

Abstract
Prostate-specific membrane antigen (PSMA) is an attractive target for the imaging of prostate cancer (PCa) due to the elevated expression on the surface of prostate tumor cells. Most PSMA-targeted low molecular weight imaging agents are inhibitors of PSMA. We have synthesized a series of substrate-based PSMA-targeted imaging agents by mimicking poly-γ-glutamyl folic acid, an endogenous substrate of PSMA. In vitro the γ-linked polyglutamate conjugates proved to be better substrates than the corresponding α-linked glutamates. However, in vivo imaging studies of γ-ray-emitting and γ-linked glutamates did not demonstrate selective uptake in PSMA-pos-itive over PSMA-negative tumors. Subsequent chromatographic studies and in silico molecular dynamics simulations indicated that hydrolysis of the substrates is slow and access to the enzymatic active site is limited. These results inform the design of future substrate-based imaging agents for PSMA.
Keywords: molecular imaging, prodrug, prostate cancer, SPECT, PSMA
Introduction
Prostate-specific membrane antigen (PSMA), a type II integral membrane metalloprotease, is an attractive target for imaging of prostate cancer (PCa) because it is highly expressed on the surface of prostate carcinomas, particularly those associated with castration-resistant and metastatic disease.1 The extracellular active site of PSMA is readily accessed by a variety of chemical structures, many of which have been fashioned into imaging agents for a variety of modalities.2–7
PSMA is active as the dimer, each monomer of which is comprised of 750 amino acids. Two zinc ions are found at the active site of PSMA and are involved in the hydrolytic cleavage of endogenous substrates, including N-acetylaspartylglutamate (NAAG) in the brain and poly-γ-glutamyl folic acid in the intestine. Accordingly, PSMA has been referred to as glutamate carboxypeptidease II (GCPII) in the brain and folate hydrolase I (FOLH1) in the intestine. In the brain PSMA modulates the concentration of glutamate in the synaptic cleft through cleavage of NAAG. Abnormal concentrations of glutamate in the brain may be associated with a variety of disorders including ischemia, traumatic brain injury, amyotrophic lateral sclerosis, and schizophrenia.8–12 FOLH1 hydrolyzes glutamates from the C-terminus of poly-γ-glutamyl folate to produce folic acid, a methyl donor in the synthesis of DNA.13 PSMA is also found in the neovasculature of a variety of non-prostate solid tumors.14 These diverse effects of PSMA render it an important target for the study of both central nervous system disease and cancer.1,15
Over the past decade, X-ray crystal structures of PSMA in complex with a variety of inhibitors or substrates have provided detailed information on the active site,16,17 which has enabled rational ligand design. According to those crystal structures, the active site is composed of two distinct pockets, a so-called ‘glutamate-sensing’ pocket (S1’) and the non-pharmacophore pocket (S1). The S1’pocket is highly sensitive to structural modification of putative ligands, having a nearly absolute requirement for a glutamate moiety, which make strong salt bridges with Arg 210 and Lys 699. Recent reports of P1’-modified inhibitors and substrates have demonstrated that the lipophilic alkyl chain of the P1’moiety also provides hydrophobic interactions with lipophilic amino acids in the S1’-site.18,19 In the S1 pocket the side chains of amino acids are flexible and can undergo rearrangement upon interaction with various functional groups. In the absence of ligand, three arginines (Arg463, Arg534, and Arg536) are in a stacked conformation, which forms an additional subpocket, known as an “arginine patch,” upon productive ligand binding.20 In addition to the S1’ and S1 sites, a flexible cylinder-like, 20 Å-deep tunnel exists adjacent to the S1 pocket and undergoes a conformational change upon ligand binding.3,21 The tunnel region extends the hydrophilic surface area of the enzyme and can accommodate bulky and hydrophilic scaffolds. The tunnel region is much more flexible than the two aforementioned pockets and undergoes an inducedfit to accommodate the bulky linker of a variety of conjugates that bind to PSMA. We and others have used the tunnel region to develop inhibitor-based imaging agents conjugated with bulky functional groups, including radiometal-coordinating species and optical dyes.5–7,21
But apart from ligand-mediated internalization of PSMA,22,23 which may regenerate the protein on the cell surface available for binding by another ligand, there is no amplification mechanism for concentration of targeted imaging agents within PSMA-expressing cells. Such amplification can increase the sensitivity of detection of those cells, as has been demonstrated for other enzyme- and transporter-based imaging agents.24–26 The enzymatic activity of PSMA has been exploited for the targeted delivery of therapeutic agents. Mhaka and coworkers have targeted PSMA with toxic species by linking them to poly-γ-glutamates and poly-glutamate-asparates, with various permutations of aspartate and glutamate in α-or γ-linkages.27 Anderson and coworkers reported a number of N-acyldipeptide-derived PSMA substrates to monitor PSMA activity by identifying the cleaved product by highperformance liquid chromatography (HPLC).28
In an effort to leverage the amplification possible with enzymatic turnover of suitably functionalized substrates, we describe the development of potential substrate-based imaging agents for PSMA. The agents designed mimic the endogenous substrate, poly-γ-glutamyl folate. We have designed α- or γ-linked glutamate conjugates linked to aminostrylpyridinium (ASP) dyes, on which 125I and a lipophilic alkyl chain have been introduced (Figure 1). The ASP dye was intended to enable intercalation within the cell membrane of the radioactive species liberated upon cleavage.29–31 Amphiphilic ASP dye was expected to orient with the polar head protruding from and the lipophilic tail embedded within, the cell membrane. It also provides the capacity to study cellular uptake with optical methods.30,31 In order to modulate the hydrophilicity/lipophilicty of the substrates, we introduced penta- or hexa-glutamates. Our goal is to uncover a conjugate, the radiolabeled, cleaved product of which can be incorporated into the cell membrane with concurrent signal amplification in vivo. Here we focus on validating the concept of substrate-based imaging agents for PSMA and obtaining basic information on substrate properties for further structural modification.
Figure 1.
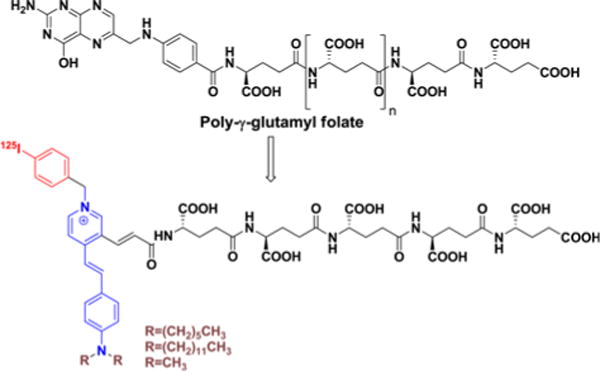
Design of aminostrylpyridium (ASP)-polyglutamate conjugates.
Experimental
Materials
The protected amino acids and Fmoc-Glu(OtBu)-COOH preloaded Wang resin were purchased from ChemImpex (Chicago, IL). Chemicals and solvents for chemical reactions and for high-performance liquid chromatography (HPLC) were of reagent grade, were acquired from Sigma-Aldrich (Milwaukee, WI) or Fisher Scientific (Pittsburgh, PA), and were used without further purification. [125I]NaI was purchased from MP Biomedicals (Costa Mesa, CA).
HPLC Analysis
The purity of final products was analyzed using a Waters (Milford, MA) HPLC apparatus using a NovaPak C18 reverse phase column (7.8 mm×300 mm, pore size: 6 μm). Separation of the products was achieved using an isocratic method of water (0.1% TFA) and acetonitrile or methanol (0.1% TFA). Reverse phase radio-HPLC purifications of 125I-labeled compounds were performed using a Waters 510 pump, Waters 490E variable wavelength UV/vis detector at 254 nm and a Bioscan Flow Count PMT radioactivity detector (Washington, DC).
NMR and Mass Spectrometry
Mass spectra of positive ions were recorded with an ESI Bruker Esquire 3000 plus mass spectrometer (BrukerDaltonics, Billerica, MA) and 1H NMR and 13C NMR spectra were obtained on a Bruker Avance 400 MHz Spectrometer (Bruker Daltonics, Billerica, MA).
Chemistry
General Procedure for the Syntheses of 3a–3b (Scheme I)
Scheme I.
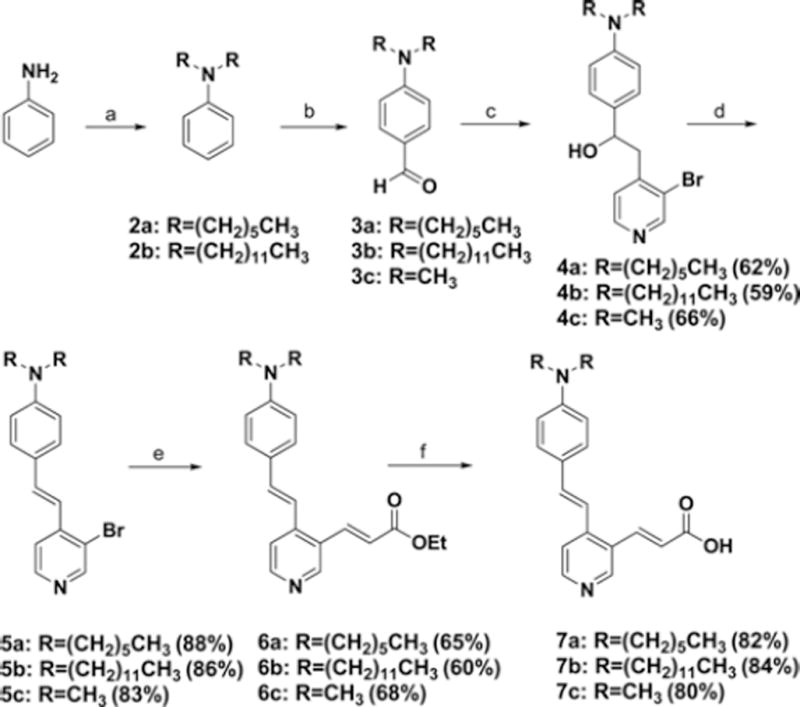
Synthetic route to aminostrylpyridine moieties. Reagents and conditions: (a) K2CO3, DMF, 1-iodohexane (2a) or 1-iodo-dodecane (2b), (b) POCl3, DMF, (c) 3-bromo-4-methylpyridine, LDA, Et2O, (d) 3N-HCl, dioxane, 60 °C, (e) ethylacrylate, PPh3, Pd(OAc)2, TEA, DMF, 130 °C, (f) NaOH, H2O/EtOH/i-PrOH.
The syntheses of 3a–3b were accomplished from aniline by following published procedures.32,33
General Procedure for the Syntheses of 4a–4c (Scheme I)
To a solution of 3-bromo-4-methylpyridine (1 mmol) in anhydrous diethylether (4 mL) was added LDA (1.1 mmol, 0.55 mL, 2 M) at −78 °C. The suspension was stirred for 30 min at the same temperature. To the reaction mixture was added a solution of aldehyde 3a–3c (1 mmol) in anhydrous THF (2 mL) at −78 °C. The reaction mixture stirred for additional 30 min at the same temperature and for 12 h at room temperature. Diethylether (50 mL) was added and the ether layer was washed with NH4Cl solution (1 M, 25 mL×2). The organic layer was dried over Na2SO4, filtered, and evaporated. The residue was purified by silica gel column chro-matography (hexane:ethylacetate=3:2) to give 4a–4c in 62%, 59%, and 66%, respectively.
2-(3-Bromopyridin-4-yl)-1-(4-(dihexylamino)phenyl)ethanol (4a)
1H NMR (CDCl3): 8.62 (s, 1H), 8.34 (d, 1H, J=4.9 Hz), 7.20 (d, 2H, J=8.7 Hz), 7.19 (d, 1H, J=4.9 Hz) 6.60 (d, 2H, J=8.7 Hz), 4.88–4.92 (m, 1H), 3.25 (t, 4H, J=7.8 Hz), 3.12–3.15 (m, 2H), 1.55–1.59 (m, 4H), 1.29–1.33 (m, 12H), 0.90 (t, 6H, J=6.6 Hz), 13C NMR (CDCl3): 151.5, 150.3, 147.6, 147.4, 131.4, 126.8, 126.7, 123.4, 112.4, 72.3, 44.9, 40.6, Theoretical MS C25H37BrN2O: 460.21, observed: [M+H]+; 461.2.
2-(3-Bromopyridin-4-yl)-1-(4-(didodecylamino)phenyl) ethanol (4b)
1H NMR (CDCl3): 8.66 (s, 1H), 8.36 (d, 1H, J=4.5 Hz), 7.23 (d, 2H, J=8.2 Hz), 7.21 (d, 1H, J=4.5 Hz) 6.61 (d, 2H, J=8.2 Hz), 4.89–4.93 (m, 1H), 3.26 (t, 4H, J=7.6 Hz), 3.11–3.14 (m, 2H), 1.54–1.58 (m, 4H), 1.27–1.33 (m, 36H), 0.89 (t, 6H, J=6.7 Hz), 13C NMR (CDCl3): 151.69, 148.04, 147.80, 147.36, 129.53, 126.87, 126.68, 123.47, 111.43, 72.62, 51.05, 44.65, 31.86, 29.65, 29.55, 29.52, 29.30, 27,15, 22.65, 14.10, Theoretical MSC37H61BrN2O: 628.40, observed: [M+H]+; 629.5.
2-(3-Bromopyridin-4-yl)-1-(4-(dimethylamino)phenyl) ethanol (4c)
1H NMR (CDCl3): 8.52 (s, 1H), 8.24 (d, 1H, J=4.5 Hz), 7.24 (d, 2H, J=8.2 Hz), 7.11 (d, 1H, J=4.5 Hz) 6.69 (d, 2H, J=8.2 Hz), 4.87–4.90 (m, 1H), 3.08–3.11 (m, 2H), 2.94 (s, 6H), 13C NMR (CDCl3): 151.5, 150.3, 147.6, 147.4, 131.4, 126.8, 126.7, 123.4, 112.4, 72.3, 44.9, 40.6, Theoretical MSC15H17BrN2O: 320.05, observed: [M+H]+; 320.8.
General Procedure for the Syntheses of 5a–5c (Scheme I)
To a solution of carbinol (0.5 mmol) in dioxane (10 mL) was added a 3 N-HCl solution (2 mL) at room temperature. The reaction mixture was stirred at 60 °C for 6 h. The progress of the reaction was monitored by thin layer chromatography (TLC). After removing the excess dioxane, NaHCO3 solution (3 N, 2 mL) was added to adjust pH ~7. The aqueous layer was extracted with dichloromethane (50 mL). The organic layer was dried over Na2SO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane:ethylacetate=3:1, 0.1% triethylamine) to give aminostrylpyridines 5a–5c, respectively.
(E)-4-(2-(3-Bromopyridin-4-yl)vinyl)-N,N-dihexylaniline (5a)
1H NMR (CDCl3): 8.65 (s, 1H), 8.37 (d, 1H, J=5.2 Hz), 7.48 (d, 1H, J=5.2 Hz), 7.44 (d, 2H, J=8.8 Hz) 7.24 (d, 1H, J=16.1 Hz), 7.15 (d, 1H, J=16.1 Hz), 6.62 (d, 2H, J=8.8 Hz), 3.30 (t, 4H, J=7.8 Hz), 1.56–1.60 (m, 4H), 1.31–1.35 (m, 12H), 0.91 (t, 6H, J=6.8 Hz), Theoretical MSC25H35BrN2: 442.20, observed: [M+H]+; 443.3.
(E)-4-(2-(3-Bromopyridin-4-yl)vinyl)-N,N-didodecyl-aniline (5b)
1H NMR (CDCl3): 8.65 (s, 1H), 8.38 (d, 1H, J=5.2 Hz), 7.49 (d, 1H, J=5.2 Hz), 7.44 (d, 2H, J=8.8 Hz), 7.20 (d, 1H, J=16.1 Hz) 7.10 (d, 1H, J=16.1 Hz), 6.62 (d, 2H, J=8.8 Hz), 3.30 (t, 4H, J=7.4 Hz), 1.57–1.61 (m, 4H), 1.27–1.32 (m, 36H), 0.88 (t, 6H, J=7.0 Hz), 13C NMR (CDCl3): 152.41, 148.86, 147.80, 144.99, 135.56, 128.93, 122.82, 121.17, 119.51, 118.71, 111.41, 51.02, 31.86, 29.62, 29.53, 29.49, 29.29, 27.24, 27.10, 22.65, 14.10, Theoretical MSC37H59BrN2: 610.39, observed: [M+H]+; 611.4.
(E)-4-(2-(3-Bromopyridin-4-yl)vinyl)-N,N-dimethyl-aniline (5c)
1H NMR (CDCl3): 8.66 (s, 1H), 8.39 (d, 1H), 7.46–7.50 (m, 3H), 7.22 (d, 1H) 7.13 (d, 1H), 6.71 (d, 2H), 3.02 (s, 6H), Theoretical MSC15H15BrN2: 302.04, observed: [M+H]+; 303.1.
General Procedure for the Syntheses of 6a–6c (Scheme I)
To a solution of substituted aminostrylpyridine (5a–5c, 1 mmol) in anhydrous DMF (10 mL) was added Pd(OAc)2 (2.5 mol%), PPh3 (5 mol%), TEA (5 mmol) and ethylacrylate (1.5 mmol) under nitrogen purge. The reaction mixture was stirred at 120 °C for 48 h. The mixture was cooled down and diluted with 10 mL of water. The aquous layer was extracted with ethylacetate (30 mL). The organic layer was washed with brine, dried over Na2SO4, and filtered through Celite. After evaporating the excess solvent, the residue was purified by silica gel column chromatography (hexane:ethylacetate=3:1) to give aminostrylpyridine acrylate (6a–6c).
(E)-Ethyl 3-(4-(4-(dihexylamino)styryl)pyridin-3-yl)acry-late (6a)
1H NMR (CDCl3): 8.67 (s, 1H), 8.45 (d, 1H, J=5.3 Hz), 8.04 (d, 1H, J=16.0 Hz), 7.44 (d, 1H, J=5.4 Hz) 7.41 (d, 2H, J=8.8 Hz), 7.14 (d, 1H, J=16.0 Hz), 7.02 (d, 1H, J=16.0 Hz), 6.61 (d, 2H, J=8.8 Hz), 6.41 (d, 1H, J=16.0 Hz), 4.29 (q, 2H, J=J=7.1 Hz), 3.30 (t, 4H, J=7.8 Hz), 1.56–1.60 (m, 4H), 1.31–1.37 (m, 15H), 0.90 (t, 6H, J=6.7 Hz), 13C-NMR (CDCl3): 167.51, 150.96, 149.84, 145.90, 140.84, 137.10, 129.89, 128.71, 124.02, 122.35, 120.23, 117.72, 112.45, 61.70, 52.05, 32.71, 28.24, 27.80, 23.68, 15.33, 15.06, Theoretical MS C30H42N2O2: 462.32, observed: [M+H]+; 463.4.
(E)-Ethyl 3-(4-(4-(didodecylamino)styryl)pyridin-3-yl) acrylate (6b)
1H NMR (CDCl3): 8.68 (s, 1H), 8.47 (d, 1H, J=5.2 Hz), 8.04 (d, 1H, J=16.0 Hz), 7.45 (d, 1H, J=5.2 Hz), 7.41 (d, 2H, J=8.8 Hz), 7.14 (d, 1H, J=15.9 Hz) 7.03 (d, 1H, J=15.9 Hz), 6.62 (d, 2H, J=8.8 Hz), 6.42 (d, 1H, J=16.0 Hz), 4.29 (q, 2H, J=7.1Hz), 3.27–3.31 (m, 4H), 1.57–1.61 (m, 4H), 1.26–1.33 (m, 39H), 0.88 (t, 6H, J=7.0 Hz), 13C NMR (CDCl3): 166.5, 149.9, 148.9, 148.8, 144.9, 139.8, 136.0, 128.8, 127.7, 123.0, 121.3, 119.2, 116.7, 111.4, 60.7, 51.0, 31.9, 29.6, 29.6, 29.5, 29.3, 27.2, 27.1, 22.7, 14.3, 14.1, Theoretical MSC42H66N2O2: 630.51, observed: [M+H]+; 631.7.
(E)-Ethyl-3-(4-(4-(dimethylamino)styryl)pyridin-3-yl)acry-late (6c)
1H NMR (CDCl3): 8.69 (s, 1H), 8.48 (d, 1H), 8.05 (d, 1H), 7.44–7.47 (m, 3H), 7.17 (d, 1H) 7.07 (d, 1H), 6.71 (d, 2H), 6.43 (d, 1H), 4.29 (q, 2H), 3.02 (s, 6H), 1.35 (t, 3H) Theoretical MSC20H22N2O2: 322.17, observed: [M+H]+; 323.1.
General Procedure for the Syntheses of 7a–7c (Scheme I)
To a solution of substituted aminostrylpyridine acrylate (6a–6c, 0.5 mmol) in ethanol (8 mL) and 2-propanol (8 mL) was added a solution of NaOH (1.5 mmol) in water (4 mL). In the case of 6c only ethanol and water were used. The reaction mixture was stirred at room temperature for 2 h. At the end of the reaction the excess ethanol and 2-propanol were removed under reduced pressure. A solution of 3 N-HCl (3 mL) was added to the reaction mixture to neutralize the solution. The aqueous layer was extracted with CH2Cl2 (30 mL). The organic layer was combined, dried over Na2SO4, and filtered. Without purification, the amphiphilic compounds 7a–7c were used for the next step. 1H NMR showed the crude material in high purity (>95%).
(E)-3-(4-(4-(Dihexylamino)styryl)pyridin-3-yl)acrylic acid (7a)
1H NMR (CD3OD): 8.72 (s, 1H), 8.40 (d, 1H, J=6.1 Hz), 8.05 (d, 1H, J=6.1 Hz), 8.03 (d, 1H, J=16.1 Hz) 7.67 (d, 1H, J=15.7 Hz), 7.58 (d, 2H, J=8.9 Hz), 7.17 (d, 1H, J=15.7 Hz), 6.73 (d, 2H, J=8.9 Hz), 6.58 (d, 1H, J=16.1 Hz), 3.38 (t, 4H, J=7.8 Hz), 1.58–1.62 (m, 4H), 1.32–1.36 (m, 12H), 0.91 (t, 6H, J=6.8 Hz), Theoretical MS C28H38N2O2: 434.29, observed: [M+H]+; 435.3.
(E)-3-(4-(4-(Didodecylamino)styryl)pyridin-3-yl)acrylic acid (7b)
1H NMR (CD3OD): 8.57 (s, 1H), 8.31 (d, 1H, J=5.4 Hz), 7.80 (d, 1H, J=16.0 Hz), 7.65 (d, 1H, J=5.4 Hz), 7.50 (d, 2H, J=8.8 Hz), 7.31 (d, 1H, J=15.7 Hz), 7.22 (d, 1H, J=15.7 Hz), 6.67 (d, 2H, J=8.8 Hz), 6.46 (d, 1H, J=16.0 Hz), 3.30–3.35 (m, 4H), 1.57–1.61 (m, 4H), 1.27–1.34 (m, 36H), 0.86–0.90 (m, 6H), Theoretical MSC40H62N2O2: 602.48, observed: [M+H]+; 603.5.
(E)-3-(4-(4-(dimethylamino)styryl)pyridin-3-yl)acrylic acid (7c)
1H NMR (CD3OD): 9.06 (s, 1H), 8.72 (d, 1H), 8.43 (d, 1H), 8.06 (d, 1H), 7.99 (d, 2H), 7.89 (d, 2H), 7.74 (d, 2H), 7.66 (d, 1H), 6.72 (d, 1H), 3.29 (s, 6H), 13C NMR (CD3OD): 166.9, 152.9, 140.6, 140.2, 139.7, 135.2, 131.8, 130.0, 126.9, 123.0, 121.6, 120.2, 44.9, Theoretical MSC18H18N2O2: 294.14, observed: [M+H]+; 295.0.
General Procedure for the Syntheses of 11a–11c (Scheme II)
Scheme II.
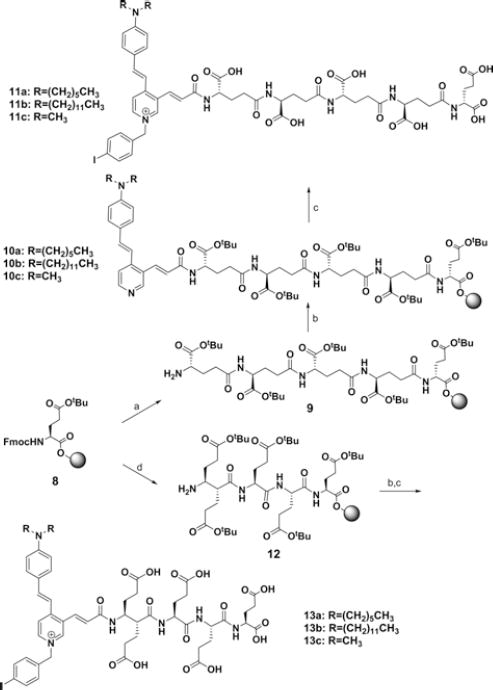
Synthetic route for resin-bound polyglutamates. Reagents and conditions: (a) i: 20% piperidine in DMF, rt, 60 min, ii: Fmoc-Glu-OtBu, HATU, HOBt, DIPEA, DMF, (b) HATU, HOBt, DIPEA, DMF, 7a (for 10a), 7b (for 10b), or 7c (for 10c), (c) i: 4-iodobenzyl chloride, CH3CN, ii: TFA/CH2Cl2, (d) 20% piperidine in DMF, rt, 60 min, ii: Fmoc-Glu(OtBu)-OH, HATU, HOBt, DIPEA, DMF.
To a suspension of compound 10a–10c (30 mg) in CH3CN was added 4-iodobenzylbromide (5 mg). The reaction mixture was refluxed for 12 h with gentle stirring. After monitoring formation of resin-bound pyridinium compound by mass spectroscopy, the excess solvent and reagent were removed by filtration. The resin-bound compound was combined and diluted in TFA/CH2Cl2 solution (1:1, 3 mL). The reaction mixture was stirred at room temperature for 2 h and was filtered. The combined solution was concentrated under reduced pressure and was subjected to HPLC to give the final compounds 11a–11c.
4-(4-(Dihexylamino)styryl)-3-((5S,10S,15S,20S,25S,30S,E)-5,10,15,20,25,30,32-heptacarboxy-3,8,13,18,23,28-hexaoxo-4,9,14,19,24,29-hexaazadotriacont-1-enyl)-1-(4-iodoben-zyl)pyridinium (11a)
HPLC: MeOH/CH3CN/H2O=94/2/4 (0.1% TFA, 4 mL/min), retention time: 20 min, 1H NMR (CD3OD): 9.10 (s, 1H), 8.41 (d, 1H, J=7.0 Hz), 8.16 (d, 1H, J=7.0 Hz), 7.87 (d, 1H, J=15.7 Hz), 7.82 (d, 1H, J=15.9 Hz), 7.81 (d, 2H, J=8.4 Hz), 7.64 (d, 2H, J=9.0 Hz), 7.33 (d, 2H, J=8.4 Hz), 7.25 (d, 1H, J=15.9 Hz), 6.92 (d, 1H, J=15.7 Hz), 6.71 (d, 2H, J=9.0 Hz), 5.66 (s, 2H), 4,45–4.50 (m, 1H), 4.25–4.32 (m, 5H), 3.36–3.40 (m, 4H), 2.27–2.33 (m, 12H), 2.18–2.23 (m, 6H), 1.98–2.07 (m, 6H) 1.57–1.63 (m, 4H), 1.32–1.36 (m, 12H), 0.91 (m, 6H), Theoretical MS: 1425.50, observed: [M]+; 1425.6, [M+H]2+; 724.3.
4-(4-(Didodecylamino)styryl)-3-((5S,10S,15S,20S,25S,E)-5,10,15,20,25,27-hexacarboxy-3,8,13,18,23-pentaoxo-4,9,14,19,24-pentaazaheptacos-1-enyl)-1-(4-iodobenzyl)pyridinium (11b)
HPLC: MeOH/CH3CN/H2O=94/2/4 (0.1% TFA, 4 mL/min), retention time: 32 min, 1H NMR (CD3OD): 9.03 (s, 1H), 8.40 (d, 1H, J=6.7 Hz), 8.15 (d, 1H, J=6.7 Hz), 7.79–7.89 (m, 4H) 7.63 (d, 2H, J=8.8 Hz), 7.30 (d, 2H, J=8.4 Hz), 7.22 (d, 1H, J=15.4 Hz), 6.71 (d, 2H, J=8.8 Hz), 6.68 (d, 1H, J=14.9 Hz), 5.63 (s, 2H), 4,45–4.50 (m, 1H), 4.29–4.36 (m, 4H), 3.36–3.40 (m, 4H), 2.31–2.36 (m, 10H), 2.12–2.18 (m, 5H), 1.97–2.08 (m, 5H) 1.58–1.63 (m, 4H), 1.22–1.36 (m, 36H), 0.89 (m, 6H), Theoretical MSC72H103IN7O17+: 1464.64, observed: [M]+; 1464.8, [M+H]2+; 732.9.
4-(4-(Dimethylamino)styryl)-3-((5S,10S,15S,20S,25S,E)-5,10,15,20,25,27-hexacarboxy-3,8,13,18,23-pentaoxo-4,9,14,19,24-pentaazaheptacos-1-enyl)-1-(4-iodobenzyl)pyridinium (11c)
HPLC: MeOH/CH3CN/H2O=40/30/30 (0.1% TFA, 4 mL/min), retention time: 16 min, 1H NMR (CD3OD): 8.91 (s, 1H), 8.51 (d, 1H, J=6.8 Hz), 8.22 (d, 1H, J=6.8 Hz), 7.95 (d, 1H, J=15.7 Hz), 7.81–7.87 (m, 3H) 7.63 (d, 2H, J=9.0 Hz), 7.28 (d, 2H, J=8.4 Hz), 7.22 (d, 1H, J=15.9 Hz), 6.75–6.79 (m, 3H), 5.62 (s, 2H), 4,45–4.50 (m, 1H), 4.40–4.44 (m, 4H), 3.36 (s, 6H), 2.31–2.40 (m, 10H), 2.15–2.25 (m, 5H), 1.98–2.08 (m, 5H),Theoretical MS C50H59IN7O17+: 1156.30, observed: [M]+; 1156.4 [M+H]2+; 578.6.
Radiosynthesis of [125I]15a and [125I]15c (Scheme III)
Scheme III.
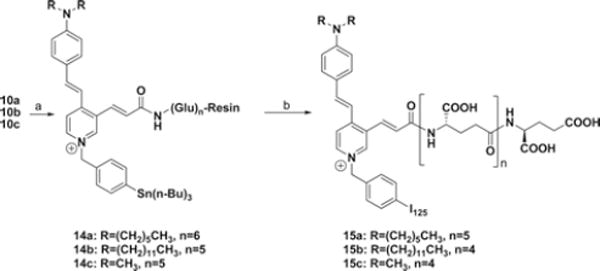
Synthetic route to [125I]15a and [125I]15c. Reagents and conditions: (a) i: 4-((bromomethyl)phenyl)tributylstannane, CH3CN, reflux, 24 h, (b) i: Na[125I]I, N-chlorosuccinimide, methanol (0.1% acetic acid), rt, 2 h, ii: TFA/CH2Cl2 (1:1), rt, 90 min.
To a solution of compound 14a (6 mg) suspended in 300 μL in MeOH was added 300 μL of NCS solution (0.8 mg in 1 mL of MeOH). To this was added a solution of [125I]NaI (2.78 mCi, MP Biomedicals, Costa Mesa, CA) diluted with 15 μL of phosphate buffered saline. After 60 min at room temperature, the excess methanol was removed with the resin in the vial. The radioactivity of the remained resin was 2.38 mCi while the methanol solution was 300 μCi. To the reaction vial was added 200 μL of anhydrous dichloromethane and 200 μL of TFA. The reaction mixture was stirred very slowly at room temperature for 90 min. The dichloromethane and TFA were removed under a gentle stream of nitrogen. The reaction mixture was diluted with 500 μL of MeOH/H2O (80/20, 0.5% TEA). HPLC conditions for 15a and 15c were 38% MeOH/62% H2O/0.3% TEA and 15% MeOH/85% H2O/ 0.3% TEA with a flow rate of 1.5 mL/min on a Waters Radial Pak analytical column (8×10 mm). About 1.2 mCi of 15a and 15c (retention time: 11.5 and 7.2 min) were collected and evaporated to dryness under vacuum (43% RCY, 1500 mCi/ μmol). They were formulated in 250 μL saline for subsequent imaging.
Biology
PSMA Substrate Specificity
Glutamate release from the synthesized conjugates was determined using a fluorescence-based assay. Lysates of LNCaP cell extracts were incubated with the substrates at different concentrations. ASP-polyglutamate conjugates and NAAG were incubated with PSMA-expressing LNCaP cell extracts in 0.1 M Tris-HCl (pH 7.5) at 37 °C for 2 h. The amount of glutamate released by the hydrolysis of C-terminal glutamates was measured by incubating with a working solution of the Amplex Red glutamic acid kit (Molecular Probes Inc., Eugene, OR, USA). The fluorescence was measured with a VICTOR3V multilabel plate reader (Perkin Elmer Inc., Waltham, MA, USA) with excitation at 530 nm and emission at 560 nm.
Cell Lines and Mouse Models
PC3 PIP (PSMA+) and PC3 flu (PSMA−) cell lines were obtained from Dr. Warren Heston (Cleveland Clinic, USA) and were maintained as previously described. All cells were grown to 80%–90% confluence before trypsinization and formulation in Hank’s Balanced Salt Solution (HBSS, Sigma, St. Louis, MO) for implantation into mice. All animal studies were undertaken in full compliance with institutional guidelines related to the conduct of animal experiments. Male severe combined immunodeficient (SCID) mice (Charles River Laboratories, Wilmington, MA) were implanted subcutaneously with 1–5 ×106 cells forward of each flank. PSMA+ PC3 PIP cells were implanted behind the left flankand PSMA− PC3 flu cells were implanted behind the right flank. Mice were imaged or used in biodistribution assays when the tumor xenografts reached 3–5 mm in diameter.
Single Photon Emission Computed Tomography-Computed Tomography (SPECT-CT) Imaging of [125I]15a and [125I]15c
A single SCID mouse implanted with both a PSMA+PC3 PIP and a PSMA-PC3 flu xenograft was injected intravenously with ~700 μCi (25.9 MBq) of [125I]15a–15c in saline. The mouse was positioned on the X-SPECT (Gamma Medica, Northridge, CA) gantry and was scanned using two low energy, high-resolution pinhole collimators (Gamma Medica) rotating through 360° in 6° increments for 45 s per increment. Images were reconstructed using LumaGEM software (Gamma Medica, Northridge, CA). Immediately following SPECT acquisition the mice were then scanned by CT (X-SPECT) over a 4.6 cm field-of-view using a 600 A, 50 kV beam. The SPECT and CT data were then coregistered using the supplier’s software (Gamma Medica) and displayed using AMIDE (http://amide.sourceforge.net/). Data were reconstructed using the ordered subsets-expectation maximization (OS-EM) algorithm.
Biodistribution of [125I]15a and 15c
Blood was collected immediately after sacrifice by cardiac puncture and heart, lung, liver, stomach, pancreas, spleen, white fat, kidney, muscle, small intestine, large intestine, urinary bladder, and tumor xenografts were harvested, weighed, and counted in an automated gamma counter (LKB Wallace 1282 Compugamma CS Universal Gamma Counter). Animals were sacrificed at 24 and 144 h postinjection for 15c and 15a, respectively. Tissue radiopharmaceutical uptake values were calculated as percent injected dose per gram (% ID/g).
HPLC of Tumor Cell Extracts
Compound 11c in LNCaP cell extracts (50 μM in 100 μL) was incubated at 37 °C for 30 min, 3 h, and 20 h. The mixture was diluted with HPLC solvent system (MeOH/CH3CN/H2O=40/30/30, 0.1% TFA, 300 μL) and the crude solution (50 μL) was subjected to was subjected to the analytical HPLC system. The peaks were monitored at 254 nm and confirmed by mass spectrometer.
Molecular Modeling
Molecular modeling studies of 11c were performed with Discovery Studio 2.1 (Accelrys Inc. San Diego, USA) implemented on a Windows XP2 system. The PSMA crystal structure (PDB ID: 2C6C) was downloaded from the Protein Data Bank. The 3-D structure of 11c was superimposed upon GPI-18431 by tethering four attachment points (N-CH(COO-)-CH2 in glutamate). The coordinates of superimposed 11c were transferred into the apoprotein of 2C6C and merged in the system. The 2C6C/11c complex system was minimized by modifying default settings (max steps: 20000, max RMS gradient: 0.01, implicit solvent model: Generalized Born with a simple switching (GBSW), dielectric constant: 1, implicit solvent dielectric constant: 80). Consecutive simulations of the obtained complex system from minimization were carried out using a standard simulation cascade protocol, which performs several consecutive simulations (minimization, heating, equilibration, production) for a 2C6C/11c complex system. For 2C6C/11c dynamics with constraints, amino acid residues within 7 Å of 11c remained flexible while all other amino acids were fixed. Dynamics simulation was performed by slightly modifying default settings [minimization algorithm: steepest descent (max steps: 500, minimization RMS gradient: 0.1), minimization algorithm2: conjugate gradient (max steps: 500, RMS gradient: 0.0001, heating (steps: 2000, initial temperature: 50.0, target temperature: 300.0), equilibration (steps: 1000, target temperature: 300.0), implicit solvent model: GBSW, dielectric constant: 1, solvent dielectric constant: 80)].
Results and Discussion
Chemistry
The synthetic routes for γ- and α-linked ASP-pentaglutamates or ASP-hexaglutamates are outlined in Schemes I and II. Key intermediate aminostrylpyridines 7a–7c were prepared in four steps from 4-(dialkylamino) benzaldehydes 3a–3c which were synthesized by reacting N,N-dialkylaniline 2a–2c with POCl3 in DMF.32,33 Lithiation of 3-bromo-4-methylpyridine using lithium diisopropylamide (LDA) and subsequent addition of 3a–3c gave a racemic mixture of the erythro and threo forms of carbinols 4a–4c in 59%–66% yield. Dehydration of 4a–4c under acidic condition afforded strylpyridines 5a–5c in 83%–88% yield with the E-isomer as the major product. Palladium-catalyzed Heck coupling of 5a–5c with ethylacrylate in DMF at 130 °C using palladium acetate and triphenylphosphine gave aminostryl-pyridine acrylates 6a–6c in 60%–68% yield. The less lipophilic dimethyl analog 7a was obtained by reacting 6a with NaOH in a mixture of ethanol/water (2:1). However, hydrolysis of the more lipophilic dihexyl analog 6b and didodecyl analog 6c occurred by treating NaOH in a mixture of 2-propanol/EtOH/water (2:2:1). 2-Propanol improved the solubility of the lipophilic ethylesters 6b–6c.
The other key polyglutamate intermediates, poly-γ-glutamate 9 and poly-α-glutamate 12 were prepared by utilizing solid phase chemistry. Reaction of the Fmoc-Glu(OtBu)-preloaded Wang resin with 20% piperidine in DMF for 60 min, followed by coupling with Fmoc-Glu-OtBu under peptide coupling conditions (HBTU, HOBt, DIPEA, DMF, 2 h) gave Fmoc-Glu(OtBu)-Glu(OtBu)-resin. ubsequent deprotection of the Fmoc group and coupling with Fmoc-Glu-OtBu in 3 or 4 consecutive reactions afforded compounds 9a and 9b, respectively. To prepare resin-bound α-linked penta-glutamate 12, Fmoc-Glu(OtBu)-OH was used instead of Fmoc-Glu-OtBu in each reaction cycling step.
Conjugations of 7a–7c with 9a–9b under peptide coupling conditions (TBTU, HOBt, and DIPEA) gave compounds 10a–10c. Reaction of 10a–10c with 4-iodobenzyl bromide in acetonitrile under reflux for 12 h, followed by deprotection of the tert-butyl (tBu) groups and removal of Wang resin by treating trifluoroacetic acid (TFA), afforded the γ-linked ASP-glutamates 11a–11c. The α-linked ASP-glutamate conjugates 13a–13c were prepared in the same synthetic strategy as that for 11a–11c. The syntheses of 125I-labeled, γ-linked ASP-glutamates 15a–15c for in vivo imaging studies are shown in Scheme III. Reaction of 10a–10c with 4-(tributylstannyl)benzyl bromide, which was obtained from 4-bromobenzyl alcohol in 3 steps, gave the radioiodination precursors 14a–14c. Radioiodination-destannylation of 14a–14c with Na[125I]I and N-chlorosuccinimide (NCS), followed by removal of tBu groups and Wang resin with TFA in dichloromethane, afforded the [125I]I-labeled γ-linked ASP-glutamate conjugates 15a–15c. The radiochemical purity of each compound was > 97%, with minimum specific activity >42.5 GBq/μmol.
Biology
In order to investigate the ability of the synthesized compounds to serve as substrates for PSMA, we determined the release of glutamate from ASP-polyglutamate conjugates by incubating them with PSMA-expressing LNCaP cell extracts (Figure 2). The fluorescence intensity of γ-linked ASP-pentaglutamate conjugates 11b and 11c increased in a concentration-dependent manner while compounds 13a (not shown) and 13b, the α-linked ASP-pentaglutamate conjugates, demonstrated background levels of fluorescence at the same concentrations. Incubation with NAAG provided a pattern similar to that produced by the γ-linked ASP-glutamate conjugates. These data indicate that γ-linked ASP-penta-glutamate conjugates are substrates for PSMA while the α-linked ASP-pentaglutamate conjugates are not.
Figure 2.
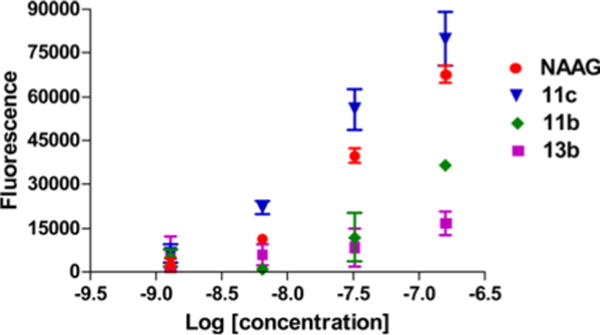
Glutamate production of 11b, 11c, 13b, and NAAG.
We attempted imaging of 125I-labeled 15a–15c in SCID mice harboring experimental models of PCa. Isogenic PSMA+ PIP and PSMA− flu tumors were grown in opposite flanks, and animals were imaged with SPECT-CT (Figure 3). Imaging showed that lipophilic conjugates 15a and 15b had extended blood pool residence times and significant accumulation in the liver. However, selective uptake within PSMA+ PIP tumors over PSMA− flu tumors was not observed. Preliminary biodistribution studies of 15a showed 3.14%ID/g for PIP and 3.02%ID/g for flu tumors at 6 days post-injection (Table I).
Figure 3.
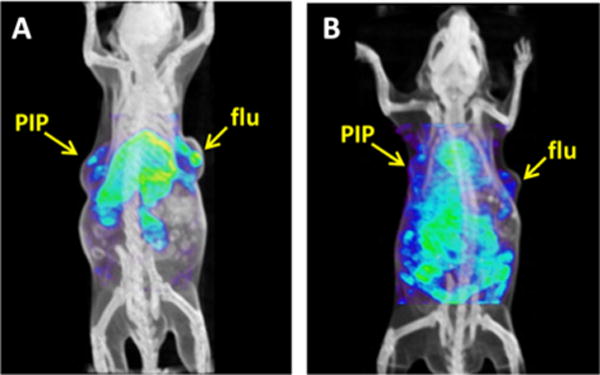
SPECT-CT images of 15a at 6 days (A) and of 15c at 24 h post-injection (B). PIP=PSMA+ PC3 tumors; flu=PSMA− PC3 tumors.
Table I.
Ex vivo Biodistribution Studies of 15a at 6 Days and 15c at 24 h Post-Injection
| Organ | 15a | 15c |
|---|---|---|
| Blood | 0.35 | 0.31 |
| Heart | 2.06 | 0.12 |
| Lung | 3.01 | 0.30 |
| Liver | 15.15 | 0.49 |
| Stomach | 1.33 | 0.07 |
| Pancreas | 0.66 | 0.06 |
| Spleen | 23.79 | 0.32 |
| Kidney | 6.02 | 0.50 |
| Muscle | 0.52 | 0.04 |
| Small Intestine | 2.13 | 0.06 |
| Large Intestine | 2.98 | 0.07 |
| Bladder | 2.35 | 0.14 |
| PIP-Tumor | 3.14 | 0.15 |
| Flu-Tumor | 3.02 | 0.12 |
To investigate further the nonspecific uptake of 15a−15c in the PSMA-expressing tumors, the cleavage of the polyglutamate moiety of the γ-linked ASP conjugates was studied. HPLC studies of 11c (50 μM) in LNCaP cell extracts (100 μL) exhibited slow cleavage of the C-terminal glutamate (24 h), indicating that the designed substrates are recognized by but only very slowly cleaved by PSMA (Figure 4). Computational docking studies of 11c with the crystal structure of PSMA were undertaken to elucidate how 11c binds to the active site of PSMA and why glutamate liberation is not as effective as for poly-γ-glutamyl folate. As shown in Figure 5, the ASP moiety is almost perpendicular to the pentaglutamate moiety, suggesting that the bulky ASP region of the T-shape 11c appears to prevent the C-terminus glutamate moiety from gaining access to the enzymatic catalytic regions (Glu 424 and zinc ions) of PSMA. Quite possibly that T-shape pattern prevents the cleaved product of 11c from binding to the active site more productively than 11c itself.
Figure 4.
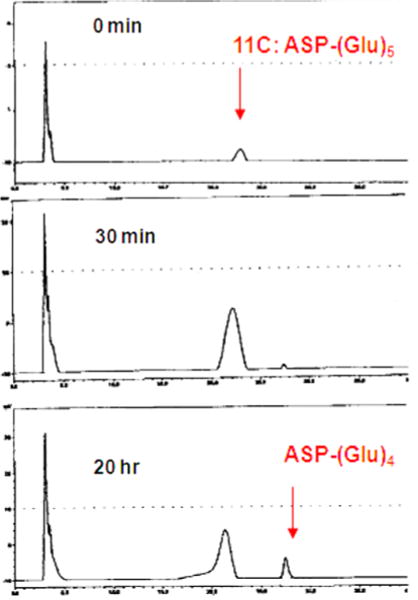
HPLC trace of 11c.
Figure 5.
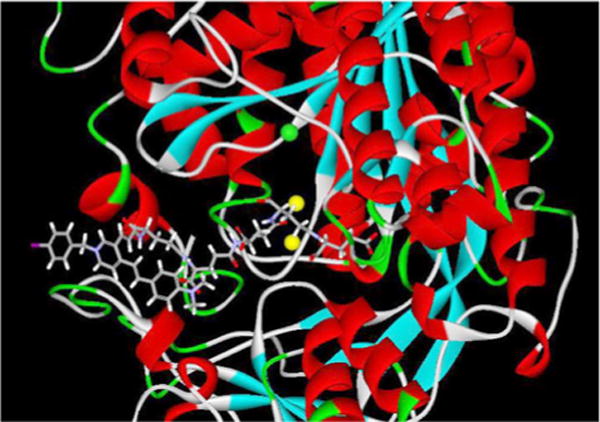
Binding mode of 11c in the active site of PSMA.
Conclusions
We designed, synthesized, and evaluated substrate-based PSMA-targeting imaging agents for in vivo animal studies. We successfully introduced 125I into the ASP-dyes using solid-phase chemistry and confirmed that γ-linked ASP conjugates are better PSMA substrates as compared to the corresponding α-linked ASP conjugates. Although in vivo SPECT-CT imaging of the designed ASP-polyglutamate conjugates (15a and 15c) did not show selective uptake in PSMA-positive over PSMA-negative tumors, the more lipophilic 15c circulated in the body for up to six days and accumulated in the tumor higher than 3%ID/g. In vitro HPLC studies and molecular modeling with 11 c exhibited the limitation of ASP-conjugates with a T-shape, indicating that a comprehensive structural modification of the current PSMA-based substrates will be needed to identify better substrates of PSMA to achieve the successful substrate-based imaging agents for this target.
Acknowledgments
The authors are grateful for financial support from National Institutes of Health Grants MH080580 and CA134675.
References
- 1.Zhou J, Neale JH, Pomper MG, Kozikowski AP. Nat Rev Drug Discov. 2005;4:1015. doi: 10.1038/nrd1903. [DOI] [PubMed] [Google Scholar]
- 2.Serda RE, Adolphi NL, Bisoffi M, Sillerud LO. Mol Imaging. 2007;6:277. [PubMed] [Google Scholar]
- 3.Banerjee SR, Foss CA, Castanares M, Mease RC, Byun Y, Fox JJ, Hilton J, Lupold SE, Kozikowski AP, Pomper MG. J Med Chem. 2008;51:4504. doi: 10.1021/jm800111u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Y, Foss CA, Byun Y, Nimmagadda S, Pullambahatla M, Fox JJ, Castanares M, Lupold SE, Babich JW, Mease RC, Pomper MG. J Med Chem. 2008;51:7933. doi: 10.1021/jm801055h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Y, Dhara S, Banerjee SR, Byun Y, Pullambhatla M, Mease RC, Pomper MG. Biochem Biophys Res Commun. 2009;390:624. doi: 10.1016/j.bbrc.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banerjee SR, Pullambhatla M, Byun Y, Nimmagadda S, Green G, Fox JJ, Horti A, Mease RC, Pomper MG. J Med Chem. 2010;53:5333. doi: 10.1021/jm100623e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banerjee SR, Pullambhatla M, Byun Y, Nimmagadda S, Foss CA, Green G, Fox JJ, Lupold SE, Mease RC, Pomper MG. Angew Chem Int Ed. 2011;50:9167. doi: 10.1002/anie.201102872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang W, Slusher B, Murakawa Y, Wozniak KM, Tsukamoto T, Jackson PF, Sima AAF. J Neurol Sci. 2002;194:21. doi: 10.1016/s0022-510x(01)00670-0. [DOI] [PubMed] [Google Scholar]
- 9.Tiffany CW, Cai NS, Rojas C, Slusher BS. Eur J Pharm. 2001;427:91. doi: 10.1016/s0014-2999(01)01236-5. [DOI] [PubMed] [Google Scholar]
- 10.Ghadge GD, Slusher BS, Bodner A, Dal Canto M, Wozniak K, Thomas AG, Rojas C, Tsukamoto T, Majer P, Miller RJ, Monti AL, Roos RP. Proc Natl Acad Sci USA. 2003;100:9554. doi: 10.1073/pnas.1530168100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guilarte TR, Hammoud DA, McGlothan JL, Caffo BS, Foss CA, Kozikowski AP, Pomper MG. Schizophr Res. 2008;99:324. doi: 10.1016/j.schres.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sacha P, Zamecnik J, Bacrinka C, Hlouchova K, Vicha A, Mlcochova P, Hilgert I, Eckschlager T, Konvalinka J. J Neurosci. 2007;144:1361. doi: 10.1016/j.neuroscience.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 13.Duthie SJ, Narayanan S, Brand GM, Pirie L, Grant G. J Nutr. 2002;132:2444s. doi: 10.1093/jn/132.8.2444S. [DOI] [PubMed] [Google Scholar]
- 14.Chang SS, Reuter VE, Heston WDW, Bander NH, Grauer LS, Gaudin PB. Cancer Res. 1999;59:3192. [PubMed] [Google Scholar]
- 15.Tsukamoto T, Wozniak KM, Slusher BS. Drug Discov Today. 2007;12:767. doi: 10.1016/j.drudis.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 16.Davis MI, Bennett MJ, Thomas LM, Bjorkman PJ. Proc Natl Acad Sci USA. 2005;102:5981. doi: 10.1073/pnas.0502101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mesters JR, Barinka C, Li WX, Tsukamoto T, Majer P, Slusher BS, Konvalinka J, Hilgenfeld R. EMBO J. 2006;25:1375. doi: 10.1038/sj.emboj.7600969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plechanovova A, Byun Y, Alquicer G, Skultetyova L, Mlcochova P, Nemcova A, Kim HJ, Navratil M, Mease R, Lubkowski J, Pomper MG, Konvalinka J, Rulisek L, Barinka C. J Med Chem. 2011;54:7535. doi: 10.1021/jm200807m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang HF, Byun Y, Barinka C, Pullambhatla M, Bhang HEC, Fox JJ, Lubkowski J, Mease RC, Pomper MG. Bioorg Med Chem Lett. 2010;20:392. doi: 10.1016/j.bmcl.2009.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barinka C, Byun Y, Dusich CL, Banerjee SR, Chen Y, Castanares M, Kozikowski AP, Mease RC, Pomper MG, Lubkowski J. J Med Chem. 2008;51:7737. doi: 10.1021/jm800765e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang AX, Murelli RP, Barinka C, Michel J, Cocleaza A, Jorgensen WL, Lubkowski J, Spiegel DA. J Am Chem Soc. 2010;132:12711. doi: 10.1021/ja104591m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu TC, Wu LY, Kazak M, Berkman CE. Prostate. 2008;68:955. doi: 10.1002/pros.20753. [DOI] [PubMed] [Google Scholar]
- 23.Liu H, Rajasekaran AK, Moy P, Xia Y, Kim S, Navarro V, Rahmati R, Bander NH. Cancer Res. 1998;58:4055. [PubMed] [Google Scholar]
- 24.Johnson M, Karanikolas BDW, Priceman SJ, Powell R, Black ME, Wu HM, Czernin J, Huang SC, Wu L. J Nucl Med. 2009;50:757. doi: 10.2967/jnumed.108.058438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu DX, Tanhehco YC, Chen JM, Foss CA, Fox JJ, Lemas V, Chong JM, Ambinder RF, Pomper MG. Clin Cancer Res. 2007;13:1453. doi: 10.1158/1078-0432.CCR-06-2295. [DOI] [PubMed] [Google Scholar]
- 26.Zhang ZX, Liu YY, Jhiang SM. J Clin Endocrinol Metab. 2005;90:6131. doi: 10.1210/jc.2005-0895. [DOI] [PubMed] [Google Scholar]
- 27.Mhaka A, Gady AM, Rosen DM, Lo KM, Gillies SD, Denmeade SR. Cancer Biol Ther. 2004;3:551. doi: 10.4161/cbt.3.6.846. [DOI] [PubMed] [Google Scholar]
- 28.Anderson MO, Wu LY, Santiago NM, Moser JM, Rowley JA, Bolstad ES, Berkman CE. Bioorg Med Chem. 2007;15:6678. doi: 10.1016/j.bmc.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambert C, Mease RC, Avren L, Le T, Sabet H, McAfee JG. Nucl Med Biol. 1996;23:417. doi: 10.1016/0969-8051(95)02101-9. [DOI] [PubMed] [Google Scholar]
- 30.Albright JW, Mease RC, Lambert C, Albright JF. Mech Ageing Dev. 1998;101:197. doi: 10.1016/s0047-6374(97)00145-0. [DOI] [PubMed] [Google Scholar]
- 31.Mease RC, Lambert C, McAfee JG. J Nucl Med. 1999;40:318. [Google Scholar]
- 32.Raimundo JM, Blanchard P, Gallego-Planas N, Mercier N, Ledoux-Rak I, Hierle R, Roncali J. J Org Chem. 2002;67:205. doi: 10.1021/jo010713f. [DOI] [PubMed] [Google Scholar]
- 33.Ozturk T, Klymchenko AS, Capan A, Oncul S, Cikrikci S, Taskiran S, Tasan B, Kaynak FB, Ozbey S, Demchenko AP. Tetrahedron. 2007;63:10290. [Google Scholar]