Biochim. Biophys. Acta 1668, 175–189. [DOI] [PubMed] [Google Scholar]
- Haug B. E.; Stensen W.; Kalaaji M.; Rekdal O.; Svendsen J. S. (2008) Synthetic antimicrobial peptidomimetics with therapeutic potential. J. Med. Chem. 51, 4306–4314. [DOI] [PubMed] [Google Scholar]
- Raguse T. L.; Porter E. A.; Weisblum B.; Gellman S. H. (2002) Structure-activity studies of 14-helical antimicrobial β-peptides: probing the relationship between conformational stability and antimicrobial potency. J. Am. Chem. Soc. 124, 12774–12785. [DOI] [PubMed] [Google Scholar]
- Castrucci A. M. L.; Hadley M. E.; Sawyer T. K.; Hruby V. J. (1984) Enzymological Studies of Melanotropins. Comp. Biochem. Physiol. 78B, 519–524. [DOI] [PubMed] [Google Scholar]
- Nguyen L. T.; Chau J. K.; Perry N. A.; de Boer L.; Zaat S. A.; Vogel H. J. (2010) Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PloS One 5, e12684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele T. E.; van Dijk G.; Yagaloff K. A.; Fisher S. L.; Schwartz M.; Burn P.; Seeley R. J. (1998) Central infusion of melanocortin agonist MTII in rats: assessment of c-Fos expression and taste aversion. Am. J. Physiol. 274, R248–R254. [DOI] [PubMed] [Google Scholar]
- Hagan M. M.; Benoit S. C.; Rushing P. A.; Pritchard L. M.; Woods S. C.; Seeley R. J. (2001) Immediate and prolonged patterns of Agouti-related peptide-(83-132)-induced c-Fos activation in hypothalamic and extrahypothalamic sites. Endocrinology 142, 1050–1056. [DOI] [PubMed] [Google Scholar]
- Wirth M. M.; Giraudo S. Q. (2000) Agouti-related protein in the hypothalamic paraventricular nucleus: effect on feeding. Peptides 21, 1369–1375. [DOI] [PubMed] [Google Scholar]
- Kaiser E.; Colescott R. L.; Bossinger C. D.; Cook P. I. (1970) Color Test for Detection of Free Terminal Amino Groups in the Solid-Phase Synthesis of Peptides. Anal. Biochem. 34, 595–598. [DOI] [PubMed] [Google Scholar]
- Thirumoorthy R.; Holder J. R.; Bauzo R. M.; Richards N. G. J.; Edison A. S.; Haskell-Luevano C. (2001) Novel Agouti-related Protein (AGRP) Based Melanocortin-1 Receptor Antagonist. J. Med. Chem. 44, 4114–4124. [DOI] [PubMed] [Google Scholar]
- Wüthrich K. (1986) NMR of Proteins and Nucleic Acids, John Wiley & Sons, New York. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.