

Abstract
Chemokines comprise a family of secreted proteins that activate G protein-coupled chemokine receptors and thereby control the migration of leukocytes during inflammation or immune surveillance. The positional information required for such migratory behavior is governed by the binding of chemokines to membrane-tethered glycosaminoglycans (GAGs), which establishes a chemokine concentration gradient. An often-observed but incompletely understood behavior of chemokines is the ability of unrelated chemokines to enhance the potency with which another chemokine subtype can activate its cognate receptor. This phenomenon has been demonstrated to occur between many chemokine combinations and across several model systems, and has been dubbed “chemokine cooperativity”. Here, we have used GAG binding-deficient chemokine mutants and cell-based functional (migration) assays to demonstrate that chemokine cooperativity is caused by competitive binding of chemokines to GAGs. This mechanistic explanation of chemokine cooperativity provides insight into chemokine gradient formation in the context of inflammation, where multiple chemokines are secreted simultaneously.
Introduction
Chemokines are 8–12 kDa-sized secreted proteins that mediate the directed migration (chemotaxis) of leukocytes. The chemokine family encompasses nearly fifty members, which are classified based on the relative position of their conserved N-terminal cysteine residues (CC, CXC, CX3C and C). Chemokines elicit intracellular responses via G protein-coupled receptors (GPCRs). Upon ligand binding, chemokine receptors activate G proteins of the Gαi family, leading to inhibition of adenylyl cyclases and mobilization of Ca2+ from intracellular stores. Furthermore, activated chemokine receptors bind to the scaffolding protein β-arrestin (1–3). Chemokine receptor activation mediates leukocyte chemotaxis towards lymphoid organs or sites of inflammation along a chemokine gradient that is established by binding of chemokines to membrane-tethered and extracellular matrix-associated glycosaminoglycans (GAGs) (4). GAGs represent a heterogenous population of unbranched polysaccharides, with heparin, heparan sulphate, dermatan sulphate, chondroitin sulphate and hyaluronic acid comprising the largest groups.
An interesting feature of chemokine biology is the ability of distantly related chemokines to enhance each other's function (5–12). This behavior is referred to as “chemokine cooperativity” or “chemokine synergy”, but a clear mechanistic understanding of this phenomenon is lacking. We became interested in chemokine cooperativity while testing the ability of several chemokines to activate the chemokine receptor CCX-CKR. CCX-CKR is an atypical chemokine receptor in that it does not activate conventional G protein-mediated signaling pathways (3, 13–14), but it does recruit the scaffolding protein β-arrestin2 upon activation by CCL19, CCL21 and CCL25 (3). We noted that several chemokines, that did not activate or bind CCX-CKR directly, enhanced the potency of CCL19 and CCL21 to activate CCX-CKR. We subsequently observed that multiple chemokine combinations can synergistically enhance the activation of typical (G protein-coupled) chemokine receptors, such as CCR7 and CXCR5. Through analysis of chemokine mutants that are deficient in GAG-binding, we discovered that chemokine cooperativity is mediated by competitive binding of chemokines to GAGs.
Materials and methods
Reagents
All wildtype chemokines were obtained from PeproTech (London, Great Britain). 125I-labeled CCL19 (125I-CCL19) was from Perkin Elmer (Boston, MA, USA). Forskolin was from Sigma-Aldrich (Steinheim, Germany). Production of mtCXCL12 (15), mtCXCL11 (16), CXCL121 (17) and CXCL122 (18) has been described previously.
Cell culture
The generation of the CHO-CCX-CKR cells has been described previously (3). These cells were maintained in DMEM F12 (PAA Laboratories, Cölbe, Germany) supplemented with 10% v/v Bovine Calf Serum (BCS; Hyclone, Logan, UT), 250 μg/ml hygromycin, 800 μg/ml geneticin, 100 U/ml penicillin and 100 μg/ml streptomycin (all from Invitrogen, Carlsbad, CA). CHO cells stably expressing the β-arrestin2-EA fusion protein and GPCRs C-terminally extended with a complementary β-galactosidase mutant (Prolink) (i.e. CHO-CCR7, CHO-CXCR5 and CHO-CCR5 cells) were purchased from DiscoveRx (Fremont, CA, USA). CHOCXCR3 cells were kindly provided by Toon van der Doelen (MSD, Oss, The Netherlands). These cell lines were cultured in the medium mentioned above. HEK293T and CHO cells were purchased from the American Type Culture Collection (ATCC) and maintained in DMEM F12 supplemented with 10% v/v BCS, 100 U/ml penicillin and 100 μg/ml streptomycin. L1.2 cells (ATCC) were cultured in RPMI (Sigma-Aldrich) containing 10% v/v heat-inactivated BCS, 100 U/ml penicillin and 100 μg/ml streptomycin, 25 mM Hepes, 2 mM L-glutamine, 1% non-essential amino acids, 1 mM sodium pyruvate (all from PAA Laboratories) and 50 μM β-mercapthoethanol (Invitrogen).
Enzyme fragment complementation assays
EFC assays were performed as described previously (3). Briefly, cells were dispensed in a 384-well white CulturPlate (Perkin Elmer, Boston, MA) with 1×104 cells in 15 μl OPTImem (Invitrogen) containing 1% BCS, 100 U/ml penicillin and 100 μg/ml streptomycin. Following overnight culturing in a humidified incubator (37°C, 5% CO2, 95% humidity), cooperative and receptor-engaging chemokines were added in 5 μl OPTImem each, followed by 90 min incubation at 37°C. Subsequently, cells were disrupted by addition of 12.5 μl of PathHunter Detection Reagent (DiscoveRx) in the formulation specified by the manufacturer. Following 1 hr incubation at room temperature in the dark, luminescence was measured using a Victor3 multilabel plate reader (Perkin Elmer).
Radioligand binding
Radioligand binding experiments were performed as described previously (3). For saturation binding experiments, 25×104 CHO cells in 100 μl culture medium were dispensed in a poly-L-lysine coated 96-well culture plate (Greiner) and incubated overnight in a humidified incubator. Medium was replaced by 50 μl non-radiolabeled chemokine in 20 mM Hepes, 5 mM MgCl2, 1 mM CaCl2, 100 mM NaCl, pH 7.4 at 4°C (assay medium), followed by addition of 50 μl 125I-CCL19 in assay medium. Plates were incubated for 3 hrs at 4°C and then washed with ice-cold assay medium supplemented with 500 mM NaCl. Cells were disrupted by addition of 150 μl RIPA buffer (Sigma-Aldrich) and radioactivity in lysates was measured with a Compugamma gamma counter (Perkin Elmer).
Bare-filter migration (chemotaxis) assays
30 μl of chemokine in RPMI (Sigma-Aldrich) containing 0.1% BSA (assay buffer) was added to the lower compartment of a 96-well ChemoTx plate (5 μm pore size; Neuroprobe Inc., Gaithersburg, MD). 25×104 L1.2 cells in 20 μl assay buffer were administered to the upper compartment of the plate. Cells were then allowed to migrate through the filter for 5 hrs in a humidified incubator (37°C). Twenty-five microliter assay buffer from the lower well was then transferred to a white 96-well CulturPlate Plate (Perkin Elmer), followed by addition of 25 μl CalceinAM (1 μg/ml final concentration) in assay buffer. Fluorescence intensities were measured on a Victor3 plate reader.
Data analysis
Sigmoidal dose-response curves were plotted using Graphpad Prism 6.0 software. All data are presented as averages ± standard error in the mean (SEM). Statistical significance of observed differences was determined using Student's t-test or two-way ANOVA as indicated in the figure legend and indicated in the figures with asterisks (*). p<0.05 was regarded as statistically significant.
Results
CXCL13 enhances the ability of CCL19, CCL21 and CCL25 to activate CCX-CKR
Previous studies have suggested that CXCL13 is a ligand for CCX-CKR (13, 19). We therefore tested the ability of this chemokine to activate or inhibit CCX-CKR in Chinese Hamster Ovary (CHO) cells genetically engineered to express complementary β-galactosidase mutants coupled to CCX-CKR and the scaffolding protein β-arrestin2 (CHO-CCX-CKR cells). The recruitment of β-arrestin2 to activated CCX-CKR enables enzyme fragment complementation (EFC) between the two β-galactosidase mutants, thus allowing measurement of receptor activation by assessing β-galactosidase activity (3, 20). In accordance, when CHO-CCX-CKR cells were treated with CCL19, CCL21 and CCL25, they responded with a concentration-dependent increase in β-galactosidase activity ((3) and Fig. 1A). In contrast, CXCL13 did not activate CCX-CKR in these cells (Fig. 1A, filled circles (●)). CXCL13 was also unable to displace radiolabeled CCL19 (125I-CCL19) from CHO cells expressing CCX-CKR (Fig. S1A, filled circles (●)). Thus, CXCL13 is unlikely to be a ligand for CCX-CKR. Nonetheless, we found that CXCL13 concentration-dependently enhanced the potency with which CCL19 activated CCX-CKR (Fig. 1A,B). This effect was not limited to activation of CCX-CKR by CCL19, as similar results were observed in experiments using CCL21 and CCL25 (Fig. S1B,C).
Fig. 1. CXCL13 increases the ability of CCL19 to activate CCX-CKR.

(A) CHO cells stably expressing CCX-CKR fused to a peptide fragment of β-galactosidase and β-arrestin2 coupled to a complementary β-galactosidase mutant (CHO-CCX-CKR cells) were treated with increasing concentrations of CCL19 (□) or CXCL13 (●) before measurement of β-galactosidasee activity. In addition, CHO-CCX-CKR cells were treated with increasing concentrations of CXCL13 in the presence of 2 nM CCL19 (▼). Asterisks (*) indicate statistically significant differences (Two-way ANOVA, p<0.05) between cells treated with 2 nM CCL19 in the presence and absence of CXCL13. (B) CHO-CCX-CKR cells were treated with increasing concentrations of CCL19 in the presence of 1000 nM CXCL13, 100 nM CXCL13 or corresponding vehicle, followed by measurement of β-galactosidase activity. pEC50 values were: Vehicle: 8.6±0.2 (4), 100 nM CXCL13: 9.1±0.2 (4) and 1000 nM CXCL13: 9.4±0.1 (4) (average±SD (n)). Differences in pEC50 values between vehicle-treated cells and cells treated with either 100 nM or 1000 nM CXCL13 reached statistical significance (Student's T-test, p<0.05).
Secondly, we measured β-arrestin2 recruitment to CCX-CKR using a bioluminescence resonance energy transfer (BRET) approach (3). For this, we transiently transfected HEK293T cells with vectors encoding CCX-CKR genetically extended with Renilla luciferase (CCX-CKR-Rluc) and β-arrestin2 fused to YFP (β-arr2-YFP). Similar to results for the β-galactosidase complementation assay, CXCL13 increased the potency with which CCL19 activated CCX-CKR (Fig. S1D). Thus, CXCL13 cooperates with CCL19, CCL21 and CCL25 in activating CCX-CKR.
Chemokine cooperativity is specific for certain chemokine combinations
We then tested whether the cooperative effect of CXCL13 was specific for certain chemokine pairs or chemokine receptors. To this end, we used CHO cells that complement β-galactosidase following recruitment of β-arrestin2 to the Gαi-coupled chemokine receptors CCR7 (CHO-CCR7), CCR5 (CHO-CCR5) and CXCR5 (CHO-CXCR5). CXCL13 enhanced the potency with which CCL19 induced β-arrestin recruitment and Gαi-activation by CCR7 in CHO-CCR7 cells (Fig. 2A,B). Reciprocally, CCL19 enhanced the potency of CXCL13 towards activation of CXCR5 (Fig. 2C). However, both CCL19 and CXCL13 were incapable of potentiating CCL3-induced activation of CCR5 (Fig. 2D).
Fig. 2. Distinct chemokine combinations display cooperativity.

(A) CHO-CCR7 cells were treated with increasing concentrations of CCL19 in the presence of 1000 nM CXCL13, 100 nM CXCL13 or corresponding vehicle, followed by measurement of β-galactosidase activity. (B) CHO-CCR7 cells were treated with 0.5 μM forskolin and increasing concentrations of CCL19 in the presence of 1000 nM or 100 nM CXCL13, followed by measurement of cAMP levels. (C) CHO-CXCR5 cells were treated with increasing concentrations of CXCL13 in the presence of 1000 nM CCL19, 100 nM CCL19 or corresponding vehicle, followed by measurement of β-galactosidase activity. (D) CHO-CCR5 cells were treated with ascending concentrations of CCL3 and 1000 nM CCL19, 1000 nM CXCL13 or corresponding vehicle, followed by measurement of β-galactosidase activity. (E) CHO-CCX-CKR cells were treated with 2 nM CCL19 in the absence or presence of 1000 nM of the indicated chemokines (x-axis), followed by measurement of β-galactosidase activity. Asterisks (*) indicate statistically significant differences (Student's T-test, p<0.05).
Next, a panel of chemokines was tested for their ability to cooperate with CCL19 in activation of CCX-CKR. Several chemokines other than CXCL13, including CXCL12 and CXCL11, were found to cooperate with CCL19, whereas several others (CCL3, CCL4 and CCL23) did not (Fig. 2E). Essentially the same pattern was observed when CCX-CKR was activated with CCL21: CXCL13, CXCL12 and CXCL11 cooperated with CCL21, whereas CCL3 and CCL4 did not (Fig. S2).
CXCL13 enhances CCL19-mediated chemotaxis
Next, we used a transwell migration assay to test whether the enhanced receptor activity translated to a more potent chemotactic response. We used murine L1.2 cells, a pre-B lymphoma cell line, which migrated towards a gradient of CCL19 (Fig. 3A), implying that these cells endogenously express CCR7. However, the L1.2 cells did not migrate towards CXCL13 (Fig. 3A). In accordance with our functional assays, 1 μM of CXCL13 significantly potentiated CCL19-mediated chemotaxis in these cells (Fig. 3B).
Fig. 3. CXCL13 enhances the ability of CCL19 to induce chemotaxis.

(A) Murine pre-B lymphoma (L1.2) cells were tested for their ability to migrate towards ascending concentrations of CCL19 or CXCL13 in a bare-filter migration assay. (B) L1.2 cells were tested for their ability to migrate towards ascending concentrations of CCL19 in the absence or presence of 1 μM CXCL13. Asterisks (*) indicate statistically significant differences (Two-way ANOVA, p<0.05) between vehicle- and CXCL13-treated cells.
Chemokine cooperativity is dependent on GAG binding
Our analysis revealed that CCL3 and CCL4 did not induce chemokine cooperativity (Fig. 2E). These chemokines bind to several GAGs with relatively low affinity compared to other chemokines (e.g. CXCL8, CCL2 and CCL5) (21–22). To investigate the importance of GAG binding for chemokine cooperativity, we tested the ability of GAG binding-deficient chemokine mutants to cooperate in CCX-CKR activation. Such mutants have been described for CXCL12 and CXCL11 (15–16, 23). In sharp contrast to wildtype CXCL12 (wtCXCL12), a GAG binding-deficient CXCL12 mutant (CXCL12K24S,H25S,K27S, hereafter referred to as mtCXCL12) did not cooperate with CCL21 in the EFC assay (Fig. 4A). Likewise, we found that mtCXCL12 did not cooperate with CCL21 in activating CCX-CKR in the BRET assay (Fig. S3). Similarly, a GAG binding-deficient CXCL11 mutant (CXCL11 50s or CXCL11K57A,K59A,R62A, hereafter referred to as mtCXCL11) was impaired in its ability to cooperate with CCL21 in activating CCX-CKR (Fig. 4B). Both mtCXCL12 and mtCXCL11 were capable of activating their cognate receptors with potencies and efficacies comparable to wildtype chemokines (Fig. S4).
Fig. 4. Mutant chemokines that do not bind to glycosaminoglycans (GAGs) do not display chemokine cooperativity.
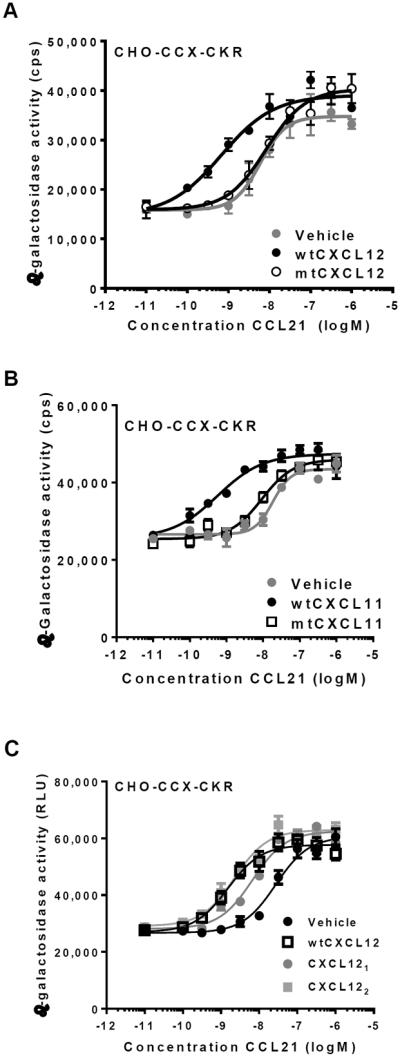
(A) CHO-CCX-CKR cells were treated with wildtype CXCL12 (wtCXCL12) and a CXCL12 mutant that is impaired in its ability to bind to GAGs (mtCXCL12) in the presence of increasing concentrations of CCL21. Subsequently, β-galactosidase activity was determined. pEC50 values were: Vehicle: 7.7±0.3 (5), wtCXCL12: 8.7±0.4 (5) and mtCXCL12: 7.7±0.2 (5) (average±SD (n)). Values for vehicle-treated and mtCXCL12-treated cells were not statistically different, whereas wtCXCL12 significantly differed from the other treatments (Student's T-test, p<0.01). (B) CHO-CCX-CKR cells were treated with CCL21 and 1 μM wildtype CXCL11 (wtCXCL11) or 1 μM mutant CXCL11 that is impaired in its ability to bind GAGs (mtCXCL11), followed by measurement of β-galactosidase activity. pEC50 values were: Vehicle: 7.8±0.2 (3), wtCXCL11: 9.6±0.3 (3) and mtCXCL11: 8.3±0.4 (3) (average±SD (n)). Values for vehicle-treated and mtCXCL11-treated cells were not statistically different, whereas wtCXCL11 significantly differed from the other treatments (Student's T-test, p<0.01). (C) CHO-CCX-CKR cells were treated either with 1 μM wtCXCL12, constitutively monomeric (CXCL121) or dimeric CXCL12 (CXCL122) in the presence of increasing concentrations of CCL21, followed by β-galactosidase measurement. pEC50 values were: Vehicle: 7.7±0.2 (6), wtCXCL12: 8.5±0.2 (6), CXCL121: 8.3±0.3 (6) and CXCL122: 8.5±0.5 (6) (average±SD (n)). Differences between values for vehicle-treated cells and cells treated with all three CXCL12 proteins reached statistical significance (Student's T-test, p<0.01). pEC50 values for wtCXCL12, CXCL121 and CXCL122 were not statistically different.
Chemokine cooperativity is mediated by both chemokine monomers and dimers
Chemokine heterodimerization has previously been suggested to underlie chemokine cooperativity (10). GAG binding and dimerization of chemokines are intimately linked processes (18, 22). Indeed, the CXCL12-GAG binding interface significantly overlaps with the CXCL12 (homo)dimerization surface, and mtCXCL12 is not only impaired in its ability to bind to GAGs, but also in its ability to dimerize (18). To differentiate between dimerization and GAG binding as potential mechanisms for chemokine cooperativity, we used a CXCL12 mutant that forms obligate monomers (termed CXCL121) and a constitutively dimeric mutant of CXCL12 (CXCL122) (–18, 24–25). CXCL121 and CXCL122 are strictly monomeric and dimeric at concentrations up to 10 μM (17, 24). Both mutants bound to heparin, although CXCL121 did so with an approximately 10-fold lower potency than wtCXCL12 (17–18). When tested in the EFC assay on CHO-CCX-CKR cells, both CXCL121 and CXCL122 cooperated with CCL21 in the activation of CCX-CKR (Fig. 4C). In conclusion, both monomeric and dimeric CXCL12 proteins can mediate chemokine cooperativity.
Chemokine cooperativity is caused by competitive GAG binding
The simplest model that explains the necessity for GAG binding in chemokine cooperativity is one in which the cooperative chemokines displace the receptor-engaging chemokine from binding to GAGs. This scenario would result in elevated concentrations of soluble receptor-engaging chemokines that are available to bind and activate receptors. To test whether CXCL12 and CXCL13 compete with CCL19 for binding to GAGs on the cell surface, we monitored the binding of 125I-CCL19 to CHO cells. The CHO cells did not express CCX-CKR or CCR7, as determined by RT-PCR (data not shown), whereas they abundantly express GAGs (23, 26). Incubation of CHO cells with 125I-CCL19 led to a concentration-dependent increase in cell-associated radioactivity, a large portion of which could be competed for by adding excess (1 μM) non-labeled CCL19 (Fig. 5A). wtCXCL12 was also capable of displacing 125I-CCL19 from CHO cells, whereas mtCXCL12 was not (Fig. 5A). Similarly, CXCL13 displaced 0.5 nM 125I-CCL19 from both CHO and HEK293T cells, whereas the non-cooperative chemokine CCL3 did not affect 125I-CCL19 binding to these cells (Fig. 5B and data not shown). These data indicate that cooperative chemokines compete with CCL19 for GAG binding on cells, whereas non-cooperative chemokines do not.
Fig. 5. Cooperative chemokines displace membrane-associated CCL19.

(A) CHO cells were treated with increasing concentrations of radiolabeled CCL19 (125I-CCL19) in the presence of 1 μM CCL19, wtCXCL12, mtCXCL12 or vehicle, followed by determination of cell-associated radioactivity. (B) CHO cells were incubated with 0.5 nM 125I-CCL19 in the presence of 1 μM CCL19, CXCL13 and CCL3, followed by determination of cell-associated radioactivity. Asterisks (*) indicate statistically significant differences (Student's T-test, p<0.01).
Discussion
Chemokine cooperativity has previously been suggested to be caused by convergence of intracellular signaling pathways downstream of receptor activation (5–7, 11–12). Our data indicate that chemokine cooperativity is not strictly dependent on the receptor of the cooperative chemokine. For example, mtCXCL12 and mtCXCL11 fully activate their respective G protein-coupled receptors (Fig. S4), but do not cooperate with CCL21 (Fig. 4). Furthermore, RT-PCR analyses and cAMP measurements indicated that CHO-CCX-CKR cells do not express CXCR5 or CXCR4, the cognate Gαi-coupled receptors for CXCL13 and CXCL12, respectively (data not shown). Our data concur with previous evidence suggesting that activation of the receptor for the cooperative chemokine is not necessary for cooperativity (8–9).
Alternatively, chemokine cooperativity has been suggested to originate from chemokine heterodimerization (9–10). Our observation that both monomeric and dimeric CXCL12 species can induce chemokine cooperativity argues against this mode of action.
Here, we demonstrate a determining role for GAG binding in chemokine cooperativity. Our data suggest that cooperative chemokines compete for binding to GAGs, thereby raising the effective “free” concentration of chemokine that can engage its receptor (Fig. 6). Other researchers performed experiments on heparinase-treated cells to test whether GAGs influence chemokine cooperativity, and observed no effect of heparinase treatment on chemokine cooperativity (8, 10). Similarly, pretreatment of CHO-CCX-CKR cells with a cocktail of heparinases did not influence the ability of CXCL13 to cooperate with CCL19 (data not shown). A flaw in this approach is that GAGs form a large heterogeneous population of molecules, and it is experimentally challenging to ascertain that all of the GAGs relevant to chemokine cooperativity are removed by treatment with heparinase and chondroitinase cocktails. Our analysis with GAG binding-deficient mutants represents a method of analysis that lacks this pitfall. A remaining challenge will be to deduce which GAG subtypes are actually involved in chemokine cooperativity. Such GAGs may be identified by studying the ability of chemokines to compete for binding on arrays or columns containing immobilized synthetic or purified GAGs (21, 27).
Fig. 6. A model for chemokine cooperativity.

(A) In the absence of cooperative chemokines, CCL19/CCL21 binds to its seven-transmembrane receptors (CCX-CKR or CCR7) or to membrane-tethered GAGs. (B) In the presence of cooperative chemokines, CCL19/CCL21 is competed from the GAGs, raising the “free” concentration of CCL19, which results in enhanced association of CCL19/CCL21 with its receptors and consequent receptor activation.
Another outstanding question is whether chemokine cooperativity occurs in vivo. While systemic, homeostatic levels of CCL19, CCL21, CXCL13 and CXCL12 are generally much lower than the chemokine concentrations required to induce chemokine cooperativity in our functional assays, local concentrations of chemokine may well exceed the concentrations necessary for cooperativity to occur. Of note, CCL19, CCL21, CXCL12 and CXCL13 have exceptionally broad and overlapping expression patterns, which would provide many potential opportunities for synergistic interactions. For instance, CCL19, CCL21 and CXCL12 are expressed on the luminal side of high endothelial venules (HEVs) (28), and CCL21, CXCL12 and CXCL13 are expressed in lymph nodes (29–31). Indeed, CXCL12 has been shown to enhance the CCR7-mediated recruitment of naïve T cells from HEVs into the underlying lymphoid tissue (5). Furthermore, CCL19, CCL21, CXCL12 and CXCL13 are expressed locally during chronic inflammation (32–34), and individual chemokine concentrations within these inflamed sites may very well exceed the threshold for cooperativity to occur. Additionally, previous work has suggested that chemokine cocktails can concertedly induce cooperativity at low individual chemokine concentrations (10). Since multiple chemokines are expressed concomitantly during inflammation and homeostatic trafficking (35–37), the concentration of several cooperative chemokines combined may be sufficient for chemokine cooperativity to occur.
How would chemokine cooperativity influence chemokine function, and how will chemokine cooperativity be altered by (patho)physiological processes? Since chemokine cooperativity would allow chemokines to activate their cognate receptors at lower chemokine concentrations, it is likely to extend the range from which chemokines can induce recruitment of leukocytes. Another implication from our data is that alterations in GAG composition or abundance, as have been observed to occur in several pathologies (38–41), might alter chemokine cooperativity. Lastly, our data present a new perspective on therapeutic targeting of the chemokine system. Through modulation of cooperativity, therapeutic strategies aimed at inhibiting chemokine-GAG interactions might establish a broader effect on leukocyte trafficking than targeting chemokine receptors. For instance, chemokine cooperativity is likely to be modulated by administration of heparin or GAG derivatives, which have previously been shown to affect chemokine gradient formation (42–43). Furthermore, small molecules or antibodies targeting chemokines, such as those described for CXCL12 (44–46), may have applicability as cooperativity-disrupting drugs. Future endeavors will have to elucidate how chemokine cooperativity is modulated in such instances.
Acknowledgements
The authors thank Marloes van der Zwam (University of Groningen, The Netherlands), Winfried Mulder and Toon van der Doelen (MSD, Oss, The Netherlands) for providing reagents. The authors gratefully acknowledge Azra Mujic-Delic, Maarten Bebelman, Quinte Braster, Danny Scholten and Sabrina de Munnik for expert technical assistance. We thank Danny Scholten, Saskia Nijmeijer and Laura Heitman (Leiden University, The Netherlands) for helpful discussions.
TH acknowledges support from the NIH/NIAID R01 AI37113. DA is supported by an NIH/NIGMS K12 GM068524 IRACDA Grant. AR and JvdV were funded by the Dutch Kidney Foundation, grants KJPB 09.01 and consortium grant CP09.03 (GLYCOREN). FV and JvO were funded by the Dutch Top Institute Pharma initiative (program D1-105).
Abbreviations
- BRET
Bioluminesence resonance energy transfer
- CHO
Chinese hamster ovary
- EFC
Enzyme fragment complementation
- GAG
Glycosaminoglycan
- GPCR
G protein-coupled receptor
- HEV
High endothelial venule
- Rluc
Renilla luciferase
- YFP
Yellow fluorescent protein
References
- 1.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 2.Canals M, Scholten DJ, de Munnik S, Han MK, Smit MJ, Leurs R. Ubiquitination of CXCR7 controls receptor trafficking. PLoS One. 2012;7:e34192. doi: 10.1371/journal.pone.0034192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watts AO, Verkaar F, van der Lee MM, Timmerman CA, Kuijer M, van Offenbeek J, van Lith LH, Smit MJ, Leurs R, Zaman GJ, Vischer HF. beta-Arrestin Recruitment and G Protein Signaling by the Atypical Human Chemokine Decoy Receptor CCX-CKR. J Biol Chem. 2013;288:7169–7181. doi: 10.1074/jbc.M112.406108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TN, Kosco-Vilbois MH. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci U S A. 2003;100:1885–1890. doi: 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai Z, Hayasaka H, Kobayashi M, Li W, Guo Z, Jang MH, Kondo A, Choi BI, Iwakura Y, Miyasaka M. CXC chemokine ligand 12 promotes CCR7-dependent naive T cell trafficking to lymph nodes and Peyer's patches. J.Immunol. 2009;182:1287–1295. doi: 10.4049/jimmunol.182.3.1287. [DOI] [PubMed] [Google Scholar]
- 6.Gouwy M, Struyf S, Catusse J, Proost P, Van Damme J. Synergy between proinflammatory ligands of G protein-coupled receptors in neutrophil activation and migration. J. Leukoc. Biol. 2004;76:185–194. doi: 10.1189/jlb.1003479. [DOI] [PubMed] [Google Scholar]
- 7.Gouwy M, Struyf S, Noppen S, Schutyser E, Springael JY, Parmentier M, Proost P, Van Damme J. Synergy between coproduced CC and CXC chemokines in monocyte chemotaxis through receptor-mediated events. Mol. Pharmacol. 2008;74:485–495. doi: 10.1124/mol.108.045146. [DOI] [PubMed] [Google Scholar]
- 8.Kuscher K, Danelon G, Paoletti S, Stefano L, Schiraldi M, Petkovic V, Locati M, Gerber BO, Uguccioni M. Synergy-inducing chemokines enhance CCR2 ligand activities on monocytes. Eur.J.Immunol. 2009;39:1118–1128. doi: 10.1002/eji.200838906. [DOI] [PubMed] [Google Scholar]
- 9.Sebastiani S, Danelon G, Gerber B, Uguccioni M. CCL22-induced responses are powerfully enhanced by synergy inducing chemokines via CCR4: evidence for the involvement of first beta-strand of chemokine. Eur.J.Immunol. 2005;35:746–756. doi: 10.1002/eji.200525800. [DOI] [PubMed] [Google Scholar]
- 10.Paoletti S, Petkovic V, Sebastiani S, Danelon MG, Uguccioni M, Gerber BO. A rich chemokine environment strongly enhances leukocyte migration and activities. Blood. 2005;105:3405–3412. doi: 10.1182/blood-2004-04-1648. [DOI] [PubMed] [Google Scholar]
- 11.Krug A, Uppaluri R, Facchetti F, Dorner BG, Sheehan KC, Schreiber RD, Cella M, Colonna M. IFN-producing cells respond to CXCR3 ligands in the presence of CXCL12 and secrete inflammatory chemokines upon activation. J Immunol. 2002;169:6079–6083. doi: 10.4049/jimmunol.169.11.6079. [DOI] [PubMed] [Google Scholar]
- 12.Vanbervliet B, Bendriss-Vermare N, Massacrier C, Homey B, de Bouteiller O, Briere F, Trinchieri G, Caux C. The inducible CXCR3 ligands control plasmacytoid dendritic cell responsiveness to the constitutive chemokine stromal cell-derived factor 1 (SDF-1)/CXCL12. J Exp Med. 2003;198:823–830. doi: 10.1084/jem.20020437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gosling J, Dairaghi DJ, Wang Y, Hanley M, Talbot D, Miao Z, Schall TJ. Cutting edge: identification of a novel chemokine receptor that binds dendritic cell- and T cell-active chemokines including ELC, SLC, and TECK. J.Immunol. 2000;164:2851–2856. doi: 10.4049/jimmunol.164.6.2851. [DOI] [PubMed] [Google Scholar]
- 14.Townson JR, Nibbs RJ. Characterization of mouse CCX-CKR, a receptor for the lymphocyte-attracting chemokines TECK/mCCL25, SLC/mCCL21 and MIP-3beta/mCCL19: comparison to human CCX-CKR. Eur.J.Immunol. 2002;32:1230–1241. doi: 10.1002/1521-4141(200205)32:5<1230::AID-IMMU1230>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 15.O'Boyle G, Mellor P, Kirby JA, Ali S. Anti-inflammatory therapy by intravenous delivery of non-heparan sulfate-binding CXCL12. FASEB J. 2009;23:3906–3916. doi: 10.1096/fj.09-134643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Severin IC, Gaudry JP, Johnson Z, Kungl A, Jansma A, Gesslbauer B, Mulloy B, Power C, Proudfoot AE, Handel T. Characterization of the chemokine CXCL11-heparin interaction suggests two different affinities for glycosaminoglycans. J Biol Chem. 2010;285:17713–17724. doi: 10.1074/jbc.M109.082552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ziarek JJ, Getschman AE, Butler SJ, Taleski D, Stephens B, Kufareva I, Handel TM, Payne RJ, Volkman BF. Sulfopeptide Probes of the CXCR4/CXCL12 Interface Reveal Oligomer-Specific Contacts and Chemokine Allostery. ACS Chem Biol. 2013 doi: 10.1021/cb400274z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ziarek JJ, Veldkamp CT, Zhang F, Murray NJ, Kartz GA, Liang X, Su J, Baker JE, Linhardt RJ, Volkman BF. Heparin oligosaccharides inhibit chemokine (CXC motif) ligand 12 (CXCL12) cardioprotection by binding orthogonal to the dimerization interface, promoting oligomerization, and competing with the chemokine (CXC motif) receptor 4 (CXCR4) N terminus. J Biol Chem. 2013;288:737–746. doi: 10.1074/jbc.M112.394064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng LY, Ou ZL, Wu FY, Shen ZZ, Shao ZM. Involvement of a novel chemokine decoy receptor CCX-CKR in breast cancer growth, metastasis and patient survival. Clin.Cancer Res. 2009;15:2962–2970. doi: 10.1158/1078-0432.CCR-08-2495. [DOI] [PubMed] [Google Scholar]
- 20.Verkaar F, van Rosmalen JW, Blomenrohr M, van Koppen CJ, Blankesteijn WM, Smits JF, Zaman GJ. G protein-independent cell-based assays for drug discovery on seven-transmembrane receptors. Biotechnol.Annu.Rev. 2008;14:253–274. doi: 10.1016/S1387-2656(08)00010-0. [DOI] [PubMed] [Google Scholar]
- 21.Kuschert GS, Coulin F, Power CA, Proudfoot AE, Hubbard RE, Hoogewerf AJ, Wells TN. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry. 1999;38:12959–12968. doi: 10.1021/bi990711d. [DOI] [PubMed] [Google Scholar]
- 22.Hoogewerf AJ, Kuschert GS, Proudfoot AE, Borlat F, Clark-Lewis I, Power CA, Wells TN. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry. 1997;36:13570–13578. doi: 10.1021/bi971125s. [DOI] [PubMed] [Google Scholar]
- 23.Amara A, Lorthioir O, Valenzuela A, Magerus A, Thelen M, Montes M, Virelizier JL, Delepierre M, Baleux F, Lortat-Jacob H, Arenzana-Seisdedos F. Stromal cell-derived factor-1alpha associates with heparan sulfates through the first beta-strand of the chemokine. J Biol Chem. 1999;274:23916–23925. doi: 10.1074/jbc.274.34.23916. [DOI] [PubMed] [Google Scholar]
- 24.Veldkamp CT, Seibert C, Peterson FC, De la Cruz NB, Haugner JC, 3rd, Basnet H, Sakmar TP, Volkman BF. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci Signal. 2008;1:ra4. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drury LJ, Ziarek JJ, Gravel S, Veldkamp CT, Takekoshi T, Hwang ST, Heveker N, Volkman BF, Dwinell MB. Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc Natl Acad Sci U S A. 2011;108:17655–17660. doi: 10.1073/pnas.1101133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, Lawrence R, Frazier BA, Esko JD. CHO glycosylation mutants: proteoglycans. Methods Enzymol. 2006;416:205–221. doi: 10.1016/S0076-6879(06)16013-9. [DOI] [PubMed] [Google Scholar]
- 27.de Paz JL, Moseman EA, Noti C, Polito L, von Andrian UH, Seeberger PH. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem Biol. 2007;2:735–744. doi: 10.1021/cb700159m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okada T, Ngo VN, Ekland EH, Forster R, Lipp M, Littman DR, Cyster JG. Chemokine requirements for B cell entry to lymph nodes and Peyer's patches. J.Exp.Med. 2002;196:65–75. doi: 10.1084/jem.20020201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gunn MD, Tangemann K, Tam C, Cyster JG, Rosen SD, Williams LT. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc Natl Acad Sci U S A. 1998;95:258–263. doi: 10.1073/pnas.95.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ansel KM, Ngo VN, Hyman PL, Luther SA, Forster R, Sedgwick JD, Browning JL, Lipp M, Cyster JG. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 31.Hargreaves DC, Hyman PL, Lu TT, Ngo VN, Bidgol A, Suzuki G, Zou YR, Littman DR, Cyster JG. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med. 2001;194:45–56. doi: 10.1084/jem.194.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mazzucchelli L, Blaser A, Kappeler A, Scharli P, Laissue JA, Baggiolini M, Uguccioni M. BCA-1 is highly expressed in Helicobacter pylori-induced mucosa-associated lymphoid tissue and gastric lymphoma. J Clin Invest. 1999;104:R49–54. doi: 10.1172/JCI7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hjelmstrom P, Fjell J, Nakagawa T, Sacca R, Cuff CA, Ruddle NH. Lymphoid tissue homing chemokines are expressed in chronic inflammation. Am J Pathol. 2000;156:1133–1138. doi: 10.1016/S0002-9440(10)64981-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pablos JL, Amara A, Bouloc A, Santiago B, Caruz A, Galindo M, Delaunay T, Virelizier JL, Arenzana-Seisdedos F. Stromal-cell derived factor is expressed by dendritic cells and endothelium in human skin. Am J Pathol. 1999;155:1577–1586. doi: 10.1016/S0002-9440(10)65474-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams DH, Lloyd AR. Chemokines: leucocyte recruitment and activation cytokines. Lancet. 1997;349:490–495. doi: 10.1016/s0140-6736(96)07524-1. [DOI] [PubMed] [Google Scholar]
- 36.Mahalingam S, Karupiah G. Chemokines and chemokine receptors in infectious diseases. Immunol Cell Biol. 1999;77:469–475. doi: 10.1046/j.1440-1711.1999.00858.x. [DOI] [PubMed] [Google Scholar]
- 37.Luster AD. Chemokines--chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 38.Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J. 2006;20:9–22. doi: 10.1096/fj.05-4682rev. [DOI] [PubMed] [Google Scholar]
- 39.Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nat Rev Cancer. 2002;2:521–528. doi: 10.1038/nrc842. [DOI] [PubMed] [Google Scholar]
- 40.Lortat-Jacob H. The molecular basis and functional implications of chemokine interactions with heparan sulphate. Curr Opin Struct Biol. 2009;19:543–548. doi: 10.1016/j.sbi.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 41.Rops AL, van der Vlag J, Lensen JF, Wijnhoven TJ, van den Heuvel LP, van Kuppevelt TH, Berden JH. Heparan sulfate proteoglycans in glomerular inflammation. Kidney Int. 2004;65:768–785. doi: 10.1111/j.1523-1755.2004.00451.x. [DOI] [PubMed] [Google Scholar]
- 42.Johnson Z, Kosco-Vilbois MH, Herren S, Cirillo R, Muzio V, Zaratin P, Carbonatto M, Mack M, Smailbegovic A, Rose M, Lever R, Page C, Wells TN, Proudfoot AE. Interference with heparin binding and oligomerization creates a novel anti-inflammatory strategy targeting the chemokine system. J Immunol. 2004;173:5776–5785. doi: 10.4049/jimmunol.173.9.5776. [DOI] [PubMed] [Google Scholar]
- 43.Severin IC, Soares A, Hantson J, Teixeira M, Sachs D, Valognes D, Scheer A, Schwarz MK, Wells TN, Proudfoot AE, Shaw J. Glycosaminoglycan analogs as a novel anti-inflammatory strategy. Front Immunol. 2012;3:293. doi: 10.3389/fimmu.2012.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hachet-Haas M, Balabanian K, Rohmer F, Pons F, Franchet C, Lecat S, Chow KY, Dagher R, Gizzi P, Didier B, Lagane B, Kellenberger E, Bonnet D, Baleux F, Haiech J, Parmentier M, Frossard N, Arenzana-Seisdedos F, Hibert M, Galzi JL. Small neutralizing molecules to inhibit actions of the chemokine CXCL12. J Biol Chem. 2008;283:23189–23199. doi: 10.1074/jbc.M803947200. [DOI] [PubMed] [Google Scholar]
- 45.Blanchetot C, Verzijl D, Mujic-Delic A, Bosch L, Rem L, Leurs R, Verrips CT, Saunders M, de Haard H, Smit MJ. Neutralizing nanobodies targeting diverse chemokines effectively inhibit chemokine function. J Biol Chem. 2013;288:25173–25182. doi: 10.1074/jbc.M113.467969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gonzalo JA, Lloyd CM, Peled A, Delaney T, Coyle AJ, Gutierrez-Ramos JC. Critical involvement of the chemotactic axis CXCR4/stromal cell-derived factor-1 alpha in the inflammatory component of allergic airway disease. J Immunol. 2000;165:499–508. doi: 10.4049/jimmunol.165.1.499. [DOI] [PubMed] [Google Scholar]