

Abstract
The tumor microenvironment consists of stromal cells, extracellular matrix (ECM), and signaling molecules that communicate with cancer cells. As tumors grow and develop, the tumor microenvironment changes. In addition, the tumor microenvironment is not only influenced by signals from tumor cells, but also stromal components contribute to tumor progression and metastasis by affecting cancer cell function. One of the mechanisms that cancer cells use to invade and metastasize is mediated by actin-rich, proteolytic structures called invadopodia. Here, we discuss how signals from the tumor environment, including growth factors, hypoxia, pH, metabolism, and stromal cell interactions, affect the formation and function of invadopodia to regulate cancer cell invasion and metastasis. Understanding how the tumor microenvironment affects invadopodia biology could aid in the development of effective therapeutics to target cancer cell invasion and metastasis.
Keywords: invadopodia, podosomes, tumor microenvironment, hypoxia, metastasis
Background
The tumor microenvironment
The tumor microenvironment encompasses a complex meshwork of non-malignant cells, structural components, molecules, and chemicals that surround cancer cells. The non-malignant cells, including endothelial cells, pericytes, fibroblasts, and immune cells, together with the surrounding extracellular matrix (ECM), comprise the supportive stroma of the tumor and modulate the tumor microenvironment. Both tumor cells and stromal cells secrete ECM components, such as fibronectin, collagens, proteoglycans, glycoproteins, growth factors, and matrix metalloproteinases (MMPs).1 The production of both tumor-promoting and tumor-suppressing signals from these various cell types influence the tumor microenvironment. Communication between epithelial and mesenchymal/stromal cells is critical for tumor growth and progression.2,3
Two key hallmarks of cancer, angiogenesis, and metastasis are modulated by the composition of the tumor microenvironment.3 The normal cellular microenvironment inhibits tumor cell growth, but alterations within the tumor microenvironment affect the regulation of both cancer and stromal cells.4 These changes ultimately affect tumor cell proliferation and tumor growth. The tumor microenvironment acts as a tumor and metastasis promoter, for example, by accumulating MMPs and activating growth factors that facilitate autocrine and paracrine signaling. Understanding how the tumor microenvironment affects both malignant and non-malignant cells is critical for designing effective cancer therapeutics.
The tumor vasculature exhibits abnormalities both in structure and in function, including leakiness, a heterogeneous basement membrane, irregular vessel branching, and poor pericyte coverage, which ultimately contribute to a hypoxic tumor microenvironment.5 A balance between pro-angiogenic and anti-angiogenic growth factors controls the formation of new blood vessels. When secretion of pro-angiogenic factors, such as vascular endothelial growth factor (VEGF), MMPs, transforming growth factor-β (TGF-β), and platelet-derived growth factor (PDGF), exceeds the amount of anti-angiogenic factors, endothelial cells are activated, and initiate new blood vessel formation, known as the “angiogenic switch.”6 Tumor-associated endothelial cells exhibit differences in their signaling pathways and gene expression that influence the vasculature of the tumor microenvironment.7 For example, tumor-associated endothelial cells upregulated genes associated with ECM degradation, such as MMP9, and downregulated anti-proliferative genes, such as cyclin-dependent kinase inhibitor 2A (CDKN2A), which enhanced their cellular invasion in comparison to normal endothelial cells.7
Another important cell type present in the tumor microenvironment responsible for the initiation of angiogenesis and survival of endothelial cells are pericytes. Percityes are perivascular cells that are embedded in the basement membrane and wrap around endothelial cells. Through direct contact and paracrine signaling, pericytes control the differentiation, proliferation, angiogenic capacity, and survival of endothelial cells.3 The recruitment and association of pericytes affects the development of the tumor endothelium; tumor vasculature lacking in pericytes is more prone to cancer cell intravasation.5 Similarly, in breast cancer, a decreased number of pericytes correlates with decreased survival and is associated with increased hypoxia and metastasis, which are factors that contribute to drug resistance.8 Thus, these differences in the tumor microenvironment affect the function of pericytes and their potential responsiveness to therapies.
One of the main constituents of the tumor stroma is fibroblasts.3 Fibroblasts facilitate ECM remodeling. Cancer-associated fibroblasts (CAFs) comprise two distinct groups: (1) fibroblasts that contribute to the structural components of the microenvironment and (2) myofibroblasts that accumulate at sites of chronic inflammation.3 CAFs secrete growth factors and cytokines that promote cancer cell proliferation, angiogenesis, invasion, and metastasis.3 Studies have suggested that epigenetic and genetic changes, which occur in stromal fibroblasts, can alter the composition of the tumor microenvironment and influence tumor progression by promoting epithelial cell proliferation.9 For example, loss of the phosphatase and tensin homolog (PTEN) tumor suppressor in stromal fibroblasts facilitates breast cancer metastasis by increasing collagen deposition and deregulating genes involved in inflammation, angiogenesis, and ECM remodeling.10,11
Lacking normal physiological controls, the tumor microenvironment exhibits similar inflammatory characteristics to a wound that will not heal.12 Whereas in a normal wound inflammatory cells transiently appear and then disappear, they persist in chronic conditions, such as cancer, leading to molecular and cellular changes within the tumor microenvironment. Inflammatory cells found within the tumor exhibit both tumor-promoting and tumor-suppressing roles. Tumor-promoting inflammatory cells, such as tumor-associated macrophages (TAMs), neutrophils, and T and B lymphocytes, release growth factors to amplify the inflammatory state, and/or produce pro-angiogenic/invasive matrix-degrading enzymes such as MMP-9 and cysteine cathepsin proteases.1,3,13 Angiogenesis, cancer cell proliferation, invasion, and metastasis are all enhanced by the presence of tumor-promoting inflammatory cells, which override the ability of tumor-killing immune cells to disrupt the metastatic process. These tumor-killing immune cells are a prime focus for immunotherapies in cancer.14
Through both autocrine and paracrine signaling mechanisms, the diverse cell types found in the tumor microenvironment affect the ability of tumor cells to proliferate, migrate, invade, and metastasize. Conversely, tumor cells alter the structure and composition of the tumor microenvironment. These reciprocal interactions can transform normal tissues to high-grade disease.
Invadopodia and podosomes
Invasive migration is a key feature of cancer cells that disseminate during the process of metastasis. In order for cancer cells to metastasize successfully, they must cross barriers imposed by blood vessels, tissues, and the ECM. Cancer cells can migrate using amoeboid movement through gaps in the ECM, such as movement within the tumor. Alternatively, a mesenchymal-type movement that relies on proteolytic degradation of the ECM is most likely required for passage through dense ECM, such as basement membrane.15-19 One mechanism by which cancer cells proteolytically invade through the ECM is using distinct structures called invadopodia.
Invadopodia, and their corresponding counterparts found in normal cells called podosomes, are actin-rich protrusions of the plasma membrane that coordinate adhesive proteins and proteolytic enzymes to facilitate cell adhesion and focal proteolysis.15,20,21 Because both structures control ECM attachment and degradation, podosomes and invadopodia have been grouped under the term “invadosomes.”15,22 However, for this review, we will refer to “podosomes” in normal cells and “invadopodia” in cancer cells in order to distinguish the two structures. Here, we will briefly describe important characteristics of these structures, but many reviews are available that extensively describe their formation, regulation, and function both in normal and in cancer cells.15,20,23,24
Podosomes and invadopodia have been identified in a variety of cell types. Podosomes are found in cells of the monocytic lineage such as macrophages, monocytes, dendritic cells, synovial cells, and osteoclasts.25-28 In these cells, podosomes, which often group together to form clusters, rings, and belts, control matrix remodeling. Defects in podosome function in these cell types have pathophysiological outcomes in diseases, such as Wiskott-Aldrich Syndrome and Frank-Ter Haar syndrome.29 Podosomes can also form in vascular smooth muscle cells (VSMCs) and endothelial cells. Podosome formation is triggered by signals from the extracellular environment. For example, stimulation with phorbol esters induces podosome formation in VSMCs.30 Phorbol esters and TGF-β induce podosome super-structures, termed rosettes, in endothelial cells to control vascular remodeling.31-34
Podosomes and invadopodia are dynamic structures with a half-life of actin turnover ranging from minutes to hours, which are controlled by a variety of proteins, including kinases, GTPases, and scaffolds that are involved in processes such as adhesion, actin polymerization, proteolysis, and signaling.20,24,35 Key constituents of these structures include the tyrosine kinase substrate four/five SH3 domains (Tks4/5) adaptor proteins, Src, the actin regulators cofilin and cortactin, and neural Wiskott-Aldrich syndrome protein (N-WASP), and the protease transmembrane type I MMP (MT1-MMP), which are critical for the initiation, assembly, and maturation of functional invadopodia.25,36-43 Several reviews have compiled known invadopodia/podosome proteins and what their respective functions are in the regulation of these structures.20,35,44,45 High-throughput screening strategies, using either siRNA/shRNA or chemical libraries, have identified other novel invadopodia regulators.46,47 Although podosomes and invadopodia share strong similarities in their structure, subtle differences in their actin dynamics, signaling scaffolds, and signaling inputs have been reported. This result likely reflects the fact that acquisition of pro-invasive capabilities by cancer cells is an imperfect mimicry of a normal cellular function.
Both podosomes and invadopodia localize or control the activity of proteolytic enzymes required for ECM degradation and invasive migration. Invadopodia readily form and degrade ECM in a variety of cancer cells, including breast, melanoma, and head and neck.37,48,49 Podosomes in endothelial cells, VSMCs, and dendritic cells also have this capability, but they are more dependent on input signals and ECM substrate.20 The final maturation process of invadopodia requires the recruitment and activation of proteases to initiate ECM degradation. The types of proteases that are present at invadopodia include MMPs, ADAM (a disintegrin and metalloproteinase) sheddases, cysteine cathepsin proteases, and serine proteases.20,35
Invadopodia utilize protease-driven, ECM remodeling to promote cellular invasion. The ability of cancer cells to form invadopodia in vitro correlates with their invasive capacity as typically assayed by ECM degradation and cellular invasion assays.15,20,37,39,50 In addition, invadopodia have been observed in 3D ECM and ex vivo culture systems that more closely mimic physiological conditions.19 For example, the use of intravital imaging demonstrated the presence of invadopodia-like protrusions in metastatic breast cancer cells during intravasation.51 In tumor sections of mammary xenografts, the key invadopodia marker Tks5 was detected in invadopodia.52 N-WASP-deficient tumors also lacked the ability to form invasive, degradative protrusions.53 Although no concrete evidence has yet demonstrated the presence of invadopodia in vivo, several key invadopodia proteins, such as Tks5, cortactin, and N-WASP, are necessary for tumor growth and metastasis in mouse cancer models.52-56
Here, we assess the role of the tumor microenvironment in the regulation of invadopodia formation and function and how this relates to cancer progression and metastasis. We discuss recent data demonstrating how inputs from the tumor microenvironment, such as stromal-cell interactions, growth factors, hypoxia, pH, and metabolism, alter invadopodia biology and the implications for the surrounding components subject to these signaling changes.
Signals from the Tumor Microenvironment
Tumor–stromal cell interactions
Cancer cells influence, and are influenced by, their surrounding tuomr microenvironment, resulting in the release of growth factors and the creation of a hypoxic, acidic milieu, which promotes invadopodia formation, invasion, and metastasis. Cancer cells can communicate to stromal cells via paracrine signaling mechanisms (i.e., growth factor secretion) to influence the invasive phenotype (Fig. 1). In pancreatic cancer, tumor cells induce expression of palladin, a cytoskeletal protein, in neighboring fibroblasts via paracrine signaling, which transforms them into CAFs or myofibroblasts.57,58 These myofibroblasts then facilitate invasion by elaborating invadopodia, which generates migratory tracks through the ECM.57 In addition, palladin activates the small GTPase Cdc42, a known regulator of invadopodia activity, in CAFs, which links invadopodia to CAF-induced ECM remodeling in pancreatic cancer.59-61

Figure 1. Signals from the tumor microenvironment that affects invadopodia formation and function. The signals from the tumor microenvironment that regulate invadopodia formation and function include: (1) growth factors, (2) hypoxia, (3) pH, (4) metabolism, and (5) stromal–cell interactions. Growth factors (GF) signal through receptors on the cell surface to initiate downstream signaling pathways mediated by Src, PI3K, and GTPases. Hypoxia initiates signaling through the HIF-1α transcription factor in the nucleus. NHE1 controls intracellular and extracellular pH. These hypoxic and acidic conditions lead to an altered cellular metabolism. Finally, interactions with surrounding stromal cells through paracrine signaling mechanisms affect tumor cell signaling. All of these factors promote invadopodia formation and increased proteolytic activity.
Interestingly, physical contact between cells can also initiate the invasive response. A recent studied showed that hypoxia activates the cell contact-dependent Notch signaling pathway to induce invadopodia formation in cancer cells.62 But can stromal cells influence invadopodia formation in tumor cells? Studies have shown that macrophages induce invadopodia formation in cancer cells through direct cell contact, and facilitate in vitro intravasation via increased RhoA signaling both in breast cancer cell lines and patient-derived tumor cells.63 This study suggests that invadopodia are required for tumor cells to penetrate through the basement membrane of blood vessels to initiate intravasation, mediated by heterotypic RhoA signaling between macrophages and tumor cells.63
Growth factors
Another means by which the tumor microenvironment communicates with tumor cells is through growth factor signaling, which modulates cellular growth and differentiation (Fig. 1). Both epithelial and mesenchymal cells release growth factors into the microenvironment to serve as a means of communication. Growth factors such as TGF-β and EGF are synthesized in inactive proforms that are processed by MMPs and other proteases into active, soluble ligands, suggesting a possible role for invadopodia in growth-factor processing.64 On the other hand, the formation of both podosomes and invadopodia are regulated by growth factors, such as colony stimulating factor-1 (CSF-1), TGF-β, VEGF, PDGF, epidermal growth factor (EGF), heparin-binding EGF (HB-EGF), hepatocyte growth factor/scatter factor (HGF), and stromal cell-derived factor 1α (SDF1α).23 Many of these growth factors act through similar signaling pathways involving Src, phosphoinositide 3-kinase (PI3K), and Rho family GTPases, which control the initiation, assembly, and maturation of invadopodia.20,65
Upon binding of EGF or related ligands, the epidermal growth factor receptor (EGFR) signaling pathway mediates a wide variety of cellular responses including proliferation, migration, and invasion, and is typically hyperactive in a wide variety of human cancers.66 EGF is a well-characterized inducer of invadopodia formation and an important regulator of the tumor microenvironment.41,67 In normal breast epithelial cells, upregulation of a cell-surface glycoprotein, CD147, increases EGFR activation leading to invadopodia formation and upregulation of MT1–MMP expression.68 This effect is mediated by increased hyaluronan-CD44 signaling and interactions between CD147, CD44, and EGFR, which facilitates invadopodia formation and cancer cell migration.68,69 Here, localization of growth factor receptors, which generate the necessary invadopodia-forming signals, with cell-surface proteins such as CD147 is critical for invadopodia formation.
Invadopodia can also affect the production of these growth factor signals by localizing the enzymes required for growth factor processing, thus generating “growth factor pools,” to affect cancer cell invasiveness. For example, in an experimental overexpression system, ADAM12, a protease which processes growth factor ligands and localizes to invadopodia, induces clusters of invadopodia where ectodomain shedding of EGFR ligands occurs.70,71 This result suggests that invadopodia may provide spatio-temporal control of growth factors to control the invasive and metastatic potential of cancer cells in addition to their role of proteolyzing ECM.
Growth factors can either be secreted by the same cells that express their receptor (autocrine) or from neighboring cells (paracrine) (Fig. 2). Recent studies have demonstrated how invadopodia can be regulated by growth factor signaling using these two different mechanisms. For example, secretion of HB–EGF drives invadopodia formation and matrix degradation in both an autocrine and paracrine manner.62,72 In head and neck squamous cell carcinoma (HNSCC) cells, inhibition of Abl or Arg tyrosine kinases stimulates HB–EGF shedding. Elevated HB–EGF then leads to hyperactivation of EGFR signaling and Src activity, which results in cortactin phosphorylation and upregulation of invadopodia in an autocrine manner.72 In contrast, under conditions of low oxygen levels, which are typically present within tumors (hypoxia), Notch-induced cell contact-dependent signaling in HNSCC and lung cancer cells acts in a paracrine manner to increase ADAM12 expression and HB–EGF shedding to promote invadopodia formation.62 This study also suggests that paracrine secretion of HB–EGF may induce invadopodia in nearby normoxic (normal oxygen) cells to enhance the invasive phenotype. These studies reveal how autocrine and paracrine cell–cell communication in the tumor microenvironment not only potentiate the invasive phenotype in other cancer cells but also in the surrounding stromal cells.
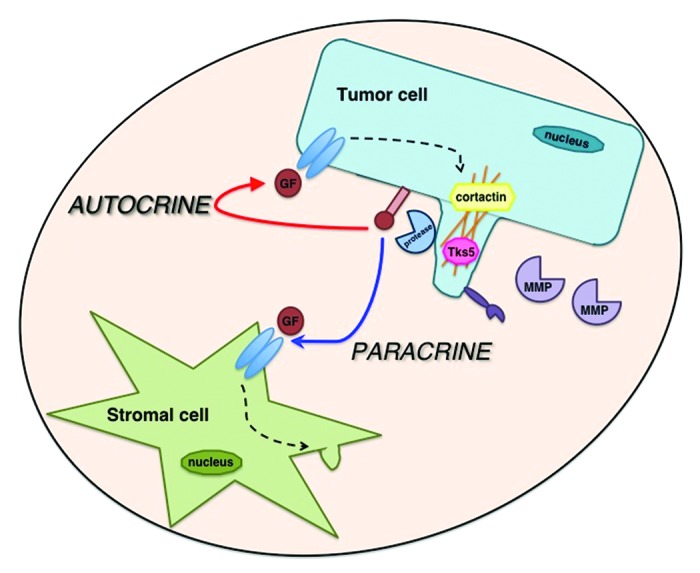
Figure 2. Tumor cells and stromal cells communicate with each other through both autocrine and paracrine signaling mechanisms. Invadopodia localize proteolytic machinery that is necessary for ECM degradation and growth factor processing. Growth factors secreted by the tumor cell can either activate receptors on the tumor cell itself (autocrine signaling) or receptors on nearby stromal cells (paracrine signaling) to affect invadopodia formation and function.
Disruption in growth factor-mediated, cell–cell communication in the tumor microenvironment also alters cancer cell signaling. In breast cancer, a paracrine loop exists between cancer cells and macrophages, in which tumor cells secrete CSF-1 to stimulate TAMs, and TAMs release EGF to activate tumor cells.73,74 This paracrine mechanism of cell–cell communication between TAMs and cancer cells promotes podosome and invadopodia formation in macrophages and tumor cells, respectively, as well as ECM degradation, and invasion.73,75 However, increased autocrine expression of HB–EGF in cancer cells disrupts this paracrine loop with macrophages and promotes invasion and metastasis through Src-dependent invadopodia formation and matrix degradation in tumor cells independent of TAMs.73 Persistent, autocrine, growth factor activation enhances the invasive phenotype mediated by invadopodia in a cell-autonomous manner, revealing how amplification of signaling pathways in the tumor microenvironment enhances malignancies.
Hypoxia
Tumors are often subject to lower oxygen levels than the surrounding normal tissue. Hypoxia in the tumor microenvironment promotes angiogenesis via upregulation of several angiogenic growth factors such as PDGF, VEGF, and HGF and can also promote invasion via upregulation of MMPs.76,77 The primary regulator of the hypoxic response is hypoxia inducible factor 1α (HIF-1α).77,78 HIF-1α is regulated post-translationally based on the concentration of oxygen in the environment. Under normoxic conditions, HIF-1α is ubiquitinated and degraded, terminating downstream gene expression. However, during hypoxia, HIF-1α is stabilized and translocates to the nucleus where it initiates the transcription of genes involved in tumor growth, angiogenesis, and metastasis.79 HIF-1α levels are increased in a variety of human cancers, correlating with a decreased patient survival.79 Both hypoxia-induced genetic and epigenetic changes in the tumor microenvironment facilitate metastatic progression.80
Does hypoxia regulate invadopodia? Hypoxia potentiates invadopodia formation in cancer cells in a HIF-1-dependent manner (Fig. 1).62,81,82 In the study using HNSCC, lung, and pancreatic cancer cells mentioned earlier, hypoxia-induced invadopodia formation and ECM degradation were shown to be dependent upon HIF-1α.62 In another study, both HIF-1α and HIF-2α increase melanoma invasion by promoting invadopodia formation through activation of PDGF receptor-α (PDGFRα) and Src, respectively, and coordinating ECM degradation through MT1–MMP and MMP-2 expression.82 Finally, in breast cancer cells, invadopodia formation and function downstream of HIF-1α is dependent upon expression of the Rho family activator, p-21 activated protein kinase-interacting exchange factor (β-PIX).81 HIF-1α also regulates invadopodia through epithelial–mesenchymal transition (EMT)-promoting transcription factors, such as Twist. Twist induces invadopodia formation through PDGFRα-mediated activation of Src and is directly regulated by HIF-1α to promote metastasis.52,83 Hypoxia also upregulates caveolin-1, a major structural component of lipid rafts that is required for invadopodia formation and MT1–MMP recruitment, which promotes ligand-independent activation of EGFR signaling.84,85 The tightly regulated localization of invadopodia components and growth factor receptors by the hypoxic tumor microenvironment allows for coordinated control of cellular invasion.
Recently, a role for epigenetic regulators, such as histone deactylases (HDACs), has been implicated in invadopodia control under hypoxia. Deacetylation of cortactin by HDAC6 is required for invadopodia formation.86 Similarly, in breast cancer cells, HDAC6 is required for ECM degradation and MT–MMP-1-dependent invasion.87,88 Hypoxia activates TGF-β and EGF signaling pathways to induce responsive genes and promote HDAC6 activity, respectively, to enhance invadopodia production.89 Thus, the hypoxic tumor microenvironment affects invadopodia formation and function by controlling known regulatory pathways of invadopodia.
pH
The hypoxic tumor microenvironment not only facilitates tumor invasion and metastasis but also affects cellular metabolism by increasing the rate of glycolysis. HIF1-α mediates this effect in part by transcriptionally upregulating glycolytic enzymes.90 This altered metabolism in cancer cells increases lactate production, generating an excess of protons and acidifying the surrounding tumor microenvironment.91 pH homeostasis is maintained by sodium–proton exchangers (NHEs), sodium-dependent and -independent HCO3-/Cl- exchanges, H+/lactate co-transporters, and V-ATPases. The expression and activity of these transporters is frequently upregulated in cancer cells.92 Normal cells typically have an intracellular pH ranging from 6.9–7.2 and extracellular pH of 7.2–74. However, this gradient is reversed in cancer cells, which have an intracellular pH ranging from 7.2–7.7 and an extracellular pH ranging from 6.2–6.8. The low pH of the tumor microenvironment increases VEGF transcription to facilitate angiogenesis and promotes the release/activity of proteases, such as cathepsins and MMPs, to degrade the ECM, thus amplifying invasion and metastasis.93
Interestingly, early studies in avian osteoclasts demonstrated a role for pH-mediated regulation of podosomes.94 The bone resorption process mediated by osteoclast podosomes is tightly controlled by pH regulators and intracellular pH. Intracellular acidification by butyric acid increases podosome formation, whereas alkalinization reduces podosome formation.94 NHE-1 can function in the control of cytosolic pH, but a role in osteoclast podosome function remains to be investigated.92 In contrast, in cancer cells, increased NHE-1 activity is known to facilitate tumor growth and metastasis.95,96
NHE-1 is a key transporter involved in the pH-dependent activation of cathepsin B, MMP-9, MMP-2, and MT1–MMP, and is upregulated and overexpressed in a wide variety of cancers, including cervical, heptocellular carcinoma, glioma, and breast.92,97 NHE-1 activity drives invadopodia formation. In hypoxic cancer cells, NHE-1 localizes to invadopodia, and its expression and activation by p90 ribosomal S6 kinase (p90RSK) is required for invadopodia formation.98,99 In breast cancer cells, NHE-1 promotes invadopodia formation by acidifying the extracellular invadopodia environment and alkalinizing the invadopodia cytosol to release cofilin from cortactin (Fig. 1).100,101 Dissociation of cofilin from cortactin increases the amount of free barbed ends available for actin polymerization, which is necessary for invadopodia formation.102 Cofilin can act as a pH biosensor, and pH-mediated changes in its activity affect actin cytoskeletal dynamics.103 The phosphorylation state of cortactin regulates the dynamics of invadopodia formation and maturation by recruiting Nck1 (non-catalytic region of tyrosine kinase adaptor protein 1), N-WASP, and cofilin, but it also recruits the pH regulator NHE-1 to invadopodia.100 Cortactin’s recruitment of NHE-1 to invadopodia is akin to its recruitment of additional proteolytic machinery (the MMPs), suggesting that cortactin functions as a master coordinator of invasive components to invadopodia.104 These studies have demonstrated a clear role for NHE-1 activity in the dynamics of invadopodia formation.
NHE-1 also modulates invadopodia function by controlling pericellular proteolytic activity. In HT1080 fibrosarcoma cells, exposure to acidic pH increases ECM degradation, which is augmented by hypoxia.98 Alteration of the tumor microenvironmental pH affects the expression, secretion, and activation of proteases, including MT1-MMP, MMP-9, and MMP-2, which are essential for the ECM-degrading activity of invadopodia. Acidic extracellular pH increases MMP-9 expression, and pharmacological inhibition of NHE-1 downregulates MT1–MMP, a key activator of MMP-2.105,106 Intracellular trafficking via lyosomes and vesicles targets these proteases to invadopodia, a process that is dependent upon tumor microenvironmental pH.107,108 In prostate cancer cells, NHE-1 activity promotes lysosomal fusion to invadopodia; these lysosomes then release cathepsin B to facilitate ECM proteolysis.109 This lysosomal-trafficking mechanism is not restricted to invadopodia; similar effects have been observed in osteoclast and phagocytic podosomes, which could also be modulated by extracellular pH.110,111 Thus, the pH of the tumor microenvironment regulates the proteolytic activity of invadopodia/podosomes through direct (protease expression and trafficking) and indirect (protease activation) mechanisms to affect cancer cell migration and invasion.
Metabolism
Cancer cells have an altered metabolism that provides them with a growth advantage in the low oxygen, low glucose, and high acidic conditions found in the tumor microenvironment. Under standard growth conditions, a normal cell converts glucose to pyruvate (glycolysis) in the cytosol, which is subsequently metabolized to carbon dioxide (oxidative phosphorylation) in the mitochondria to produce ATP. However, in hypoxic conditions, little pyruvate is sent to the mitochondria. As opposed to utilizing oxidative phosphorylation, cancer cells shift their metabolism toward aerobic glycolysis, a phenomenon known as the “Warburg effect” (Fig. 1).3 Constitutive glycolysis results in acidification of the tumor microenvironment, and as discussed earlier, increases the invasive ability of cancer cells. Thus, tumor metabolism drives the cancer phenotype and could potentially affect invadopodia formation and function.
A proteomic analysis of an enriched invadopodia fraction from human melanoma cells revealed additional links between metabolism and invadopodia.44 Three glycolytic enzymes, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), pyruvate kinase (PK-M2), and lactate dehydrogenase A (LDH-A) localized to invadopodia.44 To determine the roles of oxidative phosphorylation and glycolysis in invadopodia regulation, each pathway was inhibited in melanoma, breast, and glioma cancer cell lines.112 To differentiate between the glycolytic and oxidative pathway, cells were grown in either glucose or galactose media, respectively, because galactose is more efficiently used in oxidative phosphorylation than glucose.112,113 Inhibition of oxidative phosphorylation with oligomycin did not affect invadopodia formation, whereas reduction in glycolysis by growth in galactose media varied in a cell type-dependent manner, indicating different metabolic requirements for different cancer cell types.112 Interestingly, when pyruvate was omitted from galactose media, all three cancer cell types exhibited reduced ECM degradation, suggesting the importance of pyruvate in promoting invadopodia function.112 It is perhaps not surprising that metabolic enzymes would be associated with invadopodia, whose actin dynamics rely on the readily available supply of ATP. Similarly, metabolic enzymes are upregulated in stimulated macrophages, which typically form large numbers of podosomes.114
Another important pathway in cancer cell metabolism is de novo fatty acid synthesis, in which fatty acids are generated from acetyl-CoA and malonyl-CoA by fatty acid synthase (FASN).115 These fatty acids are primarily used for phospholipids in cellular membranes and lipid raft components.115,116 Interestingly, lipid rafts and caveolin-1 are required for invadopodia formation and ECM degradation in breast cancer cells, linking this metabolic pathway to invadopodia function.84,117 Invadopodia themselves are membrane protrusions, and key components of these structures, Tks4 and Tks5, have lipid-binding phox (PX) domains at their N-termini, underscoring the necessity of lipid production in the formation of invadopodia.37,38,118,119 Inhibition of the rate-limiting step in fatty acid synthesis, catalyzed by acetyl-CoA carboxylase 1 (ACC1), decreases invadopodia formation and function, and addition of exogenous fatty acids restores invadopodia function in cancer cells lacking ACC1 activity.120 Similarly, inhibition of AMP-activated kinase (AMPK), an upstream activator of ACC1, reduces fatty acid synthesis and coincidentally, invadopodia formation.120 These data demonstrate that modulation of fatty acid synthesis affects invadopodia formation and function in cancer cells.
By affecting cancer cell metabolism, the tumor microenvironment regulates cancer invasion. Hypoxia and low pH alter metabolism, and subsequently, affect invadopodia formation and function. Future work is needed to delineate how metabolic changes within the tumor, generated by the tumor microenvironment, control the dynamics of invadopodia to subsequently affect invasion and metastasis.
Conclusions and Perspectives
The tumor microenvironment is not a passive bystander but an active player during tumor progression. Hypoxic, acidic conditions and stromal cell-mediated signaling promote the formation and activity of invadopodia in cancer cells, leading to increased invasion and metastasis. In turn, invadopodia-mediated activity in cancer cells enhances growth factor production and paracrine signaling mechanisms to influence both cancer cells and stromal cells to perpetuate the metastatic process. This reciprocal heterotypic signaling within the tumor microenvironment, mediated by invadopodia, is constantly evolving as the tumor environment changes and the tumor progresses. Therefore, a complete understanding of the mechanisms that mediate malignant progression is necessary for developing therapies that will target both primary and metastatic tumors.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank all of the members of the Courtneidge laboratory for useful discussions and Dr Begoña Diaz and Christopher Abdullah for comments on the manuscript. Research was supported by the National Cancer Institute to Courtneidge SA and the American Cancer Society to Gould CM.
Glossary
Abbreviations:
- ACC1
acetyl-CoA carboxylase 1
- ADAM
a disintegrin and metalloproteinase
- AMPK
AMP-activated kinase
- CAF
cancer-associated fibroblast
- CDKN2A
cyclin-dependent kinase inhibitor 2A
- CSF-1
colony stimulating factor-1
- ECM
extracellular matrix
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- EMT
epithelial-mesenchymal transition
- FASN
fatty acid synthase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GLUT-1
glucose transporter-1
- HB-EGF
heparin-binding epidermal growth factor
- HDAC
histone deacetylase
- HGF
hepatocyte growth factor/scatter factor
- HIF-1α
hypoxia-inducible factor-1α
- HNSCC
head and neck squamous cell carcinoma
- LDH-A
lactate dehydrogenase A
- MMP
matrix metalloproteinase
- MT1-MMP
transmembrane type I-matrix metalloproteinase
- Nck1
non-catalytic region of tyrosine kinase adaptor protein 1
- NHE
sodium-proton exchanger
- N-WASP
neural Wiskott-Aldrich syndrome protein
- PDGF
platelet-derived growth factor
- PDGFRα
platelet-derived growth factor receptor α
- PI3K
phosphoinositide 3-kinase
- β-PIX
p21 activated protein kinase-interacting exchange factor
- PK
pyruvate kinase
- PTEN
phosphatase and tensin homolog
- PX
phox
- p90RSK
p90 ribosomal S6 kinase
- SDF1α
stromal cell-derived factor 1α
- TAM
tumor-associated macrophage
- TGF-β
transforming growth factor-β
- Tks4/5
tyrosine kinase substrate with four/five SH3 domains
- VSMC
vascular smooth muscle cell
- VEGF
vascular endothelial growth factor
References
- 1.Weber CE, Kuo PC. The tumor microenvironment. Surg Oncol. 2012;21:172–7. doi: 10.1016/j.suronc.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Hu M, Polyak K. Microenvironmental regulation of cancer development. Curr Opin Genet Dev. 2008;18:27–34. doi: 10.1016/j.gde.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Flaberg E, Markasz L, Petranyi G, Stuber G, Dicso F, Alchihabi N, Oláh È, Csízy I, Józsa T, Andrén O, et al. High-throughput live-cell imaging reveals differential inhibition of tumor cell proliferation by human fibroblasts. Int J Cancer. 2011;128:2793–802. doi: 10.1002/ijc.25612. [DOI] [PubMed] [Google Scholar]
- 5.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–27. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 6.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–10. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 7.Du XL, Jiang T, Zhao WB, Wang F, Wang GL, Cui M, Wen ZQ. Gene alterations in tumor-associated endothelial cells from endometrial cancer. Int J Mol Med. 2008;22:619–32. [PubMed] [Google Scholar]
- 8.Cooke VG, LeBleu VS, Keskin D, Khan Z, O’Connell JT, Teng Y, Duncan MB, Xie L, Maeda G, Vong S, et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell. 2012;21:66–81. doi: 10.1016/j.ccr.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30–8. doi: 10.1016/j.tig.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 10.Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, Thompson JC, Caserta E, Wang H, Chong JL, et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461:1084–91. doi: 10.1038/nature08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallace JA, Li F, Leone G, Ostrowski MC. Pten in the breast tumor microenvironment: modeling tumor-stroma coevolution. Cancer Res. 2011;71:1203–7. doi: 10.1158/0008-5472.CAN-10-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–9. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 13.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao S, Zhu Y, Chen L. Advances in targeting cell surface signalling molecules for immune modulation. Nat Rev Drug Discov. 2013;12:130–46. doi: 10.1038/nrd3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linder S, Wiesner C, Himmel M. Degrading devices: invadosomes in proteolytic cell invasion. Annu Rev Cell Dev Biol. 2011;27:185–211. doi: 10.1146/annurev-cellbio-092910-154216. [DOI] [PubMed] [Google Scholar]
- 16.Sabeh F, Shimizu-Hirota R, Weiss SJ. Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J Cell Biol. 2009;185:11–9. doi: 10.1083/jcb.200807195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sahai E. Mechanisms of cancer cell invasion. Curr Opin Genet Dev. 2005;15:87–96. doi: 10.1016/j.gde.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer. 2003;3:921–30. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- 19.Yamaguchi H. Pathological roles of invadopodia in cancer invasion and metastasis. Eur J Cell Biol. 2012;91:902–7. doi: 10.1016/j.ejcb.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 20.Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12:413–26. doi: 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caldieri G, Ayala I, Attanasio F, Buccione R. Cell and molecular biology of invadopodia. Int Rev Cell Mol Biol. 2009;275:1–34. doi: 10.1016/S1937-6448(09)75001-4. [DOI] [PubMed] [Google Scholar]
- 22.Destaing O, Block MR, Planus E, Albiges-Rizo C. Invadosome regulation by adhesion signaling. Curr Opin Cell Biol. 2011;23:597–606. doi: 10.1016/j.ceb.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 23.Hoshino D, Branch KM, Weaver AM. Signaling inputs to invadopodia and podosomes. J Cell Sci. 2013;126:2979–89. doi: 10.1242/jcs.079475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mueller S, Artym VV, Kelly T. Invadopodia: Interface for Invasion. In: Dylan Edwards GH-H, Francesco Blasi, and Boonie F. Sloane, ed. The Cancer Degradome: Springer New York, 2008:403-31. [Google Scholar]
- 25.Linder S, Nelson D, Weiss M, Aepfelbacher M. Wiskott-Aldrich syndrome protein regulates podosomes in primary human macrophages. Proc Natl Acad Sci U S A. 1999;96:9648–53. doi: 10.1073/pnas.96.17.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burns S, Thrasher AJ, Blundell MP, Machesky L, Jones GE. Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation. Blood. 2001;98:1142–9. doi: 10.1182/blood.V98.4.1142. [DOI] [PubMed] [Google Scholar]
- 27.Destaing O, Saltel F, Géminard JC, Jurdic P, Bard F. Podosomes display actin turnover and dynamic self-organization in osteoclasts expressing actin-green fluorescent protein. Mol Biol Cell. 2003;14:407–16. doi: 10.1091/mbc.E02-07-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lauzier A, Charbonneau M, Harper K, Jilaveanu-Pelmus M, Dubois CM. Formation of invadopodia-like structures by synovial cells promotes cartilage breakdown in collagen-induced arthritis: involvement of the protein tyrosine kinase Src. Arthritis Rheum. 2011;63:1591–602. doi: 10.1002/art.30305. [DOI] [PubMed] [Google Scholar]
- 29.Cejudo-Martin P, Courtneidge SA. Podosomal proteins as causes of human syndromes: a role in craniofacial development? Genesis. 2011;49:209–21. doi: 10.1002/dvg.20732. [DOI] [PubMed] [Google Scholar]
- 30.Burgstaller G, Gimona M. Podosome-mediated matrix resorption and cell motility in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2005;288:H3001–5. doi: 10.1152/ajpheart.01002.2004. [DOI] [PubMed] [Google Scholar]
- 31.Moreau V, Tatin F, Varon C, Génot E. Actin can reorganize into podosomes in aortic endothelial cells, a process controlled by Cdc42 and RhoA. Mol Cell Biol. 2003;23:6809–22. doi: 10.1128/MCB.23.19.6809-6822.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tatin F, Varon C, Génot E, Moreau V. A signalling cascade involving PKC, Src and Cdc42 regulates podosome assembly in cultured endothelial cells in response to phorbol ester. J Cell Sci. 2006;119:769–81. doi: 10.1242/jcs.02787. [DOI] [PubMed] [Google Scholar]
- 33.Varon C, Tatin F, Moreau V, Van Obberghen-Schilling E, Fernandez-Sauze S, Reuzeau E, Kramer I, Génot E. Transforming growth factor beta induces rosettes of podosomes in primary aortic endothelial cells. Mol Cell Biol. 2006;26:3582–94. doi: 10.1128/MCB.26.9.3582-3594.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gimona M, Buccione R, Courtneidge SA, Linder S. Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol. 2008;20:235–41. doi: 10.1016/j.ceb.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 35.Klemke RL. Trespassing cancer cells: ‘fingerprinting’ invasive protrusions reveals metastatic culprits. Curr Opin Cell Biol. 2012;24:662–9. doi: 10.1016/j.ceb.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma VP, Eddy R, Entenberg D, Kai M, Gertler FB, Condeelis J. Tks5 and SHIP2 regulate invadopodium maturation, but not initiation, in breast carcinoma cells. Curr Biol. 2013;23:2079–89. doi: 10.1016/j.cub.2013.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seals DF, Azucena EF, Jr., Pass I, Tesfay L, Gordon R, Woodrow M, Resau JH, Courtneidge SA. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell. 2005;7:155–65. doi: 10.1016/j.ccr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 38.Buschman MD, Bromann PA, Cejudo-Martin P, Wen F, Pass I, Courtneidge SA. The novel adaptor protein Tks4 (SH3PXD2B) is required for functional podosome formation. Mol Biol Cell. 2009;20:1302–11. doi: 10.1091/mbc.E08-09-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowden ET, Barth M, Thomas D, Glazer RI, Mueller SC. An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene. 1999;18:4440–9. doi: 10.1038/sj.onc.1202827. [DOI] [PubMed] [Google Scholar]
- 40.Chen WT, Wang JY. Specialized surface protrusions of invasive cells, invadopodia and lamellipodia, have differential MT1-MMP, MMP-2, and TIMP-2 localization. Ann N Y Acad Sci. 1999;878:361–71. doi: 10.1111/j.1749-6632.1999.tb07695.x. [DOI] [PubMed] [Google Scholar]
- 41.Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, Segall J, Eddy R, Miki H, Takenawa T, et al. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–52. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bravo-Cordero JJ, Oser M, Chen X, Eddy R, Hodgson L, Condeelis J. A novel spatiotemporal RhoC activation pathway locally regulates cofilin activity at invadopodia. Curr Biol. 2011;21:635–44. doi: 10.1016/j.cub.2011.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beaty BT, Sharma VP, Bravo-Cordero JJ, Simpson MA, Eddy RJ, Koleske AJ, Condeelis J. β1 integrin regulates Arg to promote invadopodial maturation and matrix degradation. Mol Biol Cell. 2013;24:1661–75, S1-11. doi: 10.1091/mbc.E12-12-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Attanasio F, Caldieri G, Giacchetti G, van Horssen R, Wieringa B, Buccione R. Novel invadopodia components revealed by differential proteomic analysis. Eur J Cell Biol. 2011;90:115–27. doi: 10.1016/j.ejcb.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 45.Artym VV, Matsumoto K, Mueller SC, Yamada KM. Dynamic membrane remodeling at invadopodia differentiates invadopodia from podosomes. Eur J Cell Biol. 2011;90:172–80. doi: 10.1016/j.ejcb.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quintavalle M, Elia L, Price JH, Heynen-Genel S, Courtneidge SA. A cell-based high-content screening assay reveals activators and inhibitors of cancer cell invasion. Sci Signal. 2011;4:ra49. doi: 10.1126/scisignal.2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winograd-Katz SE, Brunner MC, Mirlas N, Geiger B. Analysis of the signaling pathways regulating Src-dependent remodeling of the actin cytoskeleton. Eur J Cell Biol. 2011;90:143–56. doi: 10.1016/j.ejcb.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Diaz B, Shani G, Pass I, Anderson D, Quintavalle M, Courtneidge SA. Tks5-dependent, nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Sci Signal. 2009;2:ra53. doi: 10.1126/scisignal.2000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamaguchi H, Yoshida S, Muroi E, Yoshida N, Kawamura M, Kouchi Z, Nakamura Y, Sakai R, Fukami K. Phosphoinositide 3-kinase signaling pathway mediated by p110α regulates invadopodia formation. J Cell Biol. 2011;193:1275–88. doi: 10.1083/jcb.201009126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weaver AM. Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis. 2006;23:97–105. doi: 10.1007/s10585-006-9014-1. [DOI] [PubMed] [Google Scholar]
- 51.Yamaguchi H, Wyckoff J, Condeelis J. Cell migration in tumors. Curr Opin Cell Biol. 2005;17:559–64. doi: 10.1016/j.ceb.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 52.Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, Yang J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell. 2011;19:372–86. doi: 10.1016/j.ccr.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gligorijevic B, Wyckoff J, Yamaguchi H, Wang Y, Roussos ET, Condeelis J. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J Cell Sci. 2012;125:724–34. doi: 10.1242/jcs.092726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blouw B, Seals DF, Pass I, Diaz B, Courtneidge SA. A role for the podosome/invadopodia scaffold protein Tks5 in tumor growth in vivo. Eur J Cell Biol. 2008;87:555–67. doi: 10.1016/j.ejcb.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clark ES, Brown B, Whigham AS, Kochaishvili A, Yarbrough WG, Weaver AM. Aggressiveness of HNSCC tumors depends on expression levels of cortactin, a gene in the 11q13 amplicon. Oncogene. 2009;28:431–44. doi: 10.1038/onc.2008.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li CM, Chen G, Dayton TL, Kim-Kiselak C, Hoersch S, Whittaker CA, Bronson RT, Beer DG, Winslow MM, Jacks T. Differential Tks5 isoform expression contributes to metastatic invasion of lung adenocarcinoma. Genes Dev. 2013;27:1557–67. doi: 10.1101/gad.222745.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brentnall TA. Arousal of cancer-associated stromal fibroblasts: palladin-activated fibroblasts promote tumor invasion. Cell Adh Migr. 2012;6:488–94. doi: 10.4161/cam.21453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brentnall TA, Lai LA, Coleman J, Bronner MP, Pan S, Chen R. Arousal of cancer-associated stroma: overexpression of palladin activates fibroblasts to promote tumor invasion. PLoS One. 2012;7:e30219. doi: 10.1371/journal.pone.0030219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goicoechea SM, García-Mata R, Staub J, Valdivia A, Sharek L, McCulloch CG, Hwang RF, Urrutia R, Yeh JJ, Kim HJ, et al. Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer-associated fibroblasts. Oncogene. 2013 doi: 10.1038/onc.2013.68. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakahara H, Otani T, Sasaki T, Miura Y, Takai Y, Kogo M. Involvement of Cdc42 and Rac small G proteins in invadopodia formation of RPMI7951 cells. Genes Cells. 2003;8:1019–27. doi: 10.1111/j.1365-2443.2003.00695.x. [DOI] [PubMed] [Google Scholar]
- 61.Albiges-Rizo C, Destaing O, Fourcade B, Planus E, Block MR. Actin machinery and mechanosensitivity in invadopodia, podosomes and focal adhesions. J Cell Sci. 2009;122:3037–49. doi: 10.1242/jcs.052704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Díaz B, Yuen A, Iizuka S, Higashiyama S, Courtneidge SA. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol. 2013;201:279–92. doi: 10.1083/jcb.201209151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roh-Johnson M, Bravo-Cordero JJ, Patsialou A, Sharma VP, Guo P, Liu H, Hodgson L, Condeelis J. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene. 2013 doi: 10.1038/onc.2013.377. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoshino D, Jourquin J, Emmons SW, Miller T, Goldgof M, Costello K, Tyson DR, Brown B, Lu Y, Prasad NK, et al. Network analysis of the focal adhesion to invadopodia transition identifies a PI3K-PKCα invasive signaling axis. Sci Signal. 2012;5:ra66. doi: 10.1126/scisignal.2002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang X, Nie D, Chakrabarty S. Growth factors in tumor microenvironment. Front Biosci (Landmark Ed) 2010;15:151–65. doi: 10.2741/3612. [Landmark Ed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mader CC, Oser M, Magalhaes MA, Bravo-Cordero JJ, Condeelis J, Koleske AJ, Gil-Henn H. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 2011;71:1730–41. doi: 10.1158/0008-5472.CAN-10-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grass GD, Tolliver LB, Bratoeva M, Toole BP. CD147, CD44, and the epidermal growth factor receptor (EGFR) signaling pathway cooperate to regulate breast epithelial cell invasiveness. J Biol Chem. 2013;288:26089–104. doi: 10.1074/jbc.M113.497685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bourguignon LY, Gunja-Smith Z, Iida N, Zhu HB, Young LJ, Muller WJ, Cardiff RD. CD44v(3,8-10) is involved in cytoskeleton-mediated tumor cell migration and matrix metalloproteinase (MMP-9) association in metastatic breast cancer cells. J Cell Physiol. 1998;176:206–15. doi: 10.1002/(SICI)1097-4652(199807)176:1<206::AID-JCP22>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 70.Albrechtsen R, Stautz D, Sanjay A, Kveiborg M, Wewer UM. Extracellular engagement of ADAM12 induces clusters of invadopodia with localized ectodomain shedding activity. Exp Cell Res. 2011;317:195–209. doi: 10.1016/j.yexcr.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 71.Abram CL, Seals DF, Pass I, Salinsky D, Maurer L, Roth TM, Courtneidge SA. The adaptor protein fish associates with members of the ADAMs family and localizes to podosomes of Src-transformed cells. J Biol Chem. 2003;278:16844–51. doi: 10.1074/jbc.M300267200. [DOI] [PubMed] [Google Scholar]
- 72.Hayes KE, Walk EL, Ammer AG, Kelley LC, Martin KH, Weed SA. Ableson kinases negatively regulate invadopodia function and invasion in head and neck squamous cell carcinoma by inhibiting an HB-EGF autocrine loop. Oncogene. 2013;32:4766–77. doi: 10.1038/onc.2012.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou ZN, Sharma VP, Beaty BT, Roh-Johnson M, Peterson EA, Van Rooijen N, Kenny PA, Wiley HS, Condeelis JS, Segall JE. Autocrine HBEGF expression promotes breast cancer intravasation, metastasis and macrophage-independent invasion in vivo. Oncogene. 2013 doi: 10.1038/onc.2013.363. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, Condeelis J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022–9. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 75.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 76.Lu X, Kang Y. Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clin Cancer Res. 2010;16:5928–35. doi: 10.1158/1078-0432.CCR-10-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 80.Subarsky P, Hill RP. The hypoxic tumour microenvironment and metastatic progression. Clin Exp Metastasis. 2003;20:237–50. doi: 10.1023/A:1022939318102. [DOI] [PubMed] [Google Scholar]
- 81.Md Hashim NF, Nicholas NS, Dart AE, Kiriakidis S, Paleolog E, Wells CM. Hypoxia-induced invadopodia formation: a role for β-PIX. Open Biol. 2013;3:120159. doi: 10.1098/rsob.120159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hanna SC, Krishnan B, Bailey ST, Moschos SJ, Kuan PF, Shimamura T, Osborne LD, Siegel MB, Duncan LM, O’Brien ET, 3rd, et al. HIF1α and HIF2α independently activate SRC to promote melanoma metastases. J Clin Invest. 2013;123:2078–93. doi: 10.1172/JCI66715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10:295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- 84.Yamaguchi H, Takeo Y, Yoshida S, Kouchi Z, Nakamura Y, Fukami K. Lipid rafts and caveolin-1 are required for invadopodia formation and extracellular matrix degradation by human breast cancer cells. Cancer Res. 2009;69:8594–602. doi: 10.1158/0008-5472.CAN-09-2305. [DOI] [PubMed] [Google Scholar]
- 85.Wang Y, Roche O, Xu C, Moriyama EH, Heir P, Chung J, Roos FC, Chen Y, Finak G, Milosevic M, et al. Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factor-mediated upregulation of caveolin-1. Proc Natl Acad Sci U S A. 2012;109:4892–7. doi: 10.1073/pnas.1112129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kaluza D, Kroll J, Gesierich S, Yao TP, Boon RA, Hergenreider E, Tjwa M, Rössig L, Seto E, Augustin HG, et al. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 2011;30:4142–56. doi: 10.1038/emboj.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Castro-Castro A, Janke C, Montagnac G, Paul-Gilloteaux P, Chavrier P. ATAT1/MEC-17 acetyltransferase and HDAC6 deacetylase control a balance of acetylation of alpha-tubulin and cortactin and regulate MT1-MMP trafficking and breast tumor cell invasion. Eur J Cell Biol. 2012;91:950–60. doi: 10.1016/j.ejcb.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 88.Rey M, Irondelle M, Waharte F, Lizarraga F, Chavrier P. HDAC6 is required for invadopodia activity and invasion by breast tumor cells. Eur J Cell Biol. 2011;90:128–35. doi: 10.1016/j.ejcb.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 89.Arsenault D, Brochu-Gaudreau K, Charbonneau M, Dubois CM. HDAC6 deacetylase activity is required for hypoxia-induced invadopodia formation and cell invasion. PLoS One. 2013;8:e55529. doi: 10.1371/journal.pone.0055529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marín-Hernández A, Gallardo-Pérez JC, Ralph SJ, Rodríguez-Enríquez S, Moreno-Sánchez R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem. 2009;9:1084–101. doi: 10.2174/138955709788922610. [DOI] [PubMed] [Google Scholar]
- 91.McCarty MF, Whitaker J. Manipulating tumor acidification as a cancer treatment strategy. Altern Med Rev. 2010;15:264–72. [PubMed] [Google Scholar]
- 92.Brisson L, Reshkin SJ, Goré J, Roger S. pH regulators in invadosomal functioning: proton delivery for matrix tasting. Eur J Cell Biol. 2012;91:847–60. doi: 10.1016/j.ejcb.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 93.Xu L, Fukumura D, Jain RK. Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: mechanism of low pH-induced VEGF. J Biol Chem. 2002;277:11368–74. doi: 10.1074/jbc.M108347200. [DOI] [PubMed] [Google Scholar]
- 94.Teti A, Grano M, Teitelbaum SL, Hruska KA, Colucci S, Zambonin Zallone A. Regulation of podosomes by intracellular pH in avian osteoclasts. Boll Soc Ital Biol Sper. 1989;65:597–601. [PubMed] [Google Scholar]
- 95.Rotin D, Steele-Norwood D, Grinstein S, Tannock I. Requirement of the Na+/H+ exchanger for tumor growth. Cancer Res. 1989;49:205–11. [PubMed] [Google Scholar]
- 96.Amith SR, Fliegel L. Regulation of the Na+/H+ Exchanger (NHE1) in Breast Cancer Metastasis. Cancer Res. 2013;73:1259–64. doi: 10.1158/0008-5472.CAN-12-4031. [DOI] [PubMed] [Google Scholar]
- 97.Reshkin SJ, Cardone RA, Harguindey S. Na+-H+ exchanger, pH regulation and cancer. Recent Pat Anticancer Drug Discov. 2013;8:85–99. doi: 10.2174/1574892811308010085. [DOI] [PubMed] [Google Scholar]
- 98.Lucien F, Brochu-Gaudreau K, Arsenault D, Harper K, Dubois CM. Hypoxia-induced invadopodia formation involves activation of NHE-1 by the p90 ribosomal S6 kinase (p90RSK) PLoS One. 2011;6:e28851. doi: 10.1371/journal.pone.0028851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lucien F, Brochu-Gaudreau K, Arsenault D, Harper K, Dubois CM. Hypoxia-induced invadopodia formation involves activation of NHE-1 by the p90 ribosomal S6 kinase (p90RSK) PLoS One. 2011;6:e28851. doi: 10.1371/journal.pone.0028851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Magalhaes MA, Larson DR, Mader CC, Bravo-Cordero JJ, Gil-Henn H, Oser M, Chen X, Koleske AJ, Condeelis J. Cortactin phosphorylation regulates cell invasion through a pH-dependent pathway. J Cell Biol. 2011;195:903–20. doi: 10.1083/jcb.201103045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Busco G, Cardone RA, Greco MR, Bellizzi A, Colella M, Antelmi E, Mancini MT, Dell’Aquila ME, Casavola V, Paradiso A, et al. NHE1 promotes invadopodial ECM proteolysis through acidification of the peri-invadopodial space. FASEB J. 2010;24:3903–15. doi: 10.1096/fj.09-149518. [DOI] [PubMed] [Google Scholar]
- 102.Oser M, Yamaguchi H, Mader CC, Bravo-Cordero JJ, Arias M, Chen X, Desmarais V, van Rheenen J, Koleske AJ, Condeelis J. Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J Cell Biol. 2009;186:571–87. doi: 10.1083/jcb.200812176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Frantz C, Barreiro G, Dominguez L, Chen X, Eddy R, Condeelis J, Kelly MJ, Jacobson MP, Barber DL. Cofilin is a pH sensor for actin free barbed end formation: role of phosphoinositide binding. J Cell Biol. 2008;183:865–79. doi: 10.1083/jcb.200804161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Clark ES, Whigham AS, Yarbrough WG, Weaver AM. Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 2007;67:4227–35. doi: 10.1158/0008-5472.CAN-06-3928. [DOI] [PubMed] [Google Scholar]
- 105.Kato Y, Lambert CA, Colige AC, Mineur P, Noël A, Frankenne F, Foidart JM, Baba M, Hata R, Miyazaki K, et al. Acidic extracellular pH induces matrix metalloproteinase-9 expression in mouse metastatic melanoma cells through the phospholipase D-mitogen-activated protein kinase signaling. J Biol Chem. 2005;280:10938–44. doi: 10.1074/jbc.M411313200. [DOI] [PubMed] [Google Scholar]
- 106.Lin Y, Chang G, Wang J, Jin W, Wang L, Li H, Ma L, Li Q, Pang T. NHE1 mediates MDA-MB-231 cells invasion through the regulation of MT1-MMP. Exp Cell Res. 2011;317:2031–40. doi: 10.1016/j.yexcr.2011.05.026. [DOI] [PubMed] [Google Scholar]
- 107.Poincloux R, Lizárraga F, Chavrier P. Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J Cell Sci. 2009;122:3015–24. doi: 10.1242/jcs.034561. [DOI] [PubMed] [Google Scholar]
- 108.Caldieri G, Buccione R. Aiming for invadopodia: organizing polarized delivery at sites of invasion. Trends Cell Biol. 2010;20:64–70. doi: 10.1016/j.tcb.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 109.Steffan JJ, Snider JL, Skalli O, Welbourne T, Cardelli JA. Na+/H+ exchangers and RhoA regulate acidic extracellular pH-induced lysosome trafficking in prostate cancer cells. Traffic. 2009;10:737–53. doi: 10.1111/j.1600-0854.2009.00904.x. [DOI] [PubMed] [Google Scholar]
- 110.Luxenburg C, Geblinger D, Klein E, Anderson K, Hanein D, Geiger B, Addadi L. The architecture of the adhesive apparatus of cultured osteoclasts: from podosome formation to sealing zone assembly. PLoS One. 2007;2:e179. doi: 10.1371/journal.pone.0000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cougoule C, Carréno S, Castandet J, Labrousse A, Astarie-Dequeker C, Poincloux R, Le Cabec V, Maridonneau-Parini I. Activation of the lysosome-associated p61Hck isoform triggers the biogenesis of podosomes. Traffic. 2005;6:682–94. doi: 10.1111/j.1600-0854.2005.00307.x. [DOI] [PubMed] [Google Scholar]
- 112.van Horssen R, Buccione R, Willemse M, Cingir S, Wieringa B, Attanasio F. Cancer cell metabolism regulates extracellular matrix degradation by invadopodia. Eur J Cell Biol. 2013;92:113–21. doi: 10.1016/j.ejcb.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 113.Kalckar HM, Ullrey D, Kijomoto S, Hakomori S. Carbohydrate catabolism and the enhancement of uptake of galactose in hamster cells transformed by polyoma virus. Proc Natl Acad Sci U S A. 1973;70:839–43. doi: 10.1073/pnas.70.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Oikawa T, Oyama M, Kozuka-Hata H, Uehara S, Udagawa N, Saya H, Matsuo K. Tks5-dependent formation of circumferential podosomes/invadopodia mediates cell-cell fusion. J Cell Biol. 2012;197:553–68. doi: 10.1083/jcb.201111116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Baron A, Migita T, Tang D, Loda M. Fatty acid synthase: a metabolic oncogene in prostate cancer? J Cell Biochem. 2004;91:47–53. doi: 10.1002/jcb.10708. [DOI] [PubMed] [Google Scholar]
- 116.Swinnen JV, Van Veldhoven PP, Timmermans L, De Schrijver E, Brusselmans K, Vanderhoydonc F, Van de Sande T, Heemers H, Heyns W, Verhoeven G. Fatty acid synthase drives the synthesis of phospholipids partitioning into detergent-resistant membrane microdomains. Biochem Biophys Res Commun. 2003;302:898–903. doi: 10.1016/S0006-291X(03)00265-1. [DOI] [PubMed] [Google Scholar]
- 117.Yamaguchi H, Oikawa T. Membrane lipids in invadopodia and podosomes: key structures for cancer invasion and metastasis. Oncotarget. 2010;1:320–8. doi: 10.18632/oncotarget.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Oikawa T, Itoh T, Takenawa T. Sequential signals toward podosome formation in NIH-src cells. J Cell Biol. 2008;182:157–69. doi: 10.1083/jcb.200801042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Oikawa T, Takenawa T. PtdIns(3,4)P2 instigates focal adhesions to generate podosomes. Cell Adh Migr. 2009;3:195–7. doi: 10.4161/cam.3.2.7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Scott KE, Wheeler FB, Davis AL, Thomas MJ, Ntambi JM, Seals DF, Kridel SJ. Metabolic regulation of invadopodia and invasion by acetyl-CoA carboxylase 1 and de novo lipogenesis. PLoS One. 2012;7:e29761. doi: 10.1371/journal.pone.0029761. [DOI] [PMC free article] [PubMed] [Google Scholar]