

Abstract
Retinal ganglion cells (RGCs) are the only afferent neurons that can transmit visual information to the brain. The death of RGCs occurs in the early stages of glaucoma, diabetic retinopathy, and many other retinal diseases. Autophagy is a highly conserved lysosomal pathway, which is crucial for maintaining cellular homeostasis and cell survival under stressful conditions. Research has established that autophagy exists in RGCs after increasing intraocular pressure (IOP), retinal ischemia, optic nerve transection (ONT), axotomy, or optic nerve crush. However, the mechanism responsible for defining how autophagy is induced in RGCs has not been elucidated. Accumulating data has pointed to an essential role of reactive oxygen species (ROS) in the activation of autophagy. RGCs have long axons with comparatively high densities of mitochondria. This makes them more sensitive to energy deficiency and vulnerable to oxidative stress. In this review, we explore the role of oxidative stress in the activation of autophagy in RGCs, and discuss the possible mechanisms that are involved in this process. We aim to provide a more theoretical basis of oxidative stress-induced autophagy, and provide innovative targets for therapeutic intervention in retinopathy.
Keywords: autophagy, axon, mitophagy, oxidative stress, retinal ganglion cells
Introduction
Retinal ganglion cells differentiate earliest among all retinal neurons, and they can convey visual information to the visual cortex. By contrast, their death is associated with various visual diseases and conditions.1 Oxidative stress can induce apoptosis by increasing mitochondrial membrane permeability transition, by inhibiting the mitochondrial respiratory chain, and by damaging mitochondrial DNA. RGCs are the longest-surviving cells in the retina.2 Their long axons are sensitive to a lack of energy, and for this reason, they depend more on mitochondria, and are more vulnerable to mitochondrial dysfunction3 and sensitive to oxidative stress.
Oxidative stress and cellular accumulation of ROS play a vital role in the stimulation of autophagy under conditions of nutrient deficiency,4 hypoxia,5 ischemia/reperfusion injury,6 and other cellular stress responses.7 By their very nature, ROS are unstable molecules, which have the capacity to readily convert to many different viable and active forms. For example hydrogen peroxide (H2O2)8 and oxy-radicals (O2−) are 2 major forms of ROS, both of which mediate the induction of autophagy.9
Research has shown that after increasing IOP, retinal ischemia, ONT, axotomy, or optic nerve crush, the levels of autophagy increase in RGCs. Considering that ischemia/hypoxia can provoke oxidative stress, the axon is responsible for material transportation, and has high densities of mitochondria. Traumatic injury to the central nervous system (CNS) often results in secondary degeneration, which involves oxidative stress. Thus, we hypothesize that oxidative stress may exert important influences on the induction of autophagy in RGCs.
Autophagy and its Role in RGCs
Autophagy is a highly conserved metabolic process that permits the degradation and recycling of cellular constituents (Fig. 1), including long-lived proteins and organelles, which are induced by various forms of stress, including hypoxia, growth factor withdrawal, starvation, or increased production of ROS.10 In mammalian cells, there are predominantly 3 autophagic pathways that have been recognized: 1) macroautophagy (hereafter referred to as autophagy), 2) microautophagy, and 3) chaperone-mediated autophagy.11
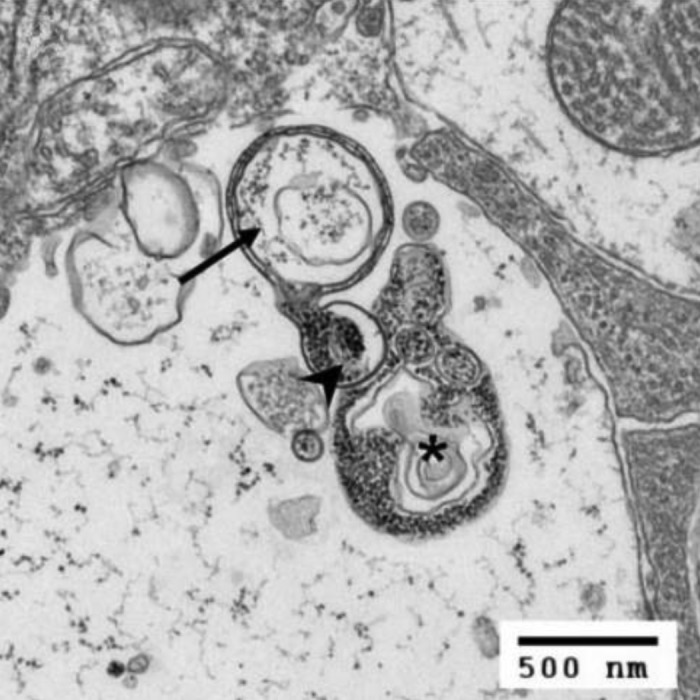
Figure 1. Early and late stage autophagy in retinal ganglion cell layers. In the earlier stage, the double-membrane-bound structure (see arrow) is fusing with a lysosome (shown by the arrowhead). At the later stages (see asterisk), fusion with the lysosome has occurred, and the structure (an autolysosome) is undergoing proteolysis. Scale bar: 500 nm.
Autophagy is primarily a nonselective degradation pathway, but evidence has confirmed the existence of different kinds of selective autophagy,12 such as mitophagy,13 reticulophagy,14 pexophagy,15 xenophagy,16 and nucleophagy17 that respectively refer to the selective removal of mitochondria, endoplasmic reticulum (ER), peroxisomes, intruding microorganisms, and nuclei.
The Molecular Mechanism of Autophagy
Our molecular understanding of autophagy was originally based on the identification of autophagy-related (ATG) genes in yeast, and the subsequent finding of orthologs in other eukaryotes.18 Currently, 38 different ATG genes have been discovered primarily through S. cerevisiae genetics. Many of these genes were subsequently found to be conserved and to have parallel functions in multicellular eukaryotes.19 Among all of the ATG proteins, one subgroup plays a central role in the molecular mechanisms of autophagy, which involves the formation, expansion and fusion of autophagosomes.
The complex molecular machinery of autophagy makes its regulation further complicated, and involves numerous signaling pathways that might also interact at multiple levels. The target of rapamycin is one of the central regulators of autophagy, and MTOR (mechanistic target of rapamycin) integrates upstream activating signals and subsequently inhibits autophagy. Class I phosphoinositide 3-kinase mediates autophagy, and does so predominantly through the regulation of TOR activity in response to insulin-like and other growth factors.20 The AMP-activated protein kinase (AMPK) is an intracellular energy sensor, which specifically responds to energy depletion and positively regulates autophagy. The BCL2 (B-cell CLL/lymphoma 2) family proteins also play important roles in the regulation of autophagy. For example, BCL2 binds to BECN1 (Beclin 1, autophagy-related) and disorganizes the formation of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, and thus disrupts the induction of autophagy. In addition to BECN1, ER-located BCL2 may also inhibit autophagy by regulating free ER Ca2+ homeostasis.21
Autophagy Exists Extensively in the Central Nervous System
Mizushima22 sub-classified autophagy into “baseline” and “induced,” depending on its role. Neuronal autophagy, at basal levels, is protective, and it prevents the aggregation of damaged organelles and the accumulation of proteins in neurons, thus promoting axonal homeostasis and efficient clearance of cellular corpses. However, with the progression of autophagy, neurons subsequently undergo further stimulation, which deregulates the autophagic mechanism, making basal autophagy convert to induced autophagy.23
In the CNS, autophagic imbalance is seen in various chronic neurodegenerative diseases, including Parkinson,24,25 Alzheimer,26,27 and Huntington diseases,28,29 and in acute neural disorders, including cerebral ischemia,30-33 neurotoxin injury,34-36 brain trauma,37,38 and stroke.39-41 The accumulation and aggregation of toxic or mutated proteins are common features of many neurodegenerative diseases, and autophagy appears to serve as an adaptive response in these chronic pathogenic processes, which degrades toxic or mutated proteins and does not appear to modulate neuronal cell death.42 Additionally, the enhanced appearance of autophagy may act as a compensatory degradation system for proteasomal impairment.43 By using pharmacological agents, the consensus opinions posit that the beneficial effects of autophagy in neurodegenerative disorders are associated with proteinopathies.28,29,44 Autophagy in acute neuronal injury has been extensively described. However, the role of autophagy in these processes remains uncertain and controversial. The most relevant study for a deleterious effect of autophagy after neonatal hypoxic/ischemic brain injury is from Koike et al.,45 in which they used neonatal mice in a model of specific Atg7 knockout in the CNS, and found that Atg7 deficiency prevents hippocampal neuronal death after hypoxia-ischemia injury. The chronic process of disrupted autophagy is well tolerated at the beginning, while longer durations and acute processes are not. It remains unknown precisely what duration of autophagy is not tolerated, and clearly further explorations of the molecular mechanisms that are involved in autophagy are also needed to gain greater understanding of the specific roles of autophagy under different conditions.
Roles of Autophagy in RGCs
Retinal ganglion cells are myelinated long-axon neurons in the CNS, and their axons constitute the optic nerves that transmit retinal signals to higher regions in the brain. Unlike other neurons in the CNS, the blood supply of RGC axons is different from that supplying cell bodies,46 making it sensitive and vulnerable to different stress-induced damage. Autophagy is an adaptive process in response to various stressful conditions. In the next section, we summarize the role of induced autophagy in RGCs.
1) Elevated intraocular pressure
Autophagy is originally activated in the dendrites of RGCs to promote cellular protection (Fig. 2A and B). Thereafter, autophagy is predominantly activated in the cytoplasm where it provokes cell death. BECN1 is a component of several class III PtdIns3K complexes and contributes to the early formation of autophagic vesicles47 in the retina, which predominantly locate to the ganglion cell layer (GCL).48 In a rat model of chronic hypertensive glaucoma,49 the expression of BECN1 and MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3)-II/LC3-I proteins are increased throughout the 8-wk period following increased IOP. 3-methyladenine (3-MA) inhibits autophagy, and by contrast, it decreases RGC apoptosis. By electron microscopy, it was determined that activation of autophagy predominantly appears in the dendrites of RGCs during the first 4 wk. Thereafter, activated autophagy is essentially found in the cytoplasm of RGCs. Coincidentally, the clear loss of RGCs in the GCL also occurs during this time period. An explanation for these observations is that with the progressive increase in IOP, the neuroprotective effect of autophagy is dampened after 4 wk, and then triggers autophagic cell death. Russo et al.,48 also found that dysregulation of basal autophagy occurs under conditions of retinal transient ischemia that is induced by acutely increasing IOP in adult rats. In addition, expression of both retinal LC3-II and BECN1 in retina are significantly decreased after 24 h of reperfusion. It was also found that calpain activity is responsible for BECN1 cleavage, which is mediated by the activation of the N-methyl-D-aspartate receptor. In the same study, under starvation conditions, silencing of Becn1 prevents induction of autophagy, and increases cell death, which indicates a prosurvival role for autophagy in RGC-5 cells. In a rat retinal ischemia/reperfusion model elicited by acute increases in IOP, Piras et al.,50 found that the number of neurons in the GCL decreases significantly after 24 h, an observation that can be partially prevented by 3-MA by potently inhibiting the formation of autophagosomes. After 12 h and 24 h of increasing IOP in the retina, the enhancement of both autophagosome formation and lysosomal activity are observed in GCL-neurons, which indicates an improvement in the autophagic flux.

Figure 2. Autophagy is activated in RGCs. (A and B) Comparison of normal retinas (A) with experimentally-induced glaucomatous retinas (Exp GL, B). LC3 immunoreactivity is observed as clusters of small, intensely stained green granules. A markedly increased number of positive granules are visible in the cytosol of the RGC in the GCL (white arrows) and in the dendrites of the RGC in the IPL (red arrows). (C and D) RGCs in control retina and at 6 d after ONT. Cells from control retinas display a diffuse and homogeneous GFP-LC3 green fluorescence in the cytosol, whereas the axotomized RGCs in the right panel show visible GFP-LC3 dots in the cytosol that are identified as autophagosomes, and indicated by arrowheads. (A and B) Scale bar: 20 μm; (C and D) scale bar: 10 μm.
In these experiments, we found that with progressive increases in IOP, autophagy is originally activated in the dendrites of RGCs and protects the cells. Subsequently, autophagy is predominantly activated in the cytoplasm and causes cell death. We speculate that axons contain a large number of mitochondria, which make them more sensitive to chronic ischemia, which triggers autophagy to first maintain intracellular homeostasis. The protective effect of autophagy in axons might be prolonged; however, with the progressive increase in IOP, autophagy in the neuronal soma is activated and this process disrupts homeostasis, which may decrease cell viability. If the IOP increases acutely, then autophagy in the retina would be dysregulated, and the level of autophagy in GCL would be enhanced immediately. Such autophagic processes occur mostly in cell bodies, and induce neuronal cell death in a relatively short time.
2) Traumatic injury
In traumatically injured RGCs, enhanced autophagy often starts before cell death for neuroprotection. However, the increasing post-lesional autophagy will aggravate axonal degradation, which may be calcium dependent. By electron microscopy, Sternberg et al.,51 found that axotomy induces ultrastructural signs of both apoptosis and autophagy in neonatal rat RGCs for 24 h, and inhibition of autophagy by 3-MA decreases cell viability, which suggests a neuroprotective role of autophagy. Following optic nerve transection, the upregulation of autophagy-related genes such as Lc3 (3 d after ONT) and Becn1 (1 d after ONT) occur prior to RGC death (5 d after ONT). Treatment of RGC-5 cells with inhibitors of autophagy under serum deprivation decreases cell viability by nearly 40%, which indicates that activation of autophagy is neuroprotective and enhances survival of RGCs in adverse conditions.52 In another study of RGCs in mice,53 augmented autophagy also occurs on the third day of ONT and before the peak in cell death (Fig. 2C and D). Moreover, genetic downregulation of autophagy in an atg5flox/flox mouse model (specific deletion of Atg5 in RGCs) or in an atg4b−/− mouse model reduces the number of living cells after ONT. Pharmacological augmentation of autophagy increases cell viability in vivo. These observations collectively support the notion that autophagy constitutes a protective response after traumatic injury in RGCs.
Following optic nerve crush, the RGC axons degenerate and electron microscopy analysis reveals severe ultrastructural changes and autophagosome formation within the first hour after formation of the lesion.54 Ninety min after the application of an autophagy inhibitor, axonal degeneration is significantly delayed. In the same study, researchers observed the intra-axonal calcium levels with the application of calcium ionophore and calcium channel inhibitors, and demonstrated that the increase of postcrush autophagy is calcium dependent. They hypothesized that mechanical injury to RGCs induces a rapid increase of intraaxonal calcium ions via calcium channels, resulting in a secondary accumulation of autophagosomes, which participate in the degeneration of axons.
Axonal dystrophy is a hallmark of axonopathy, which can be triggered by various neurodegenerative conditions, excitotoxicity and neuronal injuries.55 It has been previously suggested that autophagy is associated with axonal dystrophic swellings and axonopathy. Following excitotoxic or axotomy injury, accumulated double-membrane vacuoles resembling autophagosomes are detected in dystrophic axon terminals.56,57 Traumatic injury destroys the integrity of the axons, and we speculate that the observation of accumulated autophagosome-like vacuoles in these injured axons reflects the stimulated synthesis of autophagosomes, which is crucial for maintaining neuronal survival following axonal injury. Autophagic flux in axons may increase at one point after the formation of the lesion to protect neuronal soma from axonal damage, and prolonging neuronal soma survival. However, with the progression of the injury, autophagy may be inhibited or return to normal levels.
Interactions between Oxidative Stress and Autophagy in RGCs
Oxidative stress is recognized as a central contributor to neuronal injury. Axons are more sensitive to oxidative stress than cell bodies, and the elevation of ROS levels in axons occurs much earlier than is found in somas.58 RGCs have numerous mitochondria in the intraretinal portion of their axons,59 resulting in greater demand for energy and vulnerability to hypoxia-ischemia. A frequently used model for retinal ischemia is established by using elevated IOP.60 Both acute and chronic ischemia contribute to oxidative stress, brought on by an imbalanced metabolic demand and associated with production of free radicals or reactive oxygen species.
The induction of autophagy is seen in adult and neonatal mouse striatum and cortex after hypoxic-ischemic injury,30,61 and oxidative stress following cerebral hypoxia-ischemia may induce autophagy, which initially aborts apoptosis by eliminating damaged mitochondria and preventing necrosis via catabolic energy production.62 However, it is unknown how autophagy participates in the execution of neuronal death following hypoxic-ischemic injury.63
Traumatic injury to the CNS often accompanies the progressive impairment of secondary degeneration, leading to greater reductions in neurons and cell function.64 In axotomied or crushed phrenic nerves, oxidative stress might be directly produced via an increase in lipid peroxidation, and a decrease in antioxidant enzyme activities.65 After partial optic nerve injury, RGCs undergo secondary death, but it is unclear how these nonviable cells distribute, and which cell death pathways are involved.66 It has also been demonstrated that oxidative stress spreads from injured axons to surrounding RGC somata via the astrocytic network, which occurs in the early stages of secondary degeneration.67 Blockade of calcium channels can protect RGCs from secondary death. We hypothesize that oxidative stress may play a leading role in the induction of autophagy in RGCs and present possible mechanisms as described in the sections below.
ROS Induces ATG4 Inactivation
In response to starvation, oxidative stress is activated and induces the formation of ROS, especially H2O2, which serve as signaling molecules in the induction of autophagosomes by regulating the activity of ATG4. H2O2 could bind to the cysteine 81 site of ATG4,68 forming reversible sulfenic acid or disulfide bonds that shield cysteine 77 that can directly activate ATG4, and drive lipidation of LC3 (a mammalian ATG8 homolog), priming it for subsequent conjugation with phosphatidylethanolamine (Fig. 3). This process is essential for autophagosome formation. After autophagosomes mature and fuse with lysosomes, the local environment changes to H2O2− deficiency, where ATG4 turns to an active state, and thus LC3 is delipidated and recycles once again.68

Figure 3. Regulation of autophagy by oxidative stress in RGCs. Various injuries cause oxidative stress in RGCs by primary and secondary degeneration, and thereby induce autophagy. ATG4 has 2 effects: A) it primes ATG8 homologs for conjugation with PE, allowing for lipidated LC3 and GABARAP family protein incorporation into the phagophore membrane, and B) deconjugation of LC3 and GABARAP protein from phosphatidylethanolamine apparently permits recycling of these proteins. Increased concentrations of ROS inactivate the second function of ATG4. In response to oxidative stress, HIF-1 is activated, which indirectly augments NMNAT via induction of IL6. Additionally, NMNAT may act on the upstream pathways of autophagy. Oxidative stress and increasing intracellular Ca2+ levels will activate AMPK, which promotes the formation of a TSC1-TSC2 complex, leading to MTOR inactivation and initiation of autophagy. Under oxidative stress, mitophagy is activated. BNIP3L localizes to the mitochondrial outer membrane, it interacts directly with LC3 or GABARAP, and mediates the recruitment of damaged mitochondria to phagophores. BNIP3L also competes with BECN1 for binding to BCL2 and thereby releases BECN1 to induce autophagy. PINK1 recruits the E3 ligase PARK2 to damaged mitochondria, which then mediates mitochondrial fragmentation and targeting to phagophores, which is followed by ubiquitination of VDAC1 and binding to SQSTM1. Then SQSTM1 targets this complex to the autophagosome by interacting with the autophagic protein LC3.
ATG4 exists in the CNS, and so oxidative stress may influence the activity of ATG4 and induce autophagy. Atg4b/ATG4B has been cloned as a mammalian homolog of yeast ATG4 in humans and mice. Yoshimura et al.,69 found that mRNA levels of Atg4B are high in brain tissues, especially in the olfactory bulb, cerebellum, and retina. Rodriguez-Muela et al.,53 found that retinas from atg4b−/− mice show a clear reduction in LC3 lipidation and an increase in the expression of SQSTM1 (sequestosome 1) levels. Compared with the control group, the number of living RGCs in atg4b−/− mice is also lower after axotomy, suggesting that ATG4 exerts a crucial function in the autophagy of retinas and RGCs.
HIF-1 Mediates NMNAT Transcription
The elevation of IOP often causes ocular ischemia, and ischemia/reperfusion injury induces ROS generation. In response to hypoxia, hypoxia-inducible factor (HIF-1) is activated. HIF-1 is an important regulator that facilitates rapid adaptation and survival of cells under conditions of relative hypoxia (~1% O2) to normoxia (~21% O2). HIF-1 is composed of a constitutively expressed ARNT/HIF-1β subunit and an oxygen-dependent HIF1A subunit.70,71 Ali et al.,72 showed that HIF1A indirectly induces IL6/HSF (interleukin 6) and upregulates NMNAT (nicotinamide nucleotide adenylyltransferase) under hypoxia in Drosophila heads. Current research in Drosophila has revealed the neuroprotective effects of NMNAT against neuronal excitotoxicity- or injury-induced axonal degeneration and polyglutamine protein-induced neurodegeneration.73-75
Studies in mammalian neurons have also shown that overexpression of NMNAT protects against injury- or stress-induced axonal degeneration.76-78 Previous findings also showed that NMNAT can inhibit ROS accumulation79 and may suppress autophagy by inhibiting ROS accumulation under hypoxic conditions in the neuron. NMNAT3 is present in the optic nerve axon and is located in the mitochondria.80 Kitaoka et al.,81 has observed that NMNAT3 is present in RGC bodies and nerve fibers in rat retina. They transfected an Nmnat3–enhanced green fluorescent protein plasmid into RGC bodies and observed a significant increase in NMNAT3 protein levels in the optic nerve. Nmnat3 transfection increases autophagic flux and decreases SQSTM1 protein levels in RGC-5 cells. Overexpression of NMNAT3 exerts a significant protective effect against axonal loss after IOP elevation, but this effect is suppressed by 3-MA, an autophagy inhibitor. This observation suggests that the protective effect of NMNAT3 might be involved in the autophagy machinery in optic nerve axons. Autophagy plays a self-destructive role in hypoxia, and NMNAT may act on the upstream elements or mechanisms of autophagy in a HIF1A-IL6 transcriptional pathway to protect optic nerve damage induced by hypoxic stress (Fig. 3).
AMPK-Dependent Signaling and Autophagy
Oxidative stress will destroy the mitochondrial structure, which will further cause an energy imbalance. AMPK regulates metabolic pathways and acts as a crucial sensor of cellular energy.82 When nutrients are scarce or when AMP/ATP ratio rises, AMPK will be induced.83 Active AMPK promotes the formation of tuberous sclerosis complex (TSC1-TSC2). TSC1-TSC2 can decrease the GDP-bound form of RHEB (Ras homolog enriched in brain) and increase its GDPase activity. Many studies have shown that RHEB-GTP strongly stimulates the activity of MTOR, and does so both in vitro and in vivo.84
Thus, in an AMPK-TSC1-TSC2-RHEB pathway, MTOR is inhibited and subsequently autophagy is activated (Fig. 3). Furthermore, AMPK is activated by 3 mammalian upstream kinases: 1) MAP3K7/TAK1, 2) STK11 (serine/threonine kinase 11) and 3) CAMK2B (calcium/calmodulin-dependent protein kinase II β).85,86 Traumatic injury increases the intracellular Ca2+ levels, following which CAMK2B will be activated. Thus, the secondary death of RGCs may be associated with the activation of AMPK, which ultimately leads to engagement of autophagy.
Oxidative Stress-Induced Mitophagy
Accumulating evidence reveals that in addition to its distinct regulation in neurons, autophagy may also display unique functions in axons, and independently from the soma and dendrites.23 In mammalian cells, axons are rich in mitochondria, where ROS are predominantly produced. The acute burst of ROS in mitochondria may be a crucial activator of mitophagy,87 which is a process of mitochondria-selective autophagy in response to various signals, including oxidative stress, starvation, and modification of mitochondrial proteins. The selectivity of mitophagy is adjusted by various proteins like BNIP3 (BCL2/adenovirus E1B 19 kDa interacting protein 3), PARK2, PINK1 (PTEN-induced putative kinase 1), and the fusion and fission processes of mitochondria.88-90
BNIP3L/NIX (BCL2/adenovirus E1B 19 kDa interacting protein 3-like), localizes to mitochondria, and regulates the aggregation of damaged mitochondria to autophagosomes by directly interacting with LC3 or GABARAP (GABA[A] receptor-associated protein) (Fig. 3). In vivo studies showed that HIF1A is stably expressed throughout the hypoxic retina, in the inner nuclear layer, and in RGCs.91 Among the downstream targets affected by HIF1A, BNIP3 is one of the primary death-promoting proteins, which competes with BECN1 for binding to BCL2, and hence releases “free” BECN1 to induce autophagy.92 In a hypoxic model made by CoCl2,93 significant levels of autophagic cell death are observed in RGC-5 cells and the levels of HIF1A and BNIP3 are also increased. N-acetylcysteine94 and pilocarpine95 both protect RGC-5 cells against CoCl2-induced autophagy and apoptosis by targeting the HIF1A pathway via their effects on BNIP3.
Additionally, PARK2 is an E3 ubiquitin ligase, and studies have shown that PINK1 regulates the recruitment of PARK2 to depolarized fragmented mitochondria, which then activates the ligase activity of PARK2.96-100 This process involves the ubiquitination of VDAC1 (voltage-dependent anion channel 1) and its combination with SQSTM1, which subsequently interacts with LC3 and directs this complex to the autophagosome (Fig. 3).92 In cultured dopaminergic neurons of the substantia nigra, PINK1 helps maintain standard mitochondrial morphology and mitochondrial membrane potential (ΔΨm), which plays a neuroprotective role in inhibiting ROS formation.101 It has been reported that PARK2 is distributed throughout the brain, including the cerebral cortex, midbrain, cerebellum, and basal ganglia.102-105 A recent study demonstrated that PARK2 exists in rodent, bovine, and primate retinas, and the location of PARK2 is quite ubiquitous, and is found in almost all of the major retinal neuronal types.106 Cai et al.,107 found that in mature cortical neurons, PARK2-tagged and depolarized mitochondria are predominantly detected in the soma and proximal dendrites, a site where mature lysosomes are located. The activity of PARK2 can be induced by ROS. Additionally, in a cortical neuron model, Joselin et al.,108 showed that PARK2 translocates to the mitochondria, and this translocation only occurs in culture media that lacks antioxidants, which implies an important role for ROS in this system.
Conclusion
Autophagy operates at the basal level in various cell types for constitutive intracellular clearance under normal conditions. Moreover, autophagy can be upregulated in response to oxidative stress, and the accumulation of ROS would further induce autophagy and mitophagy. After increasing IOP, retinal ischemia, ONT, axotomy, and optic nerve crush, the levels of autophagy in RGCs all increase. This phenomenon is associated with the activation of intracellular oxidative stress. However, it remains controversial whether the induction of autophagy in RGCs is actually beneficial.
We considered that the axons of RGCs contain a large number of mitochondria; they have a greater energy demand, and are more sensitive to oxidative stress. Thus, at the early stages of chronic injury, the oxidative stress-induced autophagy is predominantly concentrated in axons, and exerts a neuroprotective effect.
Axotomy and ONT destroy the integrity of RGC axons, and oxidative stress may initially activate autophagy in axons to avoid axonal degeneration and neuron cell death. Traumatic injury also provokes secondary degeneration of RGCs, which involves oxidative stress and leads to additional autophagy. However, with the development of oxidative stress, the increased degree of autophagy and the accumulation of autophagosomes not only exist in axons, but also in somas. We thus speculate that it is just at this time that the harmful effects of autophagy become evident.
Considering that the influence of individual differences and environmental factors might play important roles in the progression of various optic neuropathies, the extent of oxidative stress and autophagy are different in RGCs. If we could increase autophagy during the early stages of neuropathy and promote the level of autophagy in axons, the death of RGCs might be decreased. However, it remains a challenge to decide what exactly constitutes the “early stage” and the subsequent induction of autophagy, which might aggravate cell death. Drug-dependent methods lack specificity in regulating autophagy. If we could use genetic engineering techniques, and specifically achieve an increase in the levels of autophagy in RGC axons, and inhibit the activity of autophagy in somas, the protection of RGCs would be achieved regardless of the progression of various optic neuropathies.
In this review, we have summarized current opinion with regard to the relationship among RGCs, autophagy, and oxidative stress, and also discussed the possible mechanism of the activation of autophagy and mitophagy that is induced by oxidative stress in RGCs. In future studies, searching for new targets could specifically induce autophagy in axons or in somas, and thus derive genetic interventions that might represent a new method to protect RGCs from oxidative stress. More research is needed to explore the responsible molecular pathways about how autophagy is induced by oxidative stress in RGCs. This will be helpful in determining the differences between autophagy that is induced in axons and in somas, with the intention of designing novel genetic targets in the future for the early protection of RGCs in various retinopathies.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed by any of the authors.
Acknowledgments
The Natural Science Foundation of Heilongjiang Province (NO.D201253), The Traditional Chinese Medicine Scientific Research of Heilongjiang Province (NO.ZHY12-Z132) and a Science and Technology Project from the education department of Heilongjiang Province (NO.2511234) supported this work.
Glossary
Abbreviations:
- AMPK
AMP-activated protein kinase
- ATG
autophagy-related
- BCL2
B-cell CLL/lymphoma 2
- BECN1
Beclin 1, autophagy-related
- CNS
central nervous system
- ER
endoplasmic reticulum
- GCL
ganglion cell layer
- H2O2
hydrogen peroxide
- HIF-1
hypoxia-inducible factor
- IOP
intraocular pressure
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MTOR
mechanistic target of rapamycin
- NMNAT
nicotinamide nucleotide adenylyltransferase
- ONT
optic nerve transection
- PE
phosphatidylethanolamine
- PINK1
PTEN-induced putative kinase 1
- PtdIns3K
phosphatidylinositol 3-kinase
- RGCs
retinal ganglion cells
- RHEB
ras homolog enriched in brain
- ROS
reactive oxygen species
- SQSTM1
sequestosome 1
- TSC
tuberous sclerosis complex
- 3-MA
3-methyladenine
References
- 1.Ren R, Li Y, Liu Z, Liu K, He S. Long-term rescue of rat retinal ganglion cells and visual function by AAV-mediated BDNF expression after acute elevation of intraocular pressure. Invest Ophthalmol Vis Sci. 2012;53:1003–11. doi: 10.1167/iovs.11-8484. [DOI] [PubMed] [Google Scholar]
- 2.Caporale N, Kolstad KD, Lee T, Tochitsky I, Dalkara D, Trauner D, Kramer R, Dan Y, Isacoff EY, Flannery JG. LiGluR restores visual responses in rodent models of inherited blindness. Mol Ther. 2011;19:1212–9. doi: 10.1038/mt.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen H, Chan DC. Critical dependence of neurons on mitochondrial dynamics. Curr Opin Cell Biol. 2006;18:453–9. doi: 10.1016/j.ceb.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Russell FD, Hamilton KD. Nutrient deprivation increases vulnerability of endothelial cells to proinflammatory insults. Free Radic Biol Med. 2014;67:408–15. doi: 10.1016/j.freeradbiomed.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 5.Cimini S, Rizzardini M, Biella G, Cantoni L. Hypoxia causes autophagic stress and derangement of metabolic adaptation in a cell model of amyotrophic lateral sclerosis. J Neurochem. 2014;129:413–25. doi: 10.1111/jnc.12642. [DOI] [PubMed] [Google Scholar]
- 6.Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2011;14:2179–90. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Essick EE, Sam F. Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxid Med Cell Longev. 2010;3:168–77. doi: 10.4161/oxim.3.3.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.She C, Zhu LQ, Zhen YF, Wang XD, Dong QR. Activation of AMPK protects against hydrogen peroxide-induced osteoblast apoptosis through autophagy induction and NADPH maintenance: new implications for osteonecrosis treatment? Cell Signal. 2014;26:1–8. doi: 10.1016/j.cellsig.2013.08.046. [DOI] [PubMed] [Google Scholar]
- 9.Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441:523–40. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao J, Ying M, Xie N, Lin G, Dong R, Zhang J, Yan H, Yang X, He Q, Yang B. The Oxidation States of DJ-1 Dictate the Cell Fate in Response to Oxidative Stress Triggered by 4-HPR: Autophagy or Apoptosis? Antioxid Redox Signal. 2014;••• doi: 10.1089/ars.2013.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69–79. doi: 10.1038/cr.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P. Autophagy: for better or for worse. Cell Res. 2012;22:43–61. doi: 10.1038/cr.2011.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Müller M, Reichert AS. Mitophagy, mitochondrial dynamics and the general stress response in yeast. Biochem Soc Trans. 2011;39:1514–9. doi: 10.1042/BST0391514. [DOI] [PubMed] [Google Scholar]
- 14.Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal. 2011;14:2201–14. doi: 10.1089/ars.2010.3482. [DOI] [PubMed] [Google Scholar]
- 15.Oku M, Sakai Y. Peroxisomes as dynamic organelles: autophagic degradation. FEBS J. 2010;277:3289–94. doi: 10.1111/j.1742-4658.2010.07741.x. [DOI] [PubMed] [Google Scholar]
- 16.Knodler LA, Celli J. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol. 2011;13:1319–27. doi: 10.1111/j.1462-5822.2011.01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mijaljica D, Prescott M, Devenish RJ. The intricacy of nuclear membrane dynamics during nucleophagy. Nucleus. 2010;1:213–23. doi: 10.4161/nucl.1.3.11738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. doi: 10.1007/978-3-642-00302-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stanley RE, Ragusa MJ, Hurley JH. The beginning of the end: how scaffolds nucleate autophagosome biogenesis. Trends Cell Biol. 2014;24:73–81. doi: 10.1016/j.tcb.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 22.Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 2005;12(Suppl 2):1535–41. doi: 10.1038/sj.cdd.4401728. [DOI] [PubMed] [Google Scholar]
- 23.Yue Z, Friedman L, Komatsu M, Tanaka K. The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim Biophys Acta. 2009;1793:1496–507. doi: 10.1016/j.bbamcr.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, et al. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–88. doi: 10.1172/JCI3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One. 2009;4:e5515. doi: 10.1371/journal.pone.0005515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–91. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 27.Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28:6926–37. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 29.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–17. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 30.Zhu C, Wang X, Xu F, Bahr BA, Shibata M, Uchiyama Y, Hagberg H, Blomgren K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005;12:162–76. doi: 10.1038/sj.cdd.4401545. [DOI] [PubMed] [Google Scholar]
- 31.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 32.Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–9. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- 33.Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66:378–89. doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- 34.Borsello T, Croquelois K, Hornung JP, Clarke PG. N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur J Neurosci. 2003;18:473–85. doi: 10.1046/j.1460-9568.2003.02757.x. [DOI] [PubMed] [Google Scholar]
- 35.Guimarães CA, Benchimol M, Amarante-Mendes GP, Linden R. Alternative programs of cell death in developing retinal tissue. J Biol Chem. 2003;278:41938–46. doi: 10.1074/jbc.M306547200. [DOI] [PubMed] [Google Scholar]
- 36.Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–60. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26:86–93. doi: 10.1016/j.nbd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 38.Erlich S, Shohami E, Pinkas-Kramarski R. Neurodegeneration induces upregulation of Beclin 1. Autophagy. 2006;2:49–51. doi: 10.4161/auto.2156. [DOI] [PubMed] [Google Scholar]
- 39.Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen JL, et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169:566–83. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tian F, Deguchi K, Yamashita T, Ohta Y, Morimoto N, Shang J, Zhang X, Liu N, Ikeda Y, Matsuura T, et al. In vivo imaging of autophagy in a mouse stroke model. Autophagy. 2010;6:1107–14. doi: 10.4161/auto.6.8.13427. [DOI] [PubMed] [Google Scholar]
- 41.He S, Wang C, Dong H, Xia F, Zhou H, Jiang X, Pei C, Ren H, Li H, Li R, et al. Immune-related GTPase M (IRGM1) regulates neuronal autophagy in a mouse model of stroke. Autophagy. 2012;8:1621–7. doi: 10.4161/auto.21561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puyal J, Ginet V, Grishchuk Y, Truttmann AC, Clarke PG. Neuronal autophagy as a mediator of life and death: contrasting roles in chronic neurodegenerative and acute neural disorders. Neuroscientist. 2012;18:224–36. doi: 10.1177/1073858411404948. [DOI] [PubMed] [Google Scholar]
- 43.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 44.Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–77. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–69. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernstein SL, Guo Y, Kelman SE, Flower RW, Johnson MA. Functional and cellular responses in a novel rodent model of anterior ischemic optic neuropathy. Invest Ophthalmol Vis Sci. 2003;44:4153–62. doi: 10.1167/iovs.03-0274. [DOI] [PubMed] [Google Scholar]
- 47.Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem. 2009;284:35973–86. doi: 10.1074/jbc.M109.051086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Russo R, Berliocchi L, Adornetto A, Varano GP, Cavaliere F, Nucci C, Rotiroti D, Morrone LA, Bagetta G, Corasaniti MT. Calpain-mediated cleavage of Beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis. 2011;2:e144. doi: 10.1038/cddis.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park HY, Kim JH, Park CK. Activation of autophagy induces retinal ganglion cell death in a chronic hypertensive glaucoma model. Cell Death Dis. 2012;3:e290. doi: 10.1038/cddis.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piras A, Gianetto D, Conte D, Bosone A, Vercelli A. Activation of autophagy in a rat model of retinal ischemia following high intraocular pressure. PLoS One. 2011;6:e22514. doi: 10.1371/journal.pone.0022514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sternberg C, Benchimol M, Linden R. Caspase dependence of the death of neonatal retinal ganglion cells induced by axon damage and induction of autophagy as a survival mechanism. Braz J Med Biol Res. 2010;43:950–6. doi: 10.1590/S0100-879X2010007500082. [DOI] [PubMed] [Google Scholar]
- 52.Kim SH, Munemasa Y, Kwong JM, Ahn JH, Mareninov S, Gordon LK, Caprioli J, Piri N. Activation of autophagy in retinal ganglion cells. J Neurosci Res. 2008;86:2943–51. doi: 10.1002/jnr.21738. [DOI] [PubMed] [Google Scholar]
- 53.Rodríguez-Muela N, Germain F, Mariño G, Fitze PS, Boya P. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ. 2012;19:162–9. doi: 10.1038/cdd.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knöferle J, Koch JC, Ostendorf T, Michel U, Planchamp V, Vutova P, Tönges L, Stadelmann C, Brück W, Bähr M, et al. Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc Natl Acad Sci U S A. 2010;107:6064–9. doi: 10.1073/pnas.0909794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, Stefanis L, Tolkovsky A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1:11–22. doi: 10.4161/auto.1.1.1513. [DOI] [PubMed] [Google Scholar]
- 56.Dixon JS. “Phagocytic” lysosomes in chromatolytic neurones. Nature. 1967;215:657–8. doi: 10.1038/215657a0. [DOI] [PubMed] [Google Scholar]
- 57.Matthews MR, Raisman G. A light and electron microscopic study of the cellular response to axonal injury in the superior cervical ganglion of the rat. Proc R Soc Lond B Biol Sci. 1972;181:43–79. doi: 10.1098/rspb.1972.0040. [DOI] [PubMed] [Google Scholar]
- 58.Zherebitskaya E, Akude E, Smith DR, Fernyhough P. Development of selective axonopathy in adult sensory neurons isolated from diabetic rats: role of glucose-induced oxidative stress. Diabetes. 2009;58:1356–64. doi: 10.2337/db09-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res. 2012;31:152–81. doi: 10.1016/j.preteyeres.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 60.Hein TW, Ren Y, Potts LB, Yuan Z, Kuo E, Rosa RH, Jr., Kuo L. Acute retinal ischemia inhibits endothelium-dependent nitric oxide-mediated dilation of retinal arterioles via enhanced superoxide production. Invest Ophthalmol Vis Sci. 2012;53:30–6. doi: 10.1167/iovs.11-8753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu C, Xu F, Wang X, Shibata M, Uchiyama Y, Blomgren K, Hagberg H. Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxia-ischaemia. J Neurochem. 2006;96:1016–27. doi: 10.1111/j.1471-4159.2005.03639.x. [DOI] [PubMed] [Google Scholar]
- 62.Adhami F, Schloemer A, Kuan CY. The roles of autophagy in cerebral ischemia. Autophagy. 2007;3:42–4. doi: 10.4161/auto.3412. [DOI] [PubMed] [Google Scholar]
- 63.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fitzgerald M, Bartlett CA, Payne SC, Hart NS, Rodger J, Harvey AR, Dunlop SA. Near infrared light reduces oxidative stress and preserves function in CNS tissue vulnerable to secondary degeneration following partial transection of the optic nerve. J Neurotrauma. 2010;27:2107–19. doi: 10.1089/neu.2010.1426. [DOI] [PubMed] [Google Scholar]
- 65.Sayır F, Kavak S, Meral I, Demir H, Cengiz N, Cobanoğlu U. Effects of crush and axotomy on oxidative stress and some trace element levels in phrenic nerve of rats. Brain Res Bull. 2013;92:84–8. doi: 10.1016/j.brainresbull.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 66.Fitzgerald M, Payne SC, Bartlett CA, Evill L, Harvey AR, Dunlop SA. Secondary retinal ganglion cell death and the neuroprotective effects of the calcium channel blocker lomerizine. Invest Ophthalmol Vis Sci. 2009;50:5456–62. doi: 10.1167/iovs.09-3717. [DOI] [PubMed] [Google Scholar]
- 67.Fitzgerald M, Bartlett CA, Harvey AR, Dunlop SA. Early events of secondary degeneration after partial optic nerve transection: an immunohistochemical study. J Neurotrauma. 2010;27:439–52. doi: 10.1089/neu.2009.1112. [DOI] [PubMed] [Google Scholar]
- 68.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoshimura K, Shibata M, Koike M, Gotoh K, Fukaya M, Watanabe M, Uchiyama Y. Effects of RNA interference of Atg4B on the limited proteolysis of LC3 in PC12 cells and expression of Atg4B in various rat tissues. Autophagy. 2006;2:200–8. doi: 10.4161/auto.2744. [DOI] [PubMed] [Google Scholar]
- 70.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–7. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 71.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ali YO, McCormack R, Darr A, Zhai RG. Nicotinamide mononucleotide adenylyltransferase is a stress response protein regulated by the heat shock factor/hypoxia-inducible factor 1α pathway. J Biol Chem. 2011;286:19089–99. doi: 10.1074/jbc.M111.219295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhai RG, Cao Y, Hiesinger PR, Zhou Y, Mehta SQ, Schulze KL, Verstreken P, Bellen HJ. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:e416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, Bellen HJ. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–91. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron. 2006;50:869–81. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 76.Sasaki Y, Vohra BP, Lund FE, Milbrandt J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci. 2009;29:5525–35. doi: 10.1523/JNEUROSCI.5469-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yan T, Feng Y, Zheng J, Ge X, Zhang Y, Wu D, Zhao J, Zhai Q. Nmnat2 delays axon degeneration in superior cervical ganglia dependent on its NAD synthesis activity. Neurochem Int. 2010;56:101–6. doi: 10.1016/j.neuint.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 78.Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Press C, Milbrandt J. Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J Neurosci. 2008;28:4861–71. doi: 10.1523/JNEUROSCI.0525-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–41. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- 81.Kitaoka Y, Munemasa Y, Kojima K, Hirano A, Ueno S, Takagi H. Axonal protection by Nmnat3 overexpression with involvement of autophagy in optic nerve degeneration. Cell Death Dis. 2013;4:e860. doi: 10.1038/cddis.2013.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee Y, Kim EK. AMP-activated protein kinase as a key molecular link between metabolism and clockwork. Exp Mol Med. 2013;45:e33. doi: 10.1038/emm.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–95. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 84.Avruch J, Long X, Lin Y, Ortiz-Vega S, Rapley J, Papageorgiou A, Oshiro N, Kikkawa U. Activation of mTORC1 in two steps: Rheb-GTP activation of catalytic function and increased binding of substrates to raptor. Biochem Soc Trans. 2009;37:223–6. doi: 10.1042/BST0370223. [DOI] [PubMed] [Google Scholar]
- 85.Poels J, Spasić MR, Callaerts P, Norga KK. Expanding roles for AMP-activated protein kinase in neuronal survival and autophagy. Bioessays. 2009;31:944–52. doi: 10.1002/bies.200900003. [DOI] [PubMed] [Google Scholar]
- 86.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89:1025–78. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 87.Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy. 2012;8:1462–76. doi: 10.4161/auto.21211. [DOI] [PubMed] [Google Scholar]
- 88.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–5. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007;27:6229–42. doi: 10.1128/MCB.02246-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mowat FM, Luhmann UF, Smith AJ, Lange C, Duran Y, Harten S, Shukla D, Maxwell PH, Ali RR, Bainbridge JW. HIF-1alpha and HIF-2alpha are differentially activated in distinct cell populations in retinal ischaemia. PLoS One. 2010;5:e11103. doi: 10.1371/journal.pone.0011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–8. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 93.Naves T, Jawhari S, Jauberteau MO, Ratinaud MH, Verdier M. Autophagy takes place in mutated p53 neuroblastoma cells in response to hypoxia mimetic CoCl(2) Biochem Pharmacol. 2013;85:1153–61. doi: 10.1016/j.bcp.2013.01.022. [DOI] [PubMed] [Google Scholar]
- 94.Yang L, Tan P, Zhou W, Zhu X, Cui Y, Zhu L, Feng X, Qi H, Zheng J, Gu P, et al. N-acetylcysteine protects against hypoxia mimetic-induced autophagy by targeting the HIF-1α pathway in retinal ganglion cells. Cell Mol Neurobiol. 2012;32:1275–85. doi: 10.1007/s10571-012-9852-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu X, Zhou W, Cui Y, Zhu L, Li J, Feng X, Shao B, Qi H, Zheng J, Wang H, et al. Pilocarpine protects cobalt chloride-induced apoptosis of RGC-5 cells: involvement of muscarinic receptors and HIF-1 α pathway. Cell Mol Neurobiol. 2010;30:427–35. doi: 10.1007/s10571-009-9467-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–21. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ziviani E, Tao RN, Whitworth AJ. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A. 2010;107:5018–23. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Park J, Kim Y, Chung J. Mitochondrial dysfunction and Parkinson’s disease genes: insights from Drosophila. Dis Model Mech. 2009;2:336–40. doi: 10.1242/dmm.003178. [DOI] [PubMed] [Google Scholar]
- 100.Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107:378–83. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang HL, Chou AH, Wu AS, Chen SY, Weng YH, Kao YC, Yeh TH, Chu PJ, Lu CS. PARK6 PINK1 mutants are defective in maintaining mitochondrial membrane potential and inhibiting ROS formation of substantia nigra dopaminergic neurons. Biochim Biophys Acta. 2011;1812:674–84. doi: 10.1016/j.bbadis.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 102.Horowitz JM, Myers J, Stachowiak MK, Torres G. Identification and distribution of Parkin in rat brain. Neuroreport. 1999;10:3393–7. doi: 10.1097/00001756-199911080-00025. [DOI] [PubMed] [Google Scholar]
- 103.Shimura H, Hattori N, Kubo S, Yoshikawa M, Kitada T, Matsumine H, Asakawa S, Minoshima S, Yamamura Y, Shimizu N, et al. Immunohistochemical and subcellular localization of Parkin protein: absence of protein in autosomal recessive juvenile parkinsonism patients. Ann Neurol. 1999;45:668–72. doi: 10.1002/1531-8249(199905)45:5<668::AID-ANA19>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 104.Stichel CC, Augustin M, Kühn K, Zhu XR, Engels P, Ullmer C, Lübbert H. Parkin expression in the adult mouse brain. Eur J Neurosci. 2000;12:4181–94. doi: 10.1111/j.1460-9568.2000.01314.x. [DOI] [PubMed] [Google Scholar]
- 105.Zarate-Lagunes M, Gu WJ, Blanchard V, Francois C, Muriel MP, Mouatt-Prigent A, Bonici B, Parent A, Hartmann A, Yelnik J, et al. Parkin immunoreactivity in the brain of human and non-human primates: an immunohistochemical analysis in normal conditions and in Parkinsonian syndromes. J Comp Neurol. 2001;432:184–96. doi: 10.1002/cne.1096. [DOI] [PubMed] [Google Scholar]
- 106.Esteve-Rudd J, Campello L, Herrero MT, Cuenca N, Martín-Nieto J. Expression in the mammalian retina of parkin and UCH-L1, two components of the ubiquitin-proteasome system. Brain Res. 2010;1352:70–82. doi: 10.1016/j.brainres.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 107.Cai Q, Zakaria HM, Sheng ZH. Long time-lapse imaging reveals unique features of PARK2/Parkin-mediated mitophagy in mature cortical neurons. Autophagy. 2012;8:976–8. doi: 10.4161/auto.20218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Joselin AP, Hewitt SJ, Callaghan SM, Kim RH, Chung YH, Mak TW, Shen J, Slack RS, Park DS. ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum Mol Genet. 2012;21:4888–903. doi: 10.1093/hmg/dds325. [DOI] [PubMed] [Google Scholar]