

Abstract
Receptor-mediated mitophagy is one of the major mechanisms of mitochondrial quality control essential for cell survival. We previously have identified FUNDC1 as a mitophagy receptor for selectively removing damaged mitochondria in mammalian systems. A critical unanswered question is how receptor-mediated mitophagy is regulated in response to cellular and environmental cues. Here, we report the striking finding that BCL2L1/Bcl-xL, but not BCL2, suppresses mitophagy mediated by FUNDC1 through its BH3 domain. Mechanistically, we demonstrate that BCL2L1, but not BCL2, interacts with and inhibits PGAM5, a mitochondrially localized phosphatase, to prevent the dephosphorylation of FUNDC1 at serine 13 (Ser13), which activates hypoxia-induced mitophagy. Our results showed that the BCL2L1-PGAM5-FUNDC1 axis is critical for receptor-mediated mitophagy in response to hypoxia and that BCL2L1 possesses unique functions distinct from BCL2.
Keywords: BCL2L1/Bcl-xL, FUNDC1, hypoxia, mitophagy, PGAM5 phosphatase
Introduction
Mitophagy is a specialized and highly selective form of autophagy that involves the recognition of damaged or unwanted mitochondria for selective removal via lysosomal-dependent autophagic pathways.1 Mitophagy is regarded as a major mechanism for mitochondrial quality control which is essential for cell health.2 Defects in mitophagy have been closely associated with neurodegenerative disease, metabolic disorders, and cancers.3-5 Currently, only a handful of molecules have been found to play a role in mitophagy. PARK2, an E3 ubiquitin ligase, is recruited to mitochondria that have lost their mitochondrial membrane potential6 and subsequently ubiquitinates a number of mitochondrial membrane proteins leading to the recruitment of SQSTM1 and ULK1 for mitophagy.7,8 Mitophagy is also mediated by specific receptors, such as Atg32 in yeast9,10 as well as BNIP3L and BNIP3,11 and FUNDC112 in mammalian systems. These receptors include a microtubule-associated protein 1 light chain 3 (LC3)-interacting region (LIR) and can interact with LC3, which delivers autophagosomal membrane to the mitochondria for subsequent mitophagy. It would be interesting to understand how this receptor-mediated mitophagy is activated in response to cellular and environmental cues.
The BCL2-family proteins control both mitochondrial apoptosis and starvation-induced autophagy.13,14 The classical viewpoint is that BCL2-family proteins contain both antiapoptotic molecules such as BCL2 and BCL2L1 (which contains BH4 domain), and proapoptotic molecules such as BAX and BAK1 which share high homology and contain 3 BH domains (BH1, BH2, and BH3), and BH3 only proapoptotic proteins BIM, BAD etc.15-17 Both BCL2L1 and BCL2 function to sequester BAX and BAK1 to prevent the mitochondrial outer membrane permeabilization for cytochrome c release and subsequent apoptosis.18 In addition to their fundamental role in mitochondrial apoptosis, anti-apoptotic proteins, such as BCL2, BCL2L1, and MCL1, inhibit autophagy. They bind to BECN1/Beclin 1 (the mammmalian ortholog of yeast Vps30/Atg6 that contains a putative BH3 domain), which plays a central role in the initiation of autophagosome formation through its interaction with PIK3C3, UVRAG, and ZBTB24/BIF1.14,19-22 BECN1 weakly binds to BCL2 and BCL2L1 through its BH3 domain.23,24 The interaction between BCL2 and BCL2L1 with BECN1 is thus critical for both apoptosis and autophagy. In addition, the evolutionarily conserved BCL2 and BCL2L1 proteins can regulate other cellular processes, including mitochondrial dynamics,25 bioenergetics,26 morphology,27 and the cell cycle,28 through the interaction with various binding partners. The anti-apoptotic BCL2 and the proapoptotic BAX and BAK1 proteins have been found to regulate mitochondrial dynamics (mitochondrial fission and fusion) through their interactions with either DNM1L (mitochondrial fission molecule)25 or MFN1/229 for mitochondrial fusion of the outer membrane and OPA130 for fusion of the inner membrane. BCL2-family members interact with IP3R to modulate the release of Ca2+ transients from the ER, which affects Ca2+-related cell signaling and regulates mitochondrial bioenergetics.31-33
Despite BCL2 and BCL2L1 sharing these common properties that regulate apoptosis and mitochondrial physiology, they have distinct expression profiles during development and in response to specific stressors, highlighting BCL2L1’s specific role in the regulation of mitochondrial function. It has been found that BCL2 is expressed at similar levels in most tissues but is reduced in adult neuronal cells.34,35 In contrast, BCL2L1 is strikingly abundant in tissues containing long-lived postmitotic cells, such as the adult brain.36 Bcl2l1-deficient mice have an embryonic lethal phenotype and are not viable after embryonic d 13 due to extensive neuronal apoptosis.37 BCL2L1 is exclusively expressed in mitochondria and is able to physically interact with and to stimulate F1Fo ATPase activity, which drives ATP synthesis.26 Additionally, BCL2L1 can promote both mitochondrial fission and fusion by altering the relative rates of these opposing processes and can increase the mitochondrial biomass in neuronal cells.38 As mitochondria are the principle site by which the BCL2-family proteins regulate apoptosis, autophagy, and mitochondrial dynamics,39,40 we investigated how they regulate mitochondrial quality in response to hypoxia. Interestingly, BCL2L1, but not BCL2, strongly suppresses FUNDC1-mediated mitophagy. Moreover, we demonstrate that the BCL2L1-PGAM5-FUNDC1 axis plays a significant role in mitophagy and suggest a novel and specific function of BCL2L1 distinct from BCL2.
Results
BCL2L1 inhibits both FUNDC1- and hypoxia-induced mitophagy
Our previous work has shown that FUNDC1 functions as a mitophagy receptor to mediate hypoxia-induced mitophagy through its interaction with LC3.12 To explore the potential role of the antiapoptotic BCL2-family proteins in FUNDC1-mediated mitophagy, we cotransfected BCL2 or BCL2L1 with FUNDC1 into HeLa cells and found that BCL2L1, but not BCL2, could inhibit the appearance of LC3 puncta that colocalize with fragmented mitochondria (Fig. 1A and B). Western blot analysis further confirmed that the ectopically expressed BCL2L1, but not BCL2, inhibited FUNDC1-induced LC3-II conversion, a biochemical hallmark for autophagy, which accompanies mitochondrial protein degradation (TOMM20 and TIMM23 were used as the mitochondrial outer and inner membrane markers, respectively) (Fig. 1C). Similarly, BCL2L1, but not BCL2, inhibited FUNDC1-induced mitophagy in MCF7, HepG2, and U2OS cells (Fig. S1), suggesting that BCL2L1’s inhibition of mitophagy is not restricted to HeLa cells. As we have previously shown that hypoxia-induced mitophagy is highly dependent on FUNDC1, we next examined whether ectopically expressed BCL2L1 inhibited hypoxia-induced mitophagy. As expected, transient expression of BCL2L1, but not BCL2, inhibited hypoxia-induced mitophagy, as revealed by LC3 puncta formation (Fig. 1D and E), along with the concomitant degradation of mitochondrial proteins (Fig. 1F). The inability of BCL2 to inhibit mitophagy was not due to a functional loss, as both BCL2 and BCL2L1 retained their ability to inhibit apoptosis induced by staurosporine (Fig. S2A) and to inhibit starvation-induced autophagy when treated with Earle’s balanced salt solution (EBSS), as previously reported.14,41 The PINK1-PARK2 pathway has been reported to mediate mitophagy, which is related to the Parkinson disease. BCL2L1 fails to suppress PARK2 translocation to mitochondria and LC3 puncta colocalization with depolarized mitochondria treated by short-term FCCP. It also does not prevent mitochondrial protein degradation (Fig. S3), suggesting that BCL2L1’s inhibition is specific for receptor-mediated mitophagy. Collectively, these data thus demonstrate a specific role of BCL2L1 in FUNDC1-mediated mitophagy in addition to its general role in the prevention of apoptosis and starvation-induced autophagy.

Figure 1. BCL2L1, but not BCL2, inhibits FUNDC1- and hypoxia-induced mitophagy. (A) HeLa cells stably expressing RFP-LC3 were cotransfected with a MYC-tagged FUNDC1, GFP vector, GFP-tagged BCL2L1, or BCL2 for 24 h, and cells were fixed and immunostained with an anti-MYC antibody (blue). Representative images are shown. Scale bar: 10 μm. (B) Experiments were performed as described in (A). Data represent mean ± s.e.m of 3 independent experiments (n > 100 cells per condition in each experiment). (C) HeLa cells were cotransfected with a MYC-tagged FUNDC1, FLAG vector, FLAG-tagged BCL2L1 or BCL2 for 24 h, and then the TOMM20, TIMM23, LC3, FLAG, MYC, and ACTB protein levels were detected by western blot analysis. Grayscale values of the TOMM20, TIMM23, and LC3-II bands measured with ImageJ are shown under the corresponding bands to indicate the band intensities. (D) BCL2L1 inhibits hypoxia-induced mitophagy. HeLa cells stably expressing RFP-LC3 were transfected with a GFP vector, GFP-tagged BCL2L1 or BCL2 for 18 h and this was followed by hypoxia for 24 h. Then, cells were fixed, and images were captured with a confocal microscope. Representative images are shown. Scale bar: 10 μm. (E) Experiments were performed as described in (A). Data represent mean ± s.e.m of 3 independent experiments (n > 100 cells per condition in each experiment). (F) HeLa cells were transfected with a FLAG vector, FLAG-tagged BCL2L1 or BCL2 for 18 h and followed by hypoxia for 24 h. Then, the TOMM20, TIMM23, LC3, FLAG, and ACTB protein levels were detected by western blot analysis. Grayscale values of the TOMM20, TIMM23, and LC3-II bands measured with ImageJ are shown under the corresponding bands to indicate the band intensities.
The BH3 domain of BCL2L1 is critical for mitophagy inhibition
BCL2L1 has several functional domains mediating the protein-protein interactions that inhibit apoptosis and/or autophagy. We thus determined which BCL2L1 domain is functionally required to inhibit FUNDC1-mediated mitophagy. BH and transmembrane (TM) domain BCL2L1 deletion mutants were constructed and coexpressed with FUNDC1. Interestingly, in transfected cells, we found that the deletion of the BH3 domain abrogated BCL2L1’s prevention of FUNDC1-mediated mitophagy, as assayed by the colocalization of LC3 puncta with mitochondria (Fig. 2A and B; Fig. S4). The functional importance of the BH3 domain in mitophagy was confirmed by western blot analysis examining the protein levels of the mitochondrial markers TOMM20 and TIMM23 (Fig. 2C). The BCL2L1 TM domain deletion mutant failed to localize to mitochondria and had no activity, which suggests that the site of action is at the surface of the mitochondrial outer membrane.

Figure 2. The BH3 domain of BCL2L1 is critical for mitophagy inhibition. (A) HeLa cells stably expressing RFP-LC3 were cotransfected with MYC-tagged FUNDC1 and GFP-tagged BCL2L1 or BCL2L1 deletion mutants. At 24 h post-transfection, the fixed cells were immunostained with an anti-MYC antibody (blue). Representative confocal microscopy images are presented. Scale bar: 10 μm. (B) Experiments were performed as described in (A), and the percentage of MYC-tagged FUNDC1-positive cells with LC3 puncta was quantified. Data represent mean ± s.e.m of 3 independent experiments (n > 100 cells per condition in each experiment). (C) HeLa cells were cotransfected with MYC-tagged FUNDC1 and GFP-tagged BCL2L1 or BCL2L1 mutants. At 24 h post-transfection, the cell lysates were analyzed with the indicated antibody. Grayscale values of the TOMM20 and TIMM23 bands measured with ImageJ are shown under the corresponding bands to indicate the band intensities. (D) HeLa cells stably expressing RFP-LC3 were cotransfected with MYC-tagged FUNDC1 and the 2 swapping mutants, GFP-tagged BCL2L1BCL2 BH3 or BCL2BCL2L1 BH3. At 24 h post-transfection, the fixed cells were immunostained with an anti-MYC antibody (blue). Representative confocal microscopy images are shown. Scale bar: 10 μm. (E) Experiments were performed as described in (D), and the percentage of MYC-tagged FUNDC1-positive cells with LC3 puncta was quantified. Data represent mean ± s.e.m of 3 independent experiments (n > 100 cells per condition in each experiment). (F) HeLa cells were cotransfected with a MYC-tagged FUNDC1, a GFP vector or the 2 swapping mutants, GFP-tagged BCL2L1BCL2 BH3 or BCL2BCL2L1 BH3. At 24 h post-transfection, cell lysates were prepared and the TOMM20, TIMM23, LC3, MYC, GFP, and ACTB protein levels were detected by western blot analysis. Grayscale values of the TOMM20, TIMM23, and LC3-II bands measured with ImageJ are shown under the corresponding bands to indicate the band intensities.
To account for the deletion of the BCL2L1 BH domains resulting in conformational or functional defects and to determine the specificity of the BH3 domain to inhibit FUNDC1-mediated mitophagy, we performed BH3 domain swapping experiments and replaced the BH3 domain of BCL2L1 with the BH3 domain of BCL2 (BCL2L1BCL2 BH3) or the BH3 domain of BCL2 with the BH3 domain of BCL2L1 (BCL2BCL2L1 BH3). Overexpression of either of these 2 mutants together with FUNDC1 clearly showed that the expression of the BCL2L1BCL2 BH3 mutant resulted in the loss of the inhibition of FUNDC1-mediated mitophagy and that expressing BCL2BCL2L1 BH3 further inhibited FUNDC1-mediated mitophagy, which was assessed by the colocalization of LC3 puncta with mitochondria (Fig. 2D and E) and by western blot analysis (Fig. 2F). The BH3 domain contains 15 amino acids, and 7 of them are not conserved between BCL2L1 and BCL2. We thus created mutants that replaced these amino acids of BCL2L1 with the corresponding BCL2 residues (i.e., Lys87 Gln88 Ala89 into His87 Leu88 Thr89, Glu92 into Gln92, Glu96 into Asp96 or Glu98 Leu99 into Ser98 Arg99) and examined whether there was a functional conversion in terms of a suppression of FUNDC1-induced mitophagy when they were coexpressed. This approach failed to identify the specific residue responsible for the suppressive activity (unpublished observation), suggesting that the overall structure of this domain in BCL2L1 or interaction with its to-be-characterized partner is important for its inhibitory action. The BH3 domain deletion mutant and the BH3 domain swapped mutants (BCL2L1BCL2 BH3 and BCL2BCL2L1 BH3) did not change the antiapoptotic role of the wild-type BCL2L1 and BCL2 (Fig. S2A). However, expression of the BH3 domain alone (together with the BCL2L1 TM domain to insert into mitochondria) failed to inhibit FUNDC1-mediated mitophagy (Fig. S5A–S5D). Furthermore, the synthetic cell-permeable BH3 peptide had limited effect on hypoxia-induced mitophagy (Fig. S5E), suggesting that the BH3 domain is necessary but not sufficient for the BCL2L1 inhibition of FUNDC1-mediated mitophagy.
BECN1 is required for starvation-induced autophagy but not FUNDC1-induced mitophagy
Earlier studies have suggested that BCL2L1 and BCL2 can inhibit autophagy through their binding and inhibition of BECN1, an essential mediator of autophagy.14,42,43 We thus tested whether the BCL2L1 inhibition of FUNDC1-mediated mitophagy was BECN1-dependent. We first stably knocked down BECN1 in HeLa cells (Fig. S6A) and found that ectopically expressed FUNDC1 effectively induced mitophagy both in BECN1 knockdown cells and the control cells expressing a scrambled shRNA, although the BECN1 deficiency dramatically reduced the general autophagy induced by EBSS (Fig. S6B and S6C). As expected, BCL2L1 inhibited FUNDC1-induced mitophagy regardless of the BECN1 levels (Fig. 3A and B). To further investigate the involvement of BECN1 in FUNDC1-induced BCL2L1 inhibitable mitophagy, we performed a rescue assay and found that BCL2L1, but not BCL2, was able to block FUNDC1-induced mitophagy when BECN1 was reintroduced in BECN1 KD cells (Fig. 3C). Previous reports have shown that the Gly138 of BCL2L1 is critical for its interaction with BECN1.23,44 We thus constructed a G138A BCL2L1 mutant and found that this mutant was still able to inhibit the FUNDC1-induced mitochondrial protein decrease as well as the colocalization of LC3 puncta with fragmented mitochondria (Fig. 3D and E). These data indicate that BCL2L1 suppresses FUNDC1-mediated mitophagy in a BECN1-independent fashion.

Figure 3. BCL2L1 suppression of mitophagy is independent of BECN1. (A) Stable BECN1-knockdown (KD) HeLa cells or HeLa cells stably expressing a scrambled shRNA (SC) were cotransfected with the RFP-LC3, MYC-tagged FUNDC1 and a GFP-tagged BCL2L1, BCL2, or GFP vector for 24 h. The fixed cells were immunostained with an anti-MYC antibody (blue), and the representative confocal microscopy images are shown. Scale bar: 10 μm. (B) Experiments were performed as described in (A), and the percentage of FUNDC1-positive cells with LC3 puncta was quantified. Data represent mean ± s.e.m of 3 independent experiments (n > 100 cells per condition in each experiment). (C) Stable BECN1 KD HeLa cells were transfected with plasmids as indicated for 24 h and this was followed by hypoxia for 12 h. The indicated proteins were immunoblotted with their respective antibodies. Grayscale values of the TOMM20, TIMM23, and LC3-II bands measured with ImageJ are shown under the corresponding bands to indicate the band intensities. (D) HeLa cells stably expressing RFP-LC3 were cotransfected with a MYC-tagged FUNDC1, GFP vector or plasmids encoding BCL2L1 or the BCL2L1-G138A mutant for 24 h. Cells were then fixed and immunostained with anti-MYC antibody (blue). Representative confocal microscopy images are shown. Scale bar: 10 μm. (E) HeLa cells were cotransfected with MYC-tagged FUNDC1, FLAG vector or plasmids encoding BCL2L1 or the BCL2L1-G138A mutant for 24 h. TOMM20, TIMM23, MYC, FLAG, and ACTB were immunoblotted with their respective antibodies. Grayscale values of the TOMM20 and TIMM23 bands measured with ImageJ are shown under the corresponding bands to indicate the band intensities.
BCL2L1 regulates hypoxia-induced FUNDC1-mediated mitophagy
As FUNDC1 plays a critical role in hypoxia-induced mitophagy, we further addressed the physiological significance of BCL2L1 in the suppression of hypoxia-induced mitophagy. To avoid the undesirable effects of the transient BCL2L1 expression on the cell, we constructed a tetracycline (Tet)-inducible BCL2L1 expression HeLa cells (HeLa/6TR- BCL2L1). Upon Tet addition, which induced BCL2L1 expression, hypoxia-induced mitochondrial protein degradation (TOMM20, TIMM23, and FUNDC1) was significantly suppressed (Fig. 4A), as was the colocalization of LC3 puncta with mitochondria (Fig. 4B). Similarly, we found that inducible BCL2L1 expression could also inhibit FCCP-induced mitophagy (Fig. S7A and S7B). Conversely, the degradation of the mitochondrial proteins TOMM20 and TIMM23 was significantly enhanced in the stable BCL2L1 knockdown cells during hypoxia (Fig. 4C), and there was increased colocalization of LC3 puncta and mitochondria (Fig. 4D). However, the knockdown of BCL2 did not affect mitochondrial protein degradation during hypoxia (Fig. S8). Furthermore, we found that the BCL2L1 suppression of hypoxia-induced mitophagy was FUNDC1 dependent. Knockdown of FUNDC1 by shRNA significantly blocked hypoxia-induced mitophagy even when BCL2L1 was stably knocked down (Fig. 4E). Following hypoxic treatment, knockdown of BCL2L1 had no effect on apoptosis as assessed by the phosphatidylserine exposure (Fig. S2B and S2C) or cytochrome c release from mitochondria (Fig. S2D). Collectively, these data indicate that BCL2L1 inhibition of FUNDC1-mediated mitophagy is of physiological importance.

Figure 4. BCL2L1 regulates hypoxia-induced, FUNDC1-mediated mitophagy. (A) HeLa/6TR- BCL2L1 cells were maintained under hypoxic conditions for 12 h in the presence or absence of tetracycline (Tet). The cell lysates were prepared, and the indicated proteins were detected with their respective antibodies. Grayscale values of the TOMM20 and TIMM23 bands measured with ImageJ are shown under the corresponding bands to indicate the band intensities. (B) HeLa/6TR- BCL2L1 cells were transfected with RFP-LC3 for 18 h and this was followed by hypoxia for 12 h. Cells were fixed and immunostained with an anti-TOMM20 antibody (green). Representative confocal microscopy images are shown. Scale bar: 10 μm. The data represent mean ± s.e.m of 3 independent experiments (n > 100 cells per condition in each experiment). (C) Stable BCL2L1 knockdown HeLa cells were created and subjected to hypoxia for 12 h. The cell lysates were prepared, and the indicated protein levels were detected with the respective antibodies. Grayscale values of the TOMM20 and TIMM23 bands measured with ImageJ are shown under the corresponding bands to indicate the band intensities. (D) Stable BCL2L1 knockdown HeLa cells were maintained in hypoxia for 12 h, and then the fixed cells were immunostained with an anti-TOMM20 antibody (green). Representative confocal microscopy images are shown. Scale bar: 10 μm. The data represent mean ± s.e.m of 3 independent experiments (n > 100 cells per condition in each experiment). (E) Stable BCL2L1 knockdown HeLa cells were transfected with a FUNDC1 shRNA or the negative control shRNA for 48 h and subjected to hypoxia for 12 h. The cell lysates were prepared as before, and the indicated proteins were analyzed. Grayscale values of the TOMM20 and TIMM23 bands measured with ImageJ are shown under the corresponding bands to indicate the band intensities. (F) (Left) HeLa/6TR- BCL2L1 cells were subjected to hypoxia for 12 h in the presence or absence of Tet. (Right) Scrambled (SC) and BCL2L1 knockdown (KD) HeLa cells were subjected to hypoxia for 12 h. The cell lysates were prepared, and the indicated protein levels were detected with the appropriate antibodies. Grayscale values of the FUNDC1 and p-Ser13 bands were measured with ImageJ, and the ratio of the band intensity of p-Ser13 to that of FUNDC1 (p-Ser13/FUNDC1) is shown under p-Ser13 band.
BCL2L1 modulates the PGAM5-mediated dephosphorylation of the FUNDC1 Ser13
We recently found that dephosphorylation of FUNDC1 at Ser13 resulted in the activation of hypoxia-mediated mitophagy.45 Interestingly, we noticed that inducible expression of BCL2L1 inhibited the hypoxia-induced dephosphorylation of FUNDC1 at Ser13, and knockdown of BCL2L1 enhanced this dephosphorylation in response to hypoxia (Fig. 4F). We were thus prompted to examine whether BCL2L1 inhibited FUNDC1 dephosphorylation and thereby the activation of mitophagy. This could be achieved either through direct interaction of BCL2L1 and FUNDC1 or interaction with another binding partner that activates mitophagy. However, we did not find that BCL2L1 directly interacted with FUNDC1 (data not shown). We recently determined that PGAM5, a mitochondrial-localized serine/threonine protein phosphatase, is responsible for the dephosphorylation FUNDC1 at Ser13.45 Previous studies have shown that PGAM5 acts as a BCL2L1-interacting protein, although the functional consequence of this interaction in mitochondria has yet to be determined. We thus reasoned that BCL2L1 interacts with and inhibits FUNDC1, which mediates mitophagy via the suppression of PGAM5 phosphatase activity. To test this hypothesis, we performed a coimmunoprecipitation assay and showed that exogenously expressed BCL2L1, but not BCL2, interacted with endogenous PGAM5 (Fig. 5A). Deletion of the BH3 domain and the transmembrane domain of BCL2L1 but not other functional domains remarkably impaired their interactions with PGAM5 (Fig. 5B). Furthermore, the BH3 domain swapping between BCL2L1 and BCL2 clearly showed that the BH3 domain of BCL2L1, but not of BCL2, mediated this interaction (Fig. 5B). We next addressed whether the interaction between BCL2L1 and PGAM5 was able to suppress FUNDC1 dephosphorylation by PGAM5. An in vitro phosphatase activity assay revealed that purified BCL2L1 protein, but not BCL2, inhibited the PGAM5 phosphatase activity responsible for FUNDC1 dephosphorylation at Ser13 (Fig. 5C), which was significantly impaired when the BH3 domain of BCL2L1 was deleted. Importantly, swapping of the BH3 domain of BCL2 to BCL2L1 significantly reduced this interaction and the inhibitory effects of the phosphatase. Conversely, swapping of the BH3 domain of BCL2L1 to BCL2 produced enhanced inhibitory effects. Our results thus demonstrate that BCL2L1 is a direct inhibitor of the PGAM5 phosphatase through its BH3 domain. We also found that BCL2L1 inhibited the dephosphorylation of FUNDC1 at Ser13 when PGAM5 was ectopically expressed in HeLa cells (Fig. 5D). Similarly, deletion and swapping analysis showed that BCL2L1 inhibition of the PGAM5 dephosphorylation of FUNDC1 was dependent on the BH3 domain of BCL2L1 (Fig. 5E). Conversely, the knockdown of BCL2L1 decreased the levels of phosphorylated FUNDC1 Ser13, which were further diminished by the ectopic expression of PGAM5 (Fig. 5F). To further examine whether the BCL2L1-inhibitable PGAM5-mediated dephosphorylation of the FUNDC1 Ser13 was responsible for the activation of mitophagy, we constructed a FUNDC1-S13A mutant (which retains the LIR) and found that BCL2L1 no longer inhibited the S13A mutant-induced mitochondrial protein degradation (Fig. 5G) and the colocalization of LC3 puncta with fragmented mitochondria (Fig. 5H). Taken together, these data demonstrate that BCL2L1 physically interacts with and inhibits PGAM5, which mediates the dephosphorylation of FUNDC1 at Ser13 to activate mitophagy.

Figure 5. BCL2L1 modulates the PGAM5-mediated dephosphorylation of FUNDC1 Ser13. (A) HeLa cells were transfected with a FLAG vector or plasmid encoding FLAG-tagged BCL2L1 or BCL2 for 24 h, and cell lysates were then prepared for immunoprecipitation with an anti-FLAG antibody (upper) or anti-PGAM5 antibody (lower). FLAG (for FLAG-tagged BCL2L1 or BCL2) and endogenous PGAM5 levels were detected with the appropriate antibody. (B) HeLa cells were transfected with a FLAG vector or plasmid encoding FLAG-tagged BCL2L1, BCL2, or BCL2L1 mutants, and the cell lysates were prepared for immunoprecipitation with an anti-FLAG antibody, and the FLAG and endogenous PGAM5 levels were detected with the appropriate antibody. (C) In vitro PGAM5 phosphatase activity assay. Immunoprecipitated MYC-tagged FUNDC1 was incubated with 0.5 μg recombinant His-PGAM5L protein, purified BCL2L1 protein, BCL2 protein, or mutant proteins at 30 °C for 1 h. The beads were then subjected to western blot analysis with anti-MYC and anti-p-Ser13 antibodies. (D) HeLa cells were cotransfected with PGAM5-MYC and a FLAG-tagged BCL2L1, BCL2 or FLAG vector control for 24 h. Phosphorylated FUNDC1 was detected by a specific p-Ser13 antibody. Grayscale values of the FUNDC1 and p-Ser13 bands were measured with ImageJ, and the ratio of the band intensity of p-Ser13 to that of FUNDC1 (p-Ser13/FUNDC1) is shown under p-Ser13 band. (E) HeLa cells were cotransfected with a GFP control vector or a plasmid encoding GFP-tagged BCL2L1, BCL2 or BCL2L1 mutants as well as a MYC-tagged PGAM5 for 24 h. Phosphorylated FUNDC1 was detected by a specific p-Ser13 antibody. (F) BCL2L1 SC or KD cells were transfected with a plasmid encoding MYC-tagged PGAM5, and the phosphorylated FUNDC1 levels were analyzed. Grayscale values of the FUNDC1 and p-Ser13 bands were measured with ImageJ, and the ratio of the band intensity of p-Ser13 to that of FUNDC1 (p-Ser13/FUNDC1) is shown under p-Ser13 band. (G) HeLa cells were cotransfected with plasmids encoding wild-type FUNDC1 or the FUNDC1-S13A mutant and a FLAG-tagged BCL2L1 or FLAG control vector. TOMM20 and TIMM23 were detected by western blot analysis. Grayscale values of the TOMM20 band measured with ImageJ are shown under the corresponding bands to indicate the band intensities. (H) HeLa cells stably expressing RFP-LC3 were cotransfected with a MYC-tagged FUNDC1-S13A mutant and FLAG-tagged BCL2L1 or FLAG control vector. Then FLAG and MYC antibodies were used for immunostaining, and the representative images show that BCL2L1 cannot inhibit FUNDC1-S13A-induced mitophagy.
BCL2L1 regulates the hypoxia-induced PGAM5-mediated dephosphorylation of FUNDC1
We further explored the role of the BCL2L1-PGAM5 interaction in hypoxia-induced FUNDC1-mediated mitophagy. A coimmunoprecipitation assay revealed that the enhanced interaction between PGAM5 and FUNDC1 induced by hypoxia could be reduced by inducible BCL2L1 expression (Fig. 6A). Knockdown of BCL2L1 enhanced the hypoxia-induced interaction of PGAM5 and FUNDC1 (Fig. 6B). A GST pulldown assay showed that the inducible expression of BCL2L1 inhibited the interaction between FUNDC1 and LC3 in response to hypoxia (Fig. 6C). To further ascertain the PGAM5 requirement of hypoxia-induced mitophagy, we transiently transfected PGAM5 siRNAs or the scrambled control siRNA in stable BCL2L1 knockdown cells and found PGAM5 knockdown did not affect FUNCD1 phosphorylation status under normoxic conditions (Lane 1 to 3) but had an effect under hypoxia conditions (Lane 7 to 9), PGAM5 knockdown abrogated hypoxia-induced mitochondrial protein degradation and the dephosphorylation of FUNDC1 Ser13, both in BCL2L1 control and knockdown cells (Fig. 6D). These data argue that the BCL2L1-PGAM5-FUNDC1 axis is of functional importance to the prevention of hypoxia-induced mitophagy.

Figure 6. BCL2L1 regulates the hypoxia-induced interaction of PGAM5 with FUNDC1. (A) HeLa/6TR- BCL2L1 cells were subjected to hypoxia for 6 h in the presence or absence of Tet. The cell lysates were prepared for immunoprecipitation with an anti-PGAM5 antibody, and the indicated proteins were analyzed the appropriate antibody. (B) BCL2L1 KD or SC cells were subjected to hypoxia for 6 h. The cell lysates were immunoprecipitated with an anti-PGAM5 antibody and immunoblotted with the indicated antibodies. (C) HeLa/6TR- BCL2L1 cells were subjected to hypoxia for 6 h in the presence or absence of Tet. The cell lysates were prepared and incubated with immobilized GST or GST-LC3B beads and detected by western blot analysis. Ponceau S was used to visualize GST and GST-LC3B. (D) BCL2L1 KD or SC cells were transfected with PGAM5 siRNA #1, siRNA #2, or the negative control (NC) for 48 h and followed by hypoxia for 12 h. Samples were analyzed by western blot.
Discussion
In this current study, we have uncovered a novel regulatory mechanism for hypoxia-induced mitophagy and discovered a specific function of BCL2L1 that suppresses receptor-mediated mitophagy. In normoxic conditions in particular, we showed that BCL2L1, but not BCL2, interacts with PGAM5 to inhibit its phosphatase activity and thereby prevents the dephosphorylation of FUNDC1 and the subsequent activation of mitophagy. However, under hypoxic and FCCP treatment conditions, the degradation of BCL2L1 (Fig. 4A; Fig. S7 and S9) led to PGAM5 activation that catalyzed the dephosphorylation of FUNDC1 at Ser13. The dephosphorylated form of FUNDC1 interacts with LC3 to activate mitophagy (Fig. 7).12 Our data demonstrate that the BCL2L1-PGAM5-FUNDC1 axis finely tunes mitophagy and have broadened our view of the function of BCL2L1 in selective mitophagy.
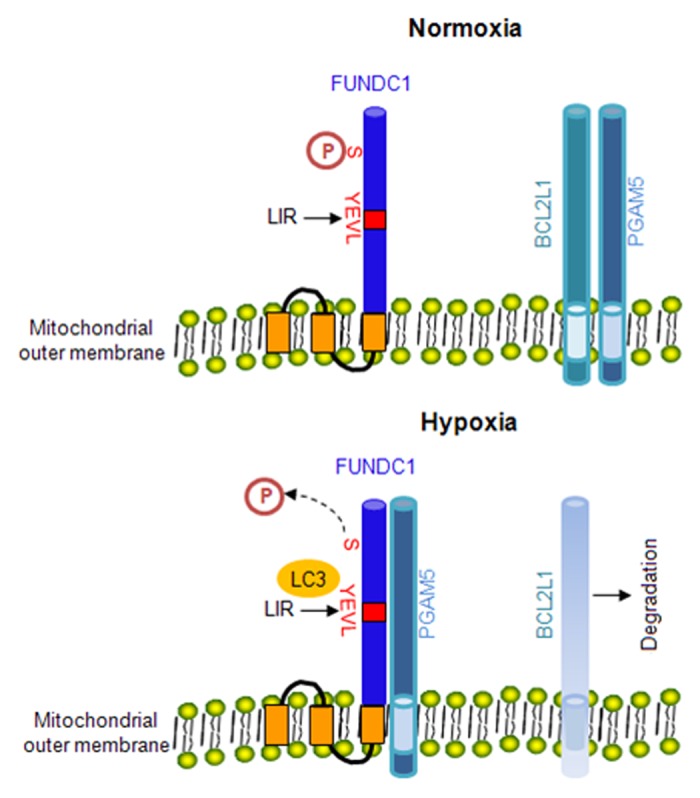
Figure 7. The hypothetical model of BCL2L1 regulation of hypoxia-induced mitophagy. In normoxic conditions, FUNDC1 maintains its phosphorylated status as BCL2L1 interacts with and suppresses the PGAM5 phosphatase, which can catalyze the dephosphorylation of FUNDC1 at Ser13. The phosphorylated FUNDC1 does not interact with LC3. During hypoxia, the degradation of BCL2L1 will release PGAM5, which leads to its activation and subsequent dephosphorylation of FUNDC1 at Ser13. The dephosphorylated form of FUNDC1 interacts with LC3 and induces mitophagy.
Earlier studies have identified that BCL2L1 interacts with the PGAM5 phosphatase.46 However, it has not been determined whether BCL2L1 is able to inhibit the phosphatase activity of PGAM5 or whether their interactions regulate mitochondrial quality. To the best of our knowledge, we are the first group to link the BCL2L1-PGAM5 axis to receptor-mediated mitophagy. The deletion and swapping analysis clearly showed that the BH3 domain of BCL2L1 is important for its interaction and inhibition of PGAM5. These results are consistent with previous findings showing that PGAM5 binds outside of the groove of BCL2L1 and that it does not directly compete with BH3-only proteins for the binding of BCL2L1.46 Our data also suggest that the level of BCL2L1 and the level and/or activity of PGAM5 determine the mitophagy in response to hypoxia. Indeed, we observed that hypoxia induced the degradation of BCL2L1 prior to the degradation of other mitochondrial marker proteins (Fig. 4A; Fig. S7). Through the manipulation of BCL2L1 by inducible expression and knockdown experiments, we clearly showed that the BCL2L1 protein levels determine the PGAM5-mediated dephosphorylation of FUNDC1 and subsequent mitophagy. Additionally, the knockdown of PGAM5 abrogated hypoxia-induced mitophagy regardless of the BCL2L1 levels. As both BCL2L1 and PGAM5 are known substrates of the KEAP1 and CUL3-dependent E3 ubiquitin ligase,47 we further speculate that this oxidative stress-activated pathway may be important for mitophagy regulation. Further studies are being directed at understanding the molecular details of their interactions and how these are regulated by oxidative stressors.
Overwhelming evidence has shown that BCL2 and BCL2L1 have overlapping and redundant roles in mitochondrial-dependent apoptosis (Fig. S2A).18,40 BCL2L1 and BCL2 proteins form a groove that interacts with and sequesters BH3-only proteins to protect the cell from fatal insults. BCL2 and BCL2L1 interact with BECN1 and BNIP3L, both putative BH3-only proteins, to inhibit starvation-induced autophagy or apoptosis.23,44 However, our results did not support the notion that BCL2L1 suppresses FUNDC1-mediated mitophagy through BECN1 or BNIP3L. Although the knockdown of BECN1 effectively prevented starvation-induced general autophagy, it did not affect FUNDC1-induced mitophagy. The BCL2L1-G138A mutant, which failed to interact with BECN1, strongly prevented FUNDC1-induced mitophagy. Elegant studies have revealed that BNIP3L could function as a mitophagy receptor and play a role in the purging of mitochondria during red blood cell maturation. However, genetic evidence does not suggest a role for BCL2L1 in BNIP3L-mediated mitochondrial clearance in reticulocytes.48 We did not find that the knockdown of BNIP3L prevented FUNDC1-induced mitophagy (unpublished observation). As both BCL2 and BCL2L1 interact with either BECN1 or BNIP3L in a similar fashion, this cannot explain the specific role of BCL2L1 in FUNDC1 mediated mitophagy.
BCL2L1 is a prominent antiapoptosis molecule. Our results thus raise an interesting question of how mitophagy, a cell-survival mechanism, is related to mitochondrial apoptosis caused by the catastrophic consequences of mitochondrial dysfunctions. It may be counterintuitive that BCL2L1 prevents mitophagy while strongly inhibiting apoptosis. One possible explanation is that BCL2L1 interacts with distinct molecules with a distinct temporal-spatial threshold. It is conceivable that in a normal unstressed situation, BCL2L1 is able to sequester and inhibit PGAM5 and the proapoptotic BCL2-family proteins through distinct and separate interactions that suppress both processes. In response to mitochondrial and metabolic stresses, one of the early cellular responses is to activate BCL2L1-PGAM5 to induce FUNDC1-dependent mitophagy, through the downregulation of BCL2L1 or the activation of PGAM5, to remove the damaged mitochondria to aid in cell survival. When the stress signal is severe and persistent, causing the activation and translocation of the proapoptotic BCL2-family proteins to the mitochondria, and is beyond the capacity of BCL2L1’s inhibitory function, the cells are destined to die either through intrinsic mitochondrial apoptosis or through PGAM5-dependent programmed necrosis.49 Thus, our results further support the proposition that BCL2L1 serves as a master regulator for mitochondrial activities through its interaction with distinct partners, such as apoptosis via other BCL2-family proteins, mitochondrial respiration via the F1Fo ATPase,26 mitochondrial dynamics via MFN1/2, and PGAM5-mediated mitophagy. As both mitophagy and apoptosis are linked with neurodegenerative and many other disease types, further dissection of the exact molecular details of the BCL2L1-PGAM5-FUNDC1 axis will be useful in understanding how mitophagy contributes to cell survival and disease development.
Materials and Methods
Reagents and antibodies
The following reagents were purchased and utilized in this study: CSNK2/casein kinase 2 inhibitor, TBB (Tocris, 2275); SRC kinase inhibitor, Su6656 (Sigma, S9692); and the uncoupler of mitochondrial oxidative phosphorylation, FCCP (Sigma, C2920).
The following antibodies were used: mouse anti-TOMM20 (BD Biosciences, 612278), mouse anti-TIMM23 (BD Biosciences, 611223), and mouse anti-BECN1 (BD Biosciences, 612113); mouse anti-MYC tag, mouse anti-GFP tag (Santa Cruz, SC-9996), rabbit anti-CSNK2A1/casein kinase 2, α 1 polypeptide (Santa Cruz, SC-6479), and rabbit anti-CSNK2A2/casein kinase 2, α prime polypeptide (Santa Cruz, SC-6481); rabbit anti-BCL2L1 (Cell Signaling Technology, 2764), rabbit anti-SRC (Cell Signaling Technology, 2108), and rabbit anti-phospho SRC (p-Tyr416) (Cell Signaling Technology, 2101); rabbit anti-PGAM5 polyclonal antibody (Abcam, ab126534); mouse anti-ACTB (Sigma, A8481), mouse anti-TUBA/tubulin (Sigma, T2200), mouse anti-FLAG (Sigma, F1804), and rabbit anti-LC3 (Sigma, L8918). The rabbit anti-FUNDC1 and rabbit anti-p-Ser13 polyclonal antibodies were generated by immunizing rabbits with a peptide or purified FUNDC1 phosphopeptides, and affinity purified (Abgent). HRP-conjugated secondary antibodies were purchased from Jackson Immuno Research Laboratories (31160). The secondary antibodies used for immunofluorescence were: goat anti-mouse IgG Alexa Fluor-488 (Molecular Probes, A11029) and -568 (Molecular Probes, A11004); anti-rabbit IgG Alexa Fluor-488 (Molecular Probes, A11008), and -568 (Molecular Probes, A11036); and anti-rat IgG Alexa Fluor-647 (Molecular Probes, A14807).
Cell culture and transfection
HeLa cells were cultured in DMEM supplemented with 10% fetal bovine serum (Hyclone, SV30087.02) and 1% penicillin-streptomycin and maintained at 37 °C in 5% CO2. For hypoxia, cells were maintained in a hypoxic chamber (Billups Rothenberg, MIC-101, Del Mar, California) flushed with a preanalyzed gas mixture of 1% O2, 5% CO2 and 95% N2. For transfection, cells were plated in a 6-well plate 24 h before transfection, and the indicated plasmids were transfected using Lipofectamine 2000 according to the manufacturer’s protocol.
cDNA constructs and mutagenesis
We are grateful to Dr Xiaodong Wang (National Institute of Biological Sciences, Beijing, China) for the MYC-tagged PGAM5L plasmid. The FLAG-, EGFP- and MYC-tagged BCL2L1 constructs were generated by the fusion of BCL2L1 wild-type cDNAs to N-terminal epitope tags. The site-specific mutants and deletion mutants were generated with the manufacturer’s (Stratagene, 200521) protocol.
Stable and tetracycline-inducible cell lines
The following PGAM5 sequences targeted by siRNA were: siRNA #1, 5′-CCATAGAGAC CACCGATAT -3′; and siRNA #2, 5′-AACCACTGTC TCTGATCAA -3′. The target sequence for BCL2L1 was 5′-GGAGAUGCAG GUAUUGGUG-3′, and the primers for short hairpin RNA were designed according to the manufacturer's instructions and cloned into the pSuper vector (OligoEngine, VEC-PRT-0002). To establish stable cell lines, HeLa cells were transfected with the appropriate vectors and placed under puromycin selection. To establish the HeLa/6TR- BCL2L1 tetracycline-inducible cell line, the HeLa/6TR cell line was transfected with a MYC-tagged BCL2L1construct and placed under zeocin (Invitrogen, R250-01) selection.
SDS-PAGE and western blotting
After the indicated treatment or transfection, cells were lysed in lysis buffer (20 mM Tris, pH 7.4, 2 mM EGTA, 1% NP-40 (Invitrogen, FNN0021), and protease inhibitors (Thermo Scientific, 88266). Equivalent protein quantities (20 μg) were subjected to SDS-PAGE and transferred to nitrocellulose membranes. Subsequently, membranes were blocked, incubated with the above-indicated primary antibodies, and followed by incubation with HRP-conjugated secondary antibodies. Immunoreactive bands were visualized with a chemiluminescence kit (ThermoFisher, 32109).
GST pulldown
GST and GST-LC3 proteins were expressed in E. coli Rosetta (DE3). GST fusion proteins were purified on glutathione-Sepharose 4 Fast Flow beads (GE Health Science, 17-0756-01). For the GST pulldown, 4 µg of GST-LC3 protein was incubated with cell lysates for 2 h at 4 °C and then washed 5 times with 1 mL PBS buffer. The precipitate complex was boiled with sample buffer containing 1% SDS for 5 min at 95 °C and subjected to SDS-PAGE. The nitrocellulose membrane was stained with Ponceau S and followed by immunoblotting with an anti-FUNDC1 antibody.
Coimmunoprecipitation
HeLa cells were transiently transfected with the indicated plasmids. At 24 h post-transfection, the cells were lysed with 0.5 mL of lysis buffer plus protease inhibitors for 30 min on ice. After a 15 min 12,000 g centrifugation step, the lysates were immunoprecipitated with specific antibodies and protein A-Sepharose (Invitrogen, 10-1041) overnight at 4 °C. Thereafter, the precipitants were washed 3 times with lysis buffer, and the immune complexes were eluted with sample buffer containing 1% SDS for 5 min at 95 °C and subjected to SDS-PAGE.
Immunofluorescence analysis
HeLa cells were transfected with the indicated plasmids or subjected to hypoxia, and 24 h post-transfection, the cells were fixed with 4% formaldehyde in DMEM for 15 min at 37 °C. The fixed cells were permeabilized with 0.2% Triton X-100 (Sigma, T8787) for 15 min on ice, subsequently blocked and incubated with indicated primary antibodies overnight on ice, and followed by the incubation of fluorescence-conjugated secondary antibodies at room temperature for 1 h. Cell images were captured with an LSM 510 Zeiss confocal microscope (Carl Zeiss Jena, Germany).
Purified BCL2L1 protein inhibits phosphatase activity of PGAM5 in vitro
For protein purification, the bacterial expression constructs pGEX-6P1 (Amersham, 27-4597-01) containing the indicated genes (the trans-membrane domain deletion mutants of BCL2L1, BCL2L1-BH3Δ, BCL2L1BCL2 BH3, BCL2, BCL2BCL2L1 BH3, and PGAM5) were transformed into BL21 cells. Cells were induced to express protein with 0.5 mM IPTG (Sigma, I6758) at 16 °C. After induction, cells were resuspended in PBS (Sigma, P3813) containing 0.5% Triton X-100, 5 mM β-mercaptoethanol, 2 mM EDTA, and 1 mM PMSF (Sigma, P7626), which was followed by ultrasonication. The proteins were purified in a single step using glutathione beads according to the manufacturer’s protocol (GE Health Science, 17-0756-01). The GST tag of the fusion proteins (BCL2L1, BCL2L1-BH3Δ, BCL2L1BCL2 BH3, BCL2, and BCL2BCL2L1 BH3) was cleaved.
For in vitro phosphatase activity of PGAM5, HeLa cells were transfected with a FUNDC1-MYC expression vector and this was followed by immunoprecipitation using an anti-MYC antibody. The beads conjugated with a MYC antibody or FUNDC1-MYC fusion protein were incubated with the PGAM5 protein, BCL2L1 protein or BCL2L1 mutants for 1 h at 30 °C. Then, the bead samples were then prepared for SDS-PAGE.
Statistical analysis
All data are expressed as the means ± s.e.m of at least 3 independent experiments. Statistical analyses were performed using the Student 2-tailed t test. Only the values of P < 0.05 were considered significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Professor Hong Zhang from the Institute of Biophysics, Chinese Academy of Sciences and Professor Li Yu from Tsinghua University, China, for their constructive suggestions and to Professor Xiaodong Wang from the National Institute of Biological Sciences, China, for generously providing the PGAM5 and shRNA plasmids. We are also grateful to Professors Shengcai Lin from Xiamen University, China, for critically reading the manuscript. This research was supported by the 973 program project (No. 2011CB910903 to QC, NO. 2013CB531200 to LL and No. 2010CB91220 to YZ) from the MOST and Natural Science Foundation of China (81130045, 31271529, 31201042).
Glossary
Abbreviations:
- ACTB
actin, beta
- ATG5
autophagy-related 5
- ATG7
autophagy-related 7
- BCL2
B-Cell CLL/lymphoma 2
- BCL2L1/Bcl-xL
BCL2-like 1
- BECN1/Beclin 1
autophagy related
- BH3 domain
BCL2 homology 3 domain
- BNIP3
BCL2/adenovirus E1B 19kDa interacting protein 3
- DNM1L
dynamin 1-like
- EBSS
Earle’s balanced salt solution
- EGFP
enhanced green fluorescent protein
- FCCP
carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- FUNDC1
FUN14 domain containing 1
- IP3R
inositol 1,4,5-trisphosphate receptor
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- LIR
LC3-interacting region
- MFN1/2
mitofusin 1/2
- OPA1
optic atrophy 1 (autosomal dominant)
- PARK2
parkin RBR E3 ubiquitin protein ligase
- PINK1
PTEN-induced putative kinase 1
- PGAM5
phosphoglycerate mutase family member 5
- RFP
red fluorescent protein
- SC
scrambled
- SQSTM1
sequestosome 1
- TM
transmembrane
- TIMM23
translocase of inner mitochondrial membrane 23 homolog (yeast)
- TOMM20
translocase of outer mitochondrial membrane 20 homolog (yeast)
- ULK1
unc-51 like autophagy activating kinase 1
References
- 1.Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- 2.Batlevi Y, La Spada AR. Mitochondrial autophagy in neural function, neurodegenerative disease, neuron cell death, and aging. Neurobiol Dis. 2011;43:46–51. doi: 10.1016/j.nbd.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He C, Song H, Yorimitsu T, Monastyrska I, Yen WL, Legakis JE, Klionsky DJ. Recruitment of Atg9 to the preautophagosomal structure by Atg11 is essential for selective autophagy in budding yeast. J Cell Biol. 2006;175:925–35. doi: 10.1083/jcb.200606084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- 5.Palikaras K, Tavernarakis N. Mitophagy in neurodegeneration and aging. Front Genet. 2012;3:297. doi: 10.3389/fgene.2012.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–31. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 8.Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci. 2012;125:1488–99. doi: 10.1242/jcs.094110. [DOI] [PubMed] [Google Scholar]
- 9.Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009;17:98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 11.Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–85. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 13.Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, Geneste O, Kroemer G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007;3:374–6. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 14.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 15.Woo JS, Jung JS, Ha NC, Shin J, Kim KH, Lee W, Oh BH. Unique structural features of a BCL-2 family protein CED-9 and biophysical characterization of CED-9/EGL-1 interactions. Cell Death Differ. 2003;10:1310–9. doi: 10.1038/sj.cdd.4401303. [DOI] [PubMed] [Google Scholar]
- 16.Conradt B, Horvitz HR. The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell. 1998;93:519–29. doi: 10.1016/S0092-8674(00)81182-4. [DOI] [PubMed] [Google Scholar]
- 17.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–6. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 18.Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–11. doi: 10.1016/S1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 19.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–99. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 20.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–96. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 21.Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2008;105:19211–6. doi: 10.1073/pnas.0810452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mulé JJ, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–51. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem. 2007;282:13123–32. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 24.Feng W, Huang S, Wu H, Zhang M. Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J Mol Biol. 2007;372:223–35. doi: 10.1016/j.jmb.2007.06.069. [DOI] [PubMed] [Google Scholar]
- 25.Sun Z, Terragni J, Borgaro JG, Liu Y, Yu L, Guan S, Wang H, Sun D, Cheng X, Zhu Z, et al. High-resolution enzymatic mapping of genomic 5-hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep. 2013;3:567–76. doi: 10.1016/j.celrep.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol. 2011;13:1224–33. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cleland MM, Norris KL, Karbowski M, Wang C, Suen DF, Jiao S, George NM, Luo X, Li Z, Youle RJ. Bcl-2 family interaction with the mitochondrial morphogenesis machinery. Cell Death Differ. 2011;18:235–47. doi: 10.1038/cdd.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janumyan Y, Cui Q, Yan L, Sansam CG, Valentin M, Yang E. G0 function of BCL2 and BCL-xL requires BAX, BAK, and p27 phosphorylation by Mirk, revealing a novel role of BAX and BAK in quiescence regulation. J Biol Chem. 2008;283:34108–20. doi: 10.1074/jbc.M806294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–87. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 30.Rolland SG, Lu Y, David CN, Conradt B. The BCL-2-like protein CED-9 of C. elegans promotes FZO-1/Mfn1,2- and EAT-3/Opa1-dependent mitochondrial fusion. J Cell Biol. 2009;186:525–40. doi: 10.1083/jcb.200905070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, Mignery GA, Roderick HL, Bootman MD, Distelhorst CW. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A. 2009;106:14397–402. doi: 10.1073/pnas.0907555106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rong YP, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, Matsuyama S, Herlitze S, Roderick HL, Bootman MD, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol Cell. 2008;31:255–65. doi: 10.1016/j.molcel.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Distelhorst CW, Bootman MD. Bcl-2 interaction with the inositol 1,4,5-trisphosphate receptor: role in Ca(2+) signaling and disease. Cell Calcium. 2011;50:234–41. doi: 10.1016/j.ceca.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jarskog LF, Gilmore JH. Developmental expression of Bcl-2 protein in human cortex. Brain Res Dev Brain Res. 2000;119:225–30. doi: 10.1016/S0165-3806(99)00176-5. [DOI] [PubMed] [Google Scholar]
- 35.Vyas S, Javoy-Agid F, Herrero MT, Strada O, Boissiere F, Hibner U, Agid Y. Expression of Bcl-2 in adult human brain regions with special reference to neurodegenerative disorders. J Neurochem. 1997;69:223–31. doi: 10.1046/j.1471-4159.1997.69010223.x. [DOI] [PubMed] [Google Scholar]
- 36.González-García M, García I, Ding L, O’Shea S, Boise LH, Thompson CB, Núñez G. bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci U S A. 1995;92:4304–8. doi: 10.1073/pnas.92.10.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urase K, Momoi T, Fujita E, Isahara K, Uchiyama Y, Tokunaga A, Nakayama K, Motoyama N. Bcl-xL is a negative regulator of caspase-3 activation in immature neurons during development. Brain Res Dev Brain Res. 1999;116:69–78. doi: 10.1016/S0165-3806(99)00076-0. [DOI] [PubMed] [Google Scholar]
- 38.Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB, 3rd, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, et al. Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol. 2009;184:707–19. doi: 10.1083/jcb.200809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 2003;15:691–9. doi: 10.1016/j.ceb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 40.Tsujimoto Y. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells. 1998;3:697–707. doi: 10.1046/j.1365-2443.1998.00223.x. [DOI] [PubMed] [Google Scholar]
- 41.Maiuri MC, Le Toumelin G, Criollo A, Rain J-C, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–88. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 2010;17:268–77. doi: 10.1038/cdd.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54:362–77. doi: 10.1016/j.molcel.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 46.Lo SC, Hannink M. PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J Biol Chem. 2006;281:37893–903. doi: 10.1074/jbc.M606539200. [DOI] [PubMed] [Google Scholar]
- 47.Lo SC, Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp Cell Res. 2008;314:1789–803. doi: 10.1016/j.yexcr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, Loyd MR, Randall MS, Waddell MB, Kriwacki RW, Ney PA. A short linear motif in BNIP3L (NIX) mediates mitochondrial clearance in reticulocytes. Autophagy. 2012;8:1325–32. doi: 10.4161/auto.20764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–43. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.