

Abstract
The cardiovascular effects of mild and overt thyroid disease include a vast array of pathological changes. As well, thyroid replacement therapy has been suggested for preserving cardiac function. However, the influence of thyroid hormones on cardiac remodeling has not been thoroughly investigated at the molecular and cellular levels. The purpose of this paper is to study the effect of hypothyroidism and thyroid replacement therapy on cardiac alterations. Thirty Wistar rats were divided into 2 groups: a control (n = 10) group and a group treated with 6-propyl-2-thiouracil (PTU) (n = 20) to induce hypothyroidism. Ten of the 20 rats in the PTU group were then treated with L-thyroxine to quickly re-establish euthyroidism. The serum levels of inflammatory markers, such as C-reactive protein (CRP), tumor necrosis factor alpha (TNF-α), interleukin 6 (IL6) and pro-fibrotic transforming growth factor beta 1 (TGF-β1), were significantly increased in hypothyroid rats; elevations in cardiac stress markers, brain natriuretic peptide (BNP) and cardiac troponin T (cTnT) were also noted. The expressions of cardiac remodeling genes were induced in hypothyroid rats in parallel with the development of fibrosis, and a decline in cardiac function with chamber dilation was measured by echocardiography. Rapidly reversing the hypothyroidism and restoring the euthyroid state improved cardiac function with a decrease in the levels of cardiac remodeling markers. However, this change further increased the levels of inflammatory and fibrotic markers in the plasma and heart and led to myocardial cellular infiltration. In conclusion, we showed that hypothyroidism is related to cardiac function decline, fibrosis and inflammation; most importantly, the rapid correction of hypothyroidism led to cardiac injuries. Our results might offer new insights for the management of hypothyroidism-induced heart disease.
Introduction
It is estimated that more than 12% of the US population will develop a thyroid condition during their lifetime, and an estimated 20 million Americans have already some form of thyroid disease [1]. Besides, some of the most prominent and common symptoms of thyroid disease are those that result from the effects of thyroid hormone on the heart and cardiovascular system [2], [3], [4], [5], [6], [7], [8], [9], [10], [11]. Both hyperthyroidism and hypothyroidism produce changes in cardiac contractility, myocardial oxygen consumption, cardiac output, blood pressure, and systemic vascular resistance [12], [13], [14], [15], [16], [17].
Several important cardiac structural and functional proteins are transcriptionally regulated by thyroid hormones. The proteins that are positively regulated by thyroid hormones include sarcoplasmic reticulum calcium ATPase (SERCA2) that uptakes calcium into the sarcoplasmic reticulum during diastole [4], [18], alpha myosin heavy chain (α-MHC) the fast myosin with higher ATPase activity as well as beta adrenergic receptors, sodium/potassium ATPase and voltage-gated potassium channels Kv1.5, Kv4.2 and Kv4.3 which together coordinate the electrochemical responses of the myocardium [3], [6], [17], [19], [20], [21], [22], [23], [24], [25]; cardiac stress markers atrial and brain natriuretic peptides (ANP and BNP) are also regulated by thyroid hormones [19], [20]. Other cardiac proteins are negatively regulated by thyroid hormones such as β-MHC the slow myosin, phospholamban the SERCA inhibitor and sodium/calcium exchanger [5], [19], [20], [26], [27]. Thus, changes in the amounts of these proteins account for the altered cardiac diastolic and systolic function induced by thyroid disease.
Non-genomic effects are also exerted by thyroid hormones on cardiac myocytes and ion transport [26], [28]. In fact, triiodothyronine (T3) exerts effects on various sodium, potassium, and calcium channels in the heart, and thus changes in intracellular levels of calcium and potassium can increase inotropy and chronotropy [5], [29], [30].
Hypertrophied and, in particular, failing hearts are characterized by an accumulation of extracellular matrix elements and a corresponding increase in cardiac muscle stiffness [31], [32], [33], [34], [35], [36], [37], [38], [39], [40]. Fibronectin and collagen types I and III are the major components of the interstitial fibrillar network [41], [42], [43], [44]; thus, it has been hypothesized that the up-regulation of fibroblasts genes encoding these components accounts, in part, for the increase in fibrosis observed during the transition to heart failure and contributes to the decline in contractile performance [31], [45], [46], [47], [48], [49], [50]. The elaboration of the extracellular matrix by fibroblasts is influenced by TGF-β1 and plays an important role in pressure-overload cardiac hypertrophy [51], [52], [53]. The cytokine TGF-β1 is expressed by and modulates myocytes, vascular cells and fibroblasts [54], [55]; its expression rises in myocardium in experimental and human heart disease [55], [56], and it promotes hypertrophy, fibrosis, apoptosis, and endothelial-mesenchymal transition [57], [58], [59], [60].
Furthermore, a generalized increase in the level of contractile proteins, such as β-MHC and myofibroblast marker alpha smooth muscle actin (α-SMA), constitutes a marker of cardiac hypertrophy [61], [62]. Shifts from the normally predominant α-MHC toward β-MHC are often observed in cardiomyocytes from hypertrophied and failing hearts [63], [64], [65], [66]. α-SMA is expressed in cardiomyocytes during early stages of heart development and in dedifferentiated cardiac fibroblasts and its reactivation is considered a potential marker of ventricular hypertrophy [67], [68], [69]. Finally, the BNP serum level is also considered to be one of several criteria indicating the initiation of a pathological response in hypertrophied failing hearts [70], [71], [72]. Cardiac and circulating pro-inflammatory markers such as CRP, interleukins and TNF-α have been also associated with cardiac disease [73].
Severe hypothyroidism caused dilated cardiomyopathy with decreased α-MHC and increased β-MHC, phospholamban and ANP, an accepted clinical marker of the diseased hypertrophic heart [74], [75]. After thyroid hormone replacement the alterations in gene expression were reversed with overall improvement in myocardial performance [75]. In keeping, thyroid replacement therapy has been used by many investigators to treat patients with heart failure [76], [77], [78], [79], [80]. However, the beneficial therapeutic effects obtained in the short term might be followed by pathological manifestations of the systemic effects of thyroid hormones during longer treatments [81], [82], [83], [84].
The 6-propyl-2-thiouracil (PTU) drug which is a thiouracil-derivative used to treat hyperthyroidism [85], [86] has been long used to induce hypothyroidism in animal models such as the rat [9], [87], [88]. Data obtained from animal models on the effect of thyroid hormone on myocardial gene expression are further supported by clinical studies on humans [74], [75].
Most studies on the cardiovascular effects of thyroid hormones have particularly targeted left ventricular improvement. Nonetheless, little is known about the effect of an excess or deficiency of thyroid hormones on the myocardium collagen gene and the responsiveness of interstitial cells to these hormones [89], [90], [91]. One study, however, showed that long term hypothyroidism caused overall cardiac muscle stiffness and left ventricle diastolic wall thickness due to increased collagen I/III-based stiffness [92]. Furthermore, to our knowledge, no studies have evaluated the combined effect of hypothyroidism and thyroid hormone therapy on cardiac inflammation. The purpose of this study was to evaluate the cardiac fibrosis, inflammation and dysfunction caused by PTU-induced hypothyroidism in adult rats. Additionally, we examined the reversibility of these changes after the establishment of a euthyroid state.
Materials and Methods
Ethical approval
The protocols of the present study were approved by the Ethical Review Committee of the Saint Joseph University, were performed in accordance with the Guiding Principles in the Care and Use of Animals approved by the Council of the American Physiological Society and adhered to the Guide for the Care and Use of Laboratory Animals published by the US National Research Council committee.
Animals and experimental design
Thirty adult male Wistar rats weighing 250±25 g were obtained from the “Centre d′Elevage R. Janvier” (Le Genest-Saint Isle, France). The animals were housed at a stable temperature (25°C) and humidity and were exposed to a 12∶12 h light-dark cycle. The rats were fed ordinary rat chow, had free access to tap water and were acclimatized for at least one week under these conditions before the start of the study. Then, all animals were weighed once per week.
As shown in Figure S1, the animals were divided randomly into 2 groups: one group of 10 rats (control group) and a second group of 20 rats treated with PTU (Sigma-Aldrich, Germany); PTU was added to the drinking water for 6 weeks at a final concentration of 0.1 g/100 ml, as previously described [9], [93], [94], [95], [96], [97], [98], [99], [100]. In fact, PTU inhibits iodine and the thyroperoxidase enzyme from their normal interactions with the hormone precursor thyroglobulin to form T4 and T3. This action decreases production of thyroid hormone. In addition to these well recognized effects on intra-thyroidal iodine metabolism, PTU inhibits the peripheral deiodination and conversion of T4 into the more potent form T3 by inhibiting the 5'-deiodinase enzyme [101], [102], [103], [104]. At the end of the 6 weeks, 10 rats from the PTU group were treated with a solution of 6 µg/ml of Euthyrox (L-thyroxine-LT4, Merck, Germany), which was added to the drinking water for one week to quickly reverse the hypothyroidism (reverse group) and recover the euthyroid state; these rats were then sacrificed five weeks later. This L-thyroxine dose was chosen as previously described to maximize left ventricular function and improve heart remodeling without the risk of possible thyrotoxicosis [105], [106], [107], [108]. The rats of the control group and the remaining PTU-treated rats (n = 10) that were not treated with LT4 were sacrificed at the same time as the reverse group. Prior to sacrifice, echocardiography and blood analysis (see below), all animals were anesthetized with a mixture of ketamine (Interchemie, Holland, 75 mg/kg) and xylazine (Rotex Medica, Germany, 10 mg/kg) and were completely non-responsiveness to toe pinching.
After the sacrifice, the heart of the rats was excised, dry-weighted then cut in half; one half was kept in a 4% formalin solution (Sigma-Aldrich, St. Louis, Missouri, USA) for histopathology assessment, and the other half was stored at −80°C for the eventual RNA extraction.
Blood analysis
The levels of free triiodothyronine (fT3), free thyroxine (fT4), thyroid-stimulating hormone (TSH), BNP, CRP, IL6, TNF-α, TGF-β1, cTnT, adiponectin and leptin were measured in the serum of all the rats. Three blood samples (0.5 ml) were collected from each anesthetized rat at three distinct times: after 6 weeks of PTU treatment, after the 1 week of LT4 treatment to check for the rapid establishment of the euthyroid state and at the time of sacrifice (Figure S1).
Blood samples were collected in EDTA tubes via the jugular vein. The blood samples were immediately centrifuged at 3500 rpm for 10 min, and the serum was frozen at −80°C for subsequent analyses. The BNP, CRP, IL6, TNF-α, TGF-β1, adiponectin and leptin ELISA kit reagents were purchased from Abcam, Cambridge - UK, and the fT3, fT4, TSH and cTnT ELISA kit reagents were purchased from CusaBio, Hubei, China. All samples were run in triplicate, and the intra- and inter-assay variations were less than 10%.
Echocardiographic assessment
The animals were anesthetized by 1.5% isoflurane gas and placed on isothermal pads with their chests shaved. Trans-thoracic two-dimensional guided M-mode echocardiography was performed via the long axis of the left ventricle at the level of the papillary muscles using a SonoScape instrument equipped with a convex 8 MHz transducer. The surgeon and echocardiographer were blinded to the rats' pre-treatments.
Histopathology
The formalin-fixed heart tissues were embedded in paraffin, and 4-µm thick sections were cut. Paraffin-embedded sections of the hearts were stained with either hematoxylin-eosin or Masson's trichrome (Sigma-Aldrich, St. Louis, Missouri, USA) for histopathological evaluation. After staining, the sections were dehydrated in ethanol/water baths with decreasing water content and finally rinsed in xylene before being mounted with a permanent mounting medium.
Gross examination and histological sections were analyzed by two independent pathologists in a blinded fashion. A semi-quantitative scoring system was used to assess the cardiac myocyte hypertrophy, tissue inflammation and fibrosis. Inflammation refers to the presence of aggregates of leukocytes in the heart muscle. Fibrosis analysis was performed using the ImageJ program by applying a threshold to the acquired pictures and creating selections of the fibrotic areas. Eight sections were analyzed for each rat in the three groups.
Real-time quantitative polymerase chain reaction
Hearts previously kept at −80°C were ground, and total cellular RNA was extracted from cells using TRIzol reagent (Life Technologies, Carlsbad, California, USA). The samples were then purified with 75% ethanol, and the purity and concentration of the RNA were determined by measuring the absorbance at 260 nm with an ND-1000 NanoDrop spectrophotometer (Thermo Scientific, Wilmington, Delaware, USA). Subsequently, the first-strand cDNA was synthesized using a Superscript III kit for RT-PCR (Life Technologies, Carlsbad, California, USA). Briefly, total RNA was denatured at 65°C for 5 min in the presence of 250 ng/µl random primers and 10 mM deoxynucleotide triphosphate. After the sample was chilled on ice for at least 1 min, reverse transcription was allowed to proceed at 25°C for 5 min in the presence of 1× first-strand buffer, 5 mM dithiothreitol, and 40 U of ribonuclease inhibitor. The reaction was then allowed to proceed at 50°C for another 60 min. The reaction was terminated by heat inactivation at 70°C for 10 min. Real-time PCR was conducted using a 7500 real-time PCR system (Applied Biosystems, USA) and SYBR Green PCR master mix (Life Technologies, Carlsbad, California, USA). Real-time Q-RT-PCR was performed in a 20-µl reaction mixture containing 250 nM forward and reverse primers, 1× SYBR Green reaction mix and various amounts of template. The reaction was performed with preliminary denaturation for 10 min at 95°C to activate Taq DNA polymerase, followed by 40 cycles of denaturation at 95°C for 15 sec and annealing/extension at 60°C for 1 min. The fluorescence emitted by SYBR Green was detected online by the ABI PRISM 7500 sequence detection system (Applied Biosystems). Melting curves were measured at the end of the amplification to confirm the specificity of the amplified products. All PCR was performed in triplicate on the same 96-well plate. Ribosomal protein L32 (Rpl32) was used as a housekeeping gene, and quantifications were conducted using the 2ΔΔCt method. Studies have shown that the initial copy number can be quantitated during real-time PCR analysis based on the threshold cycle (Ct) [109], [110]. The Ct is defined as the cycle at which fluorescence is determined to be significantly greater than the background. For quantification of the gene expression changes, the Ct method was used to calculate the relative fold changes normalized against the Rpl32 gene. To check for genomic DNA contamination, parallel samples were run without the addition of the reverse transcriptase. The primers (Eurogentec, Seraing, Belgium) used are presented in Table 1.
Table 1. Primers for the different genes.
| Gene | Forward primer (5′-3′) | Reverse primer (5′-3′) |
| Anp | CTTCCTCTTCCTGGCCTTTTG | CGCACTGTATACGGGATTTGC |
| Bnp | AAGTCCTAGCCAGTCTCCAGAACA | AGCTCCAGCAGCTTCTGCAT |
| α-Mhc | CTTCTGCTGATACCGGTGACAG | TGAGCCTTTCTTCTTGCCTCC |
| β-Mhc | ACAGGAAAGTTGGCATCTGCA | GGATTTTTCCAGAAGGTAGGTCTCT |
| α-Sma | AGAGTGGAGAAGCCCAGCCAGTC | ATCATCACCAGCAAAGCCCGCC |
| Tgf-β1 | ATGGTGGACCGCAACAACGCAATC | CACGGGACAGCAATGGGGGTT |
| cTgf | TGTGCACGGAGCGTGATCCC | TGCACCATCTTTGGCAGTGCACA |
| Il1 | CTGACAGACCCCAAAAGATTAAGG | CCTTGTCGAGATGCTGCTGTGA |
| Mcp1 | CAGCCAGATGCAGTTAATGCCCC | GCTTCTTTGGGACACCTGCTGCTG |
| Col1 | GAGAGAGCATGACCGATGGATT | GATAGCGACATCGGCAGGAT |
| Col3 | AAACGGAGAACGGGGTGGCC | TCACCAGGTGCGCCAGTAGGT |
| Rpl32 | CCAGAGGCATCGACAACA | GCACTTCCAGCTCCTTGACAT |
Forward and reverse primers used for quantitative real-time PCR. Abbreviations: Anp: Atrial natriuretic peptide; Bnp: Brain natriuretic peptide; α-Mhc: α-Myosin heavy chain; β-Mhc: β-Myosin heavy chain; α-Sma: α-Smooth muscle actin; Tgf-β1: Transforming growth factor beta 1; cTgf: Connective tissue growth factor; Il1: Interleukin 1; Mcp1: Monocyte chemoattractant protein 1; Col1: Collagen type I; Col3: Collagen type III; Rpl32: Ribosomal protein L32.
Statistical analysis
All quantitative data are reported as the mean ± SEM. Statistical analysis was performed with the SigmaPlot (v11.0) software. Kruskal-Wallis One-way ANOVA on rank tests were performed for multiple comparisons of the values because the normal distribution verified with the Shapiro-Wilk test was not met. Post hoc analysis was performed with the Dunn's multiple comparison tests to identify the group differences that accounted for the overall ANOVA results. When two independent variables were present, two-way ANOVA tests were performed, followed by post hoc Tukey's multiple comparison tests. All values with p<0.05 were considered significant.
Results
Serum levels of fT3, fT4 and TSH
Administration of PTU for 6 weeks resulted in significant decreases in fT3 and fT4 compared with the levels in the control rat group, confirming the establishment of hypothyroidism. Treatment with LT4 for 1 week raised fT3 and fT4 to levels comparable to those of the control rats, verifying the rapid reversibility of hypothyroidism (Figure 1A). After 12 weeks, at the time of sacrifice, the PTU group still presented relatively low levels of fT3 and fT4, whereas the reverse group retained levels comparable to those of the control rats (Figure 1A).
Figure 1. Serum levels of fT3 (A), fT4 (A) and TSH (B) in the different rat groups at different times after the PTU or LT4 treatments and sacrifice.

Data are represented as the mean ± SEM. n = 10 animals for each group. Statistical analysis was performed with two-way ANOVA tests followed by post hoc Tukey's multiple comparison tests. *p<0.001 vs. control; # p<0.001 vs. control and reverse; $ p<0.001 vs. 7 and 12 weeks reverse and 12 weeks PTU; § p<0.001 vs. 7 and 12 weeks reverse; + p<0.001 vs. 7 and 12 weeks reverse and 7 weeks PTU.
The TSH levels significantly increased under the PTU treatment, which also confirmed the hypothyroid state, and remained elevated at 12 weeks even with the discontinuation of PTU (Figure 1B). The LT4 treatment for 1 week decreased the TSH levels to normal values that remained stable until the end of the protocol (Figure 1B).
Plasma markers of inflammation, fibrosis and cardiac hypertrophy
Hypothyroidism resulted in significant increases in the plasma inflammatory markers CRP and TNF-α and IL6, whereas reversing the hypothyroid state further increased these levels (Figure 2A–2C). The level of adiponectin, a cardioprotective adipocyte-derived cytokine, did increase in the PTU-treated rats; however, this increase was not significant. The mean plasma levels of adiponectin returned to normal after the LT4 treatment (Figure 3A). The mean plasma level of pro-fibrotic TGF-β1 increased in the PTU rats and drastically increased after the hypothyroidism reversal (Figure 3B). Similarly, there was a significant increase in the plasma cardiac stress marker BNP in the PTU group, and this level returned to normal under the LT4 treatment (Figure 4A). In addition, the cTnT levels dramatically increased in the PTU group compared with the levels in the control group and were further induced after the hypothyroidism reversal (Figure 4B).
Figure 2. Effects of hypothyroidism and LT4 treatment on the plasma levels of inflammatory markers.

Changes in the CRP (A), TNF-α (B) and IL6 (C) levels in the different treatment groups at the time of sacrifice. Data are represented as the mean ± SEM. n = 10 animals for each group. Statistical analysis was performed with Kruskal-Wallis One-way ANOVA on rank tests followed by post hoc Dunn's multiple comparison tests. *p<0.01 vs. control; # p<0.05 vs. PTU.
Figure 3. Adiponectin (A) and TGF-β1 (B) plasma levels in the different groups of animals.
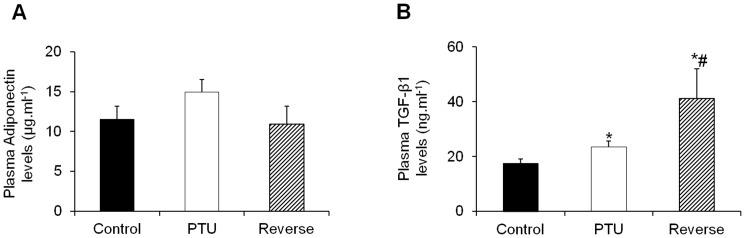
Data are represented as the mean ± SEM. n = 10 animals for each group. Statistical analysis was performed with Kruskal-Wallis One-way ANOVA on rank tests followed by post hoc Dunn's multiple comparison tests. *p<0.01 vs. control; # p<0.05 vs. PTU.
Figure 4. Effects of hypothyroidism and LT4 treatment on the plasma levels of cardiac stress markers.

Changes in the BNP (A) and cTnT (B) levels in the different groups. Data are represented as the mean ± SEM. n = 10 animals for each group. Statistical analysis was performed with Kruskal-Wallis One-way ANOVA on rank tests followed by post hoc Dunn's multiple comparison tests. *p<0.05 vs. control; # p<0.01 vs. PTU.
Gene expression of cardiac remodeling markers
Hypothyroidism induced the expression of the fetal genes for atrial and brain natriuretic peptides (Anp and Bnp, respectively) in the heart, whereas LT4 treatment abolished these expressions (Figure 5A). The expression of the cardiac muscle-specific protein α-Mhc, which is involved in active force generation, decreased in the hypothyroid rats, whereas the LT4 treatment returned this gene expression to levels comparable to those in the control rats (Figure 5B). In contrast, the slow-twitch β-Mhc isoform expression drastically increased in the hypothyroid rats and then returned to normal under LT4 treatment (Figure 5B).
Figure 5. Effects of hypothyroidism and LT4 treatment on hypertrophic, fibrotic and inflammatory gene markers in the rat heart.

Anp, Bnp (A), α-Mhc, β-Mhc (B), α-Sma (C), Tgf-β1, cTgf (D), Il1, Mcp1 (E) and Col1, Col3 (F) gene expressions relative to the housekeeping gene Rpl32. Data are represented as the mean ± SEM. n = 10 animals for each group. Statistical analysis was performed with Kruskal-Wallis One-way ANOVA on rank tests followed by post hoc Dunn's multiple comparison tests. a.u.: arbitrary units. *p<0.01 vs. control; # p<0.01 vs. PTU.
The cardiac fibroblast to myofibroblast differentiation, as shown by α-Sma expression, increased in the hypothyroid rats and then returned to normal after the euthyroid state was established (Figure 5C), whereas the cardiac extracellular matrix components Col1 and Col3 presented the same gene expression profile (Figure 5D).
Interestingly, the pro-fibrotic markers Tgf-β1 and cTgf and the inflammatory markers Il1 and Mcp1 presented expression profiles that were opposite to those of the other cardiac stress markers. In fact, Tgf-β1 and cTgf and Il1 and Mcp1 were induced by PTU and further increased under LT4 treatment (Figure 5E and 5F).
Echocardiography and cardiac hypertrophy assessment
Significant decreases in heart mass were present during the PTU treatment (Table 2) along with a reduction of the ratio of the heart weight to tibia length (Figure 6A). This heart weight loss was abolished by the LT4 treatment (Table 2, Figure 6A). In addition, the growth rate was significantly inhibited in the rats treated with PTU compared with that in the control group (Table 2), although the PTU-treated animals appeared to be reasonably healthy, with no changes in water intake and food consumption. This effect was observed very early after the beginning of the treatment. Furthermore, it was reversed after the re-establishment of a euthyroid state. At the end of the study, there was no significant difference in the mean body weight between the reverse group and the control group (Table 2). Concordantly, the “satiety hormone” leptin showed significant plasma elevations under the PTU treatment, and these elevations regressed to normal levels after hypothyroidism reversal (Figure S2). This reduction in body mass with PTU treatment affected the heart weight to body weight ratio of the rats in an opposite way as compared to the heart weight to tibia length ratio (Table 2).
Table 2. Body and heart weights in the different rat groups.
| Groups | n | Body weight (BW), g | Heart weight (HW), mg | HW/BW (mg/g) |
| Control | 10 | 404 ± 17.6 | 1582.8 ± 124 | 3.92 ± 0.3 |
| PTU | 10 | 248 ± 37.6* | 1212.4 ± 95* | 4.88 ± 0.38* |
| Reverse | 10 | 406 ± 19.2# | 1566.6 ± 156# | 3.86 ± 0.38# |
Data are represented as the mean ± SEM. Statistical analysis was performed with Kruskal-Wallis One-way ANOVA on rank tests followed by post hoc Dunn's multiple comparison tests. n: number of animals in each group. *p<0.001 vs. control; # p<0.001 vs. PTU.
Figure 6. The LT4 treatment reversed cardiac dilation and improved systolic function.

A: Heart weight to tibia length ratio (mg/cm) in the different treatment groups. Statistical analysis was performed with Kruskal-Wallis One-way ANOVA on rank tests followed by post hoc Dunn's multiple comparison tests. **p<0.01 vs. control and reverse. B and C: Echocardiographic measurements of the ejection fraction (%) and fractional shortening (%) in the control, PTU-treated and LT4-treated rats at different time points. Statistical analysis was performed with two-way ANOVA tests followed by post hoc Tukey's multiple comparison tests. *p<0.05 vs. control as well as 7 and 12 weeks reverse; # p<0.05 vs. PTU. D: M-mode echocardiographic tracings from the control, PTU and reverse groups. Echocardiography scale bars: 0.25 s and 0.5 mm. Data are represented as the mean ± SEM. n = 10 animals for each group.
Echocardiography showed significant bradycardia after the hypothyroidism induction at 6 weeks, and these decreases in heart rate were still noticeable at 12 weeks (Table 3). In the PTU group, the end-diastolic and end-systolic interventricular septum thicknesses (IVSTd and IVSTs, respectively) were significantly reduced compared with those in the control animals, whereas the end-diastolic and end-systolic left ventricular diameters (LVIDd and LVIDs, respectively) increased, indicating the presence of left ventricular dilation. Similarly, the end-diastolic and end-systolic posterior wall thicknesses (LVPWd and LVPWs, respectively) showed significant regressions in the PTU group (Table 3). All of these parameters were reversed under the LT4 treatment, demonstrating the reversal of cardiac dilation (Table 3).
Table 3. Echocardiographic parameters of the rats at different time periods after hypothyroidism induction and LT4 treatment.
| HR (bpm) | IVSTd (cm) | LVIDd (cm) | LVPWd (cm) | IVSTs (cm) | LVIDs (cm) | LVPWs (cm) | ||
| 6 weeks | Control | 475 ± 12 | 0.14 ± 0.02 | 0.65 ± 0.05 | 0.13 ± 0.02 | 0.22 ± 0.02 | 0.31 ± 0.06 | 0.21 ± 0.02 |
| PTU | 335 ± 10* | 0.11 ± 0.01* | 0.87 ± 0.07* | 0.11 ± 0.02* | 0.18 ± 0.03* | 0.48 ± 0.05* | 0.18 ± 0.01* | |
| PTU (reverse) | 327 ± 14* | 0.1 ± 0.02* | 0.84 ± 0.09* | 0.11 ± 0.01* | 0.19 ± 0.03* | 0.45 ± 0.07* | 0.17 ± 0.02* | |
| 7 weeks | Control | 490 ± 18 | 0.15 ± 0.02 | 0.68 ± 0.06 | 0.14 ± 0.02 | 0.23 ± 0.03 | 0.33 ± 0.06 | 0.21 ± 0.02 |
| PTU | 411 ± 11* | 0.12 ± 0.02* | 0.92 ± 0.1* | 0.12 ± 0.01* | 0.20 ± 0.02* | 0.49 ± 0.05* | 0.18 ± 0.01* | |
| Reverse | 500 ± 17# | 0.15 ± 0.03# | 0.78 ± 0.08# | 0.16 ± 0.02# | 0.22 ± 0.02# | 0.31 ± 0.07# | 0.23 ± 0.01# | |
| 12 weeks | Control | 480 ± 20 | 0.16 ± 0.02 | 0.67 ± 0.11 | 0.16 ± 0.01 | 0.25 ± 0.03 | 0.31 ± 0.04 | 0.23 ± 0.02 |
| PTU | 410 ± 15* | 0.13 ±0.01* | 0.85 ± 0.05* | 0.14 ± 0.01* | 0.21 ± 0.02* | 0.43 ± 0.03* | 0.19 ± 0.02* | |
| Reverse | 465 ± 22# | 0.15 ±0.01# | 0.72 ± 0.05# | 0.17 ± 0.02# | 0.26 ± 0.02# | 0.33 ± 0.05# | 0.22 ± 0.01# |
HR: heart rate; IVSTd: end-diastolic interventricular septum thickness; LVIDd: end-diastolic left ventricular diameter; LVPWd: end-diastolic posterior wall thickness; IVSTs: end-systolic interventricular septum thickness; LVIDs: end-systolic left ventricular diameter; LVPWs: end-systolic posterior wall thickness. Data are represented as the mean ± SEM. Statistical analysis was performed with two-way ANOVA tests followed by post hoc Tukey's multiple comparison tests. n = 10 rats in each group. *p<0.05 vs. control as well as 7 and 12 weeks reverse; # p<0.05 vs. PTU.
The cardiac function declined after the PTU treatment, and the ejection fraction and fractional shortening values decreased (Figure 6B and 6C). These changes were quickly reversed after just the 1 week LT4 treatment and remained unchanged until 12 weeks (Figure 6B and 6C). Representative examples of M-mode echocardiography tracings from the different groups are shown in Figure 6D.
Histopathology
Hypothyroidism resulted in a significant increase in the nucleus size of the myocardial cells as well as the development of myocardial hypertrophy (Figure 7A, 7B and 7G) and focal fibrosis lesions (Figure 7D, 7E and 7H), compared with the control group. There was no inflammatory cell infiltration and no signs of myocardial lesions or myocarditis (Figure 7A, 7B and 7I).
Figure 7. Representative microphotographs of histological slices of hearts from the different rat groups.

A and D: Heart of the control group stained with hematoxylin-eosin (A) and Masson's trichrome (D) showing a well-organized lamellar arrangement of cardiomyocytes without signs of fibrosis. B and E: Heart of the PTU group stained with hematoxylin-eosin (B) and Masson's trichrome (E) showing cardiomyocyte hypertrophy with increased size of the nuclei and presenting zones of fibrosis (blue in E). C and F: Heart of the reverse group stained with hematoxylin-eosin (C) and Masson's trichrome (F) showing a massive infiltration of lymphocytes and macrophages (arrows in C) and diminished fibrotic lesions (blue in F). G–I: Quantitative analysis of cardiomyocyte size, fibrotic areas and cellular infiltration in each of the different animal groups. Fibrosis analysis was performed using the ImageJ program by thresholding the acquired pictures and then creating selections of the fibrotic areas. Scale bars: 75 µm. Data are represented as the mean ± SEM. n = 10 animals for each group. Eight sections were analyzed for each rat in the three groups. Statistical analysis was performed with Kruskal-Wallis One-way ANOVA on rank tests followed by post hoc Dunn's multiple comparison tests. *p<0.01 vs. control and reverse; # p<0.01 vs. control and PTU.
However, in the reverse group, the myocardial hypertrophy and fibrosis regressed (Figure 7C and 7F-7H). Most importantly, there was massive infiltration of inflammatory cells more specifically macrophages and lymphocytes, whereas signs of myocardial lesions and myocarditis were clearly developed (Figure 7C and 7I).
Discussion
Our study showed that hypothyroidism in rats is related to the development of cardiac fibrosis and inflammation along with chamber dilation and function decline. Interestingly, the rapid re-establishment of euthyroidism resulted in aggravated cardiac inflammation and injury, although the fibrosis and function were contradictorily ameliorated. Thyroid replacement therapy in hypothyroid patients has been shown to improve their prognosis and reduce their cardiovascular risk [5], [108], [111], [112], [113], [114], [115]. However, some hypothyroid patients might remain at an increased risk for the morbidity associated with circulatory diseases and ischemic heart disease as well as other systemic manifestations despite treatment with LT4 [81], [116], [117], [118], [119].
After 6 weeks of PTU treatment, the fT3 and fT4 levels were depressed, whereas the TSH level drastically increased, indicating the establishment of hypothyroidism. The normal levels of fT3 and fT4 following the one week treatment with LT4 confirmed the rapid reversal of hypothyroidism. Intriguingly, the levels of fT3 and fT4 rose but still remained significantly low with high TSH 6 weeks after the PTU treatment was stopped in the animals that did not receive LT4. This apparent discrepancy in the levels of thyroid hormones and TSH has been described in the literature; in fact, the usual reverse relationship between the serum levels of TSH and T4 might not be maintained in hypothyroid patients [120]. Patients with severe or long-standing primary hypothyroidism may require three to six months of hormone replacement before the TSH levels are fully suppressed [121], [122], [123]. Conversely, the serum TSH concentration may remain low or normal for up to five weeks after the withdrawal of thyroid hormone replacement when the serum levels of T4 and T3 have already declined to values well below the lower range of normal [120], [123], [124].
Hypothyroidism significantly reduced body weight and elevated the plasma leptin concentration. These findings may be contradictory to the data in human subjects with thyroid disorders, where the majority of the hypothyroid patients suffer from an increased body weight. However, these findings are in agreement with previously reported data on rats [125], [126], [127], [128], [129], [130], [131]; those studies showed that, in hypothyroid rats, the body weight gain per week was very low (approximately one-sixth as high as the weight gain of euthyroid rats) and the body growth was strongly restricted. Additionally, the hypothyroid state was found to increase the serum leptin level; the "satiety hormone" leptin suppresses food intake in hypothyroid rats and may reduce the level of metabolism and body growth gain [132], [133], [134]. Treatment with LT4 decreased the leptin levels and re-established normal body weights.
The association of hypothyroidism with plasma pro-inflammatory markers such as TNF-α and CRP has been demonstrated in some studies [135], [136], [137], [138]. However, a few studies have reported the effect of thyroid hormone therapy on these markers with contradictory results [139], [140]. Our data clearly demonstrate the induction of inflammatory and fibrotic markers in hypothyroid rats; most importantly, this inflammation state was aggravated by the LT4 treatment. Adiponectin levels did not change during hypothyroidism or after LT4 treatment, which is consistent with previous studies [141].
In contrast, hypothyroidism stimulated the secretion of the cardiac stress markers BNP and cTnT in parallel with a global decline in cardiac function along with the development of chamber dilation. The reduced heart weight as well as septum and wall thicknesses in hypothyroid rats seemed contradictory to the cardiomyocyte hypertrophy noticed in histopathology. In fact, hypothyroidism can produce changes that resemble heart failure in many aspects [142], [143]. Indeed, studies have showed that hypothyroidism can lead to chamber dilatation from series addition of sarcomeres despite a reduction in cardiac mass [144], [145], [146], [147]. These cellular changes are recognized as components of heart failure. Furthermore, increased chamber diameter/wall thickness ratio during the decompensated phase of heart failure is reflected by a similar increase in myocyte length/width ratio [145], [148], [149]. This occurs by myocyte lengthening without a change in myocyte cross-sectional area during the transition phase [149], [150]. Similarly, early loss of cardiac mass in rats treated with PTU for 4 weeks was due to a reduction in myocyte cross-sectional area [9], [143]. Apoptosis might be another possible contributor to this cardiac mass loss since T3 treatment has been shown to inhibit cardiomyocyte apoptosis in infarcted myocardium [151]. This cardiac phenotype change was also supported by the up-regulation of hypertrophy markers, myofibroblast differentiation, extracellular matrix components, and pro-fibrotic and pro-inflammatory genes that contribute to tissue stiffness and defects in the structure and contractility of the myocardium. Nonetheless, treatment with LT4 enhanced the global cardiac function and abolished the hypertrophy and extracellular matrix markers. However, deleterious effects of this rapid reversal of hypothyroidism were noticed with the induction of the TGF-β1, IL1 and MCP1 expressions in the myocardium.
Indeed, the cardiovascular effects of hypothyroidism include changes in the expression of both structural and regulatory myocyte genes [6], [20], [152]. Thyroid hormones were shown to have opposite effects on collagen synthesis and the induction of TGF-β1. In fact, thyroid hormones induced this pro-fibrotic marker in cardiomyocyte and fibroblast cultures, whereas they decreased the expression of the collagen I gene. Similarly, cardiac hypertrophy was induced by thyroid hormones without any signs of myocardial fibrosis [40], [50], [153], [154], [155], [156], [157].
Finally, when rapidly reversing hypothyroidism, we observed the development of cardiac injury with a net elevation of the serum cTnT level; leukocyte infiltration was also noted, confirming the presence of myocarditis. This phenomenon may be explained by a direct deleterious effect of the hormone on the heart [89]. Indeed, the induction of the inflammatory IL1 and MCP1 as well as the drastic increases in TGF-β1 might be responsible for this cellular infiltration. TGF-β1 is a multi-potent cytokine that plays an important role in the onset of inflammation [158], [159].
In conclusion, we showed that hypothyroidism is related to the development of cardiac fibrosis and inflammation. Most importantly, the rapid correction of hypothyroidism led to cardiac injuries. This study might offer new insights for the management of hypothyroidism-induced heart disease.
Supporting Information
Experimental protocol with different measured parameters.
(TIF)
Plasma leptin levels in the different groups of rat treatments at the time of sacrifice. Data are represented as the mean ± SEM. n = 10 animals for each group. Statistical analysis was performed with Kruskal-Wallis One-way ANOVA on rank tests followed by post hoc Dunn's multiple comparison tests. *p<0.01 vs. control and reverse.
(TIF)
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was funded and supported by the Research Council of the Saint Joseph University - Faculty of Medicine. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.ATA (2014) Prevalence and Impact of Thyroid Disease. Available: http://www.thyroid.org/. Accessed 2014 September 29.
- 2. Biondi B, Klein I (2004) Hypothyroidism as a risk factor for cardiovascular disease. Endocrine 24: 1–13. [DOI] [PubMed] [Google Scholar]
- 3. Danzi S, Klein I (2004) Thyroid hormone and the cardiovascular system. Minerva Endocrinol 29: 139–150. [PubMed] [Google Scholar]
- 4. Dillmann WH (2002) Cellular action of thyroid hormone on the heart. Thyroid 12: 447–452. [DOI] [PubMed] [Google Scholar]
- 5. Klein I, Danzi S (2007) Thyroid disease and the heart. Circulation 116: 1725–1735. [DOI] [PubMed] [Google Scholar]
- 6. Klein I, Ojamaa K (2001) Thyroid hormone and the cardiovascular system. N Engl J Med 344: 501–509. [DOI] [PubMed] [Google Scholar]
- 7. Rhee SS, Pearce EN (2011) Update: Systemic Diseases and the Cardiovascular System (II). The endocrine system and the heart: a review. Rev Esp Cardiol 64: 220–231. [DOI] [PubMed] [Google Scholar]
- 8. Savinova OV, Liu Y, Aasen GA, Mao K, Weltman NY, et al. (2011) Thyroid hormone promotes remodeling of coronary resistance vessels. PLoS One 6: e25054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tang YD, Kuzman JA, Said S, Anderson BE, Wang X, et al. (2005) Low thyroid function leads to cardiac atrophy with chamber dilatation, impaired myocardial blood flow, loss of arterioles, and severe systolic dysfunction. Circulation 112: 3122–3130. [DOI] [PubMed] [Google Scholar]
- 10. Zhang Y, Post WS, Cheng A, Blasco-Colmenares E, Tomaselli GF, et al. (2013) Thyroid hormones and electrocardiographic parameters: findings from the third national health and nutrition examination survey. PLoS One 8: e59489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mitchell JE, Hellkamp AS, Mark DB, Anderson J, Johnson GW, et al. (2013) Thyroid function in heart failure and impact on mortality. JACC Heart Fail 1: 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weltman NY, Wang D, Redetzke RA, Gerdes AM (2012) Longstanding hyperthyroidism is associated with normal or enhanced intrinsic cardiomyocyte function despite decline in global cardiac function. PLoS One 7: e46655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Biondi B (2012) Mechanisms in endocrinology: Heart failure and thyroid dysfunction. Eur J Endocrinol 167: 609–618. [DOI] [PubMed] [Google Scholar]
- 14. Gencer B, Collet TH, Virgini V, Bauer DC, Gussekloo J, et al. (2012) Subclinical thyroid dysfunction and the risk of heart failure events: an individual participant data analysis from 6 prospective cohorts. Circulation 126: 1040–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Biondi B, Wartofsky L (2014) Treatment with thyroid hormone. Endocr Rev 35: 433–512. [DOI] [PubMed] [Google Scholar]
- 16. Biondi B, Palmieri EA, Lombardi G, Fazio S (2002) Effects of thyroid hormone on cardiac function: the relative importance of heart rate, loading conditions, and myocardial contractility in the regulation of cardiac performance in human hyperthyroidism. J Clin Endocrinol Metab 87: 968–974. [DOI] [PubMed] [Google Scholar]
- 17. Kahaly GJ, Dillmann WH (2005) Thyroid hormone action in the heart. Endocr Rev 26: 704–728. [DOI] [PubMed] [Google Scholar]
- 18. Kiss E, Jakab G, Kranias EG, Edes I (1994) Thyroid hormone-induced alterations in phospholamban protein expression. Regulatory effects on sarcoplasmic reticulum Ca2+ transport and myocardial relaxation. Circ Res 75: 245–251. [DOI] [PubMed] [Google Scholar]
- 19. Schwartz K, Lompre AM, Bouveret P, Wisnewsky C, Whalen RG (1982) Comparisons of rat cardiac myosins at fetal stages in young animals and in hypothyroid adults. J Biol Chem 257: 14412–14418. [PubMed] [Google Scholar]
- 20. Lompre AM, Nadal-Ginard B, Mahdavi V (1984) Expression of the cardiac ventricular alpha- and beta-myosin heavy chain genes is developmentally and hormonally regulated. J Biol Chem 259: 6437–6446. [PubMed] [Google Scholar]
- 21. Bahouth SW, Cui X, Beauchamp MJ, Park EA (1997) Thyroid hormone induces beta1-adrenergic receptor gene transcription through a direct repeat separated by five nucleotides. J Mol Cell Cardiol 29: 3223–3237. [DOI] [PubMed] [Google Scholar]
- 22. Gick GG, Melikian J, Ismail-Beigi F (1990) Thyroidal enhancement of rat myocardial Na,K-ATPase: preferential expression of alpha 2 activity and mRNA abundance. J Membr Biol 115: 273–282. [DOI] [PubMed] [Google Scholar]
- 23. Ojamaa K, Kenessey A, Shenoy R, Klein I (2000) Thyroid hormone metabolism and cardiac gene expression after acute myocardial infarction in the rat. Am J Physiol Endocrinol Metab 279: E1319–1324. [DOI] [PubMed] [Google Scholar]
- 24. Shao Y, Ojamaa K, Klein I, Ismail-Beigi F (2000) Thyroid hormone stimulates Na, K-ATPase gene expression in the hemodynamically unloaded heterotopically transplanted rat heart. Thyroid 10: 753–759. [DOI] [PubMed] [Google Scholar]
- 25. Ojamaa K, Sabet A, Kenessey A, Shenoy R, Klein I (1999) Regulation of rat cardiac Kv1.5 gene expression by thyroid hormone is rapid and chamber specific. Endocrinology 140: 3170–3176. [DOI] [PubMed] [Google Scholar]
- 26. Dillmann W (2010) Cardiac hypertrophy and thyroid hormone signaling. Heart Fail Rev 15: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fazio S, Palmieri EA, Lombardi G, Biondi B (2004) Effects of thyroid hormone on the cardiovascular system. Recent Prog Horm Res 59: 31–50. [DOI] [PubMed] [Google Scholar]
- 28. Davis PJ, Leonard JL, Davis FB (2008) Mechanisms of nongenomic actions of thyroid hormone. Front Neuroendocrinol 29: 211–218. [DOI] [PubMed] [Google Scholar]
- 29. Walker JD, Crawford FA, Kato S, Spinale FG (1994) The novel effects of 3,5,3'-triiodo-L-thyronine on myocyte contractile function and beta-adrenergic responsiveness in dilated cardiomyopathy. J Thorac Cardiovasc Surg 108: 672–679. [PubMed] [Google Scholar]
- 30. Davis PJ, Davis FB (1993) Acute cellular actions of thyroid hormone and myocardial function. Ann Thorac Surg 56: S16–23. [DOI] [PubMed] [Google Scholar]
- 31. Schelbert EB, Fonarow GC, Bonow RO, Butler J, Gheorghiade M (2014) Therapeutic Targets in Heart Failure: Refocusing on the Myocardial Interstitium. J Am Coll Cardiol 63: 2188–2198. [DOI] [PubMed] [Google Scholar]
- 32. Biernacka A, Frangogiannis NG (2011) Aging and Cardiac Fibrosis. Aging Dis 2: 158–173. [PMC free article] [PubMed] [Google Scholar]
- 33. Wong TC, Piehler K, Meier CG, Testa SM, Klock AM, et al. (2012) Association between extracellular matrix expansion quantified by cardiovascular magnetic resonance and short-term mortality. Circulation 126: 1206–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wong TC (2014) Cardiovascular Magnetic Resonance Imaging of Myocardial Interstitial Expansion in Hypertrophic Cardiomyopathy. Curr Cardiovasc Imaging Rep 7: 9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wong TC, Piehler KM, Kang IA, Kadakkal A, Kellman P, et al. (2014) Myocardial extracellular volume fraction quantified by cardiovascular magnetic resonance is increased in diabetes and associated with mortality and incident heart failure admission. Eur Heart J 35: 657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Venkatesh BA, Volpe GJ, Donekal S, Mewton N, Liu CY, et al. (2014) Association of Longitudinal Changes in Left Ventricular Structure and Function With Myocardial Fibrosis: The Multi-Ethnic Study of Atherosclerosis Study. Hypertension. [DOI] [PMC free article] [PubMed]
- 37. Mewton N, Liu CY, Croisille P, Bluemke D, Lima JA (2011) Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol 57: 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jugdutt B (2006) Interstitial fibrosis in heart failure. In Pr Francisco J. Villarreal, editors. Developments in Cardiovascular Medicine. Volume 253: pp 23–55. Springer New York.
- 39.Chapman E, Spinale F (2005) Interstitial fibrosis in heart failure. In Pr Francisco J. Villarreal, editors. Developments in Cardiovascular Medicine. Volume 253: pp 181–196. Springer New York.
- 40. Boluyt MO, O'Neill L, Meredith AL, Bing OH, Brooks WW, et al. (1994) Alterations in cardiac gene expression during the transition from stable hypertrophy to heart failure. Marked upregulation of genes encoding extracellular matrix components. Circ Res 75: 23–32. [DOI] [PubMed] [Google Scholar]
- 41. Frangogiannis NG (2012) Matricellular proteins in cardiac adaptation and disease. Physiol Rev 92: 635–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dobaczewski M, de Haan JJ, Frangogiannis NG (2012) The extracellular matrix modulates fibroblast phenotype and function in the infarcted myocardium. J Cardiovasc Transl Res 5: 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kong P, Christia P, Frangogiannis NG (2014) The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 71: 549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weber KT (1989) Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol 13: 1637–1652. [DOI] [PubMed] [Google Scholar]
- 45.Lal H, Ahmad F, Zhou J, Yu JE, Vagnozzi RJ, et al. (2014) Cardiac Fibroblast GSK-3beta Regulates Ventricular Remodeling and Dysfunction in Ischemic Heart. Circulation. [DOI] [PMC free article] [PubMed]
- 46. Camelliti P, Green CR, Kohl P (2006) Structural and functional coupling of cardiac myocytes and fibroblasts. Adv Cardiol 42: 132–149. [DOI] [PubMed] [Google Scholar]
- 47. Camelliti P, Borg TK, Kohl P (2005) Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res 65: 40–51. [DOI] [PubMed] [Google Scholar]
- 48. Wynn TA, Ramalingam TR (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18: 1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen W, Frangogiannis NG (2013) Fibroblasts in post-infarction inflammation and cardiac repair. Biochim Biophys Acta 1833: 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Drobnik J, Ciosek J, Slotwinska D, Stempniak B, Zukowska D, et al. (2009) Experimental hypothyroidism increases content of collagen and glycosaminoglycans in the heart. J Physiol Pharmacol 60: 57–62. [PubMed] [Google Scholar]
- 51. Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, et al. (2014) miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol Ther 22: 974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rainer PP, Hao S, Vanhoutte D, Lee DI, Koitabashi N, et al. (2014) Cardiomyocyte-specific transforming growth factor beta suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ Res 114: 1246–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Leask A (2010) Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res 106: 1675–1680. [DOI] [PubMed] [Google Scholar]
- 54. Kakkar R, Lee RT (2010) Intramyocardial fibroblast myocyte communication. Circ Res 106: 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brand T, Schneider MD (1995) The TGF beta superfamily in myocardium: ligands, receptors, transduction, and function. J Mol Cell Cardiol 27: 5–18. [DOI] [PubMed] [Google Scholar]
- 56. Li RK, Li G, Mickle DA, Weisel RD, Merante F, et al. (1997) Overexpression of transforming growth factor-beta1 and insulin-like growth factor-I in patients with idiopathic hypertrophic cardiomyopathy. Circulation 96: 874–881. [DOI] [PubMed] [Google Scholar]
- 57. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, et al. (2007) Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13: 952–961. [DOI] [PubMed] [Google Scholar]
- 58. Euler-Taimor G, Heger J (2006) The complex pattern of SMAD signaling in the cardiovascular system. Cardiovasc Res 69: 15–25. [DOI] [PubMed] [Google Scholar]
- 59. Bujak M, Frangogiannis NG (2007) The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res 74: 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, et al. (2011) Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J Clin Invest 121: 2301–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, et al. (2010) Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol 7: 30–37. [DOI] [PubMed] [Google Scholar]
- 62. de Haas HJ, Arbustini E, Fuster V, Kramer CM, Narula J (2014) Molecular imaging of the cardiac extracellular matrix. Circ Res 114: 903–915. [DOI] [PubMed] [Google Scholar]
- 63. Lompre AM, Schwartz K, d'Albis A, Lacombe G, Van Thiem N, et al. (1979) Myosin isoenzyme redistribution in chronic heart overload. Nature 282: 105–107. [DOI] [PubMed] [Google Scholar]
- 64. Miyata S, Minobe W, Bristow MR, Leinwand LA (2000) Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res 86: 386–390. [DOI] [PubMed] [Google Scholar]
- 65. Pandya K, Smithies O (2011) beta-MyHC and cardiac hypertrophy: size does matter. Circ Res 109: 609–610. [DOI] [PubMed] [Google Scholar]
- 66. Lopez JE, Myagmar BE, Swigart PM, Montgomery MD, Haynam S, et al. (2011) beta-myosin heavy chain is induced by pressure overload in a minor subpopulation of smaller mouse cardiac myocytes. Circ Res 109: 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Leslie KO, Taatjes DJ, Schwarz J, vonTurkovich M, Low RB (1991) Cardiac myofibroblasts express alpha smooth muscle actin during right ventricular pressure overload in the rabbit. Am J Pathol 139: 207–216. [PMC free article] [PubMed] [Google Scholar]
- 68. Schwartz K, de la Bastie D, Bouveret P, Oliviero P, Alonso S, et al. (1986) Alpha-skeletal muscle actin mRNA's accumulate in hypertrophied adult rat hearts. Circ Res 59: 551–555. [DOI] [PubMed] [Google Scholar]
- 69. Black FM, Packer SE, Parker TG, Michael LH, Roberts R, et al. (1991) The vascular smooth muscle alpha-actin gene is reactivated during cardiac hypertrophy provoked by load. J Clin Invest 88: 1581–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Januzzi JL, Troughton R (2013) Are serial BNP measurements useful in heart failure management? Serial natriuretic peptide measurements are useful in heart failure management. Circulation 127: 500–507 discussion 508. [DOI] [PubMed] [Google Scholar]
- 71. Savarese G, Trimarco B, Dellegrottaglie S, Prastaro M, Gambardella F, et al. (2013) Natriuretic peptide-guided therapy in chronic heart failure: a meta-analysis of 2,686 patients in 12 randomized trials. PLoS One 8: e58287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lupon J, de Antonio M, Vila J, Penafiel J, Galan A, et al. (2014) Development of a novel heart failure risk tool: the barcelona bio-heart failure risk calculator (BCN bio-HF calculator). PLoS One 9: e85466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hedayat M, Mahmoudi MJ, Rose NR, Rezaei N (2010) Proinflammatory cytokines in heart failure: double-edged swords. Heart Fail Rev 15: 543–562. [DOI] [PubMed] [Google Scholar]
- 74. McBride K, Nemer M (2001) Regulation of the ANF and BNP promoters by GATA factors: lessons learned for cardiac transcription. Can J Physiol Pharmacol 79: 673–681. [PubMed] [Google Scholar]
- 75. Ladenson PW, Sherman SI, Baughman KL, Ray PE, Feldman AM (1992) Reversible alterations in myocardial gene expression in a young man with dilated cardiomyopathy and hypothyroidism. Proc Natl Acad Sci U S A 89: 5251–5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Brenta G, Danzi S, Klein I (2007) Potential therapeutic applications of thyroid hormone analogs. Nat Clin Pract Endocrinol Metab 3: 632–640. [DOI] [PubMed] [Google Scholar]
- 77. Pingitore A, Iervasi G (2005) Thyroid (dys)function in heart failure: is it a potential target for medical treatment? Vasc Health Risk Manag 1: 97–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pingitore A, Chen Y, Gerdes AM, Iervasi G (2012) Acute myocardial infarction and thyroid function: new pathophysiological and therapeutic perspectives. Ann Med 44: 745–757. [DOI] [PubMed] [Google Scholar]
- 79. Galli E, Pingitore A, Iervasi G (2010) The role of thyroid hormone in the pathophysiology of heart failure: clinical evidence. Heart Fail Rev 15: 155–169. [DOI] [PubMed] [Google Scholar]
- 80. Coceani M, Molinaro S, Scalese M, Landi P, Carpeggiani C, et al. (2011) Thyroid hormone, amiodarone therapy, and prognosis in left ventricular systolic dysfunction. J Endocrinol Invest 34: e144–148. [DOI] [PubMed] [Google Scholar]
- 81. Flynn RW, Macdonald TM, Jung RT, Morris AD, Leese GP (2006) Mortality and vascular outcomes in patients treated for thyroid dysfunction. J Clin Endocrinol Metab 91: 2159–2164. [DOI] [PubMed] [Google Scholar]
- 82. Novitzky D, Fontanet H, Snyder M, Coblio N, Smith D, et al. (1996) Impact of triiodothyronine on the survival of high-risk patients undergoing open heart surgery. Cardiology 87: 509–515. [DOI] [PubMed] [Google Scholar]
- 83. Moruzzi P, Doria E, Agostoni PG, Capacchione V, Sganzerla P (1994) Usefulness of L-thyroxine to improve cardiac and exercise performance in idiopathic dilated cardiomyopathy. Am J Cardiol 73: 374–378. [DOI] [PubMed] [Google Scholar]
- 84. Gullo D, Latina A, Frasca F, Le Moli R, Pellegriti G, et al. (2011) Levothyroxine monotherapy cannot guarantee euthyroidism in all athyreotic patients. PLoS One 6: e22552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nakamura H, Noh JY, Itoh K, Fukata S, Miyauchi A, et al. (2007) Comparison of methimazole and propylthiouracil in patients with hyperthyroidism caused by Graves' disease. J Clin Endocrinol Metab 92: 2157–2162. [DOI] [PubMed] [Google Scholar]
- 86.Boron WaB, EL (2008) Medical Physiology, Edition 2: Elsevier Health Sciences.
- 87. Cooper DS, Kieffer JD, Halpern R, Saxe V, Mover H, et al. (1983) Propylthiouracil (PTU) pharmacology in the rat. II. Effects of PTU on thyroid function. Endocrinology 113: 921–928. [DOI] [PubMed] [Google Scholar]
- 88. Pantos C, Malliopoulou V, Mourouzis I, Sfakianoudis K, Tzeis S, et al. (2003) Propylthiouracil-induced hypothyroidism is associated with increased tolerance of the isolated rat heart to ischaemia-reperfusion. J Endocrinol 178: 427–435. [DOI] [PubMed] [Google Scholar]
- 89. Ziegelhoffer-Mihalovicova B, Briest W, Baba HA, Rassler B, Zimmer HG (2003) The expression of mRNA of cytokines and of extracellular matrix proteins in triiodothyronine-treated rat hearts. Mol Cell Biochem 247: 61–68. [DOI] [PubMed] [Google Scholar]
- 90. Winegrad S, Wisnewsky C, Schwartz K (1990) Effect of thyroid hormone on the accumulation of mRNA for skeletal and cardiac alpha-actin in hearts from normal and hypophysectomized rats. Proc Natl Acad Sci U S A 87: 2456–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gosteli-Peter MA, Harder BA, Eppenberger HM, Zapf J, Schaub MC (1996) Triiodothyronine induces over-expression of alpha-smooth muscle actin, restricts myofibrillar expansion and is permissive for the action of basic fibroblast growth factor and insulin-like growth factor I in adult rat cardiomyocytes. J Clin Invest 98: 1737–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wu Y, Peng J, Campbell KB, Labeit S, Granzier H (2007) Hypothyroidism leads to increased collagen-based stiffness and re-expression of large cardiac titin isoforms with high compliance. J Mol Cell Cardiol 42: 186–195. [DOI] [PubMed] [Google Scholar]
- 93. Ooe H, Kon J, Oshima H, Mitaka T (2009) Thyroid hormone is necessary for expression of constitutive androstane receptor in rat hepatocytes. Drug Metab Dispos 37: 1963–1969. [DOI] [PubMed] [Google Scholar]
- 94. Plateroti M, Kress E, Mori JI, Samarut J (2006) Thyroid hormone receptor alpha1 directly controls transcription of the beta-catenin gene in intestinal epithelial cells. Mol Cell Biol 26: 3204–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chen WJ, Lin KH, Lai YJ, Yang SH, Pang JH (2004) Protective effect of propylthiouracil independent of its hypothyroid effect on atherogenesis in cholesterol-fed rabbits: PTEN induction and inhibition of vascular smooth muscle cell proliferation and migration. Circulation 110: 1313–1319. [DOI] [PubMed] [Google Scholar]
- 96. Weltman NY, Ojamaa K, Savinova OV, Chen YF, Schlenker EH, et al. (2013) Restoration of cardiac tissue thyroid hormone status in experimental hypothyroidism: a dose-response study in female rats. Endocrinology 154: 2542–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Liu Y, Sherer BA, Redetzke RA, Gerdes AM (2010) Regulation of arteriolar density in adult myocardium during low thyroid conditions. Vascul Pharmacol 52: 146–150. [DOI] [PubMed] [Google Scholar]
- 98. Chen J, Ortmeier SB, Savinova OV, Nareddy VB, Beyer AJ, et al. (2012) Thyroid hormone induces sprouting angiogenesis in adult heart of hypothyroid mice through the PDGF-Akt pathway. J Cell Mol Med 16: 2726–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wang YY, Morimoto S, Du CK, Lu QW, Zhan DY, et al. (2010) Up-regulation of type 2 iodothyronine deiodinase in dilated cardiomyopathy. Cardiovasc Res 87: 636–646. [DOI] [PubMed] [Google Scholar]
- 100. Tschirgi ML, Rajapakse I, Chandra M (2006) Functional consequence of mutation in rat cardiac troponin T is affected differently by myosin heavy chain isoforms. J Physiol 574: 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Oppenheimer JH, Schwartz HL, Surks MI (1972) Propylthiouracil inhibits the conversion of L-thyroxine to L-triiodothyronine. An explanation of the antithyroxine effect of propylthiouracil and evidence supporting the concept that triiodothyronine is the active thyroid hormone. J Clin Invest 51: 2493–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Taurog A (1976) The mechanism of action of the thioureylene antithyroid drugs. Endocrinology 98: 1031–1046. [DOI] [PubMed] [Google Scholar]
- 103. Shiroozu A, Taurog A, Engler H, Dorris ML (1983) Mechanism of action of thioureylene antithyroid drugs in the rat: possible inactivation of thyroid peroxidase by propylthiouracil. Endocrinology 113: 362–370. [DOI] [PubMed] [Google Scholar]
- 104. Nakashima T, Taurog A, Riesco G (1978) Mechanism of action of thioureylene antithyroid drugs: factors affecting intrathyroidal metabolism of propylthiouracil and methimazole in rats. Endocrinology 103: 2187–2197. [DOI] [PubMed] [Google Scholar]
- 105. Johnson EO, Calogero AE, Konstandi M, Kamilaris TC, Vignera SL, et al. (2013) Effects of experimentally induced hyperthyroidism on central hypothalamic-pituitary-adrenal axis function in rats: in vitro and in situ studies. Pituitary 16: 275–286. [DOI] [PubMed] [Google Scholar]
- 106. Giannocco G, DosSantos RA, Nunes MT (2004) Thyroid hormone stimulates myoglobin gene expression in rat cardiac muscle. Mol Cell Endocrinol 226: 19–26. [DOI] [PubMed] [Google Scholar]
- 107. De Sibio MT, Luvizotto RA, Olimpio RM, Correa CR, Marino J, et al. (2013) A comparative genotoxicity study of a supraphysiological dose of triiodothyronine (T(3)) in obese rats subjected to either calorie-restricted diet or hyperthyroidism. PLoS One 8: e56913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chen YF, Weltman NY, Li X, Youmans S, Krause D, et al. (2013) Improvement of left ventricular remodeling after myocardial infarction with eight weeks L-thyroxine treatment in rats. J Transl Med 11: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Baum PD, Young JJ, Zhang Q, Kasakow Z, McCune JM (2011) Design, construction, and validation of a modular library of sequence diversity standards for polymerase chain reaction. Anal Biochem 411: 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wacker MJ, Godard MP (2005) Analysis of one-step and two-step real-time RT-PCR using SuperScript III. J Biomol Tech 16: 266–271. [PMC free article] [PubMed] [Google Scholar]
- 111. Iervasi G, Nicolini G (2013) Thyroid hormone and cardiovascular system: from basic concepts to clinical application. Intern Emerg Med 8 Suppl 1 S71–74. [DOI] [PubMed] [Google Scholar]
- 112.Grais IM, Sowers JR (2014) Thyroid and the Heart. Am J Med. [DOI] [PMC free article] [PubMed]
- 113. Nicolini G, Pitto L, Kusmic C, Balzan S, Sabatino L, et al. (2013) New insights into mechanisms of cardioprotection mediated by thyroid hormones. J Thyroid Res 2013: 264387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Gerdes AM, Iervasi G (2010) Thyroid replacement therapy and heart failure. Circulation 122: 385–393. [DOI] [PubMed] [Google Scholar]
- 115. Cini G, Carpi A, Mechanick J, Cini L, Camici M, et al. (2009) Thyroid hormones and the cardiovascular system: pathophysiology and interventions. Biomed Pharmacother 63: 742–753. [DOI] [PubMed] [Google Scholar]
- 116. Samuels MH, Schuff KG, Carlson NE, Carello P, Janowsky JS (2007) Health status, psychological symptoms, mood, and cognition in L-thyroxine-treated hypothyroid subjects. Thyroid 17: 249–258. [DOI] [PubMed] [Google Scholar]
- 117. Samuels MH, Kolobova I, Smeraglio A, Peters D, Janowsky JS, et al. (2014) The effects of levothyroxine replacement or suppressive therapy on health status, mood, and cognition. J Clin Endocrinol Metab 99: 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kahaly GJ (2000) Cardiovascular and atherogenic aspects of subclinical hypothyroidism. Thyroid 10: 665–679. [DOI] [PubMed] [Google Scholar]
- 119. Razvi S, Weaver JU, Butler TJ, Pearce SH (2012) Levothyroxine treatment of subclinical hypothyroidism, fatal and nonfatal cardiovascular events, and mortality. Arch Intern Med 172: 811–817. [DOI] [PubMed] [Google Scholar]
- 120.Refetoff S, Franklyn J, Shephard M (Revised 21 September 2000) 6E - Evaluation of Thyroid Function in Health and Disease. Leslie DeGroot, editor. Available: http://www.thyroidmanager.org/. Accessed 2014 September 29.
- 121. Brown ME, Refetoff S (1980) Transient elevation of serum thyroid hormone concentration after initiation of replacement therapy in myxedema. Ann Intern Med 92: 491–495. [DOI] [PubMed] [Google Scholar]
- 122. Aizawa T, Koizumi Y, Yamada T, Tawata M, Nagata H, et al. (1978) Difference in pituitary-thyroid feedback regulation in hypothyroid patients, depending on the severity of hypothyroidism. J Clin Endocrinol Metab 47: 560–565. [DOI] [PubMed] [Google Scholar]
- 123. Nicoloff JT, Spencer CA (1990) Clinical review 12: The use and misuse of the sensitive thyrotropin assays. J Clin Endocrinol Metab 71: 553–558. [DOI] [PubMed] [Google Scholar]
- 124. Sanchez-Franco F, Garcia MD, Cacicedo L, Martin-Zurro A, Escobar del Ray F, et al. (1974) Transient lack of thyrotropin (TSH) response to thyrotropin-releasing hormone (TRH) in treated hyperthyroid patients with normal or low serum thyroxine (T4) and triiodothyronine (T3). J Clin Endocrinol Metab 38: 1098–1102. [DOI] [PubMed] [Google Scholar]
- 125. Diffee GM, Haddad F, Herrick RE, Baldwin KM (1991) Control of myosin heavy chain expression: interaction of hypothyroidism and hindlimb suspension. Am J Physiol 261: C1099–1106. [DOI] [PubMed] [Google Scholar]
- 126. Escobar-Morreale HF, Escobar del Rey F, Morreale de Escobar G (1997) Thyroid hormones influence serum leptin concentrations in the rat. Endocrinology 138: 4485–4488. [DOI] [PubMed] [Google Scholar]
- 127. Grieve DJ, Fletcher S, Pitsillides AA, Botham KM, Elliott J (1999) Effects of oral propylthiouracil treatment on nitric oxide production in rat aorta. Br J Pharmacol 127: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Soukup T, Zacharova G, Smerdu V, Jirmanova I (2001) Body, heart, thyroid gland and skeletal muscle weight changes in rats with altered thyroid status. Physiol Res 50: 619–626. [PubMed] [Google Scholar]
- 129. Syed MA, Thompson MP, Pachucki J, Burmeister LA (1999) The effect of thyroid hormone on size of fat depots accounts for most of the changes in leptin mRNA and serum levels in the rat. Thyroid 9: 503–512. [DOI] [PubMed] [Google Scholar]
- 130. Wang JL, Chinookoswong N, Yin S, Shi ZQ (2000) Calorigenic actions of leptin are additive to, but not dependent on, those of thyroid hormones. Am J Physiol Endocrinol Metab 279: E1278–1285. [DOI] [PubMed] [Google Scholar]
- 131. Wu S, Tan G, Dong X, Zhu Z, Li W, et al. (2013) Metabolic profiling provides a system understanding of hypothyroidism in rats and its application. PLoS One 8: e55599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Cusin I, Rouru J, Visser T, Burger AG, Rohner-Jeanrenaud F (2000) Involvement of thyroid hormones in the effect of intracerebroventricular leptin infusion on uncoupling protein-3 expression in rat muscle. Diabetes 49: 1101–1105. [DOI] [PubMed] [Google Scholar]
- 133. Iossa S, Lionetti L, Mollica MP, Crescenzo R, Barletta A, et al. (2001) Fat balance and serum leptin concentrations in normal, hypothyroid, and hyperthyroid rats. Int J Obes Relat Metab Disord 25: 417–425. [DOI] [PubMed] [Google Scholar]
- 134. Leonhardt U, Gerdes E, Ritzel U, Schafer G, Becker W, et al. (1999) Immunoreactive leptin and leptin mRNA expression are increased in rat hypo- but not hyperthyroidism. J Endocrinol 163: 115–121. [DOI] [PubMed] [Google Scholar]
- 135. Dizdarevic-Bostandic A, Burekovic A, Velija-Asimi Z, Godinjak A (2013) Inflammatory markers in patients with hypothyroidism and diabetes mellitus type 1. Med Arh 67: 160–161. [DOI] [PubMed] [Google Scholar]
- 136. Tuzcu A, Bahceci M, Gokalp D, Tuzun Y, Gunes K (2005) Subclinical hypothyroidism may be associated with elevated high-sensitive c-reactive protein (low grade inflammation) and fasting hyperinsulinemia. Endocr J 52: 89–94. [DOI] [PubMed] [Google Scholar]
- 137. Enia G, Panuccio V, Cutrupi S, Pizzini P, Tripepi G, et al. (2007) Subclinical hypothyroidism is linked to micro-inflammation and predicts death in continuous ambulatory peritoneal dialysis. Nephrol Dial Transplant 22: 538–544. [DOI] [PubMed] [Google Scholar]
- 138. Zoccali C, Tripepi G, Cutrupi S, Pizzini P, Mallamaci F (2005) Low triiodothyronine: a new facet of inflammation in end-stage renal disease. J Am Soc Nephrol 16: 2789–2795. [DOI] [PubMed] [Google Scholar]
- 139. Diez JJ, Hernanz A, Medina S, Bayon C, Iglesias P (2002) Serum concentrations of tumour necrosis factor-alpha (TNF-alpha) and soluble TNF-alpha receptor p55 in patients with hypothyroidism and hyperthyroidism before and after normalization of thyroid function. Clin Endocrinol (Oxf) 57: 515–521. [DOI] [PubMed] [Google Scholar]
- 140. Aksoy DY, Cinar N, Harmanci A, Karakaya J, Yildiz BO, et al. (2013) Serum resistin and high sensitive CRP levels in patients with subclinical hypothyroidism before and after L-thyroxine therapy. Med Sci Monit 19: 210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Cinar N, Gurlek A (2013) Association between novel adipocytokines adiponectin, vaspin, visfatin, and thyroid: An experimental and clinical update. Endocr Connect 2: R30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Klein I (2002) Thyroid and the heart. Thyroid 12: 439. [DOI] [PubMed] [Google Scholar]
- 143. Liu Z, Gerdes AM (1990) Influence of hypothyroidism and the reversal of hypothyroidism on hemodynamics and cell size in the adult rat heart. J Mol Cell Cardiol 22: 1339–1348. [DOI] [PubMed] [Google Scholar]
- 144. Gerdes AM, Capasso JM (1995) Structural remodeling and mechanical dysfunction of cardiac myocytes in heart failure. J Mol Cell Cardiol 27: 849–856. [DOI] [PubMed] [Google Scholar]
- 145. Gerdes AM, Kellerman SE, Moore JA, Muffly KE, Clark LC, et al. (1992) Structural remodeling of cardiac myocytes in patients with ischemic cardiomyopathy. Circulation 86: 426–430. [DOI] [PubMed] [Google Scholar]
- 146. Graettinger JS, Muenster JJ, Checchia CS, Grissom RL, Campbell JA (1958) A correlation of clinical and hemodynamic studies in patients with hypothyroidism. J Clin Invest 37: 502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Aber CP, Thompson GS (1963) Factors Associated with Cardiac Enlargement in Myxoedema. Br Heart J 25: 421–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Gerdes AM (2002) Cardiac myocyte remodeling in hypertrophy and progression to failure. J Card Fail 8: S264–268. [DOI] [PubMed] [Google Scholar]
- 149. Onodera T, Tamura T, Said S, McCune SA, Gerdes AM (1998) Maladaptive remodeling of cardiac myocyte shape begins long before failure in hypertension. Hypertension 32: 753–757. [DOI] [PubMed] [Google Scholar]
- 150. Zimmer HG, Gerdes AM, Lortet S, Mall G (1990) Changes in heart function and cardiac cell size in rats with chronic myocardial infarction. J Mol Cell Cardiol 22: 1231–1243. [DOI] [PubMed] [Google Scholar]
- 151. Chen YF, Kobayashi S, Chen J, Redetzke RA, Said S, et al. (2008) Short term triiodo-L-thyronine treatment inhibits cardiac myocyte apoptosis in border area after myocardial infarction in rats. J Mol Cell Cardiol 44: 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Carr AN, Kranias EG (2002) Thyroid hormone regulation of calcium cycling proteins. Thyroid 12: 453–457. [DOI] [PubMed] [Google Scholar]
- 153. Chen WJ, Lin KH, Lee YS (2000) Molecular characterization of myocardial fibrosis during hypothyroidism: evidence for negative regulation of the pro-alpha1(I) collagen gene expression by thyroid hormone receptor. Mol Cell Endocrinol 162: 45–55. [DOI] [PubMed] [Google Scholar]
- 154.Weber KT, Sun Y, Campbell SE (1995) Structural remodelling of the heart by fibrous tissue: role of circulating hormones and locally produced peptides. Eur Heart J 16 Suppl N: 12–18. [DOI] [PubMed]
- 155. Yao J, Eghbali M (1992) Decreased collagen gene expression and absence of fibrosis in thyroid hormone-induced myocardial hypertrophy. Response of cardiac fibroblasts to thyroid hormone in vitro. Circ Res 71: 831–839. [DOI] [PubMed] [Google Scholar]
- 156. Diniz GP, Carneiro-Ramos MS, Barreto-Chaves ML (2007) Angiotensin type 1 (AT1) and type 2 (AT2) receptors mediate the increase in TGF-beta1 in thyroid hormone-induced cardiac hypertrophy. Pflugers Arch 454: 75–81. [DOI] [PubMed] [Google Scholar]
- 157. Diniz GP, Carneiro-Ramos MS, Barreto-Chaves ML (2009) Angiotensin type 1 receptor mediates thyroid hormone-induced cardiomyocyte hypertrophy through the Akt/GSK-3beta/mTOR signaling pathway. Basic Res Cardiol 104: 653–667. [DOI] [PubMed] [Google Scholar]
- 158. Kubiczkova L, Sedlarikova L, Hajek R, Sevcikova S (2012) TGF-beta - an excellent servant but a bad master. J Transl Med 10: 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Han G, Li F, Singh TP, Wolf P, Wang XJ (2012) The pro-inflammatory role of TGFbeta1: a paradox? Int J Biol Sci 8: 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental protocol with different measured parameters.
(TIF)
Plasma leptin levels in the different groups of rat treatments at the time of sacrifice. Data are represented as the mean ± SEM. n = 10 animals for each group. Statistical analysis was performed with Kruskal-Wallis One-way ANOVA on rank tests followed by post hoc Dunn's multiple comparison tests. *p<0.01 vs. control and reverse.
(TIF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.